

In this study, Zemke et al. show that E1A inactivates the Hippo pathway-regulated TEAD coactivators YAP and TAZ by causing their sequestration in the cytoplasm. Their findings suggest that YAP/TAZ function in a developmental checkpoint controlled by signaling from the actin cytoskeleton that prevents differentiation of a progenitor cell until it is in the correct cellular and tissue environment.
Keywords: adenovirus E1A, Hippo pathway, YAP/TAZ, dedifferentiation, mesenchymal stem cells, p300/CBP, superenhancer
Abstract
Adenovirus transformed cells have a dedifferentiated phenotype. Eliminating E1A in transformed human embryonic kidney cells derepressed ∼2600 genes, generating a gene expression profile closely resembling mesenchymal stem cells (MSCs). This was associated with a dramatic change in cell morphology from one with scant cytoplasm and a globular nucleus to one with increased cytoplasm, extensive actin stress fibers, and actomyosin-dependent flattening against the substratum. E1A-induced hypoacetylation at histone H3 Lys27 and Lys18 (H3K27/18) was reversed. Most of the increase in H3K27/18ac was in enhancers near TEAD transcription factors bound by Hippo signaling-regulated coactivators YAP and TAZ. E1A causes YAP/TAZ cytoplasmic sequestration. After eliminating E1A, YAP/TAZ were transported into nuclei, where they associated with poised enhancers with DNA-bound TEAD4 and H3K4me1. This activation of YAP/TAZ required RHO family GTPase signaling and caused histone acetylation by p300/CBP, chromatin remodeling, and cohesin loading to establish MSC-associated enhancers and then superenhancers. Consistent results were also observed in primary rat embryo kidney cells, human fibroblasts, and human respiratory tract epithelial cells. These results together with earlier studies suggest that YAP/TAZ function in a developmental checkpoint controlled by signaling from the actin cytoskeleton that prevents differentiation of a progenitor cell until it is in the correct cellular and tissue environment.
Adenovirus E1A, in addition to forcing quiescent cells into S phase by inhibiting retinoblastoma (Rb) family proteins (DeCaprio 2009) and repressing expression of CDK inhibitors (Ferrari et al. 2014), also represses cellular differentiation of various mammalian cell types (Frisch and Mymryk 2002). For example, E1A blocks differentiation of rodent myoblasts by interfering with the function of developmentally regulated cis-acting transcription control regions (Webster et al. 1988). However, how E1A represses differentiation of various cell types has not been fully clarified. While most E1A-induced transcriptional repression requires its interaction with the closely related lysine acetyltransferases CBP and p300 (Stein et al. 1990; Ferrari et al. 2014), it is unclear how particular genes, mainly cell type-specific genes, are targeted for repression.
A consequence of E1A's interaction with CBP and p300 is a dramatic reduction in acetylation of total cellular histone H3 Lys27 and Lys18 (H3K27/18ac) (Horwitz et al. 2008; Ferrari et al. 2014). H3K27/18ac are highly enriched at enhancers and promoters of active genes, and their presence correlates with enhancer activity (Creyghton et al. 2010). Enhancers are critical for establishing the diversity between different cell types necessary for the complex multitissue development of a metazoan (Carey 1998; Levine 2010; Bulger and Groudine 2011). More recently, a new classification of enhancers, termed superenhancer, was described that includes ∼1.5%–4% of enhancers in differentiated mammalian cells (Whyte et al. 2013). Superenhancers are clusters of neighboring enhancers with a high density of transcription factor (TF) binding, active chromatin histone marks, and coactivator association (Whyte et al. 2013). Superenhancers often are controlled by cell type-specific TFs and regulate key cell identity genes important for normal mammalian development.
The Hippo pathway regulates organ size across metazoans (Pan 2010; Yu et al. 2015; Zanconato et al. 2016) and is essential for the first tissue differentiation during mammalian embryonic development—the differentiation of cells on the surface of the 16- to 32-cell embryo (morula) into trophectoderm that develops into the early placenta and cells in the interior of the morula that develop into the embryo and adult (Sasaki 2017). In mammals, paralogous transcription coactivators YAP (also known as YAP1) and TAZ (also known as WWTR1) are the terminal effectors for Hippo signaling and activate target genes mainly through binding to DNA-bound TEAD family TFs (Zhao et al. 2008). YAP/TAZ are considered oncogenes because they are frequently overexpressed in a variety of human cancers and are often amplified in squamous cell carcinoma, and overexpression of YAP/TAZ target genes correlates with poor prognosis (Wang et al. 2018). YAP is indispensable for early embryonic development (Sasaki 2017) and is expressed at some point during the development of almost all mammalian cell types as they develop from the inner cell mass of the early embryo (Varelas 2014). During active Hippo signaling, a kinase cascade results in phosphorylation and activation of terminal kinases LATS1 and LATS2, which phosphorylate YAP/TAZ, leading to their cytoplasmic retention and ubiquitin-mediated degradation (Yu et al. 2015). Consequently, YAP/TAZ activities are regulated through control of their nuclear import, which occurs when they are not phosphorylated by the LATS1/2 terminal protein kinases of the Hippo pathway versus their retention in the cytoplasm through binding to 14-3-3 phospho-serine/threonine-binding proteins anchored in the cytoplasm when YAP/TAZ are phosphorylated by activated LATS1/2. Crucially, the Hippo pathway regulates expression of multiple genes in response to mechanical cues generated by interactions with neighboring cells and the extracellular matrix (ECM) (Dupont et al. 2011; Meng et al. 2018). The AMOT family proteins (AMOTs) enhance Hippo signaling by activating LATS1/2 at adherens junctions between cells in preimplantation embryos (Hirate et al. 2013). Hippo signaling is suppressed when AMOTs are sequestered away from adherens junctions by binding to filamentous actin (F-actin) (Hirate et al. 2013). AMOTs also inhibit YAP/TAZ through direct interactions (Chan et al. 2011; Zhao et al. 2011). Alternative WNT signaling also regulates YAP/TAZ (Park et al. 2015).
We began this study pursuing the mechanism of how adenovirus E1A causes preferential hypoacetylation of H3K27/18 at enhancers and superenhancers compared with promoters (transcription start sites [TSSs]) (see below). Unexpectedly, we found that most of this regulation of H3 acetylation occurs at sites of TEAD TF association, leading us to the discovery that E1A inactivates the Hippo pathway-regulated TEAD coactivators YAP and TAZ by causing their sequestration in the cytoplasm. Further analyses showed that YAP/TAZ inactivation contributes greatly to the dedifferentiated phenotype of adenovirus transformed cells. Despite hundreds of generations of E1A-induced dedifferentiation, when E1A was eliminated from HEK293 cells, they retained the ability to “redifferentiate” into cells resembling normal human mesenchymal stem cells (MSCs), the cell type from which they were likely derived. This redifferentiation was dependent on activation by both YAP and TAZ. Mechanistically, following E1A loss, YAP and TAZ translocate from the cytoplasm to the nucleus, dependent on F-actin assembly and Rho family small GTPases. In the nucleus, they associate with TEAD TFs and establish enhancers and then superenhancers that strongly activate MSC-associated genes necessary for a drastic change in cell morphology. Virtually all of the MSC-associated gene activation and enhancer establishment after removal of E1A depend on YAP/TAZ. These results, together with earlier studies, suggest that YAP/TAZ operate in a developmental checkpoint regulated by signals from the actin cytoskeleton generated through indirect interactions with adherens junctions between neighboring cells and with the surrounding ECM. Signaling generated by these cytoskeletal interactions inform the cell about its cellular and tissue environment. Such signaling from the actin cytoskeleton is required for MSC differentiation because YAP/TAZ associate with and are required for activation of virtually all MSC-associated enhancers.
Results
E1AKD in adenovirus transformed cells generates typical enhancers and superenhancers that activate MSC-specific gene expression and a dramatic change in cell morphology
Adenovirus small E1A binds with high affinity to the TAZ2 domain of CBP and the closely related nuclear lysine acetyltransferase p300 (Ferreon et al. 2009; Stein et al. 1990). This is associated with a reduction of total cell H3K27ac and H3K18ac, sites acetylated primarily by CBP and p300 (Jin et al. 2011), to ∼30% the level in uninfected cells (Horwitz et al. 2008; Ferrari et al. 2014). To determine whether this H3K27/18 hypoacetylation is reversible, we used siRNA to deplete E1A from Ad5 transformed human embryonic kidney cells (HEK293) (Graham et al. 1977). siRNA knockdown works very efficiently in HEK293 cells, and, by 4 d after siRNA transfection, E1A proteins were reduced to <1% the level in untreated HEK293 cells (Fig. 1A). This did indeed reverse H3K27/18 hypoacetylation so that the amount of acetylation at these sites was restored to relatively high levels (Fig. 1B).
Figure 1.

E1A siRNA knockdown in HEK293 cells reverses oncogenic transformation-associated gene expression and morphology. (A) Expression of E1A proteins after 1–4 d of the indicated siRNA transfections or mock transfected (−) in HEK293 cells. Nuclear protein KU86 served as a loading control. (B) Levels of total H3K18 and H3K27ac in HEK293 cells following 1 d of transfection with siE1A or siCtrl RNA (−). (C) Confocal microscopy of HEK293 cells 4 d after siRNA transfection, fixed and stained with phalloidin-iFluor or DAPI. Scale bar, 20 µM. (D,E) The percentage of significant H3K27ac ChIP-seq (chromatin immunoprecipitation [ChIP] combined with high-throughput sequencing) peaks that increased (D) or decreased (E) greater than fivefold within the indicated distance to the nearest TSS. (F,G) Averages of ChIP-seq read density for H3K27ac (left) and H3K18ac (right) centered at all significant peaks of either typical enhancers (F) or within superenhancers (G) following siRNA transfections in HEK293 cells for 1 or 4 d, as indicated. (H) Venn diagram of superenhancers in HEK293 cells and E1AKD293 cells.
E1AKD caused cell cycle arrest, as expected, and a remarkable change in the morphology of HEK293 cells (Fig. 1C). Proliferating HEK293 cells, like the control siRNA transfected HEK293 cells, contain scant cytoplasm and globular nuclei. In contrast, the arrested “E1AKD293 cells” uniformly had increased cytoplasm with extensive actin stress fibers. The E1AKD293 cells were also flattened against the fibronectin-covered glass substratum, making the nuclei appear larger when viewed from the top (Fig. 1C).
H3K18/27ac ChIP-seq (chromatin immunoprecipitation [ChIP] combined with high-throughput sequencing) in these E1AKD293 cells at 1 and 4 d after siRNA transfection revealed that acetylation at promoters changed subtly (Supplemental Fig. S1A). However, by 4 d after siRNA transfection, H3K27ac increased fivefold or more at 3657 peaks, with 90% being >5 kb from a TSS, indicative of enhancers (Fig. 1D). Only 220 H3K27ac peaks showed a greater than fivefold reduction and were associated with mostly different genes (<1% overlap) than those with increased H3K27ac peaks in their associated enhancer regions. Most of these peaks (53%) were within 5 kb of a TSS (Fig. 1E), and, of these, 85% were in the interval from 1 to 5 kb downstream. The peaks where H3K27ac decreased are highly enriched for E2F motifs (P = 1 × 10−17). Acetylation at these sites likely was reduced because of the return of Rb family protein activity when E1A was removed. The resulting Rb association with E2Fs likely repressed E2F-directed H3 acetylation and increased deacetylation by Rb-associated histone deacetylases (Dick and Rubin 2013).
To determine how superenhancers in HEK293 cells respond to the loss of E1A, we used the ranking of superenhancers (ROSE) algorithm (Hnisz et al. 2013) with H3K27ac ChIP-seq data from 4 d after siRNA transfection. This identified 272 superenhancers in HEK293 transfected with control siRNA. H3K27/18ac increased within typical enhancers after 1 d of E1AKD and then remained stable, while levels in superenhancers continued to increase for 4 d (Fig. 1F,G). Examples of superenhancers gained following E1AKD include regions near the COL1A1 and FZD2 genes (Supplemental Fig. S1B,C). Eighty-one of the 272 superenhancers in HEK293 cells were lost when E1A was removed from the cells, and 477 new superenhancers were gained (Fig. 1H).
At 8 d after siRNA transfection, E1AKD repressed 2203 genes and activated 2584 genes (Fig. 2A). E1AKD-repressed genes were highly enriched for cell cycle function (Fig. 2B) and, as mentioned above, were likely repressed by reactivation of Rb family proteins when E1A was eliminated. In contrast, the genes activated following E1AKD were enriched for mesodermal cell differentiation, glycoproteins, proteins involved in focal adhesion of cells to the substratum, and ECM receptor interactions (Fig. 2C), consistent with the flattening of E1AKD cells against the substratum (Fig. 1C).
Figure 2.

Gene expression in E1AKD293 cells is most similar to normal MSCs. (A) XY scatter plot of HEK293 genes (dots) significantly changed (q < 0.05) and more than twofold activated (yellow) or repressed (green) by 8 d of E1AKD compared with siCtrl. (B) Gene ontology (DAVID)-enriched terms of genes repressed or (C) activated by E1AKD in HEK293 cells. (D) Box plots of expression levels (FPKM) of superenhancer (SE)-associated genes following the indicated siRNA transfections in HEK293 cells. (*) P < 0.001 as compared with siCtrl of the same transfection length. (E) Gene ontology (DAVID)-enriched terms of E1AKD293-activated superenhancer-associated genes. (F) Signature visualization tool (SaVanT) analysis heat map displaying Z-scores for similarity of expression values of HEK293 siRNA transfected cells to gene signatures in various cell types. The plot was split between IMGN_MLP_FL at the bottom of the left columns and MBA_dorsal_root_ganglia at the top of the right columns. Z-scores for cell types most similar to E1AKD293 were as follows: MSC, 3.9; lymph node stroma, 3.8; cardiac myocytes, 3.3; fibroblastic reticular cell, 3.3; skin fibroblasts, 3.1; smooth muscle, 2.3; and uterus corpus, 2.0.
Genes activated by E1AKD in HEK293 are repressed by E1A in normal primary human cells, while genes repressed by E1AKD in HEK293 cells are activated by E1A in normal primary human cells (Supplemental Fig. S2A). Consequently, most genes regulated by E1A in HEK293 cells are bona fide E1A-regulated genes in acutely infected primary human cells. Since genes repressed by E1A in primary cells are activated by loss of E1A from HEK293 cells, it is probably more accurate to consider these genes to be repressed by E1A in the transformed HEK293 cells and derepressed when E1A is virtually eliminated by E1A siRNA. Superenhancer-associated genes (assigned by closest TSS of a gene expressed >1 FPKM) were significantly activated after E1AKD (Fig. 2D). The gene ontologies of the activated superenhancer-associated genes are enriched for focal adhesion, ECM, actin binding, cytoskeleton, and cell–cell adherens junctions (Fig. 2E).
RNA sequencing (RNA-seq) data from 8-d E1AKD293 cells were analyzed using the signature visualization tool (SaVanT) (Lopez et al. 2017). SaVanT compares gene expression from one data set with molecular signatures from gene expression profiles of multiple human and murine cell types and tissues. HEK293 cells did not have a gene expression profile similar to any normal human cell type (Fig. 2F). However, SaVanT scored E1AKD293 cells as most similar to mesoderm-derived tissues, with the closest match to normal MSCs (Fig. 2F). HEK293 were derived by stable transformation of cultured primary human embryonic kidney cells with fragmented Ad5 DNA (Graham et al. 1977). Consequently, the cell type originally transformed was likely one of the most abundant cell types in the developing embryonic kidney, including MSCs, the most abundant (Little and McMahon 2012). Taken together, the RNA-seq data and morphological changes suggest that following loss of E1A, the E1AKD293 cells redifferentiate into cells with properties of MSCs.
E1AKD induces YAP association with chromatin-bound TEADs, chromatin remodeling, and activation of MSC-specific enhancers
We next asked how the E1AKD-established enhancers are targeted for H3K27/18ac. To determine whether the newly established enhancers following E1AKD are in an accessible or inaccessible chromatin state prior to gaining H3 acetylation, we performed ATAC-seq (assay for transposase-accessible chromatin [ATAC] using sequencing) (Buenrostro et al. 2013), which uses a bacterial transposase to insert sequencing tags into regions of accessible chromatin in isolated nuclei. E1AKD greatly increased ATAC-seq signal at E1AKD-induced enhancers. The increase occurred ∼300 bp upstream of or downstream from E1AKD-induced H3K27ac peaks (Fig. 3A). This result suggests that in response to E1AKD, new TFs associate with these regions, generating increased chromatin accessibility and stimulating H3K27/18ac by CBP/p300. To identify the TFs involved in establishing the newly formed enhancers, we performed HOMER motif analysis (Heinz et al. 2010) on E1AKD-induced ATAC peaks (greater than fivefold increase in ATAC signal following E1AKD) and observed overwhelming enrichment for TEAD family TF-binding motifs (Fig. 3B). Using ChIP-seq, TEAD1 and TEAD4 were found to be highly enriched at the E1AKD-induced ATAC peaks (Fig. 3C,D), implicating TEAD TFs in establishment of the newly formed enhancers in E1AKD293 cells.
Figure 3.

Chromatin modification and YAP association with TEAD TFs in E1AKD293 cells. (A) Average ATAC-seq signal centered at H3K27ac peaks that increased more than fivefold after 4 d of E1AKD. (B) Sequence motif analysis (HOMER) showing the most significantly enriched TF motif at all ATAC-seq peaks that increased more than fivefold following E1AKD. (C,D) Average TEAD1 (C) and TEAD4 (D) ChIP-seq signal centered at ATAC-seq peaks that increased more than fivefold following 4 d of E1AKD. (E) Confocal microscopy of 4-d siRNA transfected HEK293 cells fixed and immunostained with anti-YAP (DH81X) and stained with phalloidin-iFluor and DAPI. Scale bars, 20 µm. (F) Venn diagram displaying the number of overlapping or nonoverlapping ATAC-seq and YAP ChIP-seq peaks. (G) Average YAP ChIP-seq signal centered at ATAC-seq peaks that increased more then fivefold following 4 d of E1AKD. (H) Genome browser plots (Integrated Genome Browser) displaying the indicated ChIP-seq, ATAC-seq, and mRNA-seq signal at superenhancers (SE; gold bars) near E1AKD-activated genes IER3 and AMOTL2. (I) Heat maps representing relative ATAC-seq or ChIP-seq read density centered at every E1AKD-induced ATAC-seq peak (increased more than fivefold compared with siCtrl) from siRNA transfected HEK293 cells. Peaks are sorted by sum of ATAC-seq peak signal in siE1A. Base pair span is ±3 kb from the center of the peak.
For TEAD family TFs to activate gene expression, they require association with the closely related coactivators YAP and/or TAZ (Vassilev et al. 2001). Since YAP and TAZ activity is controlled by their nuclear versus cytoplasmic localization (Dong et al. 2007; Zhao et al. 2007), we determined whether their subcellular localization changed following E1AKD. Indeed, in control HEK293 cells, YAP/TAZ are restricted mostly to the cytoplasm (>98% of cells) and, upon E1AKD, became concentrated in the nucleus of every cell observed (Fig. 3E; Supplemental Fig. S2B). Plouffe et al. (2018) and earlier reports indicated that E1A is nuclear in a subline of HEK293 cells called 293A when the cells are subconfluent. 293A cells were selected to be flatter and more contact-inhibited than standard 293 cells (and, consequently, less transformed) because flat cells show Ad5 plaques more clearly than standard HEK293 cells that become very dense during the course of a plaque assay that requires ∼1 wk of culture of the initially infected monolayer. All of our experiments were performed with HEK293 cells obtained directly from Graham et al. (1977) and cultured in our laboratory since then. YAP is primarily cytoplasmic in our HEK293 cells, which tend to grow in clumps on a standard cell culture plate or glass coverslip (Fig. 3E). This nuclear localization of YAP was reversible. HEK293 cells were treated with E1A targeting siRNA (siE1A) for 4 d, and then the medium was replaced with medium containing either siE1A or siCtrl (negative control siRNA) for an additional 4 d. The E1AKD cells that had the siE1A replaced with siCtrl had a return of E1A protein (Supplemental Fig. S2D), proliferated, and had mostly cytoplasmic YAP (91% of cells, SD ± 2.7%) with a reduction of F-actin filaments and a smaller surface area, similar to untreated HEK293 cells (Supplemental Fig. S2C). Cells that continued incubation in medium with siE1A remained arrested, had abundant cortical actin filaments stained with phalloidin-iFluor, and continued to exhibit nuclear YAP (Supplemental Fig. S2C).
E1A also caused cytoplasmic sequestration of YAP and TAZ in cells other than HEK293. Small E1A (243 amino acids) expressed from the E1A 12S mRNA (Perricaudet et al. 1979) contains the regions of the inherently disordered small E1A protein that fold and bind tightly to the CBP/p300 TAZ2 domain (Ferreon et al. 2009). The Ad mutant dl1500 cannot express the large E1A protein required to activate transcription from the early viral promoters (Montell et al. 1984). Consequently, dl1500 expresses small E1A but only extremely low levels of other viral proteins. When human primary fetal lung fibroblasts (IMR90 cells) were infected with dl1500 at a multiplicity of infection of 10 such that virtually every cell is expressing small E1A (Horwitz et al. 2008), it prevented YAP nuclear accumulation (Supplemental Fig. S2E), leading to a decrease in total cell YAP, since cytoplasmic YAP is subjected to ubiquitin-mediated proteasomal degradation (Yu et al. 2015). Similarly, primary baby rat kidney (BRK) cells display mostly nuclear YAP, whereas BRK cells transformed with Ad2 E1A and E1B (Yew and Berk 1992) have mostly cytoplasmic YAP (Supplemental Fig. S2F). Taken together, these results suggest that E1A inactivation of YAP/TAZ by cytoplasmic sequestration is a general occurrence during adenovirus oncogenic transformation and infection.
Remarkably, YAP ChIP-seq showed that following E1AKD, YAP peaks coincided with 91% of E1AKD-induced ATAC peaks (Fig. 3F). Conversely, control HEK293 cells had much less or no detectable YAP association at these sites (Fig. 3G). E1AKD-induced superenhancers, such as those close to E1AKD-derepressed genes IER3 and AMOTL2 (Fig. 3H), had very low H3K27/18ac, ATAC signal, or YAP association in control HEK293. However, following E1AKD, ATAC peaks were generated, all of which coincided with new peaks of YAP (Fig. 3H,I). H3K27/18ac peaks flanking YAP peaks first appeared after 1 d of E1AKD and continued to increase and spread across the superenhancers following 4 d of E1AKD (Fig. 3H,I).
TEAD4 was found at many of the E1AKD-induced enhancers prior to E1AKD (Fig. 3H,I), indicating that these enhancers were marked with DNA-bound TEAD4 prior to their activation. However, H3K27/18ac and ATAC accessibility increased greatly when YAP was associated at these sites after E1AKD (Fig. 3H,I). H3K4me1 is found at both active enhancers and inactive poised enhancers (Heintzman et al. 2009; Creyghton et al. 2010). In control HEK293 cells, H3K4me1 was enriched directly over E1AKD-induced ATAC peaks, but, in E1AKD293 cells, H3K4me1 shifted to ∼200–300 bp upstream of and downstream from the center of the ATAC peak (Fig. 4A,B). This finding suggests that chromatin remodeling occurred when YAP/TAZ associated with the DNA-bound TEAD TFs, resulting in nucleosome-depleted chromatin-accessible regions. Additionally, the level of TEAD4 and H3K4me1 correlated at E1AKD-induced ATAC peaks in control HEK293 cells (Fig. 4C), consistent with their co-occupancy at individual loci. Taken together, these data suggest that in HEK293 cells, many of the E1AKD-induced enhancers are epigenetically premarked by DNA-bound TEAD4 and H3K4me1 but undergo chromatin remodeling following YAP binding to TEAD4, which induces chromatin accessibility and recruitment of additional transcriptional activators such as TEAD1.
Figure 4.

E1AKD-induced YAP association with chromatin-bound TEADs causes chromatin remodeling and activation of MSC-specific enhancers. (A) Average ChIP-seq signal at 4 d after transfection with the indicated siRNAs centered at ATAC-seq peaks that increased more than fivefold following 4 d of E1AKD. (B) Heat map of H3K4me1 ChIP-seq reads from 4-d E1AKD cells at all ATAC-seq peaks that increased more than fivefold with E1AKD (new ATAC sites). Base pair span is ±3 kb from the center of the ATAC peak. (C) Heat maps of TEAD4 and H3K4me1 ChIP-seq reads at all new ATAC sites in HEK293 cells transfected with control siRNA for 4 d. Peaks were sorted by the amount of H3K4me1 signal in siCtrl HEK293 cells. Base pair span is ±3 kb from the center of the peak. (D) Protein levels or phosphorylation status of Hippo pathway proteins from 4-d siRNA transfected HEK293 cells. KU86 served as a loading control. (E) LATS1 protein levels from HEK293 cells transfected for 3 d with either siCtrl or siE1A and then treated with 5 µM MLN4924 or DMSO control for 8 h.
YAP activation requires F-actin and coincides with reduced Hippo signaling and AMOT relocalization
To determine whether alterations in Hippo signaling account for YAP/TAZ activation in E1AKD293 cells, we assayed the levels and phosphorylation status of proteins in the Hippo pathway. Nuclear YAP and TAZ are more stable than cytoplasmic YAP/TAZ (Yu et al. 2015). Consistent with this, the shift of YAP/TAZ from the cytoplasm into the nucleus in E1AKD293 cells was associated with substantial increases in YAP and TAZ (Fig. 4D). Unexpectedly, total LATS1 protein decreased considerably in E1AKD293 cells (Fig. 4D), while there was no significant decrease in LATS1 mRNA (Supplemental Fig. S3A). This suggests posttranscriptional down-regulation of LATS1 following loss of E1A. Consistent with this, treatment with the drug MLN4924, an inhibitor of cullin neddylation required for activity of cullin-based ubiquitin ligases (Soucy et al. 2009), prevented the decrease in LATS1 (Fig. 4E). The level of LATS1 in control HEK293 cells was unaffected by MLN4924 (Fig. 4E), suggesting ubiquitin-mediated degradation of LATS1 in E1AKD293 but not in HEK293 cells. In contrast, the protein level of LATS2 was unchanged before and after E1AKD (Fig. 4D). Activation by YAP/TAZ is promoted when LATS1/2 are in their inactive hypophosphorylated state due to low activity of upstream Hippo pathway kinases (Yu et al. 2015). The level of T1079 phosphorylated activated LATS1 in E1AKD293 cells was decreased compared with control siRNA transfected HEK293 cells (Fig. 4D); however, this may have resulted from the reduction in total LATS1 and not a change in the fraction of LATS1 phosphorylated. The decrease in total LATS1 and phospho-T1079 LATS1 probably contributes to high YAP/TAZ activity in E1AKD293 cells.
Since signals from the actin cytoskeleton activate YAP/TAZ (Dupont et al. 2011) and we observed robust F-actin assembly in E1AKD293 cells, we tested whether F-actin is necessary for YAP activation by E1AKD by treating cells with latrunculin B (LatB), an inhibitor of actin polymerization. A 30-min treatment of E1AKD293 cells caused actin filament disassembly and YAP export to the cytoplasm in >90% of observed cells (Fig. 5A). Consistent with a requirement for signaling from the actin cytoskeleton for YAP activation in E1AKD293 cells, knockdown of Ras superfamily small GTPases CDC42, RHOA, and RAC1, known to stimulate actin fiber assembly and dynamics as well as myosin-generated tension on actin filaments, prevented YAP nuclear accumulation when E1A was knocked down (Supplemental Fig. S3B). To determine whether inducing F-actin formation in HEK293 cells expressing E1A (with hypoacetylation of H3K27/18) (Fig. 1B) is sufficient to force YAP into the nucleus, we transfected cells with an expression vector for constitutively active myc-tagged RhoA(Q63L), which promotes F-actin assembly from monomeric actin (Caron and Hall 1998). Transfected HEK293 cells had a significantly higher percentage of cells with mostly nuclear YAP (14%; SD ± 4.9%) than cells transfected with an empty control vector (<1%). While virtually every cell expressed the myc-tagged RhoA(Q63L), the select cells with YAP localized in the nucleus contained a stronger YAP and phalloidin signal, indicating assembly of actin filaments (Fig. 5B). These results suggest that F-actin assembly, and not simply RhoA signaling, is required for YAP nuclear import in the presence of E1A.
Figure 5.
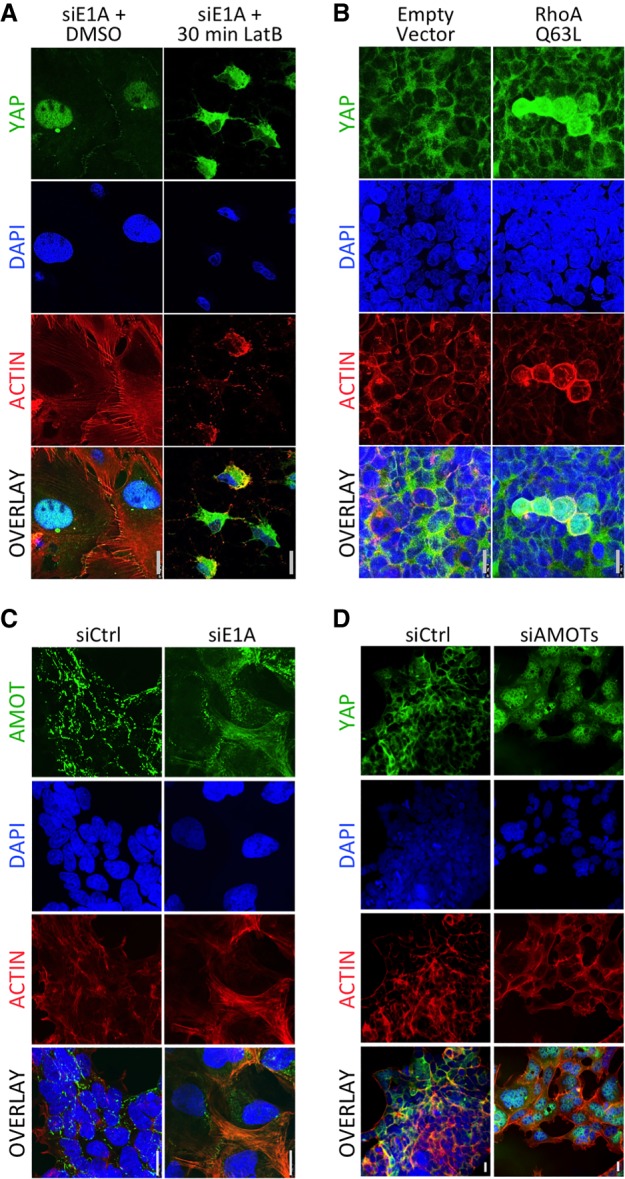
YAP nuclear localization in E1AKD293 cells requires actin polymerization and sequestration of AMOTs. (A) Four-day E1AKD293 cells treated with either DMSO or LatB for 30 min were fixed and immunostained with anti-YAP (DH81X) and stained with phalloidin-iFluor and DAPI. (B) HEK293 cells were transfected with an empty vector plasmid or plasmid expressing RhoA(Q63L) for 3 d. Cells were fixed and stained as in A. (C) Confocal microscopy of 4-d E1AKD293 and control HEK293 cells fixed and immunostained with anti-AMOT and stained with phalloidin-iFluor and DAPI. (D) HEK293 cells were transfected with control siRNA or a mixture of siRNAs for AMOT and its two human paralogs, AMOTL1 and AMOTL2. Cells were fixed and stained as in A.
During trophectoderm differentiation in the 16- to 32-cell embryo, the correlation between F-actin assembly and nuclear YAP/TAZ can be attributed to AMOT family protein binding to newly formed F-actin in the apical cortex of surface cells (Hirate et al. 2013). F-actin sequesters AMOTs, preventing them from directly inhibiting YAP/TAZ or activating LATS1/2 at adherens junctions (Hirate et al. 2013; Sasaki 2017). To test whether AMOT is relocated similarly in E1AKD293 cells, we performed AMOT immunostaining. In siCtrl HEK293 cells, AMOT was observed primarily in puncta often at cell peripheries or between cells (Fig. 5C), consistent with its association with adherens junctions, where it binds and activates LATS1/2, inhibiting YAP/TAZ. Upon E1AKD, most of the AMOT was colocalized with F-actin (Fig. 5C). Consequently, AMOT sequestration by F-actin in E1AKD293 cells likely contributes to YAP/TAZ nuclear import. Indeed, siRNA knockdown of all three AMOTs (Supplemental Fig. S3E) was sufficient to induce YAP nuclear import, with 89% (SD ± 1.7%) of cells having more nuclear than cytoplasmic YAP even though E1A was still expressed (Fig. 5D). The association of AMOTs with F-actin is inhibited by their phosphorylation by LATS1 (Chan et al. 2013). Consequently, both the reduction in active LATS1 phosphorylated at T1079 (Fig. 4D) and the great increase in polymerized actin are likely to account for the relocalization of AMOT with F-actin following E1AKD and likely contribute to YAP/TAZ nuclear accumulation.
YAP/TAZ were shown recently to be downstream effectors for alternative Wnt signaling (Park et al. 2015). To determine whether YAP/TAZ are being activated by the alternative Wnt pathway in E1AKD293 cells, we treated the cells with IWP-2, an inhibitor of Wnt processing/secretion (Chen et al. 2009). While IWP-2 prevented secretion of WNT5A/B, major activators of alternative Wnt signaling (Supplemental Fig. S3C; van Amerongen 2012), it did not prevent nuclear import of YAP (Supplemental Fig. S3D), suggesting that alternative Wnt signaling is not necessary for E1AKD-induced YAP nuclear import.
YAP/TAZ are required to establish enhancers for redifferentiating HEK293 after E1A is eliminated
To determine the extent to which E1AKD-induced gene derepression is dependent on YAP/TAZ, we used siRNA to deplete YAP and TAZ simultaneously with E1A depletion (Supplemental Fig. S4A). RNA-seq from triple knockdowns of E1A, YAP, and TAZ revealed that most of the gene derepression caused by eliminating E1A is dependent on YAP and TAZ (Fig. 6A). Out of 1890 E1AKD-derepressed genes, 1488 (∼80%; FPKM siE1A/siCtrl >2; q < 0.05) were defective (P > 0.05) for derepression in E1AKD293 cells when YAP and TAZ were also knocked down. Surprisingly, although YAP and TAZ have been considered to be largely functionally redundant (Nishioka et al. 2009; Xin et al. 2013), many of the E1AKD-derepressed genes, such as COL1A1, SERPINE1, COL3A1, CTGF, and MYOF, depend on both YAP and TAZ for derepression, indicating that YAP/TAZ have nonredundant functions required for expression of these genes (Fig. 6B, top). However, some genes rely more on YAP or TAZ for expression when E1A is removed. When YAP or TAZ were individually knocked down at the same time as E1A, a subset of genes derepressed by E1AKD was more than fivefold more dependent on TAZ than YAP (Supplemental Table S1); e.g. TGM2 (Fig. 6B, bottom left). The gene ontology of this TAZ-dependent YAP-independent subset of derepressed genes is highly enriched (P = 1.3 × 10−15) for genes involved in synthesis of molecules in the extracellular space (Supplemental Table S1; Supplemental Fig. S4B). In contrast, the subset of genes derepressed by E1AKD that were more than fivefold more dependent on YAP than TAZ was enriched for gene ontologies “positive regulation of cell proliferation” (P = 1.4 × 10−06) and “cell growth” (P = 2.7 × 10−05; e.g., FGF18) (Fig. 6B, bottom right) as well as gene ontologies related to “extracellular region” and “focal adhesions” (Supplemental Fig. S4B).
Figure 6.

YAP and TAZ are required for H3K27/18ac and redifferentiation of HEK293 cells when E1A is removed. (A) Box plots of expression (FPKM) of E1AKD-derepressed genes (more than twofold FPKM; q < 0.05) following 8 d of the indicated siRNA transfections. (E) E1A; (Y) YAP; (T) TAZ. (*) P < 1 × 10−7 as compared with siE1A. (B) Bar graphs of FPKM values of select genes that were defective for E1AKD-induced derepression when YAP and TAZ, YAP but not TAZ, or TAZ but not YAP were knocked down at the same time as E1A. (C) Total levels of H3K18ac and H3K27ac following the indicated siRNA transfections for 2 d. (YT) YAP and TAZ; (EYT) E1A, YAP, and TAZ. (D) Average ChIP-seq signal after transfection with the indicated siRNAs for 4 d centered at ATAC-seq peaks that increased more than fivefold. Base pair span is ±3 kb from the center of the peak. (E) Confocal microscopy of siRNA transfected HEK293 cells for 4 d fixed and stained for phalloidin-iFluor or DAPI. Scale bar, 20 µM.
Our observations that E1AKD in Ad transformed cells induced an increase in total H3K27/18ac and that the genomic locations for these increases overlap new YAP peaks raised the following question: How much of the increase in total cell H3K27/18ac in E1AKD293 cells results from activation by YAP and TAZ as opposed to all other activators in the cells? Western blots revealed that E1AKD-induced H3K27/18ac is entirely prevented when YAP and TAZ are additionally knocked down (Fig. 6C). H3K27/18ac ChIP-seq demonstrated that the increases in H3K18/27ac at E1AKD-induced ATAC sites were almost completely lost in E1A/YAP/TAZ triple-knockdown cells (Fig. 6D). In contrast, sites of E1AKD-induced H3 hypoacetylation (principally cell cycle gene promoters) were largely unaffected by additional knockdown of YAP and TAZ (Supplemental Fig. S5A). These data suggest that most of the H3K27/18ac and gene derepression resulting from elimination of E1A from HEK293 cells requires YAP/TAZ nuclear import to establish enhancers necessary for activating these genes.
Cohesin is necessary for many enhancer–promoter interactions (Kagey et al. 2010). Since YAP/TAZ are primarily regulating enhancers located distally from TSSs, we tested whether YAP/TAZ induce cohesin association at E1AKD-induced enhancers. The average association of cohesin subunit RAD21 with chromatin increased at E1AKD-induced ATAC sites following E1AKD but fell below siCtrl levels when YAP and TAZ were knocked down both with and without E1AKD (Supplemental Fig. S5B,D). In contrast, RAD21 association with TSSs in the same cells was only subtly affected (Supplemental Fig. S5C). These data suggest that YAP/TAZ are necessary for cohesin association at E1AKD-induced ATAC sites at enhancers such as those in the superenhancers downstream from and within AMOTL2 and upstream of COL1A1 (Supplemental Fig. S5D).
To determine the extent to which E1AKD-induced morphological changes are dependent on YAP/TAZ, we imaged DAPI- and phalloidin-stained HEK293 following siRNA knockdown. While YAP and TAZ knockdown did not have a readily apparent morphological consequence when E1A was present (Fig. 6E, si[YAP,TAZ]), YAP and TAZ knockdown prevented the induced F-actin formation and cell surface area increase that result from E1AKD (Fig. 6E, cf. siE1A and si[E1A,YAP,TAZ]). Knockdown of either YAP or TAZ plus E1A resulted in morphologies intermediate between siCtrl and E1AKD293 cells (Fig. 6E). These experiments show a codependence of the cell on YAP and TAZ for the striking change to a more differentiated morphology upon E1AKD. Consistent with this, YAP and TAZ knockdown in normal primary human bronchial/tracheal epithelial cells (HBTECs) and IMR90 cells (Supplemental Fig. S6A) caused a reduction of actin cytoskeletal fibers (Supplemental Fig. S6B). Consequently, YAP and TAZ are required for E1AKD293 cells to assume a morphology similar to MSCs and are required for maintenance of the actin cytoskeleton in primary differentiated cells. Furthermore, E1A-repressed cell type-specific genes in epithelial HBTECs, such as keratins KRT80 and KRT7 (Supplemental Fig. S6C), are repressed upon YAP and TAZ knockdown in HBTECs (Supplemental Fig. S6D). Similarly, E1A-repressed cell type-specific genes in IMR90 fibroblasts, such as collagens COL1A1 and COL3A1 (Supplemental Fig. S6C), are repressed upon YAP and TAZ knockdown in IMR90 (Supplemental Fig. S6D). Taken together, these results indicate that YAP and TAZ are required for high expression of cell type-specific genes in alternative types of primary human cells.
E1A represses principally cell type-specific genes in various cell types
To analyze E1A repression of cell type-specific genes as opposed to ubiquitously expressed genes, we performed RNA-seq on three cultured primary human cell types at 24 h after infection with the Ad5 mutant dl1500, which, as described above, expresses only the small E1A isoforms that bind the CBP/p300 TAZ2 domain (Montell and Berk 1984; Ferreon et al. 2009). Considering genes repressed to half the level in control mock-infected cells with a q-value of <0.05, 495 genes were repressed by small E1A in primary human foreskin fibroblasts (HFFs), 755 genes were repressed in primary human fetal lung fibroblasts (IMR90), and 1869 genes were repressed in primary HBTECs (Fig. 7A). Considerably more genes were repressed in HBTECs than in the IMR90 and HFFs, possibly because HBTECs are derived from the natural host tissue for Ad5. The genes repressed by E1A in the fibroblasts are expressed at higher level in fibroblasts compared with HBTECs and are enriched for fibroblast functions. In contrast, the genes repressed by E1A in HBTECs are expressed at higher levels in HBTECs than in the fibroblasts (Fig. 7B) and are enriched for gene ontologies of epithelial differentiation and development (Fig. 7C). Therefore, E1A preferentially represses different highly expressed cell type-specific genes in IMR90 and HBTECs. E1A also caused H3K27/18 hypoacetylation to a much greater extent at enhancers than at promoters in HBTECs (Fig. 7D). Since cell type-specific genes often rely on enhancers for expression (Heinz et al. 2015), E1A's preferential targeting of H3K27/18 hypoacetylation to enhancers explains why cell type-specific genes are targeted for repression and how E1A represses differentiation of multiple cell types (Frisch and Mymryk 2002). The E1A interaction with CBP/p300 is required for most E1A repression of cell type-specific genes (Ferrari et al. 2014) and is likely responsible for E1A's inhibition of YAP/TAZ. Out of 1190 derepressed genes in E1AKD293 cells that depend on YAP/TAZ for activation (by greater than twofold), 188 are also repressed by wild-type E1A in HBTECs (<0.5 mock FPKM) (Supplemental Fig. S6E) but much less so by an E1A mutant that cannot bind CBP/p300 (Supplemental Fig. S6E). These genes are enriched for focal adhesion, cytoskeleton, ECM, and Hippo pathway signaling (Supplemental Fig. S6F), all of which are functions involved in cellular differentiation and also regulate YAP/TAZ activity.
Figure 7.

E1A preferentially represses CBP/p300 acetylation at enhancers in different cell types. (A) Venn diagram representing significantly repressed genes in IMR90 (Ferrari et al. 2014), HFFs, or HBTECs (Zemke and Berk 2017) 24 h after infection with an Ad5 vector for small E1A at multiplicity of infection (MOI) 40 for IMR90 and 60 for HFFs and HBTECs. (B) Box plots of expression distributions (FPKM) in mock-infected cells of genes repressed by small E1A only in the cell types indicated at the top. (C) Gene ontology (DAVID)-enriched terms of genes uniquely repressed by small E1A in IMR90 or HBTECs. (D) Average ChIP-seq signal centered at all TSSs (top) or enhancers (H3K27ac peaks >2.5 kb from a TSS) in HBTECs 24 h after infection with an Ad5 vector for small E1A.
Discussion
Compromised cellular differentiation is generally observed as cancer progresses, contributing to invasion of surrounding tissues and metastasis. E1A inhibition of differentiation has been appreciated for decades (Webster et al. 1988) and may be a useful model for understanding molecular mechanisms regulating differentiation and causing dedifferentiation during oncogenic transformation. We found that when E1A was knocked down to <1% the level in transformed HEK293 cells (Graham et al. 1977), thousands of genes were derepressed, resulting in gene expression most similar to MSCs and slightly less so to fibroblasts, which have gene expression similar to MSCs (Fig. 2F; Driskell and Watt 2015). This result demonstrates that HEK293 cells harbor the “memory” to redifferentiate even after hundreds of generations of E1A-induced dedifferentiation. It appears that after elimination of E1A, the cells revert back to a gene expression program and morphology similar to the most abundant cell type in the originally transfected embryonic kidney culture (Graham et al. 1977), an MSC (Little and McMahon 2012) likely derived from a renal embryonic pericyte (Crisan et al. 2008).
YAP/TAZ inactivation promotes dedifferentiation of Ad transformed cells
The present study reveals that YAP and its paralog, TAZ, are inactivated by E1A via cytoplasmic retention in adenovirus-infected and transformed mammalian cells. The requirement for YAP/TAZ for maintenance of the normal morphology and actin cytoskeleton of both primary IMR90 fibroblasts and airway epithelial cells (Supplemental Fig. S6B) and for cell type-specific gene expression in these distinct cell types (Supplemental Fig. S6D) indicate that YAP/TAZ are master regulators of the differentiated state of both of these distinct cell types—one derived from the embryonic mesoderm (IMR90 fibroblasts) and the other derived from the endoderm (HBTECs). Consequently, inactivation of YAP/TAZ by E1A can explain the block to differentiation in various cell types transformed by adenovirus (Webster et al. 1988; Frisch and Mymryk 2002). This inhibition of differentiation probably requires the E1A interaction with CBP/p300 because, as discussed above (Supplemental Fig. S2A), genes derepressed in E1AKD293 cells are repressed by E1A in IMR90 fibroblasts, which have a gene expression pattern similar to MSCs (Driskell and Watt 2015). However, these genes are much less repressed by an E1A mutant that cannot bind CBP/p300 (Ferrari et al. 2014).
YAP/TAZ association with TEAD TFs establishes MSC-specific superenhancers
We observed that after release from cytoplasmic anchors by E1AKD, YAP/TAZ associated with enhancers, as reported previously (Galli et al. 2015; Stein et al. 2015). However, by comparing sites of H3K27/18ac, chromatin accessibility, and YAP association together with RNA expression in response to YAP/TAZ activation by E1AKD in HEK293 cells, we could discern that >90% of MSC-associated enhancers are bound by YAP. YAP/TAZ knockdown showed that most of the increase in transcription from these genes in response to elimination of E1A requires active YAP/TAZ (Fig. 6A). This activates MSC- and fibroblast-specific genes (Fig. 2C,E,F). Upon entry into the nucleus following E1AKD, YAP associates with DNA-bound TEAD family TFs (initially TEAD4) primarily distal to promoters and induces H3K27/18ac and chromatin opening as detected by accessibility to Tn5 transposase through ATAC-seq (Fig. 3H,I). After 1 d of E1AKD, there was a large increase in H3K27/18ac neighboring TEAD/YAP peaks, while, after 4 d, acetylation spread between closely spaced peaks, establishing superenhancers (Fig. 3H). Interestingly, TEAD4, the most highly expressed TEAD family TF in HEK293 cells, and H3K4me1 were detected at many of these YAP/TAZ-dependent enhancer sites prior to E1AKD (Figs. 3H, 4A–C). Therefore, TEAD4 and H3K4me1 mark these inactive enhancers prior to YAP association. Furthermore, the association of TEAD4 at sites of inaccessible chromatin before E1AKD suggests a possible pioneer function for TEAD4 (Zaret and Mango 2016). The gain in YAP binding and chromatin accessibility after E1AKD correlated with a shift in the peak of H3K4me1-marked nucleosomes from centered over the TEAD4 peak to adjacent sites ∼200–300 bp away, indicative of chromatin remodeling (Fig. 4A,B). We also observed YAP/TAZ-dependent cohesin association with these newly established superenhancers (Supplemental Fig. S5B,D), suggesting looping of an activated enhancer to its activated promoter.
While knockdown of E1A causes a dramatic alteration in the morphology of HEK293 cells (Fig. 1C), this is prevented when YAP and TAZ are additionally knocked down (Fig. 6E). Since YAP/TAZ are activated through sensors of the cellular physical microenvironment and control expression of genes necessary for differentiation, YAP/TAZ form a molecular link between a cell's spatial orientation within a tissue and its cellular identity. This link provides the potential for YAP/TAZ to transduce the necessary signals for a cell to undergo differentiation only after it senses a suitable physical environment (Fu et al. 2017; Sasaki 2017; Totaro et al. 2018). This provides a developmental checkpoint to prevent differentiation if a cell is not in the correct cellular and ECM environment.
Indirect regulation of YAP/TAZ nuclear import by E1A
E1A does not appear to interact with YAP or TAZ directly. We observed >100 proteins that specifically coimmunoprecipitate with E1A from infected cells; there are so many because E1A interacts with several cellular multiprotein complexes. Still, we did not detect an association between E1A and YAP or TAZ. This is consistent with the nuclear localization of E1A in HEK293 cells, while YAP is primarily cytoplasmic. Given the reliance on actin filaments for YAP activation in E1AKD293 cells, we propose that E1A inactivates YAP/TAZ indirectly by repressing genes required for assembly of the actin cytoskeleton. Previous work has reported that E1A disrupts the actin cytoskeleton (Bellett et al. 1989; Fischer and Quinlan 2000). We found that actin genes (ACTA1, ACTA2, ACTG1, ACTB, and ACTBL2) and CDC42EP3 and CDC42EP5, which stimulate actin filament assembly, are all repressed by E1A. Also, stimulation of cortical actin fiber assembly by dominant-active RhoA(Q63L) induced nuclear YAP in HEK293 cells without reducing E1A expression (Fig. 5B). Since MRTF is also regulated in response to actin fiber assembly and since knockdown of YAP plus TAZ reduces the actin cytoskeleton in primary cells (Supplemental Fig. S6B), some of the changes in gene expression observed in E1AKD293 cells may result from changes in MRTF activity as well as YAP/TAZ activity (Foster et al. 2017). However, YAP associates with TEAD sites at nearly all HEK293 cell enhancers derepressed by E1AKD (Fig. 3A,F). This repression of actin fiber assembly can explain how E1A regulates YAP/TAZ nuclear localization without interacting with them directly.
Recently, Elosegui-Artola et al. (2017) indicated that the mechanism regulating nuclear import of YAP through mechanotransduction may involve generation of force by actomyosin fibers between focal adhesions and the nucleus, causing flattening of the nucleus. They propose that this expands the cytoplasmic side of nuclear pores on the most curved portion of the flattened nuclei (Fig. 1C) and that this promotes YAP diffusion through nuclear pore complexes. Consistent with this, in E1AKD293 cells, we observed flattened nuclei that coincide with increased YAP nuclear import (Fig. 1C). Another recent report (Meng et al. 2018) ascribes regulation of YAP import by mechanotransduction to the small GTPase RAP2. At low ECM stiffness, RAP2-GTP activates LATS1/2, which phosphorylate YAP/TAZ, causing their cytoplasmic sequestration. RAP2-GTP may be very low in E1AKD293 cells because of the low level of actin filaments, resulting in very low activated phospho-LATS1 (Fig. 4D) and the resulting YAP/TAZ nuclear localization.
YAP/TAZ activity is required for most H3K27/18ac
It is remarkable that knockdown of YAP and TAZ prevented the increase in total cell H3K27/18ac in HEK293 cells when E1A was knocked down (Fig. 6C). E1A has been considered to be a direct inhibitor of p300/CBP acetyltransferase activity in part because the in vitro histone acetyltransferase activity of CBP was inhibited by recombinant E1A (Chakravarti et al. 1999). The reduction in total cell H3K27/18ac caused by E1A to ∼30% the level in control cells (Ferrari et al. 2014; Horwitz et al. 2008) also is consistent with the model of direct inhibition of CBP/p300 acetyltransferase activity by E1A. However, E1A K285 near the C terminus is acetylated by CBP (Zhang et al. 2000), so the in vitro inhibition may have been competitive rather than allosteric. Also, E1A binds to the CBP TAZ2 domain (Ferreon et al. 2009), which is far from the CBP acetyltransferase domain (Delvecchio et al. 2013). Consequently, it is unclear how the E1A–CBP TAZ2 domain interaction would inhibit CBP acetyltransferase activity. We considered the model that E1A binding to the CBP TAZ2 domain holds the E1A acetylation sites at high local concentration to the acetyltransferase catalytic domain, increasing competitive inhibition. However, mutations of K285 or all three E1A lysines to alanine or arginine had no effect on E1A's inhibition of total cell H3K27/18ac (D Gou and AJ Berk, unpubl.).
Two alternative related possible explanations for E1A-induced H3K27/18 hypoacetylation are raised by our observations: (1) Most H3K27/18ac in primary mammalian cells results from coactivator interactions with the YAP and TAZ activation domains as opposed to activation domains of all other TFs, or (2) YAP/TAZ are required to initiate differentiation programs involving additional TFs that together result in most H3K27/18ac. In either case, YAP/TAZ appear to be master regulators of MSC differentiation, since they are required for the redifferentiation of E1AKD293 cells into MSC-like cells and associate with ∼90% of the new ATAC sites and peaks of H3K27/18ac that appear when redifferentiation of HEK293 cells is induced by depletion of E1A.
Functional differences between YAP and TAZ
YAP and TAZ share 46% identical and 60% similar sequence, both interact with DNA-bound TEAD TFs through similar interfaces (Kristal Kaan et al. 2017), and both are overexpressed individually (i.e., YAP but not TAZ or TAZ but not YAP) in a variety of human cancers (Zanconato et al. 2016). However, the two proteins have distinctive features and probably activities (Wang et al. 2009). For example, both contain WW domains that mediate protein–protein interactions with LATS1/2 and AMOTs, but YAP contains two tandem WW domains, whereas TAZ has only one. An important example of the nonequivalence of YAP and TAZ was reported recently in a mouse model for basal cell carcinoma (BCC), the most common human cancer (Maglic et al. 2018). In this mouse model where BCC is initiated by abnormal Hedgehog signaling, YAP becomes overexpressed in the emerging BCC cells, but not TAZ. Importantly, YAP, but not TAZ, is required for initiation and progression of BCC in this model (Maglic et al. 2018).
Plouffe et al. (2018) reported recently on deletion of YAP, TAZ, and both genes in a subline of 293 cells (293a). The effects of these gene deletions on the response to lysophosphatidic acid (LPA) demonstrated that YAP and TAZ are not completely functionally redundant, as we observed also in response to elimination of E1A from HEK293 cells. Although far fewer genes were significantly activated greater than twofold by LPA in 293a cells (four genes) than by knockdown of E1A in HEK293 cells (2584 genes) (Fig. 2A), their results showed that LPA activation of AMOTL2 and FOSL1 was much more dependent on YAP than TAZ, as we also observed following derepression by removal of E1A (Supplemental Table S1). Moreover, far more genes (fivefold) were highly dependent on YAP and/or TAZ during redifferentiation of E1AKD293 cells (336 genes) (Supplemental Fig. S4B) than during the LPA response in 293a cells (six genes), and the extent of derepression for many genes was far greater (several hundred-fold). Consequently, the phenomena that we observed in E1AKD293 cells are very different from the LPA response in 293a cells (Plouffe et al. 2018).
The observation that genes highly dependent on YAP for their transcription in redifferentiating E1AKD293 cells and genes highly dependent on TAZ only partially overlap (Supplemental Fig. S4B) is relevant to the increased tumorigenicity of YAP compared with TAZ in the BCC model (Maglic et al. 2018). Genes strongly dependent on YAP, but not TAZ, with the gene ontology term “positive regulation of cell proliferation” include FOSL1, FGF18, and Endothelin 1, a peptide that regulates cell proliferation, survival, motility, and angiogenesis (Battistini et al. 1993). TAZ and YAP likely compete for association with DNA-bound TEAD TFs, since they bind the same surface of the TEADs. Consequently, the overexpression of YAP observed in many human cancers (Pan 2010; Zanconato et al. 2016) may contribute to tumor progression by promoting expression of these genes that stimulate cell proliferation. Increased expression of these genes is not sufficient to cause cell cycling in E1AKD293 cells, probably in part because Rb proteins become active when E1A is eliminated. However, in tumor cells with inactive RB1 (Hanahan and Weinberg 2000), overexpression of these YAP-activated genes may contribute to progression of the oncogenic phenotype.
Eliminating YAP/TAZ activity often promotes apoptosis (Plouffe et al. 2015), complicating observation of cells with inactive YAP/TAZ. However, in addition to expressing Ad5 E1A, HEK293 cells express the neighboring E1B region, and dl1500-infected primary cells express ∼20%–30% of the level in HEK293 cells (NR Zemke and AJ Berk, unpubl.). The resulting potent inhibition of apoptosis by both p53-dependent and mitochondrial-dependent mechanisms (White 2001) may have allowed us to observe E1A-induced dedifferentiation by promoting survival of the cells long enough for us to observe and analyze them.
Materials and methods
Cell culture, virus infection, RNA-seq, ChIP-seq, ATAC-seq, immunofluorescence microscopy, Western blot, and transfections were all performed using standard procedures. Detailed methods are in the Supplemental Material.
Supplementary Material
Acknowledgments
This research was supported by grants from the Professor June Lascelles Fund and the University of California at Los Angeles Jonsson Comprehensive Cancer Center. Confocal laser scanning microscopy was performed at the California NanoSystems Institute (CNSI) Advanced Light Microscopy/Spectroscopy Shared Resource Facility at University of California at Los Angeles.
Author contributions: Experiments were designed by N.R.Z., D.G., and A.J.B. and executed by N.R.Z. The manuscript was written by N.R.Z. and A.J.B.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.324814.119.
Freely available online through the Genes & Development Open Access option.
References
- Battistini B, Chailler P, D'Orléans-Juste P, Brière N, Sirois P. 1993. Growth regulatory properties of endothelins. Peptides 14: 385–399. 10.1016/0196-9781(93)90057-N [DOI] [PubMed] [Google Scholar]
- Bellett AJ, Jackson P, David ET, Bennett EJ, Cronin B. 1989. Functions of the two adenovirus early E1A proteins and their conserved domains in cell cycle alteration, actin reorganization, and gene activation in rat cells. J Virol 63: 303–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ. 2013. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods 10: 1213–1218. 10.1038/nmeth.2688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulger M, Groudine M. 2011. Functional and mechanistic diversity of distal transcription enhancers. Cell 144: 327–339. 10.1016/j.cell.2011.01.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carey M. 1998. The enhanceosome and transcriptional synergy. Cell 92: 5–8. 10.1016/S0092-8674(00)80893-4 [DOI] [PubMed] [Google Scholar]
- Caron E, Hall A. 1998. Identification of two distinct mechanisms of phagocytosis controlled by different Rho GTPases. Science 282: 1717–1721. 10.1126/science.282.5394.1717 [DOI] [PubMed] [Google Scholar]
- Chakravarti D, Ogryzko V, Kao HY, Nash A, Chen H, Nakatani Y, Evans RM. 1999. A viral mechanism for inhibition of P300 and PCAF acetyltransferase activity. Cell 96: 393–403. 10.1016/S0092-8674(00)80552-8 [DOI] [PubMed] [Google Scholar]
- Chan SW, Lim CJ, Chong YF, Pobbati AV, Huang C, Hong W. 2011. Hippo pathway-independent restriction of TAZ and YAP by angiomotin. J Biol Chem 286: 7018–7026. 10.1074/jbc.C110.212621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan SW, Lim CJ, Guo F, Tan I, Leung T, Hong W. 2013. Actin-binding and cell proliferation activities of angiomotin family members are regulated by Hippo pathway-mediated phosphorylation. J Biol Chem 288: 37296–37307. 10.1074/jbc.M113.527598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B, Dodge ME, Tang W, Lu J, Ma Z, Fan CW, Wei S, Hao W, Kilgore J, Williams NS, et al. 2009. Small molecule-mediated disruption of Wnt-dependent signaling in tissue regeneration and cancer. Nat Chem Biol 5: 100–107. 10.1038/nchembio.137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creyghton MP, Cheng AW, Welstead GG, Kooistra T, Carey BW, Steine EJ, Hanna J, Lodato MA, Frampton GM, Sharp PA, et al. 2010. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc Natl Acad Sci 107: 21931–21936. 10.1073/pnas.1016071107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crisan M, Yap S, Casteilla L, Chen CW, Corselli M, Park TS, Andriolo G, Sun B, Zheng B, Zhang L, et al. 2008. A perivascular origin for mesenchymal stem cells in multiple human organs. Cell Stem Cell 3: 301–313. 10.1016/j.stem.2008.07.003 [DOI] [PubMed] [Google Scholar]
- DeCaprio JA. 2009. How the Rb tumor suppressor structure and function was revealed by the study of Adenovirus and SV40. Virology 384: 274–284. 10.1016/j.virol.2008.12.010 [DOI] [PubMed] [Google Scholar]
- Delvecchio M, Gaucher J, Aguilar-Gurrieri C, Ortega E, Panne D. 2013. Structure of the p300 catalytic core and implications for chromatin targeting and HAT regulation. Nat Struct Mol Biol 20: 1040–1046. 10.1038/nsmb.2642 [DOI] [PubMed] [Google Scholar]
- Dick FA, Rubin SM. 2013. Molecular mechanisms underlying RB protein function. Nat Rev Mol Cell Biol 14: 297–306. 10.1038/nrm3567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong J, Feldmann G, Huang J, Wu S, Zhang N, Comerford SA, Gayyed MF, Anders RA, Maitra A, Pan D. 2007. Elucidation of a universal size-control mechanism in Drosophila and mammals. Cell 130: 1120–1133. 10.1016/j.cell.2007.07.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Driskell RR, Watt FM. 2015. Understanding fibroblast heterogeneity in the skin. Trends Cell Biol 25: 92–99. 10.1016/j.tcb.2014.10.001 [DOI] [PubMed] [Google Scholar]
- Dupont S, Morsut L, Aragona M, Enzo E, Giulitti S, Cordenonsi M, Zanconato F, Le Digabel J, Forcato M, Bicciato S, et al. 2011. Role of YAP/TAZ in mechanotransduction. Nature 474: 179–184. 10.1038/nature10137 [DOI] [PubMed] [Google Scholar]
- Elosegui-Artola A, Andreu I, Beedle AEM, Lezamiz A, Uroz M, Kosmalska AJ, Oria R, Kechagia JZ, Rico-Lastres P, Le Roux AL, et al. 2017. Force triggers YAP nuclear entry by regulating transport across nuclear pores. Cell 171: 1397–1410.e14. 10.1016/j.cell.2017.10.008 [DOI] [PubMed] [Google Scholar]
- Ferrari R, Gou D, Jawdekar G, Johnson SA, Nava M, Su T, Yousef AF, Zemke NR, Pellegrini M, Kurdistani SK, et al. 2014. Adenovirus small E1A employs the lysine acetylases p300/CBP and tumor suppressor Rb to repress select host genes and promote productive virus infection. Cell Host Microbe 16: 663–676. 10.1016/j.chom.2014.10.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreon JC, Martinez-Yamout MA, Dyson HJ, Wright PE. 2009. Structural basis for subversion of cellular control mechanisms by the adenoviral E1A oncoprotein. Proc Natl Acad Sci 106: 13260–13265. 10.1073/pnas.0906770106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer RS, Quinlan MP. 2000. While E1A can facilitate epithelial cell transformation by several dominant oncogenes, the C-terminus seems only to regulate rac and cdc42 function, but in both epithelial and fibroblastic cells. Virology 269: 404–419. 10.1006/viro.2000.0232 [DOI] [PubMed] [Google Scholar]
- Foster CT, Gualdrini F, Treisman R. 2017. Mutual dependence of the MRTF-SRF and YAP-TEAD pathways in cancer-associated fibroblasts is indirect and mediated by cytoskeletal dynamics. Genes Dev 31: 2361–2375. 10.1101/gad.304501.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisch SM, Mymryk JS. 2002. Adenovirus-5 E1A: paradox and paradigm. Nat Rev Mol Cell Biol 3: 441–452. 10.1038/nrm827 [DOI] [PubMed] [Google Scholar]
- Fu V, Plouffe SW, Guan KL. 2017. The Hippo pathway in organ development, homeostasis, and regeneration. Curr Opin Cell Biol 49: 99–107. 10.1016/j.ceb.2017.12.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galli GG, Carrara M, Yuan WC, Valdes-Quezada C, Gurung B, Pepe-Mooney B, Zhang T, Geeven G, Gray NS, de Laat W, et al. 2015. YAP drives growth by controlling transcriptional pause release from dynamic enhancers. Mol Cell 60: 328–337. 10.1016/j.molcel.2015.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham FL, Smiley J, Russell WC, Nairn R. 1977. Characteristics of a human cell line transformed by DNA from human adenovirus type 5. J Gen Virol 36: 59–74. 10.1099/0022-1317-36-1-59 [DOI] [PubMed] [Google Scholar]
- Hanahan D, Weinberg R. 2000. The hallmarks of cancer. Cell 100: 57–70. 10.1016/S0092-8674(00)81683-9 [DOI] [PubMed] [Google Scholar]
- Heintzman ND, Hon GC, Hawkins RD, Kheradpour P, Stark A, Harp LF, Ye Z, Lee LK, Stuart RK, Ching CW, et al. 2009. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature 459: 108–112. 10.1038/nature07829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK. 2010. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell 38: 576–589. 10.1016/j.molcel.2010.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Romanoski CE, Benner C, Glass CK. 2015. The selection and function of cell type-specific enhancers. Nat Rev Mol Cell Biol 16: 144–154. 10.1038/nrm3949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirate Y, Hirahara S, Inoue KI, Suzuki A, Alarcon VB, Akimoto K, Hirai T, Hara T, Adachi M, Chida K, et al. 2013. Polarity-dependent distribution of angiomotin localizes hippo signaling in preimplantation embryos. Curr Biol 23: 1181–1194. 10.1016/j.cub.2013.05.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hnisz D, Abraham BJ, Lee TI, Lau A, Saint-André V, Sigova AA, Hoke HA, Young RA. 2013. Super-enhancers in the control of cell identity and disease. Cell 155: 934–947. 10.1016/j.cell.2013.09.053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horwitz GA, Zhang K, McBrian MA, Grunstein M, Kurdistani SK, Berk AJ. 2008. Adenovirus small e1a alters global patterns of histone modification. Science 321: 1084–1085. 10.1126/science.1155544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Q, Yu LR, Wang L, Zhang Z, Kasper LH, Lee JE, Wang C, Brindle PK, Dent SYR, Ge K. 2011. Distinct roles of GCN5/PCAF-mediated H3K9ac and CBP/p300-mediated H3K18/27ac in nuclear receptor transactivation. EMBO J 30: 249–262. 10.1038/emboj.2010.318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kagey MH, Newman JJ, Bilodeau S, Zhan Y, Orlando DA, Van Berkum NL, Ebmeier CC, Goossens J, Rahl PB, Levine SS, et al. 2010. Mediator and cohesin connect gene expression and chromatin architecture. Nature 467: 430–435. 10.1038/nature09380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kristal Kaan HY, Chan SW, Tan SKJ, Guo F, Lim CJ, Hong W, Song H. 2017. Crystal structure of TAZ–TEAD complex reveals a distinct interaction mode from that of YAP–TEAD complex. Sci Rep 7: 2035 10.1038/s41598-017-02219-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine M. 2010. Transcriptional enhancers in animal development and evolution. Curr Biol 20: R754–R763. 10.1016/j.cub.2010.06.070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Little MH, McMahon AP. 2012. Mammalian kidney development: principles, progress, and projections. Cold Spring Harb Perspect Biol 4: 3 10.1101/cshperspect.a008300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez D, Montoya D, Ambrose M, Lam L, Briscoe L, Adams C, Modlin RL, Pellegrini M. 2017. SaVanT: a Web-based tool for the sample-level visualization of molecular signatures in gene expression profiles. BMC Genomics 18: 824 10.1186/s12864-017-4167-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maglic D, Schlegelmilch K, Dost AF, Panero R, Dill M, Calogero RA, Camargo FD. 2018. YAP–TEAD signaling promotes basal cell carcinoma development via a c-JUN/AP1 axis. EMBO J 37: e98642 10.15252/embj.201798642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng Z, Qiu Y, Lin KC, Kumar A, Placone JK, Fang C, Wang KC, Lu S, Pan M, Hong AW, et al. 2018. RAP2 mediates mechanoresponses of the Hippo pathway. Nature 560: 655–660. 10.1038/s41586-018-0444-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montell C, Berk AJ. 1984. Elimination of mRNA splicing by a point mutation outside the conserved GU at 5′ splice sites. Nucleic Acids Res 12: 3821–3827. 10.1093/nar/12.9.3821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montell C, Courtois G, Eng C, Berk A. 1984. Complete transformation by adenovirus 2 requires both E1A proteins. Cell 36: 951–961. 10.1016/0092-8674(84)90045-X [DOI] [PubMed] [Google Scholar]
- Nishioka N, Inoue K, Adachi K, Kiyonari H, Ota M, Ralston A, Yabuta N, Hirahara S, Stephenson RO, Ogonuki N, et al. 2009. The Hippo signaling pathway components Lats and Yap pattern Tead4 activity to distinguish mouse trophectoderm from inner cell mass. Dev Cell 16: 398–410. 10.1016/j.devcel.2009.02.003 [DOI] [PubMed] [Google Scholar]
- Pan D. 2010. The hippo signaling pathway in development and cancer. Dev Cell 19: 491–505. 10.1016/j.devcel.2010.09.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park HW, Kim YC, Yu B, Moroishi T, Mo JS, Plouffe SW, Meng Z, Lin KC, Yu FX, Alexander CM, et al. 2015. Alternative Wnt signaling activates YAP/TAZ. Cell 162: 780–794. 10.1016/j.cell.2015.07.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perricaudet M, Akusjärvi G, Virtanen A, Pettersson U. 1979. Structure of two spliced mRNAs from the transforming region of human subgroup C adenoviruses. Nature 281: 694–696. 10.1038/281694a0 [DOI] [PubMed] [Google Scholar]
- Plouffe SW, Hong AW, Guan KL. 2015. Disease implications of the Hippo/YAP pathway. Trends Mol Med 21: 212–222. 10.1016/j.molmed.2015.01.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plouffe SW, Lin KC, Moore JL, Tan FE, Ma S, Ye Z, Qiu Y, Ren B, Guan KL. 2018. The Hippo pathway effector proteins YAP and TAZ have both distinct and overlapping functions in the cell. J Biol Chem 293: 11230–11240. 10.1074/jbc.RA118.002715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki H. 2017. Roles and regulations of Hippo signaling during preimplantation mouse development. Dev Growth Differ 59: 12–20. 10.1111/dgd.12335 [DOI] [PubMed] [Google Scholar]
- Soucy TA, Smith PG, Milhollen MA, Berger AJ, Gavin JM, Adhikari S, Brownell JE, Burke KE, Cardin DP, Critchley S, et al. 2009. An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature 458: 732–736. 10.1038/nature07884 [DOI] [PubMed] [Google Scholar]
- Stein RW, Corrigan M, Yaciuk P, Whelan J, Moran E. 1990. Analysis of E1A-mediated growth regulation functions: binding of the 300-kilodalton cellular product correlates with E1A enhancer repression function and DNA synthesis-inducing activity. J Virol 64: 4421–4427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein C, Bardet AF, Roma G, Bergling S, Clay I, Ruchti A, Agarinis C, Schmelzle T, Bouwmeester T, Schübeler D, et al. 2015. YAP1 exerts its transcriptional control via TEAD-mediated activation of enhancers. PLoS Genet 11: e1005465 10.1371/journal.pgen.1005465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Totaro A, Panciera T, Piccolo S. 2018. YAP/TAZ upstream signals and downstream responses. Nat Cell Biol 20: 888–899. 10.1038/s41556-018-0142-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Amerongen R. 2012. Alternative Wnt pathways and receptors. Cold Spring Harb Perspect Biol 4: a007914 10.1101/cshperspect.a007914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varelas X. 2014. The Hippo pathway effectors TAZ and YAP in development, homeostasis and disease. Development 141: 1614–1626. 10.1242/dev.102376 [DOI] [PubMed] [Google Scholar]
- Vassilev A, Kaneko KJ, Shu H, Zhao Y, DePamphilis ML. 2001. TEAD/TEF transcription factors utilize the activation domain of YAP65, a Src/Yes-associated protein localized in the cytoplasm. Genes Dev 15: 1229–1241. 10.1101/gad.888601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K, Degerny C, Xu M, Yang X-J, Wang K, Yang X-J, Degerny C, Xu M. 2009. YAP, TAZ, and Yorkie: a conserved family of signal-responsive transcriptional coregulators in animal development and human disease. Biochem Cell Biol 87: 77–91. 10.1139/O08-114 [DOI] [PubMed] [Google Scholar]
- Wang Y, Xu X, Maglic D, Dill MT, Mojumdar K, Ng PK-S, Jeong KJ, Tsang YH, Moreno D, Bhavana VH, et al. 2018. Comprehensive molecular characterization of the Hippo signaling pathway in cancer. Cell Rep 25: 1304–1317.e5. 10.1016/j.celrep.2018.10.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster KA, Muscat GEO, Kedes L. 1988. Adenovirus E1A products suppress myogenic differentiation and inhibit transcription from muscle-specific promoters. Nature 332: 553–557. 10.1038/332553a0 [DOI] [PubMed] [Google Scholar]
- White E. 2001. Regulation of the cell cycle and apoptosis by the oncogenes of adenovirus. Oncogene 20: 7836–7846. 10.1038/sj.onc.1204861 [DOI] [PubMed] [Google Scholar]
- Whyte WA, Orlando DA, Hnisz D, Abraham BJ, Lin CY, Kagey MH, Rahl PB, Lee TI, Young RA. 2013. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell 153: 307–319. 10.1016/j.cell.2013.03.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xin M, Kim Y, Sutherland LB, Murakami M, Qi X, McAnally J, Porrello ER, Mahmoud AI, Tan W, Shelton JM, et al. 2013. Hippo pathway effector Yap promotes cardiac regeneration. Proc Natl Acad Sci 110: 13839–13844. 10.1073/pnas.1313192110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yew PR, Berk AJ. 1992. Inhibition of p53 transactivation required for transformation by adenovirus early 1B protein. Nature 357: 82–85. 10.1038/357082a0 [DOI] [PubMed] [Google Scholar]
- Yu FX, Zhao B, Guan KL. 2015. Hippo pathway in organ size control, tissue homeostasis, and cancer. Cell 163: 811–828. 10.1016/j.cell.2015.10.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanconato F, Cordenonsi M, Piccolo S. 2016. YAP/TAZ at the Roots of Cancer. Cancer Cell 29: 783–803. 10.1016/j.ccell.2016.05.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaret KS, Mango SE. 2016. Pioneer transcription factors, chromatin dynamics, and cell fate control. Curr Opin Genet Dev 37: 76–81. 10.1016/j.gde.2015.12.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zemke NR, Berk AJ. 2017. The adenovirus E1A C terminus suppresses a delayed antiviral response and modulates RAS signaling. Cell Host Microbe 22: 789–800.e5. 10.1016/j.chom.2017.11.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Yao H, Vo N, Goodman R. 2000. Acetylation of adenovirus E1A regulates binding of the transcriptional corepressor CtBP. Proc Natl Acad Sci 97: 14323–14328. 10.1073/pnas.011283598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao B, Wei X, Li W, Udan RS, Yang Q, Kim J, Xie J, Ikenoue T, Yu J, Li L, et al. 2007. Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes Dev 21: 2747–2761. 10.1101/gad.1602907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao B, Ye X, Yu J, Li L, Li W, Li S, Yu J, Lin JD, Wang CY, Chinnaiyan AM, et al. 2008. TEAD mediates YAP-dependent gene induction and growth control. Genes Dev 22: 1962–1971. 10.1101/gad.1664408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao B, Li L, Lu Q, Wang LH, Liu CY, Lei Q, Guan KL. 2011. Angiomotin is a novel Hippo pathway component that inhibits YAP oncoprotein. Genes Dev 25: 51–63. 10.1101/gad.2000111 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.