

ABSTRACT
T-cell-recruiting bispecific antibodies (T-BsAbs) have shown potent tumor killing activity in humans, but cytokine release-related toxicities have affected their clinical utility. The use of novel anti-CD3 binding domains with more favorable properties could aid in the creation of T-BsAbs with improved therapeutic windows. Using a sequence-based discovery platform, we identified new anti-CD3 antibodies from humanized rats that bind to multiple epitopes and elicit varying levels of T-cell activation. In T-BsAb format, 12 different anti-CD3 arms induce equivalent levels of tumor cell lysis by primary T-cells, but potency varies by a thousand-fold. Our lead CD3-targeting arm stimulates very low levels of cytokine release, but drives robust tumor antigen-specific killing in vitro and in a mouse xenograft model. This new CD3-targeting antibody underpins a next-generation T-BsAb platform in which potent cytotoxicity is uncoupled from high levels of cytokine release, which may lead to a wider therapeutic window in the clinic.
KEYWORDS: Bispecific antibody, T cell engager, T cells, BCMA, CD3, multiple myeloma, deep sequencing, repertoire
Introduction
In the past decade, exciting cancer treatments have emerged that leverage the potent tumor killing activity of cytotoxic T-cells, including chimeric antigen receptor T-cell (CAR-T) therapies and T-cell-engaging bispecific antibodies (T-BsAbs). CAR-T therapies, based on patient primary T-cells that are ex vivo engineered to target a specific tumor antigen and re-introduced into the patient, continue to show encouraging results but face challenges as a personalized cell-based therapy (reviewed by Pettitt et al.1). T-BsAbs are a class of T-cell-based antibody therapeutics in which one arm targets the T-cell receptor (TCR) CD3 subunit, and the other arm targets tumor cells via a tumor-associated antigen (TAA) (reviewed by Wu et al.2). One major advantage of T-BsAbs lies in their ability to elicit potent TAA-dependent tumor cell lysis by recruiting endogenous cytotoxic T-cells to the site of the tumor, thus eliminating the need to engineer and manipulate T-cells ex vivo in a patient-specific manner. 3-5 Mechanisms of T-BsAb activity are complex and may be influenced by factors such as tumor antigen density, the epitope and binding affinity of the individual targeting arms, as well as the relative affinities between the two arms. These characteristics have been shown to affect the potency, biodistribution, and specificity of T-BsAbs.6-8
While effective, first-generation T-BsAbs have encountered hurdles in the clinic related to cytokine release syndrome (CRS) and neurotoxicity.9-11 Next-generation molecules that drive effective tumor cell lysis while avoiding high levels of cytokine release may allow for wider use as single agents and in combination therapies. Previously published studies of natural T-cell activation through the interaction of the T-cell receptor and peptide MHC complex (pMHC) support the feasibility of decoupling the cytolytic activity of T-cells from high levels of cytokine release.12,13 Faroudi et al. showed that, at low levels of TCR:pMHC engagement, T-cells are able to kill target cells before stimulation of cytokine release. Therefore, with more finely tuned binding characteristics and agonist activity for the CD3-engaging arm, a T-BsAb may more closely mimic the T-cell activation induced by natural TCR:pMHC engagement.14,15
Achieving more natural T-cell engagement via T-BsAbs may be driven by development of novel CD3-binding domains. A review of first-generation of T-BsAb programs shows that nearly 75% of published CD3-engaging domains are derived from just a few hybridoma-derived antibodies, e.g., OKT3, UCHT1, TR66, that show binding affinities as low as 1nM.2 T-BsAbs using these high-affinity CD3-binding arms often show potent tumor cell killing with high levels of cytokine release. In an effort to widen the therapeutic window for the next generation of T-BsAbs, we sought to establish a platform that decouples tumor cell killing from cytokine release. Toward this goal, we discovered a novel set of anti-CD3 antibodies using next-generation sequencing (NGS)-based antibody discovery in fixed light chain humanized rats that bind to multiple epitopes on CD3 with a wide range of binding strengths and agonist activities.16 Functional evaluation in bispecific format revealed a promising new T-cell-engaging domain for the creation of T-BsAbs that elicits robust in vivo tumor cell killing and low levels of cytokine release.
Results
Discovery of novel anti-CD3 agonist monoclonal antibodies
Historically, identifying antibodies that bind to CD3 in the context of cell-surface T-cell receptors has been challenging. Traditional antibody discovery approaches, such as phage display, yeast display, and single-cell screening of primary B-cells, tend to favor high affinity binders, which complicates efforts to identify naturally occurring anti-CD3 antibodies with a range of agonist strengths.
Our team recently described a new NGS-based antibody repertoire sequencing discovery approach that was used to identify novel anti-CD3 antibodies in immunized OmniFlic rats, which are transgenic rodents expressing human fixed light chain antibodies (Figure 1(a)).16 The discovery strategy has distinct advantages for identifying agonist antibodies with broad epitope coverage and a wide variety of binding strengths and functional activities. OmniFlic animals express human IgG antibodies using a single pre-rearranged human kappa light chain transgene, and they rely on rearrangement of a transgene-based human heavy chain V-D-J gene repertoire to generate antibody diversity.17,18 Endogenous rat heavy chain, kappa and lambda loci have been knocked out.19 This approach yields very large and diverse collections of fully-human sequence-defined antibodies, and the fixed light chain format enables easy pairing with a variety of other domains to achieve bispecific binding and robust manufacturability.
Figure 1.
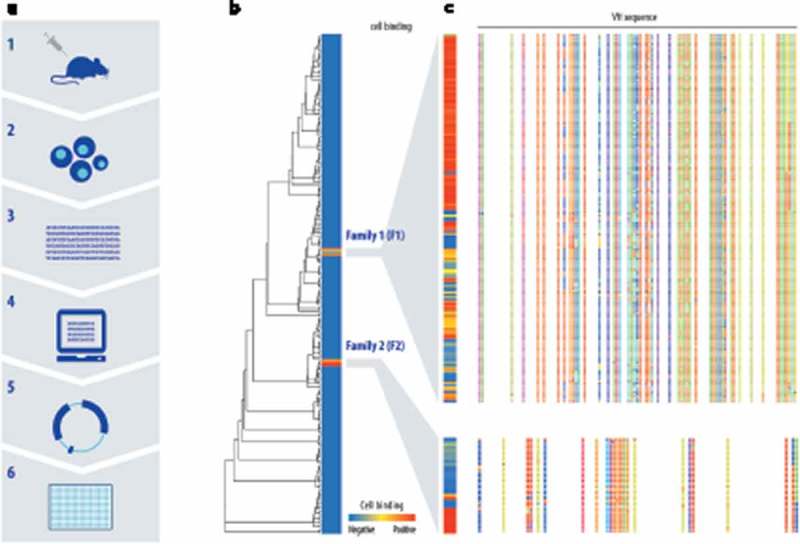
Two different CD3 cell-binding CDRH3 sequence families were identified using NGS-based discovery followed by high-throughput recombinant expression and screening. (a) The discovery workflow combines antibody repertoire deep sequencing and custom bioinformatics analysis with high-throughput gene assembly, recombinant expression and screening. OmniFlic rats express a comprehensive human VH gene repertoire with a single pre-rearranged human kappa light chain. Endogenous rat heavy chain, kappa, and lambda loci have been knocked out. (b) Based on antibody repertoire analysis, 378 heavy chain sequences were selected for expression with a fixed human kappa light chain. All fully human antibody candidates were tested by flow cytometry for the ability to bind CD3+ Jurkat cells in a primary screen, and results are shown in a heatmap in which each row is a unique heavy chain VH sequence and the degree of red indicates cell-binding strength. The dendrogram indicates the relationships between the sequences tested in the primary screen. (c) Two cell-binding VH sequence families were identified and additional family members from the repertoire were expressed and tested for cell binding in a secondary screen. Sequence variation among family members is illustrated by colored blocks, with each color representing an amino acid residue.
Applying this discovery strategy to CD3-immunized OmniFlic rats yielded >100 candidate antibody sequence families for analysis. Antibody repertoire and lineage analysis of the immunized animals yielded approximately 120 heavy chain complementarity-determining region 3 (CDR3) clonotypes (sequence families) that were strong candidates for being antigen-specific antibodies. In this context, a clonotype is defined as a group of VHs for which the CDR3 sequences show at least 80% similarity, likely representing a single original V-D-J rearrangement event.20-22
Multiple members from each clonotype family, representing 378 unique antibodies in total, were selected for high-throughput gene assembly, recombinant expression and primary functional screening. The resulting human fixed light chain antibodies (FlicAbs), in the form of transfection supernatants, were assayed for binding to human CD3δϵ protein by enzyme-linked immunosorbent assay (ELISA) and CD3-expressing Jurkat cells by flow cytometry. Approximately 20% of the candidate antibodies bound to recombinant human CD3δϵ protein, but many fewer candidates exhibited binding to CD3 in its natural context as part of the T-cell receptor on Jurkat cells (Figure 1(b)). The Jurkat-positive cell binders showed no detectable binding to CD3-negative cells. Overall, primary screening identified 11/378 novel FlicAbs with specific binding to CD3-positive Jurkat cells, representing multiple independently-isolated and naturally occurring members from two distinct CDR3 clonotype families (hereafter referred to as F1 and F2).
In pursuit of CD3-binding antibodies with a variety of agonist activities, we initiated a secondary diversity screen to survey other unique VH sequences within the two Jurkat-binding CDR3 families that contain sequence variation in CDR1, CDR2, and framework regions. Additional unique VH sequences were selected from CDR3 clonotype families F1 and F2 for further assessment. After a second round of high-throughput gene assembly and expression, 163 members from F1 and 34 members from F2 were evaluated for Jurkat binding by flow cytometry (Figure 1(c)), family members are distinguished from one another by letters appended to the end of F1 or F2). A total of 139 unique Jurkat-binding antibodies were identified from across both families, representing a wide range of cell binding strengths. This large and diverse set of novel CD3-binding antibodies enabled subsequent efforts to identify an optimal anti-CD3 binding domain for use in T-BsAb format. It is important to note that, by employing antibody repertoire analysis to identify naturally occurring sequence diversity and the associated range of binding activity, the animals’ immune systems serve to validate that the antibodies have acceptable expression levels and biophysical characteristics compatible with robust in vivo function.
Evaluation of lead anti-CD3 monoclonal antibodies
Two antibodies each from F1 and F2, representing stronger and weaker cell binders from each family, were selected for in vitro functional testing. Flow cytometry was used to evaluate relative Jurkat cell-binding strengths and ability to induce T-cell activation in samples of primary human or cynomolgus (cyno) monkey peripheral blood mononuclear cells (PBMCs) (Figure 2, Supplementary Table 1). Interestingly, maximum T-cell activation levels in human PBMCs, as measured by CD69 surface expression after 18 hours, were the same for all four tested antibodies, but significant variation in potency was observed. Cell-binding dose curves show that relative activation potency correlates with Jurkat cell-binding intensity. Subsequent studies showed that both antibodies from F1 activate primary T-cells from cynomolgus monkeys, while F2 members are not cyno-reactive, suggesting that antibodies from F1 and F2 may bind to different epitopes on CD3.
Figure 2.
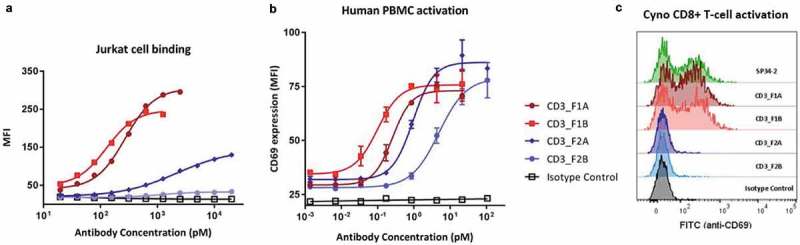
Members of both new anti-CD3 mAb families bind to and activate human T-cells. Two representative members were selected from each new anti-CD3 mAb family for purification and further functional assessment using flow cytometry. Antibodies were tested for the ability to bind human CD3-expressing Jurkat cells (a), and to activate primary T-cells in a sample of total human or cyno PBMCs as measured by T-cell surface expression of CD69 (b,c).
To further investigate CD3-binding properties of the two new antibody families, we evaluated them side-by-side with OKT3 (a well-studied anti-CD3 benchmark antibody) and a negative control antibody in a series of cell and protein binding studies (Table 1). First, binding of each antibody to human CD3-expressing Jurkat cells or cyno CD3-expressing HSC-F cells was assessed by flow cytometry, confirming that all the anti-CD3 antibodies bind to cell-surface human CD3 but only F1 antibodies are cyno-reactive (Table 1 columns 1 and 2) . Next, we used biolayer interferometry to evaluate antibody binding to recombinant human CD3ϵ, CD3δϵ or CD3γϵ (Table 1 columns 5 and 6, Supplementary Figure 1). We observed strong and roughly equivalent binding of OKT3 to CD3δϵ and CD3γϵ heterodimers. In contrast, the new F1 and F2 antibodies showed robust binding to CD3δϵ, but weaker (F1) or undetectable (F2) binding to CD3γϵ. None of the antibodies bound to CD3ϵ only.
Table 1.
CD3 binding properties vary between new anti-CD3 antibodies and OKT3.
| Cell binding by flow cytometry |
Bivalent mAb binding to immobilized CD3 protein (Octet) |
|||||
|---|---|---|---|---|---|---|
| column |
1 |
2 |
3 |
4 |
5 |
6 |
| Antibody (mAb) | Jurkat cells (human CD3+) | HSC-F cells (cyno CD3+) |
% labeled OKT3 binding to Jurkat cells pre-incubated with mAb | % labeled mAb binding to Jurkat cells pre-incubated with OKT3 | CD3δϵ KD (nM) |
CD3γϵ KD (nM) |
| OKT3 | ++ | - | 3% | 3% | 1.6 | 1.0 |
| CD3_F1F | ++ | ++ | 40% | 12% | <0.001 | 4.9 |
| CD3_F2B | + | - | 52% | 12% | 34.0 | not detectable |
| CD3_F2C | ++ | - | 43% | 12% | <0.001 | not detectable |
| Isotype ctrl | - | - | 96% | 0% | not detectable | not detectable |
CD3 binding profiles were evaluated for novel anti-CD3 mAbs CD3_F1F (F1 strong binder), CD3_F2B (F2 moderate binder), and CD3_F2C (F2 strong binder) alongside the classic anti-CD3 mAb OKT3 and an off-target isotype control mAb (anti-BCMA). Binding of individual mAbs to CD3-expressing human Jurkat cells and cyno HSC-F cells was assessed by flow cytometry. To measure the ability of mAbs to block OKT3 binding to human CD3, Jurkat cells were pre-incubated with the unlabeled mAb then treated with labeled OKT3. The ability of OKT3 to block mAb binding to human CD3 was measured in a similar manner using pre-treatment with unlabeled OKT3 followed by treatment with labeled mAb. Maximum binding (100%) was set as the mean fluorescent intensity (MFI) for cells that were not subjected to a pre-incubation step (see supplementary Table 2 for additional data). The mAb panel was also tested for CD3 subunit binding preference using biolayer interferometry with recombinantly expressed human CD3ϵ, CD3δϵ or CD3γϵ immobilized on the sensor (see Supplementary Figure 1 for details). Binding to CD3ϵ was not detectable for any of the mAbs.
Finally, we measured the ability of OKT3 and the F1 orF2 antibodies to block one another’s binding to Jurkat cells by pre-treating the cells with one unlabeled antibody, then quantifying binding of a second labeled antibody by flow cytometry (Table 1, Supplementary Table 2). We observed that, when cells are pre-saturated with F1 and F2 antibodies, subsequent OKT3 binding levels are reduced by about half (column 3). However, when OKT3 binds first, the ability of F1 and F2 antibodies to interact with Jurkat cells is almost completely abolished (column 4). Given that each T-cell receptor contains one CD3δϵ heterodimer and one CD3γϵ heterodimer,23,24 these results may be explained by preferential or exclusive binding of the F1 and F2 antibodies, respectively, to CD3δϵ. While the F1 and F2 antibodies show different cyno-reactivity properties, they compete robustly with one another for binding to Jurkat cells and may interact with overlapping epitopes (Supplementary Table 2). Taken together, these data establish that we have identified a new set of anti-CD3 antibodies characterized by distinct binding profiles.
Functional activity of novel anti-CD3 binding domains in bispecific antibody format
To facilitate characterization of the new anti-CD3 arms in bispecific format, we selected 12 different CD3-binding domains from F1 and F2, based on their wide range of binding strengths and T-cell activation profiles, for expression with a common anti-TAA arm on a silenced and stabilized human IgG4 Fc (Figure 3(a)).25-28 Previously described knobs-into-holes mutations were used to facilitate heavy chain heterodimer formation.29,30 The shared tumor-targeting arm is composed of two anti- B-cell maturation antigen (BCMA) VH binding domains arranged in tandem that binds to cell-surface BCMA with sub-nanomolar affinity. The BCMA binding arm was derived from a human heavy chain only antibody (CH1 deleted) that does not bind to kappa or lambda light chains.31,32 BCMA is a plasma cell-specific marker relevant for multiple myeloma, and is particularly well-suited for targeting by T-BsAbs because of its relatively low expression density.6,33
Figure 3.
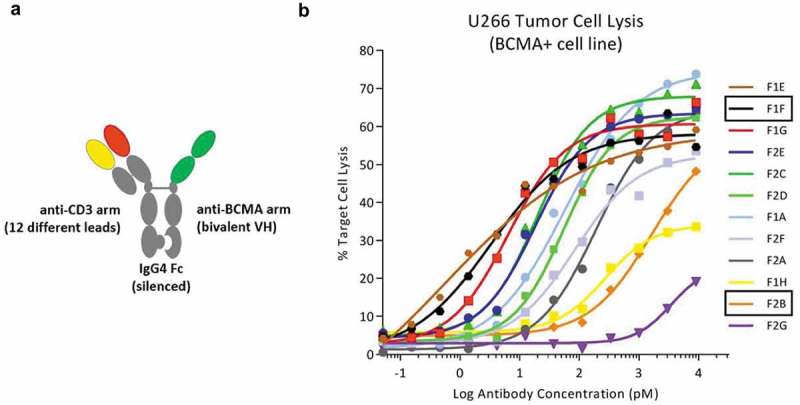
A wide range of killing potency is observed among a set of bispecific antibodies using twelve novel anti-CD3 binding arms. (a) Direct comparison of anti-CD3 functional activity was enabled by combining each of 12 new anti-CD3 arms with the same anti-BCMA arm to create bispecific proteins on a silenced Fc using a standard knob-in-hole system. The BCMA binding arm was derived from a human heavy chain only antibody (CH1 deleted) that does not bind to kappa or lambda light chains. (b) BCMAxCD3 bispecific antibodies, across a broad dose spectrum, were tested for the ability to kill BCMA+ tumor cells through redirection of activated primary T-cells. Tumor cell lysis was not observed with a BCMA- control cell line (data not shown).
To compare functional activity among the 12 different anti-CD3 arms, CD3xBCMA bispecific antibodies were expressed, purified and evaluated for antigen-dependent T-BsAb-mediated tumor cell killing activity. For in vitro cytotoxicity assays, primary human pan-T-cells were mixed with BCMA-positive (BCMA+) or BCMA-negative (BCMA-) tumor cells along with increasing amounts of T-BsAb for 18 hours. Antigen- and dose-dependent killing was observed for the BCMA+ tumor line with a striking 1,000x potency range, although all arms achieved similar levels of maximum killing (Figure 3(b), Supplementary Table 1). Molecules with the most potent activity show EC50 values similar to what has been observed for other CD3xBCMA bispecific molecules.34,35 No specific killing was observed with BCMA- tumor cells (data not shown).
In vitro characterization of lead CD3-engaging bispecific antibodies
Based on the observed range of cytotoxic potencies, two lead CD3-engaging domains were chosen for more detailed evaluation and reproducibly exhibit different functional profiles. By selecting the CD3_F1F and CD3_F2B binding arms, we sampled both ends of the potency spectrum and two different sequence families with distinct CD3 binding profiles. Each CD3-binding domain was paired with multiple tumor-targeting arms, including anti-BCMA and anti-CD19 moieties, derived from fully human heavy chain only antibodies.31,32 Functional activity was assessed by combining TAA-positive (TAA+) or TAA-negative (TAA-) tumor cell lines with primary human pan-T-cells and increasing amounts of the appropriate CD3xTAA bispecific antibody. After 18 hours of incubation, specific tumor cell lysis and cytokine release (interleukin-2 (IL-2) and interferonγ (IFNγ)) were measured for each of the tumor lines (Figure 4, Supplementary Table 1). Dose curves in the top panels of Figure 4(a and b) show that both CD3_F1F- and CD3_F2B-containing bispecific antibodies kill TAA+ tumor cells in a dose- and antigen-dependent manner, with robust maximum killing levels for TAA+ cells and little to no killing of TAA- cells. Similar antigen- and dose-dependent activity was observed in primary T-cell proliferation assays run in parallel (Supplementary Figure 2). As expected, the two different CD3-targeting arms exhibit significantly different killing potency, but achieve the same maximum percent killing levels for both tumor targets. Critically, at saturating killing doses (0.1 nM for F1F, 10 nM for F2B), the CD3_F2BxBCMA and CD3_F2BxCD19 T-BsAbs stimulate significantly less cytokine release than either of their CD3_F1F-containing counterparts. For example, at maximum killing activity doses, CD3_F1FxBCMA stimulates release of >6,000 pg/mL of IL-2 and IFNγ, while at its doses of maximum killing activity, CD3_F2BxBCMA triggers release of approximately 600 pg/mL of IL-2 and 3,500 pg/mL of IFNγ.
Figure 4.
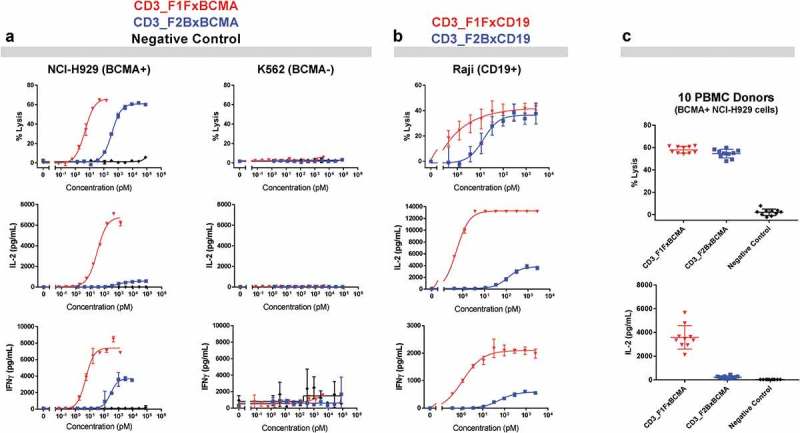
Bispecific antibody-mediated cytokine release varies significantly between CD3_F1F- and CD3_F2B-containing bispecific antibodies. (a) Levels of specific tumor cell lysis and cytokine release for IL-2 and IFNγ were measured after resting human T-cells were cultured with NCI-H929 (BCMA-positive) or K562 (BCMA-negative) cells and increasing doses of CD3xBCMA antibodies or a negative control (CD3_F1Fx[off-target arm]). (b) The same two CD3-targeting arms were combined with an anti-CD19 arm in bispecific format, and similar primary T-cell based studies were undertaken using CD19-positive Raji cells. (c) Purified pan T-cells from 10 different healthy human donors were incubated with a BCMA+ tumor cell line (NCI-H929) in the presence of plateau killing doses for CD3_F1FxBCMA and CD3_F1Fx[off-target arm] isotype control (0.1nM) as well as CD3_F2BxBCMA and CD3_F2Bx[off-target arm] isotype control (10nM).
Additional in vitro primary T-cell assays confirmed that the difference in cytokine release profiles for CD3_F1F- and CD3_F2B-containing bispecific molecules is also reproducible across independent PBMC donors. T-cell-mediated cytotoxicity assays were run with human pan-T-cells from 10 different donors, BCMA-positive cells (NCI-H929) or BCMA-negative cells (K562s), and saturating killing doses for each of the CD3xBCMA bispecific antibodies (0.1 nM for CD3_F1FxBCMA, 10 nM for CD3_F2BxBCMA). Similar antigen-dependent maximal killing levels were observed for both antibodies, but release of a representative cytokine (IL-2) was high for CD3_F1FxBCMA and minimal for CD3_F2BxBCMA (Figure 4(c)), with results being remarkably consistent across all 10 donors.
In vivo characterization of lead CD3-engaging bispecific antibodies
To confirm that the in vitro killing and cytokine release profile for CD3_F2B-containing bispecific antibodies is compatible with in vivo efficacy, a disseminated multiple myeloma xenograft mouse model was employed to compare the functional effects of CD3_F1FxBCMA and CD3_F2BxBCMA. Luciferase-expressing BCMA-positive human RPMI-8226 cells were administered to NSG mice, and the mice were subsequently treated with one dose of primary human PBMCs and three doses of CD3xBCMA bispecific antibody (Figure 5(a)). Animals treated with isotype control molecules, comprising F1F or F2B anti-CD3 arms paired with a non-targeting control arm, showed tumor progression as expected in the absence of tumor cell binding. For the CD3_F2BxBCMA molecule, robust tumor cell killing occurred at multiple dose levels with the highest tested dose (10 μg) showing near complete tumor clearance. While the strongly activating CD3_F1FxBCMA antibody also showed tumor clearance at very low to moderate doses, the tumor burden at a 10μg dose was comparable to the non-targeting negative control. Future studies will determine which mechanisms are responsible for differential function of CD3_F1F- and CD3_F2B-containing T-BsAbs at higher doses, which may include changes in biodistribution or propensity for inducing T-cell exhaustion or activation-induced cell death (AICD).
Figure 5.
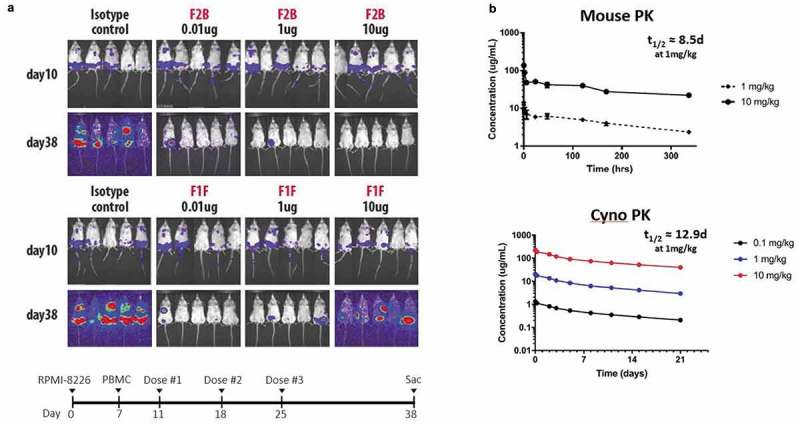
CD3_F1F and CD3_F2B bispecific antibodies kill BCMA+ tumor cells in mice and show typical PK curves. (a) NSG mice were engrafted with Luciferase-labeled RPMI-8226 BCMA+ tumor cells, and human PBMCs were injected 6 days later. Starting four days post-PBMC addition, bispecific antibodies were administered weekly for three weeks. Animals were sacrificed on day 38. Tumor burden was assessed before each antibody dose and before sacrifice using bioluminescent imaging. (b) A single IV dose of CD3_F2BxBCMA was given to BALB/c mice or cynomolgus monkeys, and serum concentrations of the bispecific molecule were measured at multiple timepoints to estimate serum half-life.
While the relatively small number of human PBMCs injected into the mice for in vivo xenograft studies precludes an accurate assessment of serum cytokine concentrations, a recent review of T-BsAbs in early clinical trials indicates that the in vitro cytokine release profile measured for CD3_F2BxBCMA is likely an accurate predictor of its effect in humans.36 In addition, our mouse xenograft results show that the CD3_F2B bispecific molecule has efficacy over a wider range of dosing than the CD3_F1F molecule, suggesting that the new CD3_F2B binding arm may exhibit a wide therapeutic window in humans.
Developability of a lead CD3-engaging bispecific antibody
Next-generation T-BsAbs created using the novel CD3-binding arms described here show favorable pharmacokinetic (PK) profiles and manufacturability characteristics. For example, CD3_F2BxBCMA has a PK profile similar to standard IgG4 antibodies in mouse and cynomolgus monkey studies with a half-life of ~12.9 days and ~8.5 days in cyno and mouse, respectively (Figure 5(b)).37 CD3_F2BxBCMA PK in Balb/c mice was linear across the dose range of 1 to 10 mg/kg following a single intravenous (IV) injection, and group mean terminal half-life (t1/2) ranged from 8.5–11.6 days. In general, CD3_F2BxBCMA PK in cynomolgus monkeys was also linear across the dose range of 0.1 to 10.0 mg/kg following a single IV injection, and t1/2 ranged from 12.9–16.1 days. Since CD3_F2BxBMCA does not cross-react with BCMA or CD3 in rodent or non-human primate species, relevant non-clinical toxicology models do not exist. The observed linear PK results were expected and are consistent with non-specific clearance mechanisms dominating PK.
The CD3_F2BxBCMA bispecific antibody shows robust expression, desirable biophysical properties and low aggregation propensity. A CD3_F2BxBCMA-expressing stable Chinese hamster ovary (CHO) cell line produces 4.7 g/L of the desired triple chain heterodimer in a bioreactor, and the purification scheme is comparable to typical antibody platform downstream processes consisting of an affinity capture step, an intermediate anion exchange step, and a final mixed mode chromatography polishing step. The bispecific antibody is stable when subjected to stresses like low pH, high pH, freeze/thaw, and elevated temperature. For example, incubation at 40°C for 3 weeks did not induce formation of higher molecular weight species, assessed by dynamic light scattering (DLS) and size-exclusion chromatography-high-performance liquid chromatography (SEC-HPLC) (Supplementary Figure 3C, 3D). To measure thermal stability, ΔHcal (kJ/mol) was measured by differential scanning calorimetry (DSC) revealing four transitions at 60.3, 64.5, 67.9 and 74.4°C, assigned to the unfolding of the VH-VH-domains of the BCMA arm, the CH2 domain, the CD3-Fab and the CH3 domain, respectively (Supplementary Figure 3A, 3B).
Discussion
T-BsAbs have shown substantial clinical efficacy by activating endogenous T-cells in a tumor antigen-dependent manner. However, the clinical utility of first-generation T-BsAbs has been tempered by narrow therapeutic windows due to dose-limiting toxicities. The therapeutic profile of a T-BsAb is highly dependent on its molecular format and the functional characteristics of each binding arm (anti-CD3 and anti-TAA). Antigen density on tumor cells and relative affinities of the binding arms of the T-BsAb are significant determinants of biodistribution, degree of T-cell activation or exhaustion, and stimulation of cytokine release.6,7 While clinicians have effective tools for managing cytokine release-related toxicities, there is hope that next-generation T-BsAbs with wider therapeutic windows may be used as single agents to address a broad range of tumor types and in combination with other approved therapies such as checkpoint inhibitors.
We developed a novel CD3-engaging antibody that serves as the foundation for a robust next-generation bispecific T-cell redirection platform. A collection of new anti-CD3 antibodies was discovered using antibody repertoire sequencing in humanized fixed light chain rats to identify naturally occurring, fully-human CD3-engaging antibodies that bind to multiple epitopes with varying affinities. By screening the various T-cell-engaging arms in bispecific antibody format, we successfully created novel T-BsAbs with low levels of cytokine release that maintain effective tumor lysis activity in vitro and in a mouse xenograft model.
Fundamental questions remain, however, about the importance of cytokine abundance in the context of effective immunotherapy, and whether the cytolytic activity of activated T-cells can be separated from cytokine release in vivo. Previous studies that quantitated native T-cell receptor engagement with MHC-displayed peptides (TCR:pMHC) have shown that two discrete thresholds exist for T-cell activation based on the number of TCR:pMHC complexes formed.12 The formation of two TCR:pMHC complexes between a T-cell and an antigen-presenting cell is sufficient to trigger T-cell-mediated cell lysis, whereas 10 TCR:pMHC complexes are required for the formation of a full immune synapse and the secretion of cytokines. Therefore, it may be possible to develop new T-BsAbs that more closely mimic the natural TCR:pMHC interaction and enable tumor cell lysis without high levels of cytokine secretion by crossing the first threshold and avoiding the second threshold.
Exact mechanisms underlying the dual thresholds of T-cell activation, by which T-cells can lyse target cells without high levels of cytokine release, are yet to be determined. The phenomenon may be related to the separate but intertwined phosphorylation signaling cascades originating at the intracellular side of the T-cell receptor that ultimately regulate gene expression and release of cytokines, as well as lytic effector molecules such as granzyme and perforin.38,39 Both epitope and affinity of CD3 agonist antibodies likely play roles in T-cell activation, so the apparent preference of our CD3_F2B antibody for CD3δϵ over CD3γϵ is intriguing, especially in light of OKT3 and our CD3_F1F antibodies showing measurable binding to both CD3δϵ and CD3γϵ heterodimers. Work by others in the field has shown that anti-CD3 antibodies induce a conformational change in the intracellular immunoreceptor tyrosine-based activation motif (ITAM) domains of the CD3 subunits.40 By binding a unique epitope exclusively on CD3δϵ with low affinity, the CD3_F2B antibody may stimulate a unique combination of phosphorylation events on the CD3 ITAM domains, which may in turn lead to differential activation of signaling cascades that regulate the expression levels of lytic effector molecules and secreted cytokines.
The differential behavior of CD3_F1F- versus CD3_F2B-containing BsAbs in the mouse xenograft model described here is also intriguing, with the more strongly activating CD3_F1FxBCMA molecule apparently losing efficacy at high doses. This loss of in vivo efficacy with strong T-cell engagement could be due to T-cell exhaustion or AICD. Alternatively, the relative binding strengths of the CD3-targeting and tumor-targeting arms of the different T-BsAbs may affect biodistribution patterns in the mouse model with subsequent effects on tumor clearance. Recent work by Mandikian et al. in human CD3ϵ transgenic mice has shown that while lower affinity CD3-containing BsAbs show robust tumor xenograft targeting via an anti-HER2 arm, high affinity CD3-containing BsAbs with the same anti-tumor arm exhibit increased localization to spleen and lymph nodes.7 It is possible that T-BsAb binding to T-cells before tumor cells, based a high-affinity anti-CD3 arm, will have significant consequences for in vivo functional activity. In contrast, a T-BsAb that binds to tumor cells before engaging T-cells, using a high affinity anti-tumor arm combined with a lower affinity CD3-targeting arm, may more closely mimic the binding and activation of a T-cell through the TCR.
Additional studies are underway to understand the relative contributions of epitope and affinity of CD3-targeting arms on in vitro killing and cytokine release levels, as well as in vivo mechanisms influencing the range of effective doses. Ultimately, clinical studies will be required to fully characterize the therapeutic window of our CD3_F2B-based bispecific platform, and multiple programs targeting both liquid and solid tumors are progressing towards the clinic.
Materials and methods
Immunization
Twelve OmniFlic rats (Ligand Pharmaceuticals) were immunized with CD3δϵ using DNA and cells (Antibody Solutions, Sunnyvale, CA; Aldevron/MFD, Freiburg, Germany). Six animals were primed with a mixture of C6 rat glioma cells expressing either human or cyno CD3δϵ, boosted with viral-based DNA immunization alternating between human and cyno CD3δϵ expression vectors, and the final boost used a mixture of C6 cells expressing either human or cyno CD3δϵ. Each animal received a total of 15 injections spaced 3–4 days apart, and terminal harvest was completed on day 44. For an alternate DNA immunization protocol, 6 animals were immunized using a combination of expression vectors containing CD3δ and CD3ϵ cDNAs, with vectors coated onto gold particles and delivered subcutaneously with a gene gun. Animals were immunized weekly for twelve weeks (prime and boosts) with terminal harvest at 13 weeks.
Variable region amplification, sequencing, and clonotype analysis
Methods for OmniFlic rat variable region amplification and sequencing have been described previously and are reviewed briefly.16 After completion of immunizations, lymphocytes from relevant draining lymph nodes were harvested, washed, pelleted and frozen. Total RNA was harvested from each cell pellet, then first strand cDNA synthesis and 5ʹRACE by PCR amplification of the full Ig heavy chain regions was performed according to previously published protocols.41,42The resulting PCR products of ~500 base pairs were isolated by gel extraction. Samples were multiplexed on a single next-generation sequencing run by adding index labels to each sample by primer extension.43 The resulting indexed samples were pooled to create a library that was sequenced using the Illumina MiSeq platform with 2 × 300 paired-end reads.
Each sample was covered by ~100,000 paired reads on average, and only those sequences that showed permissive alignment of at least 20 nucleotides to a human Ig locus were kept. Forward and reverse reads were paired when possible, and merged reads covering entire VH regions were translated into open reading frames. Framework regions and CDRs were determined using IGBLAST (https://www.ncbi.nlm.nih.gov/igblast/), then agglomerative clustering was used to cluster the full set of CDR3 protein sequences for each sample at an 80% similarity threshold, and the total number of reads in each cluster was recorded for clonotypes represented by five or more paired sequence reads. A clonotype is defined as a group of CDR3 protein sequences clustered at 80% similarity.21,22 Polarization of CDR3 clonotypes was measured by calculating the percentage of total reads in a sample that were contained in each CDR3 clonotype, then the clonotypes were ranked by abundance for each sample and those most highly represented were prioritized for functional screening.20
High-throughput recombinant antibody construction and expression of FlicAbs for primary screening
A total of 378 heavy chain variable regions were individually cloned to encompass the diversity of highly represented clonotypes in each animal, and 163 additional variable regions were subsequently cloned for a diversity screen from the two lead families. Each heavy chain variable region was cloned into an expression vector containing a leader peptide sequence and the human IgG1 Fc region, followed by validation using Sanger sequencing as described previously.42 A previously created kappa light chain expression vector contains the germline sequence of the pre-rearranged kappa light chain from the OmniFlic animal.16 Protocols for small-scale, primary screen expression have been described previously.16,42 Briefly, each expression vector was individually transformed into chemically competent E. coli growing in LB culture media, and plasmids were purified in 96-well format. Each heavy:light chain vector mix (1:1) was transfected into 293 cells in 96-well format and, after allowing for expression cell culture, supernatant was harvested and clarified by centrifugation.
Primary screening for antigen-specific binding from FlicAb supernatants
High-throughput ELISA assays were used to assay FlicAb-containing supernatants for detectable binding to recombinantly expressed CD3δϵ protein (Creative BioMart #CD3E&CD3D-219H) as described previously.16,42 Briefly, 96-well plates were coated with recombinant CD3δϵ protein in coating buffer (ThermoFisher #28382) and incubated overnight at 4°C, followed by a 1-hour incubation in blocking buffer (1% dry milk powder in 20 mM Tris, 150 mM NaCl, 0.5% Tween-20) at room temperature. After blocking, supernatants were diluted 1:100 in blocking buffer then added to the antigen-coated wells. Finally, a horseradish peroxidase-labeled secondary antibody (ThermoFisher #31413) was added, followed by an appropriate chemiluminescent substrate (ThermoFisher #37069), and luminescence quantified using a plate luminometer (SpectraMax i3X, Molecular Devices).
High-throughput flow cytometry was used to detect binding of FlicAbs in supernatant to Jurkat cells grown according to manufacturer’s instructions (ATCC TIB-152). Pelleted Jurkat cells were resuspended in flow buffer (1x phosphate-buffered saline (PBS), 1% bovine serum albumin (BSA), 0.1% NaN3) at 1 × 106 cells/mL. Antibody-containing supernatants were diluted 1:5 in flow buffer. One hundred μL of cells plus 25 μL of diluted antibody supernatant were incubated in each well in 96-well format. After washing with flow buffer, 50 μL of a phycoerythrin (PE)-conjugated secondary antibody (25 ug/mL Southern Biotech #2042–09) was added for detection and incubated at 4°C. After two wash steps, the cells were resuspended in flow buffer and analyzed using a Guava easyCyte 8HT system.
Construction, expression, and purification of lead FlicAbs on a silenced Fc
Each anti-CD3 VH of interest was subcloned onto a silenced and stabilized human IgG4 Fc.25-28 The new heavy expression vectors were co-transfected into 50 mL Expi-CHO cells with the common fixed light chain expression vector according to manufacturer’s instructions (ThermoFisher #A29133, high titer protocol). Clarified expression supernatant was harvested, the FlicAb molecules were purified using protein A, and subsequent SEC assessment showed high purity and lack of aggregation (<1%).
Cell binding dose curves
All washes and dilutions of cells, antibodies, and reagents were performed using flow buffer (1x PBS, 1% BSA, 0.1% NaN3). Human Jurkat cells or cyno HSC-F cells (NHP Reagent Resource) were pelleted and resuspended at 500,000 cells/mL in flow buffer. Then, 50 μL of cells were combined with 50 μL of test antibody and incubated on ice, followed by two wash steps using flow buffer. For in-house generated FlicAbs, 50 μL of PE-conjugated goat anti-human IgG secondary antibody, diluted to 1 ug/mL, were added to the cell mix and incubated on ice. The benchmark OKT3 mAb (BioLegend #317308) was directly conjugated to PE and required no secondary antibody. After the final staining step, cells were washed twice, resuspended in 200 uL of flow buffer, and analyzed on a Guava easyCyte 8HT system. EC90, EC50, and EC30 was calculated for each antibody using GraphPad Prism software v7 with 4PL nonlinear regression.
PBMC extraction, isolation and cryopreservation from LRS filters
PBMCs from 10 healthy, de-identified donors were isolated from LRS filters purchased from the Stanford Blood Center (Palo Alto, CA).44 Blood products were collected by flushing each LRS filter with wash buffer (RPMI-1640 + 2mM EDTA), then PBMCs were collected by Ficoll-Paque gradient centrifugation. PBMCs were washed three times with wash buffer and pelleted at 200xg for 10 minutes. The cell pellet was resuspended in 1x sterile RBC lysis buffer (BioLegend #420301), incubated for five minutes, then the reaction was quenched using a large volume of wash buffer. The remaining cells were pelleted by centrifugation at 200xg for 10 minutes, and a wash step was repeated until the supernatant was no longer cloudy (resuspension in wash buffer then centrifugation). The final cell pellet was resuspended in cryopreservation media (BioLife Solutions #210102) to a concentration of 20 × 106 cells/mL and frozen slowly in an isopropanol bath to −80°C before being transferred to vapor-phase liquid nitrogen storage.
PBMC thawing and preparation of pan-T-cells
PBMC vials were removed from liquid nitrogen storage, placed in a 37°C water bath until fully thawed but still cold, and transferred to a 50 mL conical tube with 19 mL wash buffer (RPMI-1640 + 10% fetal bovine serum (FBS)). Cells were centrifuged at 300xg for 10 minutes, then wash supernatant was carefully aspirated leaving ~1 mL of media behind. Additional wash buffer was added to bring cells to the desired concentration. T-cells were enriched from thawed PBMCs by negative selection using a bead-based pan-T-cell isolation kit (Miltenyi Biotec, Cat# 130–096-535). Purified human pan-T-cells were counted and adjusted with assay buffer (RPMI-1640 + 10% FBS) to 2x the desired final concentration (0.5x106 cells/mL).
PBMC activation and assessment of CD69 expression levels
PBMCs from a single donor were activated by positive control mAbs (OKT3: BioLegend 317304, SP34-2: BD Biosciences #551916) or anti-CD3 FlicAbs in 200 uL reactions in 96-well format. Each reaction was set up in complete culture media (RPMI-1640, 10% FBS, Penicillin-Streptomycin) to include 100,000 PBMCs, 0.5μg/mL CD28.2 anti-CD28 mAb (BioLegend #302914), 2 μg/mL goat F(ab’)2 anti-mouse or anti-human IgG Fc (Abcam #ab98644, ab98526), and antibody at the desired final concentration. Antibodies were diluted in complete culture media to create 5-fold 11-point dilution dose response curves from 2 μg/mL to 1 pg/mL. The complete reaction was incubated for 18 hours at 37°C, then cells were collected by centrifugation (500xg for 5 minutes) and stained to measure cell-surface expression of CD69. Activated PBMC pellets were resuspended in 95 μL fluorescence-activated cell sorting (FACS) buffer (1x PBS, 1% BSA, 0.1% NaN3) and 5 μL of FITC-labeled CD69 antibody (BioLegend FN50, 100 ug/mL), followed by a 30-minute incubation on ice. Labeled cells were washed twice in fresh FACS buffer by pelleting (500xg for 5 minutes) and resuspension, and final cell pellets were resuspended in FACS buffer to 0.5 × 106 cells/mL. Staining was assessed by flow cytometry using a Guava easyCyte 8HT system, and EC50s were calculated from average mean fluorescence intensities (MFIs) for each antibody using GraphPad Prism software v7 with 4PL nonlinear regression. Assessment of cyno T-cell activation followed the same protocol except primary cyno PBMCs were used (Primate Biologicals #CM-PBMC), a single mAb dose (1 ug/mL) was tested, and staining included an allophycocyanin (APC)-labeled anti-human CD8a antibody (BioLegend #301049).
Cell binding competition assay
Jurkat cells were stained with serially diluted Ax488-labeled mAbs (FlicAbs or OKT3) and relevant binding concentrations were determined (EC30 and EC90). Each unlabeled antibody was pre-incubated for 15 minutes with Jurkat cells at 4x the appropriate EC90 concentration. Then, each Ax488-labeled mAb was added at its Jurkat-binding EC30 concentration and incubated for another 15 minutes. Stained cells were washed, analyzed by flow cytometry using a BD FACS Celesta, and Ax-488 MFI was determined for each sample. Maximum binding (100%) was set as the MFI for cells that were not subjected to a pre-incubation step.
Recombinant protein binding by biolayer interferometry (Octet)
Human CD3δϵ and CD3γϵ heterodimers were expressed in ExpiCHO cells (ThermoFisher). Plasmids encoding CD3δ or CD3γ extracellular domains fused to mouse IgG1 Fc domains were co-transfected with the CD3ϵ mouse IgG1 Fc-fusion construct to express the respective heterodimer. C-terminal polyhistidine (His) tags on CD3δ and CD3γ were used for binding and imidazole-gradient elution of heterodimers from Ni-Sepharose XL (GE Healthcare). His-tagged human CD3ϵ was purchased from Creative Biomart (#CD3E-213H). Antigen-antibody binding kinetics analysis was performed on the Octet QK-384 (ForteBio). Briefly, HIS1K sensors were used to immobilize His-tagged CD3δϵ and CD3γϵ molecules. For interactions with CD3ϵ, the CD3ϵ antigen was in solution and the antibodies were immobilized using anti-human IgG Fc capture sensors (new antibodies on human Fc) or using anti-mouse IgG Fc capture sensors (OKT3 on mouse Fc). After baseline readings, sensors were dipped into antibody solutions (7-points, 2-fold dilution series). Association and dissociation were measured for 180 and 240 seconds, respectively. Data analysis was performed with Octet Data Analysis v11.0 HT (ForteBio), using a standard 1:1 binding model. Note that bivalent antibodies bind to the immobilized proteins with avidity, preventing the measurement of true kinetic rates.
Bispecific antibody construction, expression, and purification
Each anti-CD3 VH was subcloned onto a silenced and stabilized human IgG4 Fc containing a “knob” mutation.25-30 For TAA arms, anti-BCMA and anti-CD19 UniAb arms were created by cloning the appropriate UniAb VH domain(s) onto a CH1-deleted silenced and stabilized human IgG4 Fc with hole mutations. Expression constructs were combined to create each 3 chain anti-CD3xTAA bispecific molecule: 1) Anti-CD3 heavy chain with knob; 2) Common kappa light chain, which associates with only the anti-CD3 heavy chain; and 3) Anti-TAA heavy chain with hole and CH1 deleted, which does not associate with light chain. Expression vectors were co-transfected into Expi-CHO cells according to manufacturer’s instructions (ThermoFisher #A29133, high titer protocol). Clarified expression supernatant was harvested, and the bispecific molecules were purified in a two-step chromatography process using Capture Select CH1-XL (ThermoFisher) and Mono S cation exchange (GE Healthcare) columns. SEC assessment showed high purity and aggregation levels <1%.
CD3xBCMA molecules: Bispecific antibody mediated lysis of tumor cell lines and cytokine production by T-cells from a single donor in vitro
Purified pan-T-cells from a single donor were incubated with BCMA-positive (BCMA+) NCI-H929 cells and BCMA-negative (BCMA-) K562 cells (ATCC CRL-9068, CRL-3343) in the presence of varying concentrations of CD3_F1FxBCMA, CD3_F2BxBCMA or CD3_F1Fx[non-targeting control] (negative control). For labeling, tumor cells were resuspended to 1 × 106 cells/mL in 1 µM DiR (ThermoFisher # D12731 in PBS) and incubated at 37°C for 20 minutes. The reaction was quenched by adding 5x volume of assay buffer (RPMI-1640 + 10% FBS + Penicillin-Streptomycin), then DiR-labeled tumor cells were pelleted and resuspended in assay buffer at 4x the desired final concentration. Each bispecific antibody was tested in an 11-point, 3-fold titrated dose curve (n = 2 per dose point). The highest doses tested were 500 ng/mL for CD3_F1FxBCMA and 10ug/mL for CD3_F2BxBCMA and CD3_F1Fx[non-targeting control]. All dilutions of T-cells, tumor cells, and antibodies were performed in assay buffer (RPMI-1640 + 10% FBS + Penicillin-Streptomycin).
Each 96-well format biological assay was conducted in 200 μL total volume and included 100,000 pan-T-cells, 10,000 DiR-labeled tumor cells, and bispecific antibody. After an 18-hour incubation at 37°C, cells from the reactions were pelleted, and 100 µL/well of supernatant was harvested for quantification of cytokines. Each cell pellet was resuspended in 95 µL of FACS buffer (PBS + 1% BSA + 0.1% NaN3) and 5 µL of 7-AAD Viability Staining Solution (BioLegend, Cat# 420404). Cells were incubated for 10 minutes on ice, then 100 µL of FACS buffer was added to each well. Using a Guava easyCyte 8HT System, 1,000 DiR-labeled tumor cell events were collected from each sample, and the percentage of dead tumor cells was indicated by the percentage of tumor cells that were positive for 7-AAD staining. The percentage of tumor cell lysis from the no antibody control wells (T-cells + tumor) was used to set background lysis. Quantitation of IL-2 and IFNγ cytokines from the reaction supernatant was performed by ELISA according to the kit manufacturer’s protocols using harvested supernatants diluted 1:5 in 1x assay diluent (BioLegend #s 431806, 430106). Absorbance at 450 nm was read on a SpectraMax i3x multimode reader (Molecular Devices). A custom standard curve was generated for each kit and fitted to a 5PL alternative curve; curve fitting and data analysis was performed on SoftMax Pro 6.5 software (Molecular Devices).
CD3xBCMA molecules: bispecific antibody mediated T-cell proliferation from a single donor in vitro
Purified resting pan-T-cells from a single donor were loaded with carboxyfluorescein succinimidyl ester (CFSE) dye (ThermoFisher #C34554) by combining T-cells and dye in PBS at final concentrations of 1 × 107 cells/mL and 2 uM, respectively. After 5 minutes of incubation at room temperature the reaction was quenched by the addition of a 5x volume of 1xPBS + 10% FBS, then cells were pelleted and resuspended in assay buffer (RPMI-1640 + 10% FBS + Penicillin-Streptomycin). Tumor cells, BCMA-expressing NCI-H929 cells or BCMA-negative K562 cells, were counted, pelleted, washed and resuspended in assay buffer as described previously. Each 96-well format biological assay was conducted in 200 uL total volume of assay buffer and included 100,000 CFSE-labeled T-cells, 10,000 tumor cells, and varying concentrations of test article. After incubation for 5 days at 37°C, cell mixtures were pelleted and stained with an APC-conjugated anti-CD3 antibody (clone SP34-2, BD Biosciences #551916) and 7-AAD viability dye (Biolegend #420404) according to manufacturer’s instructions. Viable single T-cells were identified as APC-positive and 7-AAD negative by flow cytometry on the Guava easyCyte 8HT system. Percent proliferation was calculated as (100% – [% of cells in well that have not divided]), with non-dividing cell CFSE levels established using T-cells from wells that received no tumor cells and no test article. Using GraphPad Prism 7.0, percent proliferation was plotted against log(concentration) of test article and fitted to a 4PL sigmoidal curve.
Bispecific antibody mediated lysis of BCMA+ tumor cells and cytokine production by a panel of healthy human donor T-cells in vitro
Experimental set-up and read-outs outlined above for a single donor were used to measure tumor cell lysis and cytokine release for 10 independent donor T-cell samples, NCI-H929 cells, and CD3xBCMA bispecific molecules. A single dose sufficient for inducing maximum lysis was used for each test article: 0.1 nM CD3_F1FxBCMA and CD3_F1Fx[off-target] isotype control, 10 nM for CD3_F2BxBCMA and CD3_F2Bx[off-target] isotype control.
CD3xCD19 molecules: bispecific antibody mediated lysis of tumor cell lines and cytokine production by T-cells from a single donor in vitro
For lysis and cytokine assays, purified pan-T-cells from a single donor were incubated with CD19+ Raji cells or CD19- K562 cells (ATCC CCL-86, CCL-243) in the presence of varying concentrations of CD3_F1FxCD19 or CD3_F2BxCD19 (iQ Biosciences, Berkeley, CA). For assays measuring cytotoxicity, 100 IU/ml of IL-2 (Miltenyi Biotec #130–097-748) was supplemented in the tissue culture medium during the co-culture assay. Target cell lines were fluorescently labeled with 5 μM Calcein AM (Biolegend #425201) and incubated in with 2.5 mM probenecid (Sigma #P8761). Each biological assay was set up in 96-well format and included 10,000 target cells, 100,000 effector cells, and varying concentrations of antibody (serial semi-log dilution from a top concentration of 300 ng/ml for 10 points in triplicate). After 6 hours of incubation at 37°C, 60 μl of cell-free supernatant was transferred to a black 96-well clear-bottom plate (Sigma #CLS3614), then fluorescence was measured by plate reader (Perkin Elmer EnSpire) with excitation at 485 nm and emission at 535 nm for sample relative fluorescence units. Data was analyzed using Excel and GraphPad software to generate dose response curves by normalization where 1% saponin treatment values were used to determine maximal lysis. Medium-only condition values were used as spontaneous release values. For assays measuring cytokine expression, the biological assays were set up similarly, but T-cells were not treated with exogenous cytokines, and assays were incubated for 24 hours at 37°C. Following incubation, supernatants were harvested and cytokines measured by ELISA (BioLegend #740724).
Mouse xenograft efficacy study
Each immune-compromised NSG mouse (7–9 week-old females from Jackson Labs, Bar Harbor, ME) was injected IV with 1 × 106 luciferase-expressing BCMA-positive RPMI-8226 cells (a kind gift from Dr. Diego Acosta-Alvear at University of California, San Francisco (UCSF)). On day seven, each mouse was adoptively transferred IV with 1 × 107 human PBMCs from a single donor. On day 10, mice were randomized into 5-mouse treatment and control arms following initial bioluminescent imaging (BLI); treatment was initiated on day 10 and continued weekly for 3 weeks. The RPMI-8226 and PBMC engrafted mice were treated weekly with 10–10,000ng/animal of CD3_F1FxBCMA or CD3_F2BxBCMA and 10,000ng/animal CD3_F1Fx[off-target] negative-control antibodies. Body weight measurement and BLI were performed weekly for four weeks post-treatment initiation; the study was terminated on day 38. The study was conducted at UCSF (San Francisco, CA).
Mouse and cyno PK evaluation
The PK of CD3_F2BxBCMA was evaluated in 18 male BALB/c mice following a single tail vein injection of 1 or 10 mg/kg (n = 3/group * 6 groups) (Aragen Biosciences, Morgan Hill, CA). Serum samples were collected for 14 days post-dose. PK of CD3_F2BxBCMA was also evaluated in nine 3–4 year-old female cynomolgus monkeys (n = 3/group) following a single IV bolus dose of 0.1, 1 or 10.2 mg/kg (SNBL USA, Everett, WA). Serum samples were collected for 21 days post-dose. Serum concentrations were determined using an antigen-specific ELISA. 2-compartmental analysis was used to determine the PK of CD3_F2BxBCMA at each dose level in each animal type.
Stability assessments for CD3_F2BxBMCA
DSC was performed on a Nano DSC system (TA Instrument). A temperature ramp of 1°C/min was performed with monitoring from 25°C to 100°C. Thermograms of the blank buffer were subtracted from each antibody prior to analysis and Tm values were calculated after deconvolution using the Nano DSC software.
DLS experiments were carried out using the UNcle instrument (Unchained Labs, Pleasanton CA). The time-dependent fluctuations in the intensity of scattered light from CD3_F2BxBCMA were analyzed by DLS to yield information about the distribution and size of particles in the samples before and after thermal stress (40 oC for 3 weeks). DLS experiments were performed at a concentration of 2 mg/ml. DLS data was collected at 25°C with a 5 min equilibration time prior to data collection. The viscosity of water was used as the sample viscosity (0.8872 cP), the refractive indices (RI) of 1.450 (protein) and 1.330 (water) were utilized, and the measurement angle was set the NIBS default of 173° backscatter. Five separate DLS runs were collected for each sample using automatic measurement duration determination and these separate runs were averaged. The resulting intensity distribution plot was evaluated to reveal the measured sizes and polydispersity index (PDI) of particles in the sample.
SEC-HPLC was performed on an UltiMate TM 3000 UHPLC (ThermoFisher) equipped with a TSK-GEL UP-SW3000 column (Tosoh Bioscience). The column was equilibrated in 100 mM sodium citrate, 500 mM NaCl, 200 mM L-arginine pH 6.2. Antibody samples were filtered prior to injection (20 μg) and eluted isocratically over 10 min at 25°C. Protein elution was detected by absorption at 280 nm.
Charge heterogeneity of CD3_F2BxBCMA before and after freeze/thaw stress was assessed by imaged capillary isoelectric focusing (icIEF) in the Maurice instrument (ProteinSimple San Jose, CA). Maurice employs a prequalified, ready-to-use cartridge with automated column conditioning and whole-column detection, a five-peptide marker system suitability sample and proprietary iCE software. Samples were run according to the manufacturer’s protocol.
To assess freeze/thaw stability, CD3_F2BxBCMA was subjected to a slow freeze/thaw study and compared to a control sample stored at 2–8°C for the duration of the study. Freeze/thaw cycles were performed in a lyophilizer at 5°C. Over the course of 2 hours, the antibody was cooled from 5°C to −20°C (0.21°C/min) followed by a 2-hour hold. The antibody was then further cooled in a second stage from −20°C to −65°C (0.38°C/min) for 2 hours followed by another 2-hour hold. Following the two-hour hold the antibody was thawed from −65°C to 5°C at a rate of 0.21°C/min. This two-stage freezing cycle was performed for a total of five freeze/thaw cycles. Testing by SEC-HPLC and icIEF was performed on the samples upon conclusion of the five freeze/thaw cycles.
Funding Statement
No funding information to disclose.
Abbreviations
- APC
allophycocyanin
- Ax488
Alexa Fluor 488
- BCMA
B-cell maturation antigen
- BLI
bioluminescent imaging
- BSA
bovine serum albumin
- CAR-T
chimeric antigen receptor T-cell
- cDNA
complementary DNA
- CDR
complementarity-determining region
- CFSE
carboxyfluorescein succinimidyl ester
- CHO
Chinese hamster ovary
- CRS
cytokine release syndrome
- cyno
cynomolgus monkey
- DLS
dynamic light scattering
- DSC
differential scanning calorimetry
- ELISA
enzyme-linked immunosorbent assay
- FACS
fluorescence-activated cell sorting
- FBS
fetal bovine serum
- FlicAb
fixed light chain antibody
- icIEF
imaged capillary isoelectric focusing
- IFNγ
interferon γ
- IL-2
interleukin-2
- ITAM
immunoreceptor tyrosine-based activation motif
- IU
international units
- IV
intravenous
- kg
kilogram
- LRS
leukocyte reduction system
- MFI
mean fluorescence intensity
- mg
milligram
- mL
milliliter
- ng
nanogram
- NGS
next-generation sequencing
- nM
nanomolar
- nm
nanometer
- PBMC
peripheral blood mononuclear cell
- PBS
phosphate-buffered saline
- PE
phycoerythrin
- pg
picogram
- PK
pharmacokinetic
- pMHC
peptide MHC complex
- RACE
rapid amplification of cDNA ends
- RBC
red blood cell
- SEC-HPLC
size-exclusion chromatography-high-performance liquid chromatography
- t1/2
half-life
- TAA
tumor-associated antigen
- T-BsAb
T-cell-engaging bispecific antibody
- TCR
T-cell receptor
- μL
microliter
- μM
micromolar
Acknowledgments
The authors wish to acknowledge Dr. Byron Hann and the staff of the UCSF Preclinical Therapeutics Core Facility, supported by NIH award P30 CA082103, for their contributions to the mouse xenograft study. We also wish to thank Dr. Diego Acosta-Alvear at UCSF for the kind gift of the RPMI-8226-Luc+ cell line and the iQ Biosciences team for valuable assistance with in vitro T-cell experimental design.
Disclosure of potential conflicts of interest
P.C and A.P.W. have no competing financial interests to disclose. All other authors are employees of Teneobio Inc. with equity interests.
Author Contributions
Overall study design and analysis of results were conducted by S.F.A., N.D.T., D.P., U.S., O.V., W.v.S., and R.B.; immunizations and NGS-based repertoire analysis were completed by N.D.T., K.H., and A.B.; molecular biology was conducted by K.H. and K.D.; U.S., B.J., P.P.P., and H.S.U. contributed to expression, purification, and biophysical characterization of antibodies; ELISA binding and flow cytometry assays were run by D.P.; D.P. and H.S.U. coordinated BLI (Octet) studies; D.P., U.R., and S.C.C. were responsible for preparation of PBMCs and in vitro assays with primary T cells; B.B., D.P., P.C., and A.P.W. managed the mouse xenograft study; B.B., D.P., U.S.R., and S.I. contributed to the mouse and cyno PK studies; S.F.A., D.P., N.D.T., H.S.U., U.S., and B.B. prepared figures. The manuscript was written by N.D.T., D.P., and S.F.A.; all authors read and approved the submitted version.
Ethics Statement
PBMCs from healthy, deidentified donors were isolated from LRS filters purchased from the Stanford Blood Center (Palo Alto, CA). Human PBMCs were collected in accordance with scientific, ethical, and regulatory guidelines. Animal studies were carried out in accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. Rat maintenance and immunizations were carried out by certified animal facilities in the U.S. (Antibody Solutions, Sunnyvale, CA) and Germany (Aldevron and MfD Diagnostics GmbH, Freiburg, Germany) in accordance with national and international guidelines, with protocols reviewed by Institutional Animal Care and Use Committee (IACUC) boards in the U.S. and comparable government boards in Germany. Mouse and cyno PK studies as well as mouse xenograft studies were performed by AAALAC accredited facilities following internal approval by their IACUC boards (UCSF, San Francisco, CA; Aragen Biosciences, Morgan Hill, CA; SNBL USA, Everett, WA).
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
References
- 1.Pettitt D, Arshad Z, Smith J, Stanic T, Holländer G, Brindley D.. CAR-T cells: a systematic review and mixed methods analysis of the clinical trial landscape. Mol Ther J Am Soc Gene Ther. 2018;26:342–53. doi: 10.1016/j.ymthe.2017.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wu Z, Cheung NV.. T cell engaging bispecific antibody (T-BsAb): from technology to therapeutics. Pharmacol Ther. 2018;182:161–75. doi: 10.1016/j.pharmthera.2017.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Perez P, Hoffman RW, Shaw S, Bluestone JA, Segal DM.. Specific targeting of cytotoxic T cells by anti-T3 linked to anti-target cell antibody. Nature. 1985;316:354–56. doi: 10.1038/316354a0. [DOI] [PubMed] [Google Scholar]
- 4.Staerz UD, Kanagawa O, Bevan MJ. Hybrid antibodies can target sites for attack by T cells. Nature. 1985;314:628–31. doi: 10.1038/314628a0. [DOI] [PubMed] [Google Scholar]
- 5.Bargou R, Leo E, Zugmaier G, Klinger M, Goebeler M, Knop S, Noppeney R, Viardot A, Hess G, Schuler M, et al. Tumor regression in cancer patients by very low doses of a T cell-engaging antibody. Science. 2008;321:974–77. doi: 10.1126/science.1158545. [DOI] [PubMed] [Google Scholar]
- 6.Velders MP, van Rhijn CM, Oskam E, Fleuren GJ, Warnaar SO, Litvinov SV. The impact of antigen density and antibody affinity on antibody-dependent cellular cytotoxicity: relevance for immunotherapy of carcinomas. Br J Cancer. 1998;78:478–83. doi: 10.1038/bjc.1998.518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mandikian D, Takahashi N, Lo AA, Li J, Eastham-Anderson J, Slaga D, Ho J, Hristopoulos M, Clark R, Totpal K, et al. Relative target affinities of T-cell–dependent bispecific antibodies determine biodistribution in a solid tumor mouse model. Mol Cancer Ther. 2018;17:776–85. doi: 10.1158/1535-7163.MCT-17-0657. [DOI] [PubMed] [Google Scholar]
- 8.Mazor Y, Sachsenmeier KF, Yang C, Hansen A, Filderman J, Mulgrew K, Wu H, Dall’Acqua WF. Enhanced tumor-targeting selectivity by modulating bispecific antibody binding affinity and format valence. Sci Rep [Internet]. 2017;7 [accessed 2018 December28] http://www.nature.com/articles/srep40098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Teachey DT, Rheingold SR, Maude SL, Zugmaier G, Barrett DM, Seif AE, Nichols KE, Suppa EK, Kalos M, Berg RA, et al. Cytokine release syndrome after blinatumomab treatment related to abnormal macrophage activation and ameliorated with cytokine-directed therapy. Blood. 2013;121:5154–57. doi: 10.1182/blood-2013-02-485623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Eastwood D, Bird C, Dilger P, Hockley J, Findlay L, Poole S, Thorpe SJ, Wadhwa M, Thorpe R, Stebbings R. Severity of the TGN1412 trial disaster cytokine storm correlated with IL-2 release. Br J Clin Pharmacol. 2013;76:299–315. doi: 10.1111/bcp.12040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Velasquez MP, Bonifant CL, Gottschalk S. Redirecting T cells to hematological malignancies with bispecific antibodies. Blood. 2018;131:30–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Faroudi M, Utzny C, Salio M, Cerundolo V, Guiraud M, Müller S, Valitutti S. Lytic versus stimulatory synapse in cytotoxic T lymphocyte/target cell interaction: manifestation of a dual activation threshold. Proc Natl Acad Sci U S A. 2003;100:14145–50. doi: 10.1073/pnas.2334336100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Purbhoo MA, Irvine DJ, Huppa JB, Davis MM. T cell killing does not require the formation of a stable mature immunological synapse. Nat Immunol. 2004;5:524–30. doi: 10.1038/ni1058. [DOI] [PubMed] [Google Scholar]
- 14.Hernandez-Hoyos G, Sewell T, Bader R, Bannink J, Chenault RA, Daugherty M, Dasovich M, Fang H, Gottschalk R, Kumer J, et al. MOR209/ES414, a novel bispecific antibody targeting PSMA for the treatment of metastatic castration-resistant prostate cancer. Mol Cancer Ther. 2016;15:2155–65. doi: 10.1158/1535-7163.MCT-15-0003. [DOI] [PubMed] [Google Scholar]
- 15.Sibener LV, Fernandes RA, Kolawole EM, Carbone CB, Liu F, McAffee D, Birnbaum ME, Yang X, Su LF, Yu W, et al. Isolation of a structural mechanism for uncoupling T cell receptor signaling from peptide-MHC binding. Cell. 2018;174:672–687.e27. doi: 10.1016/j.cell.2018.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Harris KE, Aldred SF, Davison LM, Ogana HAN, Boudreau A, Brüggemann M, Osborn M, Ma B, Buelow B, Clarke SC, et al. Sequence-based discovery demonstrates that fixed light chain human transgenic rats produce a diverse repertoire of antigen-specific antibodies. Front Immunol. 2018;9:889. doi: 10.3389/fimmu.2018.00889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Osborn MJ, Ma B, Avis S, Binnie A, Dilley J, Yang X, Lindquist K, Ménoret S, Iscache A-L, Ouisse L-H, et al. High-affinity IgG antibodies develop naturally in Ig-knockout rats carrying germline human IgH/Igκ/Igλ loci bearing the rat CH region. J Immunol Baltim Md. 2013 [1950];190:1481–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ma B, Osborn MJ, Avis S, Ouisse L-H, Ménoret S, Anegon I, Buelow R, Brüggemann M. Human antibody expression in transgenic rats: comparison of chimeric IgH loci with human VH, D and JH but bearing different rat C-gene regions. J Immunol Methods. 2013;400–401:78–86. doi: 10.1016/j.jim.2013.10.007. [DOI] [PubMed] [Google Scholar]
- 19.Geurts AM, Cost GJ, Freyvert Y, Zeitler B, Miller JC, Choi VM, Jenkins SS, Wood A, Cui X, Meng X, et al. Knockout rats via embryo microinjection of zinc-finger nucleases. Science. 2009;325:433. doi: 10.1126/science.1172447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kocks C, Rajewsky K. Stepwise intraclonal maturation of antibody affinity through somatic hypermutation. Proc Natl Acad Sci U S A. 1988;85:8206–10. doi: 10.1073/pnas.85.21.8206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jung D, Giallourakis C, Mostoslavsky R, Alt FW. Mechanism and control of V(D)J recombination at the immunoglobulin heavy chain locus. Annu Rev Immunol. 2006;24:541–70. doi: 10.1146/annurev.immunol.23.021704.115830. [DOI] [PubMed] [Google Scholar]
- 22.J-P B, Dubois ARSX, Faison WJ, Farinelle S, Charpentier E, Sinner R, Wienecke-Baldacchino A, Muller CP. Functionally convergent B cell receptor sequences in transgenic rats expressing a human B cell repertoire in response to tetanus toxoid and measles antigens. Front Immunol. 2017;8:1834. doi: 10.3389/fimmu.2017.01834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Blumberg RS, Ley S, Sancho J, Lonberg N, Lacy E, McDermott F, Schad V, Greenstein JL, Terhorst C. Structure of the T-cell antigen receptor: evidence for two CD3 epsilon subunits in the T-cell receptor-CD3 complex. Proc Natl Acad Sci U S A. 1990;87:7220–24. doi: 10.1073/pnas.87.18.7220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Call ME, Pyrdol J, Wiedmann M, Wucherpfennig KW. The organizing principle in the formation of the T cell receptor-CD3 complex. Cell. 2002;111:967–79. doi: 10.1016/S0092-8674(02)01194-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Canfield SM, Morrison SL. The binding affinity of human IgG for its high affinity Fc receptor is determined by multiple amino acids in the CH2 domain and is modulated by the hinge region. J Exp Med. 1991;173:1483–91. doi: 10.1084/jem.173.6.1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xu D, Alegre ML, Varga SS, Rothermel AL, Collins AM, Pulito VL, Hanna LS, Dolan KP, Parren PW, Bluestone JA, et al. In vitro characterization of five humanized OKT3 effector function variant antibodies. Cell Immunol. 2000;200:16–26. doi: 10.1006/cimm.2000.1617. [DOI] [PubMed] [Google Scholar]
- 27.Bloom JW, Madanat MS, Marriott D, Wong T, Chan SY. Intrachain disulfide bond in the core hinge region of human IgG4. Protein Sci Publ Protein Soc. 1997;6:407–15. doi: 10.1002/pro.5560060217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Reddy MP, Kinney CA, Chaikin MA, Payne A, Fishman-Lobell J, Tsui P, Dal Monte PR, Doyle ML, Brigham-Burke MR, Anderson D, et al. Elimination of Fc receptor-dependent effector functions of a modified IgG4 monoclonal antibody to human CD4. J Immunol Baltim Md. 2000 [1950];164:1925–33. [DOI] [PubMed] [Google Scholar]
- 29.Merchant AM, Zhu Z, Yuan JQ, Goddard A, Adams CW, Presta LG, Carter P. An efficient route to human bispecific IgG. Nat Biotechnol. 1998;16:677–81. doi: 10.1038/nbt0798-677. [DOI] [PubMed] [Google Scholar]
- 30.Ridgway JB, Presta LG, Carter P. “Knobs-into-holes” engineering of antibody CH3 domains for heavy chain heterodimerization. Protein Eng. 1996;9:617–21. doi: 10.1093/protein/9.7.617. [DOI] [PubMed] [Google Scholar]
- 31.Zou X, Osborn MJ, Bolland DJ, Smith JA, Corcos D, Hamon M, Oxley D, Hutchings A, Morgan G, Santos F, et al. Heavy chain-only antibodies are spontaneously produced in light chain-deficient mice. J Exp Med. 2007;204:3271–83. doi: 10.1084/jem.20071155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Clarke SC, Ma B, Trinklein ND, Schellenberger U, Osborn MJ, Ouisse L-H, Boudreau A, Davison LM, Harris KE, Ugamraj HS, et al. Multispecific antibody development platform based on human heavy chain antibodies. Front Immunol Internet]. 2019; 9. doi: 10.3389/fimmu.2018.03037/full. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tang Y, Lou J, Alpaugh RK, Robinson MK, Marks JD, Weiner LM. Regulation of antibody-dependent cellular cytotoxicity by IgG intrinsic and apparent affinity for target antigen. J Immunol Baltim Md. 2007 [1950];179:2815–23. [DOI] [PubMed] [Google Scholar]
- 34.Hipp S, Tai Y-T, Blanset D, Deegen P, Wahl J, Thomas O, Rattel B, Adam PJ, Anderson KC, Friedrich M. A novel BCMA/CD3 bispecific T-cell engager for the treatment of multiple myeloma induces selective lysis in vitro and in vivo. Leukemia. 2017;31:2278. doi: 10.1038/leu.2017.14. [DOI] [PubMed] [Google Scholar]
- 35.Seckinger A, Delgado JA, Moser S, Moreno L, Neuber B, Grab A, Lipp S, Merino J, Prosper F, Emde M, et al. Target expression, generation, preclinical activity, and pharmacokinetics of the BCMA-T cell bispecific antibody EM801 for multiple myeloma treatment. Cancer Cell. 2017;31:396–410. doi: 10.1016/j.ccell.2017.02.002. [DOI] [PubMed] [Google Scholar]
- 36.Saber H, Del Valle P, Ricks TK, Leighton JK. An FDA oncology analysis of CD3 bispecific constructs and first-in-human dose selection. Regul Toxicol Pharmacol RTP. 2017;90:144–52. doi: 10.1016/j.yrtph.2017.09.001. [DOI] [PubMed] [Google Scholar]
- 37.Deng R, Iyer S, Theil F-P, Mortensen DL, Fielder PJ, Prabhu S. Projecting human pharmacokinetics of therapeutic antibodies from nonclinical data: what have we learned? mAbs. 2011;3:61–66. doi: 10.4161/mabs.3.1.13799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brownlie RJ, Zamoyska R. T cell receptor signalling networks: branched, diversified and bounded. Nat Rev Immunol. 2013;13:257–69. doi: 10.1038/nri3403. [DOI] [PubMed] [Google Scholar]
- 39.Gaud G, Lesourne R, Love PE. Regulatory mechanisms in T cell receptor signalling. Nat Rev Immunol. 2018;18:485–97. doi: 10.1038/s41577-018-0020-8. [DOI] [PubMed] [Google Scholar]
- 40.Martinez-Martin N, Risueno RM, Morreale A, Zaldivar I, Fernandez-Arenas E, Herranz F, Ortiz AR, Alarcon B. Cooperativity between T cell receptor complexes revealed by conformational mutants of CD3. Sci Signal. 2009;2:ra43–ra43. doi: 10.1126/scisignal.2000402. [DOI] [PubMed] [Google Scholar]
- 41.Matz M, Shagin D, Bogdanova E, Britanova O, Lukyanov S, Diatchenko L, Chenchik A. Amplification of cDNA ends based on template-switching effect and step-out PCR. Nucleic Acids Res. 1999;27:1558–60. doi: 10.1093/nar/27.6.1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Scherer EM, Smith RA, Gallego DF, Carter JJ, Wipf GC, Hoyos M, Stern M, Thurston T, Trinklein ND, Wald A, et al. A single human papillomavirus vaccine dose improves B cell memory in previously infected subjects. EBioMedicine. 2016;10:55–64. doi: 10.1016/j.ebiom.2016.06.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Son MS, Taylor RK. Preparing DNA libraries for multiplexed paired-end deep sequencing for Illumina GA sequencers. Curr Protoc Microbiol. 2011; Chapter 1: Unit1E.4.1-4.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Néron S, Thibault L, Dussault N, Côté G, Ducas E, Pineault N, Roy A. Characterization of mononuclear cells remaining in the leukoreduction system chambers of apheresis instruments after routine platelet collection: a new source of viable human blood cells. Transfusion (Paris). 2007;47:1042–49. doi: 10.1111/j.1537-2995.2007.01233.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.