

Abstract
Objective
Aging and AD are associated in some way, then it is reasonable to ask whether or not it is possible to age without AD inexorably appearing at any moment, depending on the period of life. Therefore, the goal of this review is to verify, in light of some aging theories, the prevalence of AD.
Methods
For the purpose of this manuscript, the indexers Alzheimer, aging, Alzheimer, and aging were considered; theories of aging were researched. The research was conducted using PubMed, Medline, Scopus, Elsevier, and Google Scholar.
Results
The most common subjects in the papers analyzed for this manuscript were aging and Alzheimer's disease. The association between Alzheimer and theories of aging seems inconclusive.
Conclusions
Accordingly, the general idea is that AD is associated with aging in such a way that almost all people will present this disease; however, it is plausible to consider that the increase in life expectancy will generate a high prevalence of AD. In a general sense, it seems that the theories of aging explain the origin of AD under superlative and catastrophic considerations and use more biomolecular data than social or behavioral data as the bases of analysis, which may be the problem.
1. Introduction
Alzheimer's disease (AD) was first studied and discovered by the German Psychiatrist Alois Alzheimer [1] in 1906 as the main cause of senile dementia. There is no way to definitively diagnose it during life [2] and, until now, presented an unknown, or multifactorial etiology [3–5]. AD is likely associated with environmental and genetic factors [6, 7], which generate a heterogeneous neurodegenerative disease [8], and research conducted on non-human subjects has demonstrated little potential for preclinical use [9].
Several papers have associated AD with aging [10–13], obviously because AD generally (in about 90% of cases) [14] affects individuals from the age of 65 and its prevalence doubles each 5 years, generating a time-dependent exponential increase [15].
In terms of the effects of this disease, AD is strongly associated with neurodegeneration and decreased cognition [1, 16] including language capabilities, praxis, loss of memory [8, 17] with loss of ability to recognize faces and recall names [18–20], loss of judgement and emotional stability [3, 21, 22], personality alterations [10], progressive and increased loss of neurons with presence of senile plaques, neurofibrillary tangles [10, 23], widespread neuronal network destruction [24, 25], brain [26], and evident hippocampal atrophy [27, 28]; however, several factors are associated with normal aging.
The association of AD with aging seems, at least according to some papers [14], to indicate that the majority of elderly people are subject to a high probability of having it and, considering the prevalence of other dementia, almost all elderly people have the potential, but not necessarily, to present some type of elderly disease during the third age, including Parkinson disease [29]. The above information was based on the reasoning of the theories of aging [30].
Accordingly, with the increasing average life of the population, dementia in general and, particularly, Alzheimer's, will be a public health issue [31] or, at least, a social concern [21, 32, 33]. The problem became more concerning after a recent study showed that senile dementia could potentially generate other pathologies caused by neuronal degeneration, for instance bipolar disorder [34].
Many theories were proposed in attempt to explain the process of aging and, generally, they can be divided into two groups: stochastic and non-stochastic [35]. The stochastic group is associated with molecular degradation and the actions of free radicals [36] generating a cumulative effect [37] on the cell's components [38].
The non-stochastic theories of aging are associated with the degradation of genes during the aging process, considering environmental influence [21].
Indeed, and independent of aging theories, aging and AD are associated in some way [3]. Therefore, it is reasonable to ask whether or not it is possible to age without AD inexorably appearing at any moment, depending on the period of life.
Therefore, the goal of this review is to assemble some commentary, in light of some aging theories, on the prevalence of AD.
2. Materials and Methods
For the purpose of this manuscript, 125 papers and a few books considering the indexers aging, Alzheimer disease, Alzheimer and aging, theories of aging were researched and analyzed. The research was conducted using articles from PubMed, Medline, Scopus, Elsevier, and Google Scholar under the languages of English, Spanish, and Portuguese.
The exclusion criteria for the papers eliminated those that were out of the scope this article, i.e., after establishing the goal; we sought papers that fit with the objective of associating DA with aging. As for books, only those most representative of the theme were selected, especially The Biology of Aging [39], a book we considered a classic regarding aging theories.
A lot of papers on DA, aging, and the association of both were found on the data bases but only those from best scientific journals, and some directly linked to the goal of this article, were chosen.
In order to relate the number of papers and their subjects in general terms, the qui-square test using the program StatPlus:mac AnalystSoft Inc. 2018 was applied. However, in order to improve statistical analysis, a Kruskal-Wallis ANOVA was performed using the same program.
The qui-square is considered robust for small samples and non-parametric analysis. The expected data were obtained by dividing the number of papers (totaling 100%) by the number of subjects, considering that these subjects have the same probability of being studied. For the Kruskal-Wallis ANOVA, the samples were considered independent.
Another analysis was performed associating the theories of aging and its probability of generating AD, in terms of basic probability. The theories, i.e., each one, were considered to have the same probability of generating AD, according to a qualitative analysis (see discussion below). If the theory of aging indicated at least one possibility of generating AD, it was considered positive with regard to generating AD. If the data from that theory did not present any possibility of generating AD, it was considered negative. For instance, if the theory of somatic mutations shows any possibility of generating AD, it was included in the calculation of probability; i.e., it was considered as positive. On the contrary, if an aspect of the aging theory was not linked to DA, it was removed from the calculation, i.e., was considered negative.
3. Results
The most common subjects in the papers analyzed for this manuscript were aging and Alzheimer's disease (Figure 1).
Figure 1.
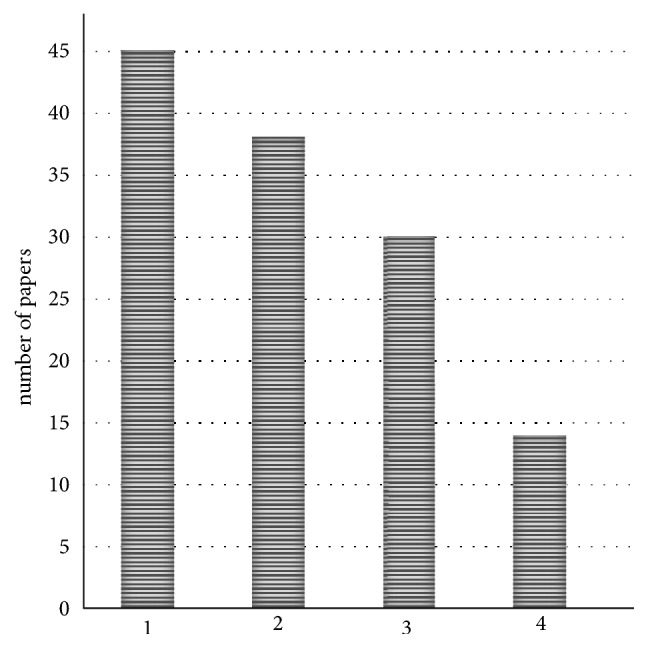
Number of papers for each subject studied for this manuscript. (1) Aging; (2) Alzheimer's disease; (3) Alzheimer's and aging; (4) other.
The qui-square test indicated a significant difference [H0 accepted] considering the analysis in conjunction with the subjects for p<0,05.
The basic probability (P) is the number of the favorable cases/number of possible cases. For the theories of aging, the total number of subgroups studied here was 11 and 8 of them presented potential cases of generating AD; therefore,
| (1) |
4. Discussion
4.1. General Data on Aging
Aging is a process of random nature, with time-dependent and chronic-degenerative aspects that all individuals are necessarily subject to [39]. It is regulated by genetic and environmental factors [3, 7] and, in general, organs and tissues age at differing rates compared to the individual because they are used differently according to the individual's life style [40].
In general, the more an organ or tissue is used the more it will age [39], for instance, the ligaments of a football player compared to the normal and non-professional athlete. However, the situation seems to be different for the brain; more educational formation has been shown to prevent AD [41]. Putatively, the aging of tissues is associated with the decrease of cell renewal [42], which is not infinite; however, this is not the case for the brain and muscles where the cells have no mitotic capacity after differentiation [39].
Thus, the average number of cell mitosis in the organism is limited [43] and, during the aging process, a reduction of cellular regeneration capacity remarkably occurs [42].
Damage to DNA generates alterations in the mitosis cycles in organisms, in some cases, diminishing the cell count in tissues [44]. Therefore, the replacement of dead cells, caused by wound healing, atrophy, reduction of vascularization, and water content in tissues, which secondarily generate a decrease in weight and organ volume, remains in a deficit and may be slowed during healthy aging [3].
In fact, physiological aging generates a series of alterations in the organic and mental functioning in the organism, decreasing the capacity to maintain normal organic functions [45].
To explain the cited facts and others, various theories of aging were proposed. In general, and arbitrarily, they can be separated into 2 main categories, i.e., stochastic and non-stochastic [35].
4.2. Theories of Aging
Stochastic theories are associated with the loss of functions during aging due to the accumulation of aleatory lesions, in part, caused by environmental factors [46].
Stochastic theory presents the subgroups known as theory of somatic mutations, theory of error-catastrophe, theory of DNA reparation, theory of the breaking of chemical bonds, theory of advanced glycosylation, and theory of oxidative stress.
In summary, the theory of somatic mutations refers to that in which sublethal radiation diminishes over the course of the life time [21], increasing the probability of acquiring diseases [47], in a way, inter alia, due to biomolecular lesions. Thus, the normal radiation that people are exposed to during life diminishes life expectancy due to the destruction of biomolecules, mainly the DNA.
The error-catastrophe theory is associated with the perpetuation of protein synthesis errors which diminish the reliability of its production, creating aberrant and/or lethal proteins [35, 48] which could affect DNA replication and increase the probability of somatic mutations [49]. The reparation of DNA theory claims that the number of DNA replications determine the life span of a species [50] and could generate a higher probability of mutations within the DNA itself, therefore impairing proteins through the process of transcription.
The theory of the breaking of biomolecular bonds cites that the modification of proteins could generate the functional failure of cell metabolism [46], because of, and for instance, the increase of chemical bonds in DNA—collagen and elastin—resulting in a decline of the physiological processes during aging [51].
The glycosylation of proteins occurs from the cross-link between glucose and protein; collagen glycosylation has been the most studied and was associated with aging, according to the theory of advanced glycosylation [52, 53]. These cross-links are caused by a high concentration of glucose in the blood and tissues and results in functional deterioration [54], cases that are usual during aging.
Free radicals, or reactive oxygen metabolites (ROMs), are the basis of the theory of oxidative stress which is associated with the reactions of biomolecules with oxides and peroxides leading to the destruction of the biomolecules, causing many degenerative alterations associated with aging [36, 38, 55, 56]. Accordingly, aging is a consequence of the actions of ROMs on biomolecules, generating disease and death [38].
Despite this theory being backed by many reputable members within the scientific community [36], as well as laymen people, some data indicates that free radicals play no significant role in aging [57] and, recently, a theory of adaptive homeostasis was proposed as a more comprehensive explanation of the aging process [58].
The adaptive homeostasis theory considers anti-stress as a form of protection that maintains homeostasis within the organism; however, because this theory is recent and lacking in data, it will not be considered in the study of theories of aging for this paper (for more details, see [58]).
The environment and genetics act together on aging, according to non-stochastic theory [35] with subgroups such as theory of cellular aging or programmed senescence, theory of telomeres, theory of intrinsic mutagenesis, neuroendocrine theory, and immunological theory.
According to Hayflic [30] and the theory of programmed senescence, aging is based on genetic programming that controls cell development. The theory refers to the existence of an organic cell program that genetically determines the life span of each of the cells, which have finite capacities, and then the organism depletes and dies [40, 59, 60].
In fact, some genes are associated with diseases in the elderly, including some alleles of the apolipoprotein E which is also associated with AD [61]. Nevertheless, genes associated only with aging have not yet been found [35].
Aging is associated with the diminishment of cell repositioning, as cited above, that, in terms of chromosomes, is linked to the modification of telomeres, which are responsible for the integrity of chromosomes during cell division during life but are constantly depleted, diminishing the size of chromosomes [62]; this is the basis of the theory of telomeres. The existence of complete telomeres is dependent on the actions of the telomerase that decline after some time, in normal cells, resulting in the shortening of the telomeres and the genes disappearing from the region [63]. According to the telomeres theory, these genes may be associated with aging. These ideas were based on the fact that the cancerogenic cells have the telomerase working constantly, permitting the continuous division of cells [64].
According to another theory, the intrinsic mutagenesis, the longevity of an animal, depends of the reliability of genetic material in its replication, i.e., depends on a minor number of errors in DNA duplication that maintain the proper functioning of restorative enzymes [46, 49, 65]. Failure in DNA replication could generate mutagenesis, indicating a loss of functions in the organism and, thereby, causing the aging. In this case, proper protein production would be corrupted.
Some studies cite the influence of melatonin, which controls circadian rhythms, and associate it with aging [66]. This observation could play a part in the neuroendocrine theory that claims the decrease of many of the hormones of the hypothalamic-pituitary-adrenal axis causes problems with metabolism [46, 67–70], thereby causing the aging phenomenon.
According to immunologic theory, immunological responses decrease with aging, a fact observed in rodents and humans [46, 71], even with the creation of a self-antibody that diminishes the responses of the T-cells, generating low resistance to infections and diseases [72–74].
4.3. Some Characteristics of AD
Aging is associated with many factors, including biological, social, intellectual, economic, functional, and chronologic [21, 22]. Therefore, one theory alone would not cover all processes associated with aging. However, AD is strongly associated with genetic [8] and environmental components. The association of these factors could explain the various processes linked to aging and, even so, the behavior, economic, and social aspects will not be adequately elucidated. In this work, the molecular analysis was prioritized.
Long life increases the probability of contracting chronic diseases and generates physical incapacity [75]. Indeed, some dementias are directly associated with aging, particularly AD [10–13].
AD is associated with two types of prevalence: familiar and sporadic, which present the same clinical and nosologic signals [33]. The sporadic type is the most common and prevails from 65 years, while the familiar type can appear more early on [76]. Interestingly, in cases of trisomy of chromosome 21, AD can begin at around 30 years [77].
The neural deficit is progressive, generating mental deterioration with neurophysiological alterations [3], and these kinds of alterations could be used as a method for distinguishing AD from normal aging as a preclinical test [77], since advanced brain aging could be, or partially be, distinct from AD [78]. Indeed, the main problem is the overlapping between the brain features of those with AD and normal elderly people. Thus, minor aspects that differentiate normal aging from AD should be largely studied in attempt to differentiate dementia apart from normal aging.
The behavior and clinical development of AD seems to be associated with the senile plates formed by the ß-amyloid protein derived from the cleavage of amyloid precursor peptide (APP) [79–81], a process that occurs as a function of gene mutation [82] and, nowadays, Positron Emission Tomography has the ability to indicate the presence of ß-amyloid in the brain [83], as well as Magnetic Resonance Imaging [84]; however, this protein also exists in brains of normal aging people.
The amyloid protein induces the formation of abnormally phosphorylated tau protein generating neuron death [23, 85]. These neurofibrillary tangles are generated by the accumulation of paired helical filaments (PHF) whose main component is the abnormal phosphorylated tau protein [38]. The normal tau protein regulates the microtubules polymerization [86, 87], but tau seems to be associated with normal aging without generating AD [88]; however, traditional opinion has essentially discarded this perspective [89]. On the other hand, the tau could be found in different patterns in the brains of both younger and older people with AD [90]. There is more than one type of data that makes the diagnosis of AD difficult.
From the cited characteristics above, problems in AD are associated with protein alterations as a function of gene mutagenesis (ß-amyloid). Indeed, the gene of presenilin is present in chromosomes 1 and 14 [6], and in chromosome 19 the gene for apolipoprotein E is situated, all with defects associated with AD, and these mutations secondarily generate protein defects (tau), or at least the mutation of apolipoprotein E [91].
In addition to biomolecular problems, recent data indicate that AD is linked to epigenetic modifications [92] caused by the methylation of DNA [22].
Another protein called kallikrein 6 seems to be associated with amyloidogenic potential since it is found in relatively elevated quantities in cerebrospinal liquid in cases of AD [93] and, recently, aquaporin was associated with edema and microvascular alterations in the brains of those with AD [94, 95]; however, the diagnosis of AD using these proteins is not completely feasible today.
Neurotransmitters also suffer alterations in AD with a decrease in the production of acetylcholine [96, 97] and the inactivation of acetylcholinesterase is the basis for AD medicine. On the other hand, recent data indicate that norepinephrine in the locus coeruleus seems to be associated with the protection of neurons during aging [98].
In addition to genetic and protein alterations, factors such as aluminum intoxication, ROMs, and neurotoxic aminoacids are some well-known agents that could lead to the generation of AD [33] and disruption of blood-brain barrier was linked to the postmortem analysis of the brains of people with AD [99], as well as to the development of neuroinflammation [100]. Uncontrolled blood pressure could generate lesions in the small vessels [13] in the white matter with consequent gray matter atrophy [12, 101, 102].
4.4. Theories of Aging Associated with AD
In many articles, AD is directly associated with aging, however, not specifically with the theories of aging in general. Consequently, it is possible to associate both AD and aging with the theories of aging and some of the morphological, genetic, and biochemical aspects shown in these theories.
However, it is difficult to separate normal aging from such dementia as AD [11] since aging is a main risk factor for acquiring AD [103, 104]. Despite and due to limitations, these theories can be associated with aspects which are peculiar to AD, since it is very difficult distinguish normal aging from AD [105].
In general, all cited theories of aging of the stochastic type explain the etiology of AD in considering at least one possibility, except for the glycosylation theory. However, if it is not possible to affirm the action of the cross-link between glucose and proteins in neurons that affect AD, it is also not plausible to deny it.
In the theories of somatic mutations, DNA reparation could be associated with the mutagenesis of presenilin, ß-amyloid, and NFG expression genes; the theory of error-catastrophe could be linked to tau abnormal phosphorylation and the break of ligations with the abnormal fracture of ß-amyloid protein. Immunological theory is associated with the immunological effects that occur in aging as inflammatory processes [106] and activated microglia and astroglia was associated with AD [107].
Non-stochastic theories are more disconnected from the prevalence of AD. Programmed senescence and intrinsic mutagenesis could explain the gene alterations found in AD; however, the neuroendocrine theory could not generate sufficient information to justify AD etiology. Telomeres theory could be associated with depletion of genes linked to AD but there are no data on the localization of specific genes associated with AD located within telomeres. This does not discard this theory, regardless of whether or not is not possible to affirm its influence on this disease.
As far as considering the same probability of theories of aging to be correct, there are eleven in total, 6 stochastic and 5 non-stochastic. Therefore, in terms of basic probability, aging with AD is about 8/11 or 72,27%.
Of course, this probability does not reflect the reality of the number of people affected by AD in the world, because the 2018 estimate is 40 million people with AD [1], therefore, 40 million/7.6 billion (approximate number of people on Earth) resulting in an approximated number of 0,52%. Indeed, the failures of the theories of aging [108] and AD's unknown etiology do not permit an accurate and precise evaluation, in both one or the other, quantitative or qualitative analysis, concluding that the aging process is not completely known [109].
The theories of aging presented until now are more targeted towards a more catastrophic analysis, in relation to possibility of acquiring AD. Factors such as life expectancy could explain the fact that not so many people present AD; however, countries with lower life expectations present a higher prevalence of AD, i.e., developing countries [31]. Indeed, the error factors for a meticulous mathematics analysis are many and very difficult to control and to put into an equation.
An important point is that the prevalence of AD in the world is increasing exponentially (for a comprehensive review, see Kirova, Bays, Lagalwar [110], and Falco et al. [111]) putatively, due to declining mortality rate around almost all of the world. Another fact is that the prevalence of AD is increasing faster than human population growth [112]. Thus, suggesting that the increase of life expectancy, at least, is linked to the prevalence of AD in the world.
5. Conclusions
From the analysis performed above, it is not possible to distinguish AD from aging; however, aging is at least deeply linked to the prevalence of AD [113]. The problem could be in the separation of normal aging, i.e., healthy elderly people, from cases of AD [114]. Therefore, a question must be asked: is it possible to age without AD or, at least, some kind of senile dementia?
In other words, will AD appear definitively at some point in life and do the elderly people that do not present AD only not present it because they have not lived long enough?
As for whether or not the increase of mutations and metabolism errors sum to the fact that aging increases the disease's progression [115], the answer could be yes.
Thus, healthy aging entails only the avoidance of AD or dementia but not their prevention. It must be a controlled process that considers social, economic, behavioral, and mental factors; preventive aspects such as nutritional care could be associated with the avoidance of AD or the extension of the life span without AD [116]. Indeed, AD has a high social impact on the patient as well as on caregivers [31] and chronic psychosocial stress is a risk factor for AD [117]; i.e., aging and AD are associated with a genotype plus lifestyle [108].
Whether or not the genotype can be directly controlled, the life style can.
In a general sense, it seems that the theories of aging explain the origin of AD under superlative and catastrophic considerations and use more biomolecular data than social or behavioral data as the bases of analysis, which may be the problem.
According to references, the general idea is that AD is associated with aging in such a way that almost all people will present this disease; however, it is plausible to consider that the increase in life expectancy will generate a high prevalence of AD [118, 119]. Interestingly, it was calculated (p<0.05) that articles on AD published over recent years do not present similarity in terms of subjects and the prevailing association between Alzheimer and aging. Furthermore, from 2009 to 2014, the main topics covered were diagnosis, disease evolution, and treatment [120]. Therefore, there is a lack of papers considering the social and behavioral aspects of the prevention of AD.
Thus, it seems that healthy aging is associated with the control of social, economic, metabolic [121], behavioral, and mental activities; however, familiar dementia, gender, depression, head trauma, smoking, high blood pressure, heart diseases, and stroke do not seem to be associated with AD, according to some studies (for more details, see Lindsay et al. [122]), therefore, generating conflict regarding these aspects cited in many papers as imperative to the acquiring of AD.
In this article, the social aspects were not discussed because the focus was on AD and the theories of aging; however, the prevalence of AD associated with patterns of life must be researched more thoroughly with an increase in the number of subjects studied because AD is a disease that there is no cure for. Therefore, prevention is the best process to avoid it, especially considering AD as a public health problem [1, 31].
Conflicts of Interest
The authors declare that there are no conflicts of interest regarding the publication of this paper.
References
- 1.Korolev I. O., Hoisington L. A., Berger K. L., Bozoki A. C. Subtypes of mild cognitive impairment differentiated based on limbic white matter integrity and cortical glucose metabolism: A pilot study using DTI and FDG-PET. Alzheimer's & Dementia. 2010;6(4):p. S10. doi: 10.1016/j.jalz.2010.05.027. [DOI] [Google Scholar]
- 2.Duboisa B., Padovanib A., Scheltensc P., Rossid A., Agnello G. D. Timely diagnosis for alzheimer's disease: A literature review on benefits and challenges. Journal of Alzheimer's Disease. 2016;49(3):617–631. doi: 10.3233/JAD-150692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Neres A. C., Rodrigues H. G., Aversi-Ferreira T. A. Alzheimer and parkinson diseases associate to aging. Bioscience Journal. 2009;25(2):139–151. [Google Scholar]
- 4.Xu Z., Xiao N., Chen Y., et al. Deletion of aquaporin-4 in APP/PS1 mice exacerbates brain Aβ accumulation and memory deficits. Molecular Neurodegeneration. 2015;10(1, article no. 56):58–74. doi: 10.1186/s13024-015-0056-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.O'Bryant S. E., Mielke M. M., Rissman R. A., et al. Blood-based biomarkers in Alzheimer disease: Current state of the science and a novel collaborative paradigm for advancing from discovery to clinic. Alzheimer’s & Dementia. 2017;13(1):45–58. doi: 10.1016/j.jalz.2016.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fridman C., Gregório S. P., Dias Neto E., Ojopi É. P. Alterações genéticas na doença de Alzheimer. Archives of Clinical Psychiatry (São Paulo) 2004;31(1):19–25. doi: 10.1590/S0101-60832004000100004. [DOI] [Google Scholar]
- 7.Nacmias B., Bagnoli S., Piaceri I., Sorbi S. Genetic heterogeneity of Alzheimer's disease: embracing research partnerships. Journal of Alzheimer's Disease. 2018;62(3):903–911. doi: 10.3233/JAD-170570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Karch C. M., Cruchaga C., Goate A. M. Alzheimer's disease genetics: from the bench to the clinic. Neuron. 2014;83(1):11–26. doi: 10.1016/j.neuron.2014.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chételat G., La Joie R., Villain N., et al. Amyloid imaging in cognitively normal individuals, at-risk populations and preclinical Alzheimer's disease. NeuroImage: Clinical. 2013;2(1):356–365. doi: 10.1016/j.nicl.2013.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.West M. J., Coleman P. D., Flood D. G., Troncoso J. C. Differences in the pattern of hippocampal neuronal loss in normal ageing and Alzheimer's disease. The Lancet. 1994;344(8925):769–772. doi: 10.1016/S0140-6736(94)92338-8. [DOI] [PubMed] [Google Scholar]
- 11.Fjell A. M., McEvoy L., Holland D., Dale A. M., Walhovd K. B., Alzheimer's Disease Neuroimaging Initiative What is normal in normal aging? Effects of aging, amyloid and Alzheimer's disease on the cerebral cortex and the hippocampus. Progress in Neurobiology. 2014;117:20–40. doi: 10.1016/j.pneurobio.2014.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lindemer E. R., Greve D. N., Fischl B. R., Augustinack J. C., Salat D. H. Regional staging of white matter signal abnormalities in aging and Alzheimer's disease. NeuroImage: Clinical. 2017;14:156–165. doi: 10.1016/j.nicl.2017.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tarantini S., Tran C. H. T., Gordon G. R., Ungvari Z., Csiszar A. Impaired neurovascular coupling in aging and Alzheimer's disease: Contribution of astrocyte dysfunction and endothelial impairment to cognitive decline. Experimental Gerontology. 2017;94:52–58. doi: 10.1016/j.exger.2016.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Herrup K. The case for rejecting the amyloid cascade hypothesis. Nature Neuroscience. 2015;18(6):794–799. doi: 10.1038/nn.4017. [DOI] [PubMed] [Google Scholar]
- 15.Charchat H., Nitrini R., Caramelli P., Sameshima K. Investigação de Marcadores Clínicos dos Estágios Iniciais da Doença de Alzheimer com Testes Neuropsicológicos Computadorizados. Psicologia: Reflexão e Crítica. 2001;14(2):305–316. doi: 10.1590/S0102-79722001000200006. [DOI] [Google Scholar]
- 16.Doan N. T., Engvig A., Zaske K., et al. Distinguishing early and late brain aging from the Alzheimer's disease spectrum: consistent morphological patterns across independent samples. NeuroImage. 2017;158:282–295. doi: 10.1016/j.neuroimage.2017.06.070. [DOI] [PubMed] [Google Scholar]
- 17.Tromp D., Dufour A., Lithfous S., Pebayle T., Després O. Episodic memory in normal aging and Alzheimer disease: insights from imaging and behavioral studies. Ageing Research Reviews. 2015;24:232–262. doi: 10.1016/j.arr.2015.08.006. [DOI] [PubMed] [Google Scholar]
- 18.Werheid K., Clare L. Are faces special in Alzheimer's disease? Cognitive conceptualisation, neural correlates, and diagnostic relevance of impaired memory for faces and names. Cortex. 2007;43(7):898–906. doi: 10.1016/S0010-9452(08)70689-0. [DOI] [PubMed] [Google Scholar]
- 19.Calabria M., Sabio A., Martin C., et al. The missing link between faces and names: Evidence from Alzheimer’s disease patients. Brain and Cognition. 2012;80(2):250–256. doi: 10.1016/j.bandc.2012.07.002. [DOI] [PubMed] [Google Scholar]
- 20.Tak S. H., Hong S. H. Face-name memory in Alzheimers disease. Geriatric Nursing. 2014;35:290–294. doi: 10.1016/j.gerinurse.2014.03.004. [DOI] [PubMed] [Google Scholar]
- 21.Aversi-Ferreira T. A., Rodrigues H. G., Paiva L. R. Efeitos do envelhecimento sobre o encéfalo. RBCEH. 2008;5:46–64. [Google Scholar]
- 22.Horvath J. C., Forte J. D., Carter O. Quantitative review finds no evidence of cognitive effects in healthy populations from single-session transcranial direct current stimulation (tDCS) Brain Stimulation. 2015;8(3):535–550. doi: 10.1016/j.brs.2015.01.400. [DOI] [PubMed] [Google Scholar]
- 23.Tarasoff-Conway J. M., Carare R. O., Osorio R. S., et al. Clearance systems in the brain - Implications for Alzheimer disease. Nature Reviews Neurology. 2015;11(8):457–470. doi: 10.1038/nrneurol.2015.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Daianu M., Jahanshad N., Nir T. M., Toga A. W., Jack Jr C. R., et al. Breakdown of brain connectivity between normal aging and Alzheimers disease: a structural k-core network analysis. Brain Connect. 2013;3:407–422. doi: 10.1089/brain.2012.0137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Miraglia F., Vecchio F., Rossini P. M. Searching for signs of aging and dementia in EEG through network analysis. Behavioural Brain Research. 2017;317:292–300. doi: 10.1016/j.bbr.2016.09.057. [DOI] [PubMed] [Google Scholar]
- 26.Pini L., Pievani M., Bocchetta M., et al. Brain atrophy in Alzheimer's Disease and aging. Ageing Research Reviews. 2016;30:25–48. doi: 10.1016/j.arr.2016.01.002. [DOI] [PubMed] [Google Scholar]
- 27.de Flores R., La Joie R., Chételat G. Structural imaging of hippocampal subfields in healthy aging and Alzheimer’s disease. Neuroscience. 2015;309:29–50. doi: 10.1016/j.neuroscience.2015.08.033. [DOI] [PubMed] [Google Scholar]
- 28.Ihara R. M. D., Vincent B. D. B. S., Baxter M. R., et al. Relative neuron loss in hippocampal sclerosis of aging and Alzheimer disease. Annals of Neurology. 2018:1–13. doi: 10.1002/ana.25344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Westerberg B., Roberson J., Stach B., Silverberg G., Heit G. The effects of posteroventral pallidotomy on balance function in patients with Parkinson’s disease. Stereotactic and Functional Neurosurgery. 2003;79(2):75–87. doi: 10.1159/000070103. [DOI] [PubMed] [Google Scholar]
- 30.Hayflick L. Human cells and aging. Scientific American. 1968;218(3):32–37. doi: 10.1038/scientificamerican0368-32. [DOI] [PubMed] [Google Scholar]
- 31.Alzheimer's Association. Alzheimer’s disease facts and figures. Alzheimers Dementia. 2017;13:325–373. [Google Scholar]
- 32.Korolev I. O. Alzheimers Disease: A clinical and basic science review. Medical Student Research Journal. 2014;4:24–33. [Google Scholar]
- 33.Smith M. A. C. Doença de Alzheimer. Revista Brasileira de Psiquiatria. 1999;21(suppl 2):03–07. doi: 10.1590/S1516-44461999000600003. [DOI] [Google Scholar]
- 34.Caixeta L., Soares V. L. D., Vieira R. T., et al. Executive function is selectively impaired in old age bipolar depression. Frontiers in Psychology. 2017;13:1–5. doi: 10.3389/fpsyg.2017.00194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mota M. P., Figueiredo P. A., Duarte J. A. Teorias biológicas do envelhecimento. Revista Portuguesa de Ciências do Desporto. 2004;2004(1):81–110. doi: 10.5628/rpcd.04.01.81. [DOI] [Google Scholar]
- 36.Krause K.-H. Aging: a revisited theory based on free radicals generated by NOX family NADPH oxidases. Experimental Gerontology. 2007;42(4):256–262. doi: 10.1016/j.exger.2006.10.011. [DOI] [PubMed] [Google Scholar]
- 37.Rita Cardoso B., Silva Bandeira V., Jacob-Filho W., Franciscato Cozzolino S. M. Selenium status in elderly: Relation to cognitive decline. Journal of Trace Elements in Medicine and Biology. 2014;28(4):422–426. doi: 10.1016/j.jtemb.2014.08.009. [DOI] [PubMed] [Google Scholar]
- 38.Harman D. The aging process: major risk factor for disease and death. Proceedings of the National Acadamy of Sciences of the United States of America. 1991;88(12):5360–5363. doi: 10.1073/pnas.88.12.5360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Arking R. Biology of Aging: Observations and Principles. 2nd. Sunderland, UK: Sinauer Associates; 1998. [Google Scholar]
- 40.Pereira A., Freitas C., Mendonça C., Marçal F., Souza J., Noronha J. P. Envelhecimento, estresse e sociedade: uma visão psiconeuroendocrinológica. Ciências & Cognição. 2004;01:34–53. [Google Scholar]
- 41.Gatz M. Educating the brain to avoid dementia: Can mental exercise prevent Alzheimer disease? PLoS Medicine. 2005;2:0038–0040. doi: 10.1371/journal.pmed.0020038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mcardle W. D., Katch F., Katch V. L. F. Fisiologia do Exercício, Energia, Nutrição e Desempenho. 5th. Rio de Janeiro, Brazil: Guanabara Koogan; 2003. [Google Scholar]
- 43.Macieira-Coelho A. The last mitoses of the human fibroblast proliferative life span, physiopathologic implications. Mechanisms of Ageing and Development. 1995;82(2-3):91–104. doi: 10.1016/0047-6374(95)01610-C. [DOI] [PubMed] [Google Scholar]
- 44.Jackson S. P., Bartek J. The DNA-damage response in human biology and disease. Nature. 2009;461(7267):1071–1078. doi: 10.1038/nature08467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Straub R. H., Cutolo M., Zietz B., Schölmerich J. The process of aging changes the interplay of the immune, endocrine and nervous systems. Mechanisms of Ageing and Development. 2001;122(14):1591–1611. doi: 10.1016/S0047-6374(01)00289-5. [DOI] [PubMed] [Google Scholar]
- 46.Cristofalo V. J., Gerhard G. S., Pignolo R. J. Molecular biology of aging. Surgical Clinics of North America. 1994;74(1):1–21. doi: 10.1016/S0039-6109(16)46225-0. [DOI] [PubMed] [Google Scholar]
- 47.Curtis H. J. Biological mechanisms underlying the aging process. Science. 1963;141(3582):686–694. doi: 10.1126/science.141.3582.686. [DOI] [PubMed] [Google Scholar]
- 48.Orgel L. E. The maintenance of accuracy of protein synthesis and its relevance to agins. Proceedings of the National Academy of Sciences of the United States of America. 1963;49:517–521. doi: 10.1073/pnas.49.4.517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Martin G. M., Hoehn H., Norwood T. H. Development and gerontological aspects of disease. In: Hil R. B., Lavia M. F., editors. Principles of Pathobiology. New York, NY, USA: Oxford University Press; 1980. pp. 287–347. [Google Scholar]
- 50.Hart R. W., Setlow R. B. Correlation between deoxyribonucleic acid excision repair and life span in a number of mammalian species. Proceedings of the National Acadamy of Sciences of the United States of America. 1974;71(6):2169–2173. doi: 10.1073/pnas.71.6.2169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lee A., Cerami A. Modifications of proteins and nucleic acids by reducing sugars: possible role in aging. In: Schneider E. L., Rowe J. W., editors. Handbook of The Biology of Aging. Academic Press; 1990. pp. 116–130. [Google Scholar]
- 52.Cerami A. Hypothesis: glucose as a mediator of aging. Journal of the American Geriatrics Society. 1985;33(9):626–634. doi: 10.1111/j.1532-5415.1985.tb06319.x. [DOI] [PubMed] [Google Scholar]
- 53.Morrison N. A., Qi J. C., Tokita A., et al. Prediction of bone density from vitamin D receptor alleles. Nature. 1994;367:284–287. doi: 10.1038/367284a0. [DOI] [PubMed] [Google Scholar]
- 54.Hayoz D., Ziegler T., Brunner H. R., Ruiz J. Diabetes mellitus and vascular lesions. Metabolism - Clinical and Experimental. 1998;47(1):16–19. doi: 10.1016/S0026-0495(98)90365-1. [DOI] [PubMed] [Google Scholar]
- 55.Fukagawa N. K. Aging: Is oxidative stress a marker or is it causal? Proceedings of the Society for Experimental Biology and Medicine. 1999;222(3):293–298. doi: 10.1046/j.1525-1373.1999.d01-146.x. [DOI] [PubMed] [Google Scholar]
- 56.Gella A., Durany N. Oxidative stress in Alzheimer disease. Cell Adhesion & Migration. 2009;3(1):88–93. doi: 10.4161/cam.3.1.7402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Viña J., Borras C., Gomez-Cabrera M. C. A free radical theory of frailty. Free Radical Biology & Medicine. 2018;124:358–363. doi: 10.1016/j.freeradbiomed.2018.06.028. [DOI] [PubMed] [Google Scholar]
- 58.Pomatto L. C. D., Davies K. J. A. Adaptive homeostasis and the free radical theory of ageing. Free Radical Biology & Medicine. 2018;124:420–430. doi: 10.1016/j.freeradbiomed.2018.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hayflick L. Methuselah secrets – cellular aging and cell immortality. Biofutur. 1990;(87):20–23. [Google Scholar]
- 60.Ikram Z., Norton T., Jat P. S. he biological clock that measures the mitotic life-span of mouse embryo fibroblast continues to function in the presence of simian virus 40 large tumor antigen. Proceedings of the National Acadamy of Sciences of the United States of America. 1994;91(14):6448–6452. doi: 10.1073/pnas.91.14.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Curtsinger J. W., Fukui H. H., Khazaeli A. A., et al. Genetic variation and aging. Annual Review of Genetics. 1995;29:553–575. doi: 10.1146/annurev.ge.29.120195.003005. [DOI] [PubMed] [Google Scholar]
- 62.Blackburn E. H. The telomere and telomerase: nucleic acid – protein complexes acting in a telomere homeostasis system. A review. Biochemistry (Moscow) 1997;62(11):1196–1201. [PubMed] [Google Scholar]
- 63.Kurenova E. V., Mason J. M. Telomere functions. A review. Biochemistry (Mosc) 1997;62:1242–1253. [PubMed] [Google Scholar]
- 64.Marx J. Chromosome ends catch fire. Science. 1994;265(5179):1656–1658. doi: 10.1126/science.7521969. [DOI] [PubMed] [Google Scholar]
- 65.Burnet M. Ntrinsic Mutagenesis: A Genetic Approach. Wiley; 1974. [DOI] [Google Scholar]
- 66.Falcón J. Cellular circadian clocks in the pineal. Progress in Neurobiology. 1999;58(2):121–162. doi: 10.1016/S0301-0082(98)00078-1. [DOI] [PubMed] [Google Scholar]
- 67.Finch C. E. Longevity, Senescence and The Genome. Chicago, Ill, USA: University of Chicago Press; 1994. [Google Scholar]
- 68.Miller R. A. The biology of aging and longevity. In: Hazzard W. R., et al., editors. Principles of Geriatric Medicine and Gerontology. Londres, UK: McGraw-Hill, Inc; 1994. pp. 3–18. [Google Scholar]
- 69.Sonntag W. E., Lynch C. D., Cefalu W. T., et al. Pleiotropic effects of growth hormone and insulin-like growth factor (IGF)-1 on biological aging: inferences from moderate caloric- restricted animals. The Journals of Gerontology. Series A, Biological Sciences and Medical Sciences. 1999;54(12):B521–B538. doi: 10.1093/gerona/54.12.B521. [DOI] [PubMed] [Google Scholar]
- 70.Levin E. R. Invited review: Cell localization, physiology, and nongenomic actions of estrogen receptors. Journal of Applied Physiology. 2001;91(4):1860–1867. doi: 10.1152/jappl.2001.91.4.1860. [DOI] [PubMed] [Google Scholar]
- 71.Walford R. L. The Immunological Theory of Aging. Stanford: Williams & Wilkins; 1969. [Google Scholar]
- 72.Ernst D. N., Hobbs M. V., Torbett B. E., et al. Differences in the expression. profiles of CD45RB, Pgp-1, and 3G11 membrane antigens and in the patterns of lymphokine secretion by splenic CD4+ T cells from Young and aged mice. The Journal of Immunology. 1990;145(5):1295–1302. [PubMed] [Google Scholar]
- 73.Miller R. A. Aging and immune response. In: Schneider E. L., Rowe J. W., editors. Handbook of The Biology of Aging. San Diego, Cali, USA: Academic Press, Inc; 1996. pp. 355–392. [Google Scholar]
- 74.Miller R. A. The aging immune system: primer and prospectus. Science. 1996;273(5271):70–74. doi: 10.1126/science.273.5271.70. [DOI] [PubMed] [Google Scholar]
- 75.Hoeman S. P. Enfermagem de reabilitação: aplicação e processos. 2nd. Lisboa, Portugal: LusociΩncia; 2000. [Google Scholar]
- 76.Bottino C. M., Carvalho I. A., Alvarez A. M., et al. Reabilitação cognitiva em pacientes com doença de Alzheimer. Arq Neuropsiquiatr. 2002;60(1):70–79. doi: 10.1590/S0004-282X2002000100013. [DOI] [PubMed] [Google Scholar]
- 77.Ojopi E. P., Bertoncini A. B., Dias Neto E. Apolipoproteína E e a doença de Alzheimer. Archives of Clinical Psychiatry (São Paulo) 2004;31(1):26–33. doi: 10.1590/S0101-60832004000100005. [DOI] [Google Scholar]
- 78.Habes M., Janowitz D., Erus G., et al. Advanced brain aging: relationship with epidemiologic and genetic risk factors, and overlap with Alzheimer disease atrophy patterns. Translational Psychiatry. 2016;6(4):e775–e775. doi: 10.1038/tp.2016.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Tariot P. N. Alzheimer Disease: an overview. Alzheimer Disease & Associated Disorders. 1994;8(2):s4–s11. doi: 10.1097/00002093-199424000-00002. [DOI] [Google Scholar]
- 80.Clark C. M., Karlawish J. H. T. Alzheimer disease: current concepts and emerging diagnostic and therapeutic strategies. Annals of Internal Medicine. 2003;138(5):400–410. doi: 10.7326/0003-4819-138-5-200303040-00010. [DOI] [PubMed] [Google Scholar]
- 81.Ryan T. M., Roberts B. R., McColl G., et al. Stabilization of nontoxic Aβ-oligomers: insights into the mechanism of action of hydroxyquinolines in Alzheimer’s disease. The Journal of Neuroscience. 2015;35(7):2871–2884. doi: 10.1523/jneurosci.2912-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Fallin M. D., Matteini A. Genetic epidemiology in aging research. The Journals of Gerontology. Series A, Biological Sciences and Medical Sciences. 2009;64(1):47–60. doi: 10.1093/gerona/gln021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Johnson K. A., Sperling R. A., Gidicsin C. M., et al. Florbetapir (F18-AV-45) PET to assess amyloid burden in Alzheimer's disease dementia, mild cognitive impairment, and normal aging. Alzheimer’s & Dementia. 2013;9(5):S72–S83. doi: 10.1016/j.jalz.2012.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Franzmeier N., Dyrba M. Functional brain network architecture may route progression of Alzheimer's disease pathology. Brain. 2017;140(12):3077–3080. doi: 10.1093/brain/awx304. [DOI] [PubMed] [Google Scholar]
- 85.Almeida O. Biologia molecular da doença de Alzheimer: uma luz no fim do túnel? Revista da Associação Médica Brasileira. 1997;43(1):77–81. doi: 10.1590/S0104-42301997000100017. [DOI] [PubMed] [Google Scholar]
- 86.Schönknecht P., Pantel J., Hartmann T., et al. Cerebrospinhal fluid tau levels in Alzheimer’s disease are elevated when compared with vascular dementia but do not correlate with measures of cerebral atrophy. Psychiatry Research. 2003;120(3):231–238. doi: 10.1016/S0165-1781(03)00197-5. [DOI] [PubMed] [Google Scholar]
- 87.Hiraoka S., Yao T.-M., Minoura K., et al. Conformational transition state is responsible for assembly of microtubule-binding domain of tau protein. Biochemical and Biophysical Research Communications. 2004;315(3):659–663. doi: 10.1016/j.bbrc.2004.01.107. [DOI] [PubMed] [Google Scholar]
- 88.Crary J. F., Trojanowski J. Q., Schneider J. A. Primary age-related tauopathy (PART): a common pathology associated with human aging. Acta Neuropathologica. 2014;128(6):755–766. doi: 10.1007/s00401-014-1349-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Price J. L., Davis P. B., Morris J. C., White D. L. The distribution of tangles, plaques and related immunohistochemical markers in healthy aging and Alzheimer's disease. Neurobiology of Aging. 1991;12(4):295–312. doi: 10.1016/0197-4580(91)90006-6. [DOI] [PubMed] [Google Scholar]
- 90.Lowe V. J., Wiste H. J., Senjem M. L., et al. Widespread brain tau and its association with ageing, Braak stage and Alzheimer’s dementia. Brain. 2018;141(1):271–287. doi: 10.1093/brain/awx320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lewczuk P., Esselmann H., Otto M., et al. Neurochemical diagnosis of Alzheimer's dementia by CSF Aβ42, Aβ42/Aβ40 ratio and total tau. Neurobiology of Aging. 2004;25(3):273–281. doi: 10.1016/S0197-4580(03)00086-1. [DOI] [PubMed] [Google Scholar]
- 92.Bustos F. J., Ampuero E., Jury N., et al. Epigenetic editing of the Dlg4/PSD95 gene improves cognition in aged and Alzheimer's disease mice. Brain. 2017;140(12):3252–3268. doi: 10.1093/brain/awx272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Diamandis E. P., Yousef G. M., Petraki C., Soosaipillai A. R. Human Kalikrein 6 as a biomarker of Alzheimer’ disease. Clinical Biochemistry. 2000;33(8):663–667. doi: 10.1016/S0009-9120(00)00185-5. [DOI] [PubMed] [Google Scholar]
- 94.Moftakhar P., Lynch M. D., Pomakian J. L., Vinters H. V. Aquaporin expression in the brains of patients with or without cerebral amyloid angiopathy. Journal of Neuropathology & Experimental Neurology. 2010;69(12):1201–1209. doi: 10.1097/NEN.0b013e3181fd252c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Zeppenfeld D. M., Simon M., Haswell J. D., et al. Association of perivascular localization of aquaporin-4 with cognition and Alzheimer disease in aging brains. JAMA Neurology. 2017;74(1):91–99. doi: 10.1001/jamaneurol.2016.4370. [DOI] [PubMed] [Google Scholar]
- 96.Almeida O. P. Tratamento da doença de Alzheimer: avaliação crítica sobre o uso de anticolinesterásticos. Arq Neuropsiquiatr. 1998;56(3B):688–696. doi: 10.1590/S0004-282X1998000400029. [DOI] [PubMed] [Google Scholar]
- 97.Hampel H., Mesulam M. M., Cuello A. C., et al. The colinergic system in the pathophysiology and treatment of Alzheimer’s disease. Brain. 2018:1–17. doi: 10.1093/brain/awy132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Mather M., Harley C. W. The Locus Coeruleus: Essential for Maintaining Cognitive Function and the Aging Brain. Trends in Cognitive Sciences. 2016;20(3):214–226. doi: 10.1016/j.tics.2016.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Montagne A., Barnes S. R., Sweeney M. D., et al. Blood-Brain barrier breakdown in the aging human hippocampus. Neuron. 2015;85(2):296–302. doi: 10.1016/j.neuron.2014.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Elahy M., Jackaman C., Mamo J. C. L., et al. Blood–brain barrier dysfunction developed during normal aging is associated with inflammation and loss of tight junctions but not with leukocyte recruitment. Immunity & Ageing. 2015;12(1) doi: 10.1186/s12979-015-0029-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kern K. C., Wright C. B., Bergfield K. L., et al. Blood pressure control in aging predicts cerebral atrophy related to small-vessel white matter lesions. Frontiers in Aging Neuroscience. 2017;9:1–10. doi: 10.3389/fnagi.2017.00132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Mutlu J., Landeau B., Gaubert M., De La Sayette V., Desgranges B., Chételat G. Distinct influence of specific versus global connectivity on the different Alzheimer's disease biomarkers. Brain. 2017;140(12):3317–3328. doi: 10.1093/brain/awx279. [DOI] [PubMed] [Google Scholar]
- 103.Gefen T., Papastefan S. T., Rezvanian A., et al. Von Economo neurons of the anterior cingulate across the lifespan and in Alzheimer's disease. Cortex. 2018;99:69–77. doi: 10.1016/j.cortex.2017.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Hargis K. E., Blalock E. M. Transcriptional signatures of brain aging and Alzheimer's disease: What are our rodent models telling us? Behavioural Brain Research. 2017;322:311–328. doi: 10.1016/j.bbr.2016.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Smith C. D., Carney J. M., Starke-Reed P. E., et al. Excess brain protein oxidation and enzyme dysfunction in normal aging and in Alzheimer disease. Proceedings of the National Acadamy of Sciences of the United States of America. 1991;88(23):10540–10543. doi: 10.1073/pnas.88.23.10540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Peña-Altamira E., Petralla S., Massenzio F., Virgili M., Bolognesi M. L., Monti B. Nutritional and pharmacological strategies to regulate microglial polarization in cognitive aging and Alzheimer's disease. Frontiers in Aging Neuroscience. 2017;9:1–15. doi: 10.3389/fnagi.2017.00175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Heneka M. T., Carson M. J., Khoury J. E. l., et al. Neuroinfl ammation in Alzheimers disease. The Lancet Neurology. 2015;14:388–405. doi: 10.1016/S1474-4422(15)70016-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.da Costa J. P., Vitorino R., Silva G. M., Vogel C., Duarte A. C., Rocha-Santos T. A synopsis on aging—Theories, mechanisms and future prospects. Ageing Research Reviews. 2016;29:90–112. doi: 10.1016/j.arr.2016.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Colin J., Thomas M. H., Gregory-Pauron L., et al. Maintenance of membrane organization in the aging mouse brain as the determining factor for preventing receptor dysfunction and for improving response to anti-Alzheimer treatments. Neurobiology of Aging. 2017;54:84–93. doi: 10.1016/j.neurobiolaging.2017.02.015. [DOI] [PubMed] [Google Scholar]
- 110.Kirova A.-M., Bays R. B., Lagalwar S. Working memory and executive function decline across normal aging, mild cognitive impairment, and Alzheimer’s disease. BioMed Research International. 2015;2015:9. doi: 10.1155/2015/748212.748212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Falco A., Cukierman D. S., Hauser-Davis R. A., Rey NA. Doença de alzheimer: hipóteses etiológicas e perspectivas de tratamento. Quim. Nova. 2016;39:63–80. [Google Scholar]
- 112.Reitz C., Mayeux R. Alzheimer disease: epidemiology, diagnostic criteria, risk factors and biomarkers. Biochemical Pharmacology. 2014;88(4):640–651. doi: 10.1016/j.bcp.2013.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Nativio R., Donahue G., Berson A., et al. Dysregulation of the epigenetic landscape of normal aging in Alzheimer's disease. Nature Neuroscience. 2018;21(4):497–505. doi: 10.1038/s41593-018-0101-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Jalili M. Graph theoretical analysis of Alzheimer's disease: Discrimination of AD patients from healthy subjects. Information Sciences. 2017;384:145–156. doi: 10.1016/j.ins.2016.08.047. [DOI] [Google Scholar]
- 115.Fiford C. M., Ridgway G. R., Cash D. M., et al. Patterns of progressive atrophy vary with age in Alzheimer's disease patients. Neurobiology of Aging. 2018;63:22–32. doi: 10.1016/j.neurobiolaging.2017.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Fenech M. Vitamins associated with brain aging, mild cognitive impairment, and alzheimer disease: Biomarkers, epidemiological and experimental evidence, plausible mechanisms, and knowledge gaps. Advances in Nutrition. 2017;8(6):958–970. doi: 10.3945/an.117.015610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Piirainen S., Youssef A., Song C., et al. Psychosocial stress on neuroinflammation and cognitive dysfunctions in Alzheimer’s disease: the emerging role for microglia? Neuroscience & Biobehavioral Reviews. 2017;77:148–164. doi: 10.1016/j.neubiorev.2017.01.046. [DOI] [PubMed] [Google Scholar]
- 118.Imtiaz B., Tolppanen A.-M., Kivipelto M., Soininen H. Future directions in Alzheimer's disease from risk factors to prevention. Biochemical Pharmacology. 2014;88(4):661–670. doi: 10.1016/j.bcp.2014.01.003. [DOI] [PubMed] [Google Scholar]
- 119.Imtiaz D., Khan A., Seelye A. A mobile multimedia reminiscence therapy application to reduce behavioral and psychological symptoms in persons with Alzheimer’s. Journal of Healthcare Engineering. 2018;2018:9. doi: 10.1155/2018/1536316.1536316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Fernandes J. S. F., Andrade M. S. Revisão sobre a doença de Alzheimer: diagnóstico, evolução e cuidados. Saúde & Doenças. 2017;18:131–140. [Google Scholar]
- 121.Yin F., Sancheti H., Patil I., Cadenas E. Energy metabolism and inflammation in brain aging and Alzheimer's disease. Free Radical Biology & Medicine. 2016;100:108–122. doi: 10.1016/j.freeradbiomed.2016.04.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Lindsay J., Laurin D., Verreault R., et al. Risk factors for Alzheimer's disease: a prospective analysis from the Canadian Study of Health and Aging. American Journal of Epidemiology. 2002;156(5):445–453. doi: 10.1093/aje/kwf074. [DOI] [PubMed] [Google Scholar]