

ABSTRACT
The construction of large libraries in mammalian cells allows the direct screening of millions of molecular variants for binding properties in a cell type relevant for screening or production. We have created mammalian cell libraries of up to 10 million clones displaying a repertoire of IgG-formatted antibodies on the cell surface. TALE nucleases or CRISPR/Cas9 were used to direct the integration of the antibody genes into a single genomic locus, thereby rapidly achieving stable expression and transcriptional normalization. The utility of the system is illustrated by the affinity maturation of a PD-1-blocking antibody through the systematic mutation and functional survey of 4-mer variants within a 16 amino acid paratope region. Mutating VH CDR3 only, we identified a dominant “solution” involving substitution of a central tyrosine to histidine. This appears to be a local affinity maximum, and this variant was surpassed by a lysine substitution when light chain variants were introduced. We achieve this comprehensive and quantitative interrogation of sequence space by combining high-throughput oligonucleotide synthesis with mammalian display and flow cytometry operating at the multi-million scale.
KEYWORDS: Mammalian display, CRISPR/Cas9, TALE nuclease, IgG antibody library, fluorescence-activated cell sorting, magnetic-activated cell sorting, affinity maturation, human therapeutic antibody discovery, gene targeting, gene editing
Introduction
The identification and engineering of recombinant binding molecules has been revolutionized by the availability of display technologies such as phage display and ribosome display.1 The basic principle of this approach, best exemplified using antibodies, relies on the linkage of the antibody product to the genetic information encoding it to allow isolation of antibody genes from libraries based on the binding properties of the encoded antibody.2
Despite the advantages of phage and ribosome display, the capacity to identify binders typically relies on enzyme-linked immunosorbent assay (ELISA) screening, which limits throughput and does not distinguish between variable levels of expression and variable affinity. In contrast, display of binding molecules on the surface of cells allows millions of cellular clones to be surveyed by flow sorting.
This approach was initially demonstrated in simple eukaryotes such as yeast cells,3 but creation of large libraries in higher eukaryotic cells would bring significant advantages. The glycosylation, expression and secretion machinery of yeast is different from that of higher eukaryotes, giving rise to antibodies with different post-translational modifications than those produced in mammalian cells. In addition, libraries of binders expressed within mammalian cells (either on the cell surface or by secretion) can be used to identify clones based on functions beyond antigen binding. Identification of binding interactions that directly affect cellular phenotype allows direct selection for biological function.4,5 Such benefits have driven the attempts described below to create a display system based in higher eukaryotes.
Construction of large libraries in mammalian cells is substantially more difficult than in yeast and bacteria. Donor DNA introduced by standard transfection methods integrates as a linear array encompassing multiple copies of transfected transgenes. Thus, the introduction of DNA encoding a repertoire of antibody genes has the potential to introduce multiple antibody genes into each cell, which reduces the relative expression of any given antibody, causes display of erroneous combinations of heavy and light chains and leads to the isolation of many passenger antibody genes, thereby reducing the rate of enrichment of specific clones.
A number of approaches have been described to introduce single antibody genes into each cell, including viral-based systems and transposons.5–8 A disadvantage of these approaches is that single copy integration is controlled by limited infection or transfection, requiring a compromise between library size and single gene insertion. In addition, integration within the genome is random, leading to potential variation in transcription level based on the transcriptional activity of the integration locus. Targeting individual antibody genes to a single locus within the population has the additional advantage of effecting transcriptional normalization across the population. Random integration also introduces the possibility of variable levels of gene silencing within the population.9 To fully realize the potential for antibody display on mammalian cells and other higher eukaryotes, there is a need for a system to create large libraries that combine accurate integration into a pre-defined site with an efficiency that allows construction of large libraries. Site-specific integration of transgenes directed by Flp recombinase using the commercial “Flp-In” system has previously been described.10,11 In direct comparison with the nuclease-directed system presented here, we found the Flp-In system to be deficient (see supplementary section), mirroring the rather limited success both in the original publications and subsequently by others.12 This is likely due to the fact that the Flp-In system is designed for accurate integration in a limited number of clones rather than large library construction. Improved integration efficiencies have been achieved using an alternative recombinase with libraries of 20,000 clones being reported.12
Homologous recombination (HR) represents an alternative means for site-directed transgene insertion, and we have previously reported the use of long homology arms to direct the integration of antibody populations into mouse embryonic stem cells, enabling direct functional selection of binders.4 It has been shown that the rate of homologous recombination can be significantly enhanced by cleavage of the target genomic locus.4,13,14 The recent development of TALE nucleases (TALENs)15 and CRISPR/Cas9 technology16 has revolutionized genome engineering by enabling the creation of bespoke site-specific nucleases. The efficiency of nuclease-directed integration creates the opportunity to introduce diverse repertoires of millions of complete antibody genes into the same genomic locus within a population of cells. The present work illustrates that gene editing through nuclease-directed integration can efficiently target dual promoter, full-length IgG-formatted antibody genes into mammalian cells to create massive repertoires of monoclonal cells encompassing one antibody gene per cell. Antibody mutagenesis by nuclease-directed integration of oligonucleotide variants has recently been demonstrated, but this was limited to modifying a single pre-integrated antibody gene in a Cas9-expressing cell line, with variation introduced at individual complementarity-determining regions (CDRs) in each transfection.17
We illustrate the potential of this approach by chain shuffling a population of variable heavy (VH) genes derived by single-chain variable fragment (scFv) phage display, reformatting into IgG-format, and creating and screening a mammalian display library. In addition, we have affinity matured a programmed cell death protein 1 (PD-1)-blocking antibody by oligonucleotide-directed mutagenesis. Random mutagenesis has routinely been used to create variant libraries from starting clones in search of improved variants. The availability of modern high throughput oligonucleotide synthesis technology, however, means that a more directed approach can be taken to library design and construction. By this approach, it becomes possible to retain much of the core sequence while still sampling variants around that sequence space. The potential of combining this approach with mammalian display was exemplified by using CRISPR/Cas9 or TALE nucleases, to create diversified libraries in an IgG format. These were subjected to stringent selection to identify critical residues involved in the interaction, resulting in higher affinity blocking antibodies.
Results
Construction of donor vectors
Antibody genes were integrated into the first intron of the protein phosphatase 1, regulatory subunit 12C (PPP1R12C) gene (Figure 1(a,b)). This locus, referred to as the “AAVS” locus has previously been used as a “safe harbor” for the stable insertion and expression of transgenes into the human genome.1,9 A pair of TALE nucleases,2,18 which cleave within the human AAVS locus, were used. We also designed guide RNAs to direct Cas9 cleavage to the same sequence as recognized by these TALE nucleases (Figure 1(a)).
Figure 1.
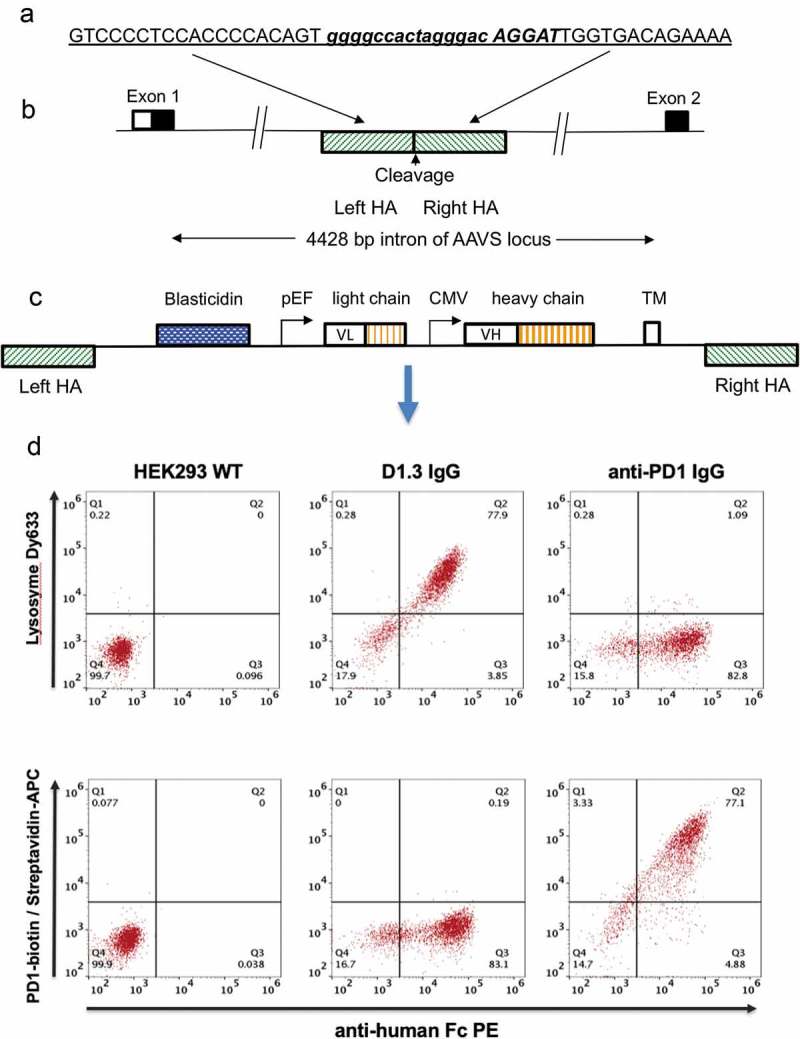
Antibody display cell line generation by nuclease mediated gene integration (a) Recognition sequence for left and right TALE nucleases are shown underlined and in upper case either side of the spacer region (shown in lower case). Emboldened and italicized sequence represents sequence of CRISPR target sequence (b) Representation of genomic AAVS locus. The AAVS cleavage site is located within a 4428 bp intron between the first and second exons of the gene encoding protein phosphatase 1, regulatory subunit 12C, PPP1R12C. TALE nucleases or CRISPR/Cas9 nucleases directed to this region are used to cleave the genome at this site. Hatched boxes on the 5ʹ and 3ʹ side of the cleavage site represent the left and right homology arms (HA), respectively (c) Representation of pD2 donor vector used to insert and display human IgG-formatted antibody genes. The transgene region is flanked by left and right homology arms (left HA, right HA) representing the sequences which flank the cleavage site in the AAVS locus. The vector encodes a promoter-less blasticidin gene, a dual promoter antibody expression cassette with antibody light chain and a human IgG2 heavy chain driven by pEF and CMV promoters, respectively. Expressed antibodies are anchored on the cell surface by a PDGFR transmembrane domain (TM) (d) Flow cytometric analysis of HEK293 cells displaying an anti-lysozyme or anti-PD1 antibody on the cell surface. Non-transfected HEK293 cells (left panel) and cells displaying either anti-lysozyme (middle panel) or anti-PD1 337_1_C08 (right panel) were stained with anti-Fc PE and either lysozyme conjugated with DyLight 633 (top row) or PD-1-biotin/streptavidin-APC.
Figure 1(c) represents the construction of “donor” plasmids for the insertion into the genome of antibody genes. In order to direct integration by homologous recombination following genomic cleavage, the expression cassette encoding the antibody gene is flanked with 800 base pair (bp) regions homologous to the flanking sequences found on either side of the genomic cleavage site (Supplementary Figure 1). The left homology arm is followed by a blasticidin gene, which lacks a promoter but is preceded by a splice acceptor site to create an in-frame fusion with the upstream endogenous exon from the AAVS1 locus (shown in Figure 1(b)). This requirement for integration and in-frame splicing to effect expression of the drug selectable marker reduces background arising from incorrect integration. The efficiency of stable nuclease-directed integration (“integration efficiency”) can be calculated by measuring the number of blasticidin-resistant colonies that arise in the presence or absence of plasmids encoding a nuclease gene. The blasticidin gene is followed in pD2 (Figure 1(c)) by pEF and a cytomegalovirus (CMV) promoter driving expression of separate light and heavy chain genes, respectively, to express an IgG formatted antibody. The sequence encoding the Fc domain is followed by an intron and an exon encoding the transmembrane (TM) domain from the platelet-derived growth factor (PDGF) receptor19 to anchor the antibody to the cell surface for cellular display. A similar vector (pD6) is used for cell surface expression of a scFv fused to a human Fc domain (scFv-Fc fusion), driven by a single CMV promoter. The design of these vectors included a transmembrane domain encoded within an exon flanked by Dre recombinase sites. This was included to enable expression of soluble antibodies following transfection of isolated cellular clones with a plasmid encoding Dre recombinase. In the absence of Dre recombinase, however, we found that soluble product was produced (approximately 2 mg/l for the D1.3 control antibody), suggesting that alternative splicing is occurring (data not shown).
An IgG-formatted anti-lysosome (humanized D1.3)20 or anti- PD1 antibody was cloned into the vector pD2, and the resulting donor vectors were transfected into HEK293 cells along with AAVS targeted TALE nuclease plasmids. After 14 days in culture in the presence of blasticidin, 77–78% of cells in the population were found to be positive for both Fc expression and for binding to the respective target antigen (Figure 1(d)). Specificity of the displayed antibodies was shown where cells expressing the anti-lysozyme antibody did not bind labeled PD1 and the anti- PD1 antibody cell line did not bind labeled lysozyme.
Nuclease-directed integration of single copy antibody genes to the AAVS locus
We have previously described the generation of scFv-formatted antibodies that specifically recognize either fibroblast growth factor (FGF) receptor 1 (αFGFR1) or FGF receptor 2 (αFGFR2).4 These two antibody genes were cloned into pD6 to create donor vectors αFGFR1-pD6 and αFGFR2-pD6. The donor vectors were electroporated by Amaxa nucleofection in the presence or absence of the AAVS-directed TALEN vector pair. Two days after transfection, selection for blasticidin resistance was initiated, and after 13 days the cells were stained with either FGFR1 labeled with Dy633 or FGFR2 labeled with Dy488. Figure 2(a,b) shows that over 96% of cells in the transfected population bound the appropriate FGFR paralogue (the ability of the antibodies themselves to distinguish between the two paralogues is demonstrated in each case by the lack of binding to the alternative FGF receptor). This demonstrates the efficient generation of a stable population of scFv-Fc fusions where the majority of cells express the antibody gene targeted through nuclease-directed integration.
Figure 2.
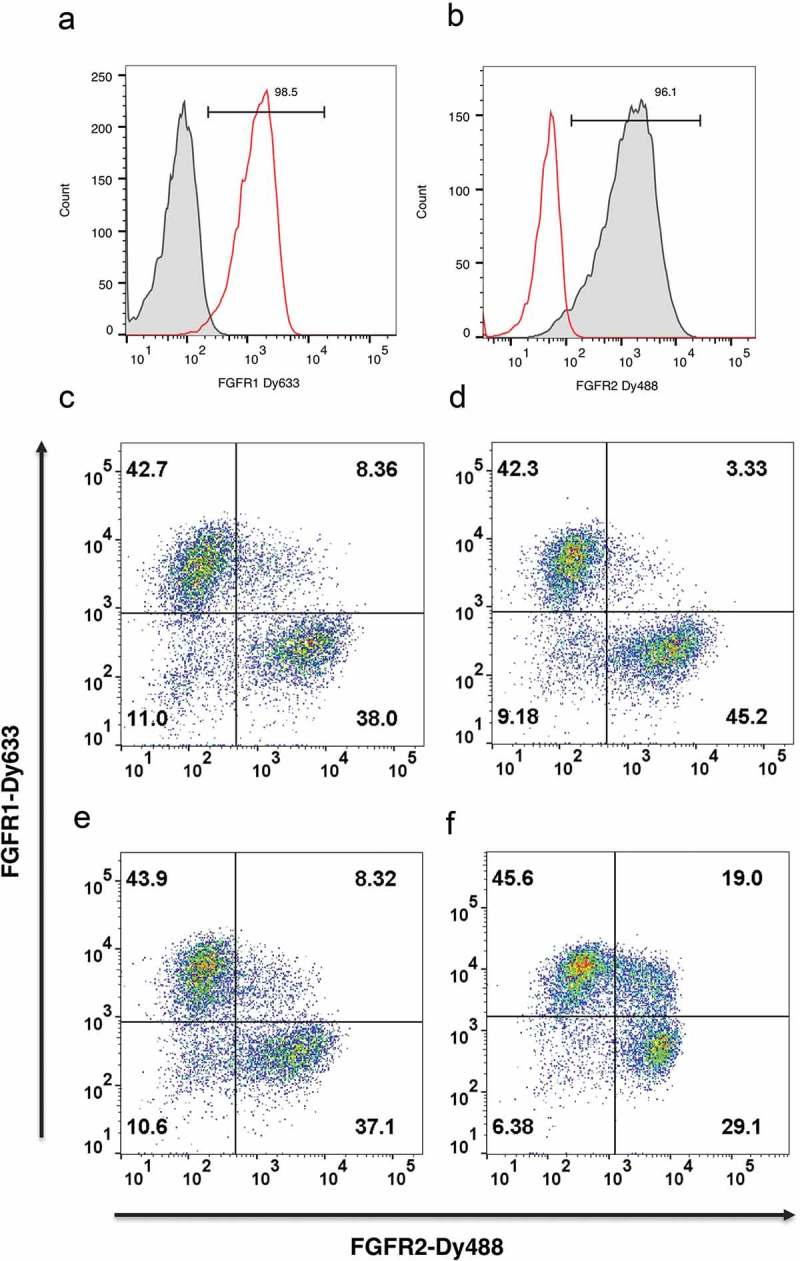
Nuclease-directed integration of single copy antibody genes. HEK293 cells transfected with either plasmid αFGFR1-pD6 or αFGFR1-pD6 (50:50 mix) were introduced via TALE nuclease into the AAVS locus of HEK293 cells, selected for blasticidin resistance for 13 days and stained with either FGFR1-Dy633 (a) or FGFR2-Dy488 (b). Plots show single color histogram overlaying anti-FGFR1 and anti-FGFR2 transfected cells. Dot plot shows dual staining to detect binding to FGFR1 (y-axis) and FGFR2 (x-axis). Samples are: (c) 300 ng donor, CRISPR/Cas9 via Amaxa; (d) 100 ng donor, CRISPR/Cas9 via Amaxa; (e) 300 ng donor, TALEN via Amaxa; (f) 300 ng donor, TALEN via PEI transfection.
The αFGFR antibodies were used in further model experiments to determine the extent to which single copy integration occurred, and also to compare the efficiency of different nucleases and transfection approaches. αFGFR1-pD6 and αFGFR2-pD6 donor plasmids were mixed in equal proportion, introduced into HEK293 cells in the presence and absence of plasmids encoding the TALE nuclease pair or a CRISPR/Cas9 encoding plasmid, and selected for blasticidin resistance.
Nuclease-directed integration into one of the alleles should lead to targeting of a single antibody gene to the AAVS locus, resulting in the majority of cells binding either labeled FGFR1 or FGFR2, but not both. Thus, dual-color flow cytometry with labeled-FGFR1 and FGFR2 allow quantitation of cells that integrate and express more than one antibody gene. Figure 2(c–f)/Table 1 shows that the proportion of double positive clones can be as low as 3.3% using CRISPR/Cas9 or TALE nucleases, suggesting that the majority of clones carry a single donor integration. While there is a formal possibility of integration of other copies of the donor into non-targeted genomic loci without expression (despite the inclusion of juxtaposed CMV and pEF promoters), this result provides sufficient comfort to proceed to library construction with the ultimate determination of success being the ability to enrich positive binders free from a background of such passenger clones.
Table 1.
Comparing gene targeting efficiency using different nucleases and transfection methods.
| Nuclease | Transfection method | Donor plasmids (ng DNA/106 cells) |
Transfection efficiency (%) | Fold improvement over “no nuclease” control | Proportion double positive | Reference to figure |
|---|---|---|---|---|---|---|
| CRISPR | Amaxa | 300 | 1.2 | 6.9x | 8.4% | 2C |
| CRISPR | Amaxa | 100 | 0.5 | 10x | 3.3% | 2D |
| TALEN | Amaxa | 300 | 1.1 | 6.1x | 8.3% | 2E |
| TALEN | PEI | 300 | 0.87 | 1.7x | 19% | 2F |
| TALEN | Maxcyte | 600 | 5.1 | 51x | 7.4% | - |
| TALEN | Maxcyte | 200 | 2.7 | 89x | 3.6% | - |
The efficiency of different transfection approaches was also compared. Using Amaxa electroporation, a gene targeting efficiency of 0.5%-1.2% was achieved (Table 1). Maxcyte electroporation gave a gene targeting efficiency of 2.7–5.1%, which was 50–90 fold above background levels found in the absence of nuclease. The poorest results were achieved by transfection using polyethylenimine (PEI) with low gene targeting efficiency and high background (0.9% efficiency representing only a 1.7-fold increase over the non-targeted background). In addition, 19% of cells were double positive, indicating that a higher degree of random integration was occurring with PEI transfection (Figure 2(f)). Thus, optimal results were achieved with the Maxcyte electroporation system, which is designed to maximize survival of transfected cells with consequent benefits for library construction. This also has “scalability” benefits by allowing flow electroporation of 1011 cells in 30 min.
Creation and selection of a stable, population of antibody-positive cells following nuclease-directed integration
Display technology can be used to improve the affinity of lead clones or populations of clones, and typically involves the creation of a library of variants that is subsequently subjected to stringent selection using limiting antigen concentrations. One simple approach to diversification is to carry out chain-shuffling by retaining a selected population of VH genes and combining with a repertoire of variable light (VL) genes (or vice versa).21 We performed two rounds of selection from a naïve phage display library on human PD-L1, and the selected VH population was amplified by polymerase chain reaction (PCR) and combined with a repertoire of VL partners. Diversification by chain shuffling was also combined with reformatting to create an IgG formatted, chain-shuffled mammalian display library.
The IgG–formatted, chain shuffled mammalian display library was electroporated into HEK293 cells and targeted to the AAVS locus using CRISPR/Cas9 to create a library of 1.8 × 106 clones. Figure 3(a) provides an overview of the workflow. At 9 days post transfection (dpt), 3% of cells were positive for binding using 10 nM PD-L1 (Figure 3(b)). Following cell sorting and culture for a further 14 days, the majority of cells remained antigen positive (Figure 3(c)), demonstrating that a stable population had been selected. Figure 3(d,e) also shows that antigen concentration can be reduced to help identify the sub-population of cells capable of binding at lower antigen concentrations. Genomic DNA from the first and second fluorescence-activated cell sorting (FACS) rounds (using 10 nM and 1 nM, respectively) was prepared, the antibody genes were recovered by PCR and cloned into a soluble IgG expression vector for production and analysis. Recovered antibodies were screened in an affinity capture assay where antibody is immobilized and captures biotinylated PD-L1 in solution. This assay further underlines the improved performance from clones derived from the more stringent selection conditions (using 1 nM versus 10 nM antigen) (Figure 3(f)).
Figure 3.
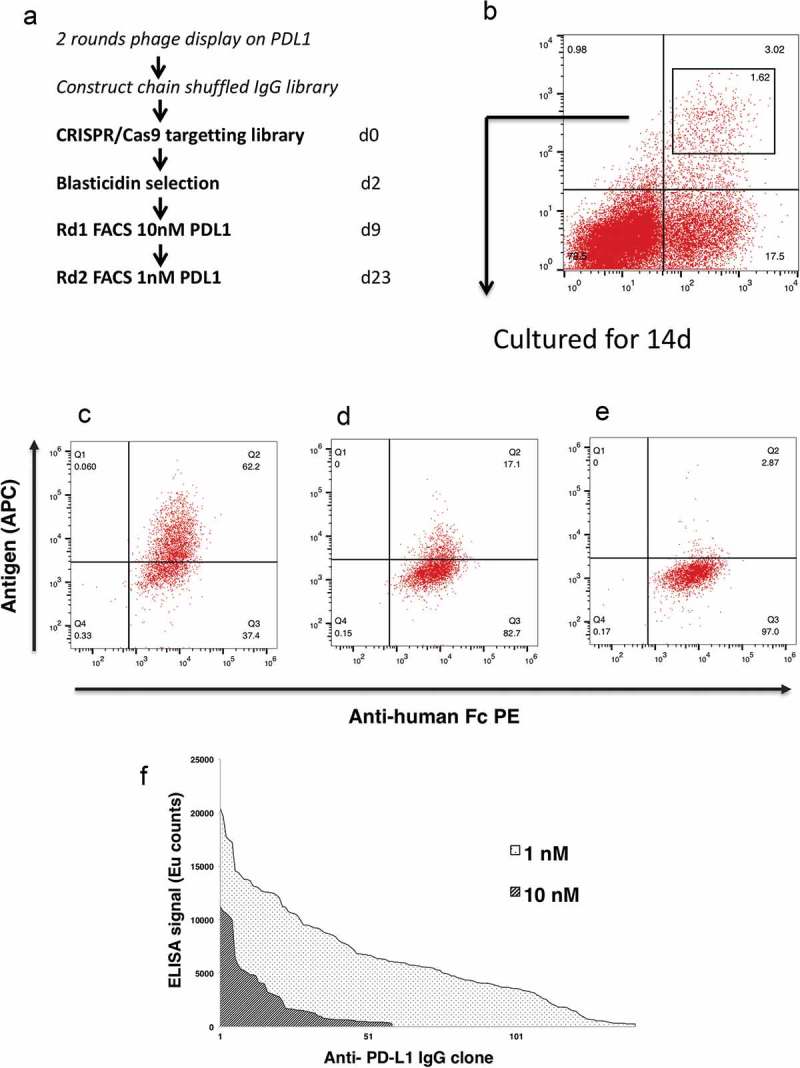
Affinity maturation using mammalian display. Schematic illustration of the experimental workflow (a). At 9dpt transfected cells were sorted for antigen binding using 10 nM PD-L1. The gate within the plot represents the population that was sorted, and the number represents percentage of the total cells (b). The sorted population was grown for an additional 14days and stained with anti-Fc PE and PD-L1-biotin at a concentration of (c) 10nM, (d) 1nM, (e) 0.1nM followed by detection with Streptavidin-APC. The dot-plot shows antibody expression (x-axis) and antigen binding (y-axis). Affinity ranking by capture ELISA (f) of anti- PD-L1 IgG, FACS selected at 10 nM (light hatch) or 1 nM PD-1 (dark hatch).
Following nuclease-directed integration, depletion of non-drug-resistant, antibody-negative cells requires 2–3 weeks of culture in the presence of blasticidin. In order to accelerate the process, we have observed that cells can be enriched at 8 dpt, using anti-human Fc antibodies and magnetic-activated cell sorting (MACS) beads (data not shown). This early MACS step has the benefit of removing non-expressing cells, thereby reducing the large culture volume following library-scale transfection and facilitating the drug selection process.
Exploring paratope sequence space by “total synthesis” and mammalian display of variant libraries
Using phage display,22 an antibody (337_1_C08) that blocked the interaction of PD-1 with PD-L1 was identified (as demonstrated both in a biochemical assay and in a functional cell co-culture assay with a Jurkat NFAT reporter cell line). The sequence of the VH and VL chains of 337_1_C08 is shown in Supplementary Figure 2. The affinity (KD) of 337_1_C08 for PD-1 is 74 nM, and here we demonstrate that it is possible to create a mutagenized library and select for variants with improved affinity by mammalian display.
Affinity maturation commonly involves creation of a mutagenic library by saturation mutagenesis where randomizing codons such as NNS (encoding all 20 amino acids within 32 codons) are included. In many cases, it is found that improved clones are related in sequence to the parental sequence.23 A more efficient and parsimonious route for mutagenesis therefore is to retain sequence information while exploring the surrounding sequence space. To achieve this, oligonucleotides were designed to a continuous stretch of eight amino acids in the CDR3 of VH and VLs such that every possible 2-mer and 1-mer variant, using all amino acids (with the exception of cysteine) were included (Figure 4(a,b)). Thus, the library was designed to retain at least six of the original eight amino acids. It was possible to accomplish this by synthesis of 9216 oligonucleotides directed to each of VH CDR3 or VL CDR3. The presence of all 9216 oligonucleotides in each set was confirmed by high throughput sequencing (Twist Bioscience). These libraries were constructed in pINT17-BSD, which is a second-generation display vector with essentially the same structure as pD2. To help reduce the size of the overall insert, the TM domain was fused directly to the end of the CH3 domain rather than being presented within an exon:intron combination. A VH antibody gene repertoire encompassing these changes was cloned into pINT17-BSD containing the original 337_1_C08 light chain. The VL CDR3 region of the anti-PD-1 clone was mutated in the same way, and was combined with the mutated VH CDR3 library (Figure 4(c)). Following nuclease-directed integration by CRISPR/Cas9, targeted integration efficiencies of 2.3% and 1.9% were achieved for the “VH only” and the “VH:VL” libraries, respectively.
Figure 4.
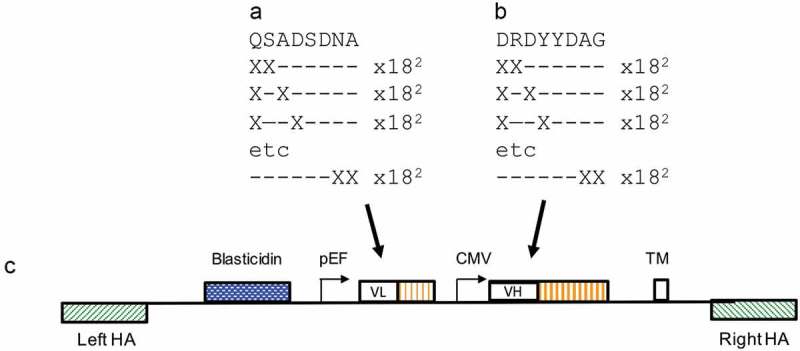
Anti- PD-1 VH and VL CDR3 library creation by “total synthesis”. Schematic diagram showing a representation of mutation of the VL CDR3 (a) and VH CDR3 (b) 8-amino acid windows by oligonucleotide “total synthesis” where every possible encoded 1-mer and 2-mer substitution was made. The libraries were cloned to the targeting vector pINT17-BSD (c) prior to transfection of HEK293 cells.
This “local search” approach was compared to complete randomization of the VH CDR3 region by NNS mutagenesis. Using mammalian display coupled with flow cytometry, it is apparent that, following complete randomization of eight amino acids of VH CDR3, the majority of clones fail to bind (although 50–55% of clones retain expression (Figure 5(b)), compared with the parental “wild-type” clone, 337_1_C08 (Figure 5(a)). In contrast, using the local search approach 21% and 11% of clones retained binding to PD-1 for the VH only and VH:VL library, respectively (Figure 5(c,d)). A similar rate of antigen binding was achieved when TALE nucleases, rather than CRISPR/Cas9, were used to direct integration (Figure 5(g,h)). Thus, despite a mutagenic “exploration” encompassing 16 amino acids with variation in 4 positions, a surprisingly high proportion of clones retained antigen binding, representing a “richer pool” in which to find improved variants.
Figure 5.
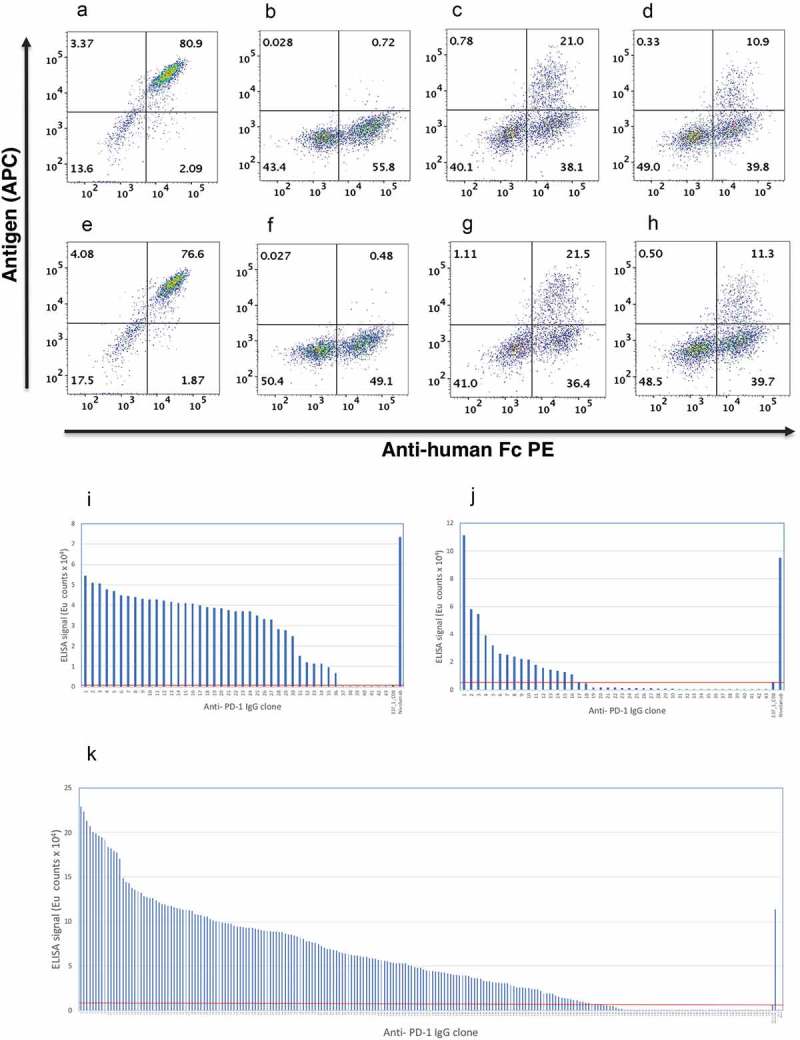
Mammalian display of variant libraries. HEK293 cells transfected with pINT17-337_1_C08 (a & e), pINT17-NNS library (b & f), pINT17-VH library (c & g) and pINT17-VH:VL combined library (d & h) were introduced via CRISPR/CAS9 (a-d) or TALE nuclease (e-h) into the AAVS locus of HEK293 cells. 14dpt cells were dual stained with 10 nM human PD-1-Avi tag antigen followed by streptavidin APC (y-axis) and anti-human Fc PE (x-axis).
Affinity ranking ELISA of the antibodies isolated from the “VH only” Selection (i), “VH:VL-1nM” selection (j) and “VH:VL-0.1nM” selection (k). 337_1_C08 and nivolumab are used as references and the line represents the antibodies with improved affinities.
A mammalian display library of 107 clones was created from the combined VH CDR3 and VL CDR3 directed oligonucleotides (“VH:VL”) and compared to a library where only the VH CDR3 was mutated. This “VH only” library was selected using three rounds of MACS enrichment culminating in a concentration of 0.1 nM biotinylated antigen. This was followed by a final round of FACS enrichment using 1 nM biotinylated PD-1 (referred as “VH only” selection). Genomic DNA was prepared for each selection, antibody genes were recovered by PCR amplification and cloned into an IgG expression vector. An indirect affinity capture ELISA was used where immobilized antibody is used to capture biotinylated antigen. This was found to be more sensitive to affinity differences than direct ELISA using immobilized antigen. In total, 43 of the resultant clones were screened, revealing that 36 clones (representing 12 unique sequences) were superior to the parental clone (Figure 5(i)).
The VH:VL library was enriched using a single round of FACS sorting with 1 nM biotinylated antigen (referred as “VH:VL-1nM” selection), resulting in a population where 16/43 clones outperformed the parental clone in affinity capture assays (Figure 5(j)). Following multiple rounds of selection (using anti-Fc MACS beads combined with 1 nM MACS at round 1, 0.1 nM MACS at round 2 and 1 nM FACS sorting, referred as “VH:VL-0.1nM” selection), 172 of 230 clones (75%) outperformed the parental clone (Figure 5(k)). Sequencing of the “VH:VL-1nM” selection and the “VH:VL-0.1nM” selection shows 40/41 (97%) and 149/230 (65%) unique sequences, respectively.
High-throughput sequencing was carried out on the populations from the “VH only” selection and the “VH:VL-0.1nM” selections, generating 71–78,000 complete sequences. In total, 1071 and 1582 sequence variants were found for VH and VH + VL, respectively. Supplementary Figure 3A shows that positions D1, D3, Y4, and D6 were highly conserved with 94–99.6% representation in the output clones from the “VH only” population. The most commonly substituted position was tyrosine at position 5 (Y5) in the center of the CDR3 region, which was replaced in 64.6% of clones by His (referred to as Y5H mutation). The “VH only” library was dominated by a particular variant clone found in 41% of clones with an additional R to E change at position 2 (hereafter VHs are named based on the replacement amino acid and its position, so this VH is referred to as VH-H5E2). A substitution of Y to K (Y5K) was found in 25.3% of clones.
Analysis of the “VH:VL” populations again revealed conservation at D1, D3, Y4, and D6, albeit to a lesser extent than the “VH only” library. A greater degree of variation was found at other positions (Supplementary Figure 3B). Looking at the frequency of specific VH sequences, there is a change in the profile of the top VHs when VL diversity is introduced. In the “VH only” library, the overall representation of Y5H mutations drops from 64.6% to 29.8% and the most frequent VH variant H5E2 drops from 41% occurrence to 1.5%.
Next, we measured the off-rate on a panel of 141 unique clones of which 132 had improved off-rates compared with the parental antibody. From the “VH only” population the highest off-rate was achieved by the most abundant H5E2 variant with an off-rate of 5.8 x 10−3/sec (compared to an off-rate in the parental clone of 19 x 10−3/sec). Within the sequences from the VH:VL library, H5E2 was found only twice with two different light chains, neither of which improved the off-rate. In fact, among 50 different Y5H-containing sequences from the VH:VL population, no clones were found with significant increase in off-rate compared to H5E2 (the best was 4.0 x 10−3/sec compared to 5.8 x 10−3/sec), suggesting a local maximum affinity with little room for improvement with this particular Y5H variant using the current mutagenesis strategy. In contrast VH-K5T8, which had a lower off-rate than VH-H5E2 (4.6 x 10−3/sec) gained significant improvements in off-rate following the introduction of VL diversity (up to 0.8 × 10−3).
A panel of variants from the “VH only” and “VH:VL” selections identified by off-rate screening were expressed, purified and the on/off rates measured to determine KD values measured (Table 2, example profiles in Supplementary Figure 5). Selected anti- PD-1 antibodies were purified and their expression yields were found to be similar to the parental sequence (23–50 mg/ml). The parental antibody and all the selected variants were specific for human and cynomolgus monkey PD-1 and failed to bind to unrelated antigens such as PD-L1, mesothelin, and β-galactosidase (Supplementary Figure 4). All variants measured were improved relative to the parental sequence, including eight with single digit nM affinities and several that had affinity equivalent to the approved anti-PD1 antibody nivolumab (measured as 2.8 nM). The parental antibody had an affinity of 74 nM, which was improved by 4.3-fold to 17 nM in the dominant VH_H5E2 variant from the “VH only” library. This sequence became less frequent in the “VH:VL” library, and the two additional VH_H5E2 variants from this library did not show such-marked improvements. The VH_K5T8 mutation had a similar affinity (16 nM), but in contrast to H5E2, improvements were found with additional VL diversity with affinities between 2.9 and 10 nM recorded. The top three clones included positively charged lysine or arginine at position 5. The light chain that gave the greatest improvement (VL9) was also found with other VHs where it contributed to an improvement in affinity. This light chain was tested with the original VH gene, and independently gave rise to a 4.3-fold improvement in affinity. Thus, a combinatorial benefit was achieved between VHK5T8 and VL9, leading to an overall 25-fold improvement. In contrast, the combination of VH-H5E2 with VL9 did not lead to any synergistic improvement.
Table 2.
Mammalian display affinity matured anti- PD-1 clones. The dissociation constant (KD), on rate (ka) and off rate (kd) were calculated from experimental data generated by a MASS2 SPR instrument (Sierra Sensors) as described in the materials and methods. The maximal response (Rmax) and a measure of the average deviation of the experimental data (χ2) is shown. The VH CDR3 window that was mutagenized is shown in the third column in single letter amino acid code for the original wild-type (WT) parental clone 337_1_C08. Each clone variant CDR3 window sequence is shown with variation from 337_1_C08 shown in single letter amino acid code and identity to parent with dot (.).
| VH | VL | VH | KD [nM] | ka (Ms−1 x105) | kd (s−1 x10−3) | Rmax | χ2 [RU2] |
|---|---|---|---|---|---|---|---|
| WT | WT | DRDYYDAG |
74 | 2.6 | 19 | 187 | 2.6 |
| H5E2 | WT | .E..H... |
17 | 3.2 | 5.8 | 134 | 1.8 |
| H5E2 | 2 | .E..H... |
30 | 2.2 | 6.6 | 111 | 1.2 |
| H5E2 | 9 | .E..H... |
17 | 2.4 | 4.0 | 132 | 0.6 |
| H5E2 | 23 | .E..H... |
54 | 2.4 | 12.8 | 175 | 1.0 |
| H5A2 | 6 | .A..H... |
18 | 3.0 | 5.2 | 247 | 1.9 |
| H5A2 | 9 | .A..H... |
24 | 2.1 | 5.1 | 137 | 0.8 |
| H5W2 | 2 | .W..H... |
28 | 2.0 | 5.7 | 175 | 1.9 |
| H5T8 | 82 | ...H..T |
14 | 1.8 | 2.5 | 126 | 0.4 |
| H5W8 | WT | ...H..W |
64 | 0.9 | 5.5 | 75 | 3.4 |
| K5T7 | WT | ...K.T. |
16 | 3.8 | 6.1 | 121 | 0.8 |
| K5T7 | 12 | ...K.T. |
14 | 3.2 | 4.5 | 127 | 0.3 |
| K5T8 | WT | ...K..T |
16 | 2.8 | 4.6 | 164 | 2.8 |
| K5T8 | 2 | ...K..T |
10 | 2.2 | 2.3 | 179 | 0.3 |
| K5T8 | 6 | ...K..T |
9 | 4.4 | 4.1 | 114 | 1.1 |
| K5T8 | 9 | ...K..T |
2.9 | 2.6 | 0.8 | 155 | 2.7 |
| K5S8 | 3 | ...K..S |
21 | 3.1 | 6.5 | 52 | 0.4 |
| K5D2 | 2 | .D..K... |
22 | 2.9 | 6.6 | 136 | 0.3 |
| K5E3 | 61 | ..E.K... |
4.8 | 3.7 | 1.8 | 107 | 1.5 |
| K5L2 | 12 | .L..K... |
24 | 2.8 | 6.8 | 200 | 2.5 |
| R5V8 | 9 | ....R..V |
3.9 | 5.2 | 2.1 | 84 | 1.3 |
| R5V8 | 2 | ....R..V |
35 | 1.8 | 6.3 | 209 | 2.3 |
| Q5W2 | 25 | .W..Q... |
7.7 | 3.0 | 2.3 | 188 | 0.8 |
| A5M7 | 13 | ....A.M. |
14 | 1.3 | 1.8 | 60 | 0.4 |
| WT | 9 | ........ |
18 | 2.1 | 3.7 | 139 | 1.0 |
| L3N7 | WT | ..L...N. |
27 | 2.1 | 5.7 | 204 | 0.8 |
| H4K4 | 123 | ...H...K |
10 | 2.7 | 2.8 | 62 | 0.5 |
| I2 | 34 | .I...... |
5.4 | 4.1 | 2.2 | 80 | 1.5 |
| I2I8 | 83 | .I....I. |
6.4 | 3.1 | 2.1 | 49 | 1.6 |
| K5T8 | 86 | ....K..T |
7.6 | 4.3 | 3.1 | 71 | 1.2 |
| Nivolumab | 2.8 | 5.0 | 1.2 | 28 | 0.1 |
Functional activity of affinity matured anti-PD-1 antibodies
Selected anti-PD-1 antibodies were expressed, purified and tested for their ability to inhibit the interaction of PD-1 with PD-L1 in a cell reporter assay. Co-culture of a Jurkat NFAT promoter luciferase reporter cell line with a Chinese hamster ovary (CHO) cell line expressing a T cell receptor (TCR) activator results in expression of luciferase. Expression of PD-L1 on the CHO cell and interaction with PD-1 on the Jurkat cell, however, results in down-regulation of luciferase expression due to PD-L1/PD-1 interaction signaling blockade. Addition of an antibody able to block the PD-L1/PD-1 interaction results in re-activation of the NFAT promoter and up-regulation of luciferase expression. When the antibodies were tested in this co-culture reporter assay at a fixed concentration of 50 nM, the original parental clone 337_1_C08 was among the least active. Several anti-PD-1 antibodies from CDR3 mutagenesis and mammalian display selection displayed increased potency compared to the original parental antibody. In fact, the majority of clones had equivalent activity compared to nivolumab (Figure 6. Antibodies are named according to the combination of VH and VL chains shown in Table 2). Two affinity matured clones were compared to nivolumab and the parental clones in a dose-response experiment as shown in Figure 7 and their half maximal response value calculated by curve fitting (Table 3). Both K5T8-9 and K5E3-61 demonstrated increased potency in PD-1 inhibition compared with the parental antibody and nivolumab, with K5T8-9 (also known as 1549_2_D06) being more than 8.3-fold more potent than the parental 337_1_C08 antibody and 2.3-fold more potent than nivolumab. An increase in clone affinity correlated with an increased potency in the cell-based assay. For example, the highest affinity clone K5T8 (KD for PD1, 2.9 nM) also possessed the greatest potency in the cell-based PD1 inhibition assay (EC50 = 5 nM). This compares to the parental antibody 337_1_C08 (KD = 74 nM, EC50 = 25 nM) and a variant with intermediate affinity, K5E3 (KD = 4.8 nM, EC50 = 6.9 nM).
Figure 6.
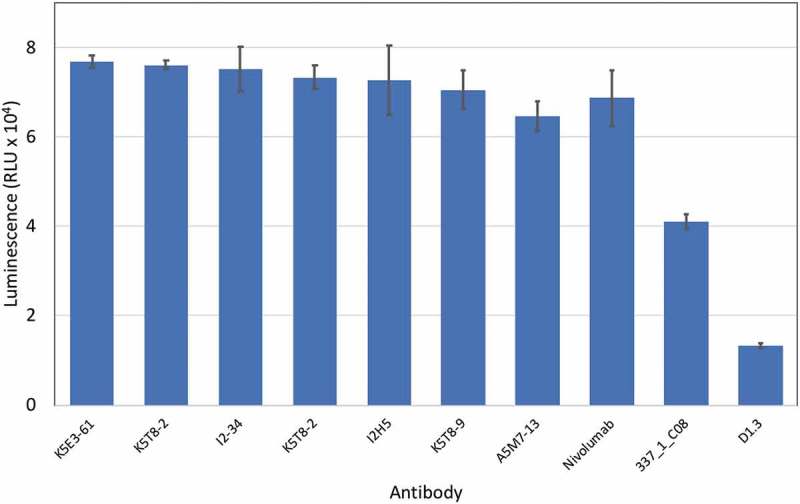
Inhibition of the interaction of PD-1 with PD-L1 in a cell reporter assay. PD-L1 artificial antigen presenting CHO-K1 (aAPC/CHO-K1) cells were co-cultured with PD-1 effector Jurkat cells, which stably express human PD-1 and a NFAT-RE-luciferase reporter, in the presence or absence of anti-PD-1 antibodies (50 nM final concentration). The luminescence signal in relative light units (RLU) is plotted on the y-axis for each antibody named on the x-axis.
Figure 7.
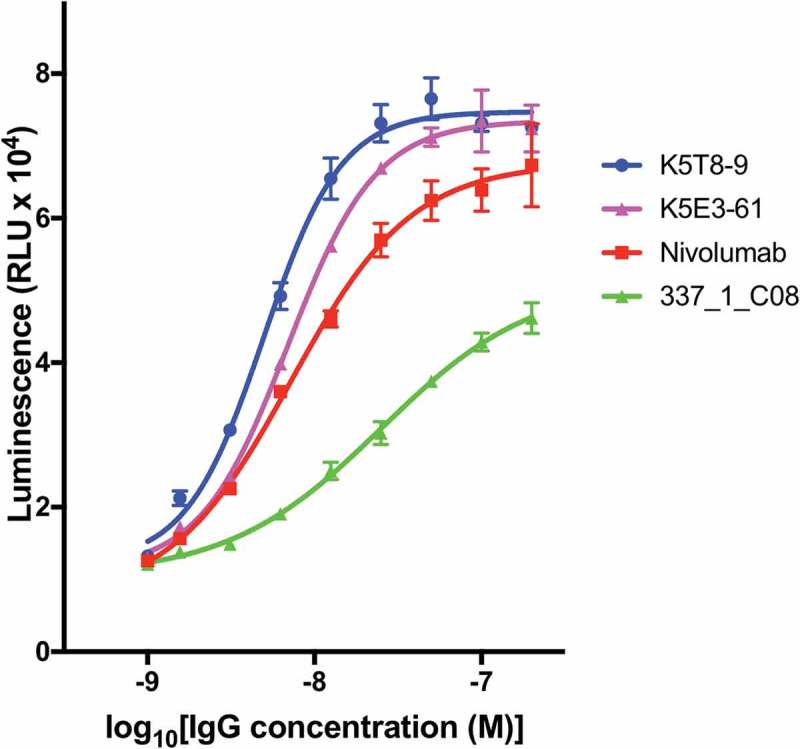
Concentration dependent inhibition of PD-1 blockade in a cell-based assay. PD-L1 artificial antigen presenting CHO-K1 (aAPC/CHO-K1) cells were co-cultured with PD-1 effector Jurkat cells, which stably express human PD-1 and a NFAT-luciferase reporter, in the presence or absence of varying concentrations of anti-PD-1 antibodies. The luminescence signal (y-axis) is plotted against the concentration of inhibitory antibody for K5T8-9, K5E3-61, nivolumab, and the parental “wild-type” clone 331_1_C08.
Table 3.
Potency of affinity matured anti- PD-1 antibodies in the inhibition of PD-1. The half-maximal response (EC50) of selected anti- PD-1 antibodies in the interaction of PD-1 with PD-L1 in a cell co-culture reporter assay was determined by fitting the data depicted in Figure 7 by non-linear regression using the dose-response (stimulation) equation (four parameter) using Prism 7 software (GraphPad Software Inc). The R-squared (R2) values statistical measure of how close the data are to the fitted regression line. VH CDR3 sequences are shown in column 3.
| VH | VL | VH | EC50 (nM) | R2 |
|---|---|---|---|---|
| 337_1_C08 | WT | DRDYYDAG |
25 | 0.982 |
| K5E3 | 61 | ..E.K... |
6.9 | 0.987 |
| K5T8 | 9 | ....K..T |
5 | 0.983 |
| Nivolumab | NDDY |
7.2 | 0.969 |
Discussion
Manufacture of antibodies for research, diagnostic and therapeutic application is typically carried out in IgG format using mammalian cells. Antibody display on mammalian cells permits rapid, high-throughput screening of millions of cells in the same format, expression environment and with the same post-translation modifications as found in cells used to manufacture proteins.
The recent development of TALEN and CRISPR/Cas9 technologies has enabled the creation of bespoke site-specific nucleases and has revolutionized genome engineering15 Engineered nucleases are routinely used to drive directed integration of transgenes, as well as site-specific modification of targeted genomic loci. Recent examples have extended this to the use of libraries of guide RNAs in genome-wide screens to identify functional perturbations arising from gene knock-outs.24,25 These nucleases have also been used to integrate single antibody genes to generate cells secreting a single antibody clone.26–28 Here, we demonstrate for the first time the successful application of site-specific nucleases to integrate repertoires of full-length IgG-formatted antibody genes at a single genomic locus in a standard cell line to create large libraries of 1–10 million cellular clones displaying antibodies on their surface. We demonstrate the functionality of these libraries by selecting novel, high affinity IgG-formatted binders recognizing PD-L1 and PD-1.
The use of nuclease-directed integration avoids the disadvantage of random integration, achieves transcriptional normalization and ensures consistent levels of display based on the inherent properties of the antibody itself. Library construction by nuclease-directed integration works because cleavage within the genome significantly increases the efficiency of DNA integration at the cleavage site through endogenous repair mechanisms, such as homologous recombination, non-homologous end joining or micro-homology-mediated end-joining.29 The current strategy targets the AAVS locus, which is considered a safe harbor for the insertion of transgenes. The AAVS locus is an intron within the PPP1R12C gene, and correct integration enables activation of an incoming promoter-less blasticidin gene through in-frame splicing to an upstream exon. Use of a splice-dependent drug resistance gene helps reduce the background from random integration.
Flow cytometry provides information on the extent of expression and the degree of antigen binding for a given level of expression for millions of clones. In contrast to phage display where hundreds of clones are screened after several rounds of selection, mammalian display has the distinct advantage that it is possible using flow cytometry to examine all members of the library for expression and binding, giving greater insight into the entire properties of the library. Thus, mammalian display coupled with flow cytometry has enabled “deep screening” of IgG-formatted antibody populations, including libraries diversified by chain shuffling and oligonucleotide-directed mutagenesis.
The underlying logic of antibody site-directed mutagenesis is that a variant of an initial binding sequence may be found where one or more amino acid substitutions within the sequence provides an improved binding solution. A number of approaches are available to search that “sequence space”, usually focused around the antibody CDR regions. Using saturation mutagenesis approaches, randomizing codons are introduced across sequence blocks (e.g., NNS covering 32 codons encoding all 20 amino acids and one stop codon). The potential diversity (32n nucleic acid and 20n protein variants with the example above) increases exponentially as the number of randomized residues (n) is increased. For example, randomization of an eight amino acid CDR region using NNS mutagenesis creates 2.5 × 1011 amino acid variants within 1012 DNA variants. Extensive randomization discards information, compromises binding for the majority of clones and creates a requirement for very large libraries for significant coverage of all variants. The ability to create libraries of billions of clones coupled with the power of phage display to extract rare variants with improved binding has enabled such extensive mutagenesis strategies to succeed.
A limitation of mammalian display has been the size of libraries that can be created, and so a more targeted approach to mutagenesis, retaining a higher proportion of the starting sequence, would enable a more efficient “local search” for variants around such a starting sequence. In fact, in many affinity maturation campaigns much of the original amino acid sequence is restored in the high affinity “solutions”.21,23 Thus, the smaller library size required by the directed mutagenic approach used here scales well with the potential size of mammalian display libraries. Such a targeted approach is possible by novel synthesis technology enabling the economic synthesis of 10,000s oligonucleotides. In total, 9,200 oligonucleotides are sufficient to explore every possible 2-mer mutation within an eight amino acid CDR region where all amino acids, with the exception of cysteine, are sampled. Combining two such libraries, e.g., VH CDR3 and VL CDR3, allows an exploration of a 16 amino acids region encompassing four amino acid changes, within a total diversity of 8 × 107 clones. In this study, we identified two main families of variants with either histidine or lysine replacing a central tyrosine residue in VH CDR3. The histidine variant appears to represent a local maximum with no additional benefits found by introduction of additional diversity within the VL CDR3. In contrast, binding of the lysine-bearing variants was supplemented by additional VL CDR3 diversity, leading to further affinity improvements.
Here, we worked with mammalian cell libraries of 107 clones that are useful for searching enriched populations, such as the output from phage display selection, affinity maturation libraries or libraries created from immunized sources. The benefits outlined above means that rather than sampling several thousand members, as occurs with phage display, these libraries can be screened in their entirety for affinity and/or specificity. It may also be possible to isolate binders directly from naïve libraries of 107 clones, and initial studies with phage display demonstrated that binders could indeed be generated directly from libraries of this size. This is not, however, an optimal size for routinely creating high affinity antibodies from non-enriched sources and larger libraries would be required. We have demonstrated transfection efficiencies of 5% using Maxcyte flow electroporation, a system that allows up to 1011 cells to be electroporated in a 30-min cycle. This makes it feasible to create libraries of up to 109 clones, albeit with a significant “overhead” compared to the construction of large phage display libraries. Library construction using nuclease-directed integration can be extended beyond antibody display libraries to include other binder formats. Antibodies and other binders secreted directly from cellular clones could be applied directly into cell-based reporter assays, eliminating extensive purification and without the complicating effects of contaminants from bacteria and yeast cells. As well as screening individual antibodies in direct binding assays, libraries of binders expressed within mammalian cells could be used in functional screens to identify clones, which directly affect cellular phenotype allowing direct selection for biological function.4,5,22 This could be done either on the cell surface (retaining a direct link between the antibody and its gene) or by secretion provided the secreted antibodies can be retained in proximity to the encoding cells (e.g., in semi-solid medium or by encapsulation of cells).
In summary, using nuclease-directed integration of antibody genes by CRISPR/Cas9 and TALE nucleases, we have constructed large libraries in mammalian cells containing a single antibody gene/cell. This permits construction of populations of millions of monoclonal stable cell lines displaying antibodies on their surface. In combination with flow cytometry or bead-based enrichment, this has permitted isolation of novel binders directly in an IgG format from mammalian cells.
Materials and methods
Vector construction
Display vectors pD2 and pD6 were created to display IgG-formatted antibodies and scFv-Fc-formatted antibodies, respectively. In initial construction of the dual promoter vector pD2, we elected to use different promoters to avoid regions of sequence duplication. Two commonly used promoters (CMV and elongation factor-1 (pEF)) were chosen. pEF was randomly assigned to drive the light chain with CMV to drive the heavy chain. In pD6, CMV promoter drives the expression of the scFv-Fc-formatted antibody. The sequence encoding the Fc domain in both vectors is followed by an intron and a separate exon encoding the TM domain from the PDGF receptor19 to anchor the antibody to the cell surface for cellular display. In this design, the exon encoding the TM domain is flanked by two Dre recombinase sites to give the option to remove this exon. Beyond this removable exon is an alternative exon encoding a stop codon giving rise to a secreted antibody. Both pD2 and pD6 plasmids are flanked on either side by a AAVS1 homology arm that facilitates the insertion of antibody-expressing cassette into the genome by homology-directed repair. An 804 bp left homology arm and an 836 bp right homology arm flanking the intended cleavage site within the AAVS locus were cloned on either side of the antibody expression cassette (Supplementary Figure 1). A promoter-less blasticidin gene preceded by a splice acceptor site is placed downstream of the left homology arm.
Subsequently, a second generation of mammalian display vectors (pINT17-BSD) with essentially the same structure was created to optimize the use of restrictions sites and co-ordinate with in house expression vectors. pINT17-BSD shares a similar topology to pD2 (Figure 1). In order to reduce the size of the construct, the TM domain was fused directly to the CH3 domain allowing the removal of the intervening intron. pINT17-BSD was constructed by PCR amplification of selected fragments from vectors previously described (WO2015166272A2) with the addition of restriction sites to enable their subsequent assembly. DNA encoding the AAVS-left homology arm, splice acceptor, blasticidin resistance gene, poly-adenylation site and the elongation factor 1 alpha promoter (pEF1α) originated from the pD2 plasmid by PCR amplification (1511 bp) with the addition of the 5ʹ AsiSI and 3ʹ BglII restriction enzymes. DNA encoding the Myc-tag and PDGFR transmembrane domain was PCR amplified from the pD2 plasmid with the addition of a 5ʹ IgG1 CH3 homology sequence and 3ʹ- HindIII site. DNA encoding the light chain BM40 leader, VL chain of the anti- lysozyme antibody D1.3, the human constant light (CL), bovine growth hormone (BGH) pA, the immediate early CMV promoter, a mouse heavy chain leader split by an intron, VH chain of the anti-lysozyme antibody D1.3 and the IgG1 antibody constant heavy domain 1 to 3 (IgG1 CH1-3) was PCR amplified from the previously described pINT3 plasmid (WO2015166272A2) with a 5ʹ BglII site and 3ʹ addition of DNA encoding the Myc-tag. The two fragments encoding the 4446 bp region of pINT17-BSD from BglII to HindIII were combined by PCR assembly to add the PDGR transmembrane domain directly to the CH3 terminus. The region from HindIII to SbfI encoding the AAVS right homology arm was PCR amplified (1168 bp) from the pD2 plasmid with the addition of the HindIII and SbfI restriction sites. The vector backbone encompassing the f1 and pUC origin of replications and kanamycin resistance gene from the SbfI to AsiSI sites originated from pSF-EF1alpha (Oxford Genetics OG43).
A pair of TALE nucleases18 that cleave within the human AAVS locus were used. AAVS1 CRISPR nuclease vector was constructed using primers to top strand 5ʹ-GGGGCCACTAGGGACAGGATGTTTT-3ʹ and to bottom strand oligo 5ʹ -ATCCTGTCCCTAGTGGCCCCCGGTG-3ʹ according to manufacturer’s instruction (A21175, Thermo Scientific). AAVS1 TALEN pair was from System Biosciences (Cat No GE601A-1).
Anti- PD-L1 library construction and chain shuffling
Construction of a human scFv phage display library (4 x 1010) and phage display selections were carried out as previously described22 using the Iontas phage display library with human PD-L1. scFv genes from a population arising from two rounds of selection on PD-L1 were recovered by PCR, digested with NcoI and Xho 1 and cloned into pD2 containing a pre-cloned light chain library.
For mammalian display, the recipient light chain library was prepared by cloning a repertoire of naturally re-arranged human kappa and lambda light chain genes from a panel of donors upstream of the CL domain into the vector pD2. This light chain repertoire can conveniently be combined with any individual VH clone or population of selected VHs to create an IgG–formatted chain–shuffled library. The light chain display library for chain-shuffling was created by cloning a repertoire of VL chain regions into the pD2 vector. VL libraries for kappa (pool of six kappa families) and lambda light chains (pool of three lambda families) were prepared separately with an average of 107 clones per library. The population of VHs selected on PD-L1 by phage display was cloned upstream of the IgG2 heavy chain constant domain to create an IgG-formatted, chain-shuffled library. VH genes from two rounds of PD-L1 selection were amplified and sub-cloned into the two light chain libraries. E. coli 10G electro-competent cells (Lucigen) were used for transformations to create a chain-shuffled library with an average of 107 clones per library.
Anti- PD-1 library construction
The anti-PD-1 antibody 337_1_C08 was identified by antibody phage display selection and screening using methods described previously.22 Geneblocks of the VH and VL where every 2-mer amino acid variation was encompassed within an 8-amino acid window (9216 variants) were synthesized by TWIST Biosciences. The VH gene was amplified using the primer pairs primers Forward: 5ʹ- CTTTCTCTCCACAGGCGCCCATGGCCGAAGTGCAGCC −3ʹ and Reverse: 5ʹ- TTTTTTCTCGAGACGGTGACCAGGGTTC −3ʹ. The VL gene was amplified using the primer pairs primers Forward: 5ʹ- TTTTTTGCTAGCTCCTATGAGCTGACTC −3ʹ and Reverse: 5ʹ- GTCACGCTTGGTGCGGCCGCGGGCTGACCTAG −3ʹ. A “stuffer” fragment encoding the CL and the CMV promoter were PCR amplified from the pINT17-BSD vector using the primer pairs: forward: 5ʹ- GGCCGCACCAAGCGTGAC −3ʹ and reverse: 5ʹ- GGCGCCTGTGGAGAGAAAG −3ʹ. The three gene fragments were first assembled in a “mock” PCR with no amplification primers to ensure no bias was introduced by partial assembly and subsequent amplification. The mock PCR was performed by mixing the three PCR products above at an equimolar ratio (90 nM each) and performing a PCR reaction with the KOD Hot Start Master Mix (Sigma, 71842) according to the manufacturer’s instructions with an annealing temperature of 60°C and elongation temperature of 68°C (45 s). The assembled product was subsequently amplified using the primer pairs primers Forward: 5ʹ- TTTTTTGCTAGCTCCTATGAGCTGACTC −3ʹ and Reverse: 5ʹ- TTTTTTCTCGAGACGGTGACCAGGGTTC −3ʹ. The assembled anti-PD-1 VH and VL CDR3 library was digested with NheI and XhoI restriction enzymes and cloned into the pINT17-BSD targeting vector and by electroporation of the E. coli strain “E. cloni 10G SUPREME” Electrocompetent Cells (Lucigen cat# 60081–1), a library size of 1.1 × 108 was created, as determined by counting individual kanamycin-resistant colonies on agar plates plated with dilutions of the transformation mix. Transfection-quality plasmid DNA was prepared and used to co-transfect HEK293 suspension cells (1.35 x 109 cells) by Maxcyte electroporation with TALE AAVS left and right arm nucleases to enable single-copy antibody gene integration. The efficiency of gene targeting was 0.8%, as determined by counting blasticidin resistant colony forming units (CFUs) in dilution plates post transfection, to yield a mammalian display library size of 10.8 million. The library was propagated for 7 days under blasticidin selection, and then selected by anti-Fc MACS.
Cell culture and transfection
HEK293 cells from LifeTech (HEK293F) were cultured in FreeStyle 293 expression medium (Invitrogen) according to the manufacturer’s instructions. Cells were transfected using either PEI-mediated transfection (Polyplus), Maxcyte electroporation or Amaxa nucleofection. Donor plasmid could be a single clone or a library, and nuclease could be either Talens or CRISPR-Cas9.
For Maxcyte electroporation, cells were seeded at 0.5 × 106 cells/ml one day before electroporation to ensure that the cells were in the log-phase. Cells were harvested by centrifugation at 200 g for 10 min and resuspended in MaxCyte Electroporation Buffer at a density of 1 × 108 cells/ml. 212ugs of total DNA (donor plasmid plus nuclease plasmid) was electroporated per 1 × 108 cells using MaxCyte STX electroporation device. Following electroporation, cells were allowed to recover at 37°C for 20 min, diluted in the culture medium and maintained at 120rpm, 37°C under 5% CO2. Amaxa nucleofection was done according to manufacturer’s instructions and PEI-mediated transfection was done as described previously.30 Blasticidin selection was started 48 h after transfection and the population was kept under selection through the whole experiment. In order to calculate the library size, transfected cells were plated in a 10 cm tissue culture dish and grown in 10% Dulbecco’s Modified Eagle Medium with blasticidin (7.5 μg/ml) for 12–14 days. Colonies were stained using 2% methylene blue (in 50% methanol) and counted using OpenCFU software.31
Cells staining and sorting
MACS was performed to enrich cells either based on surface IgG expression or antigen binding using LS column in a MidiMACS Separator according to the manufacturer’s instructions (Militenyi Biotech Ltd.). Antibody expressing cells were isolated between 6 and 8 dpt by staining with anti-human Fc PE (BioLegend, Cat. No. 409304) for 30 min at 4°C, followed by labeling with anti-PE microbeads (Militenyi Biotech Ltd.). Cells that bound antigen were isolated by staining with the biotinylated antigen at different concentrations followed by streptavidin microbeads (Militenyi Biotech Ltd.). Sorted cells were expanded and used for further rounds of sorting either by MACS or FACS.
FACS was done using BD Influx High-Speed Cell Sorter (Becton, Dickinson) where the cells are sorted based on expression using anti-human Fc PE and antigen binding using biotinylated antigen and streptavidin APC. Either DAPI or 7-AAD was used as a viability stain for dead cell exclusion.
Analytical flow cytometry was performed routinely at different time points and analyzed (Beckton Dickinson LSRII or Intellicyt). Flow cytometry data were analyzed using FlowJo software. Dead cells were excluded from the analysis.
Recovery of antibody genes
Genomic DNA was prepared from the FACS-sorted cell population; DNA encoding the IgG insert was amplified by nested PCR using KOD Hot Start DNA polymerase (Merck Millipore). Outer PCR was performed with the following genome-specific primers Forward: 5ʹ-CCGGAACTCTGCCCTCTAAC-3ʹ and Reverse: 5ʹ-TCCTGGGATACCCCGAAGAG-3ʹ. PCR product from the outer PCR was used as a template to amplify the integrated IgG insert with following primers; Forward: 5ʹ-GAGGGCCTGGATCTTCTTTCTC-3ʹ, Reverse: 5ʹ-GAAGTAGTCCTTGACCAGGCAG-3ʹ. Cycling conditions for the internal PCR were 95°C for 3 min, 30 cycles of 95°C for 20 sec, 60°C for 10 sec, 70°C for 90 sec and then 72°C for 5 min. PCR products were gel purified and digested with NheI and XhoI and cloned into the pINT3 mammalian expression vector. Clones were picked into three 96-well plates per selection, sequenced, expressed in HEK293 cells and screened for positive clones.
Next-generation sequencing analysis
VH genes were amplified using the primer pairs primers Forward: 5ʹ- TCTCTCCACAGGCGCCATGG −3ʹ and Reverse: 5ʹ- CCCTTGGTGGAGGCACTCGAG −3ʹ. VL genes were amplified using the primer pairs primers Forward: 5ʹ- CTTTGCCTGGCCGGGAGGGCTCTGGC −3ʹ and Reverse: 5ʹ- AGTCACGCTTGGTGCGGCCGC −3ʹ. Library preparation for sequencing was done using TruSeq PCR-free kit according to manufacturer’s instructions (Illumina) by the sequencing facility (Department of Biochemistry, University of Cambridge). Library pools were sequenced on the Illumina MiSeq platform using the MiSeq Nano Kit V2 (500 cycles, paired-end). Sequence analysis including the cluster generation and amino acid distribution charts were performed using the Geneious Biologics online platform.
Affinity ranking ELISA
Monoclonal ELISA was carried out as previously described22 with the following alterations. Affinity ranking with capture ELISA was carried out by immobilizing anti-Fc human antibodies (Jackson labs) on 96-well Nunc Maxisorp plates at 2.5 μg/ml overnight at 4○C. Plates were washed 3 times with phosphate-buffered saline (PBS) and blocked using 3% Marvel-PBS for 1 h at room temperature. After blocking plates were washed with PBS and cell culture supernatants containing the secreted antibodies were incubated for 1 h at room temperature, washed (3x PBS-Tween followed by 3x PBS) and probed with biotinylated antigen at various concentrations and detected with Europium-conjugated streptavidin. Absorbance was measured using PHERAstar plate reader.
To measure antigen binding, Nunc Maxisorp plates were coated with antigens at 3–5 ug/ml overnight at 4○C. Blocking and washing was performed as described above. Bound IgG was detected using Europium-labelled anti-IgG antibody (DELFIA Eu-N1 anti-human IgG, 1244–330, Perkin Elmer).
Surface plasmon resonance assays
Surface plasmon resonance (SPR) experiments were performed using a BIAcore T100 instrument and followed the protocol according to the BIAcore sensor chip protein A (29127556). The SPR experiments with the MASS2 instrument were performed according to the manufacturer’s instructions (Sierra Sensors) with all measurements performed at 25°C. Briefly, recombinant protein G (Pierce, 21193) was covalently coupled to high capacity amine sensors (SPR-AS-HCA-03) following activation with EDC/NHS. Ethanolamine was subsequently used to quench residual active groups. Approximately 5000 RU of protein G was immobilized per spot. Antibodies were captured at 10 μL/min to achieve an average response of 400 RU. Monomeric human PD-1 rCD4 antigen (see Supplementary Figure 4 legend for antigen source) was injected at a flow rate of 30 μL/min for 60 s; dissociation was set to 180 s. The chip was regenerated using 10 mM glycine-HCl, pH 1.5. Representative SPR sensorgram traces are shown in Supplementary Figure 5.
PD-1/PD-L1 co-culture cell response assay
PD-1/PD-L1 co-culture cell response assay (single point and EC50 analysis) was conducted according to the manufacturer’s instructions (Promega – J1250). In this assay, two cell types are used: CHO cells expressing PD-L1 and Jurkat T-cells expressing PD-1. The TCR complexes present on Jurkat cells are fully activated upon interaction with the TCR-activating complex, present on CHO cells, resulting in constitutive NFAT-luciferase reporter activity. Co-cultivation of the two cell lines results in PD-1/PD-L1 interaction, which prevents TCR activation and down-regulation/suppression of NFAT-luciferase reporter activity; pre-incubation of a blocking anti-PD-L1 antibody to HEK293 cells before co-culture prevents PD-1 binding to PD-L1, allowing TCR activation and resulting in downstream NFAT-luciferase reporter activity. A PD-1/PD-L1 cell reporter, anti-PD-L1 antibody concentration dose response assay was carried out and experimental data were fitted (Sigmoidal Dose Response-Variable Slope) using GraphPad Prism and EC50 values for each antibody obtained.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Abbreviations
AAVS1 adeno-associated virus integration site 1
Cas9 CRISPR associated protein 9
CDR Complementarity-determining region
CRISPR Clustered Regularly Interspaced Short Palindromic Repeats
Dpt Days post transfection
EC50 Half maximal effective concentration
ELISA Enzyme-linked immunosorbent assay
FACS Fluorescence-activated cell sorting
FGF Fibroblast growth factor
FLP Flippase recombinase
MACS Magnetic-activated cell sorting
NFAT Nuclear factor of activated T cells
PD-1 Programmed cell death protein 1
PD-L1 Programmed death-ligand 1
PDGF Platelet-derived growth factor
PEI Polyethylenimine
PPP1R12C Protein phosphatase 1, regulatory subunit 12C
scFv Single-chain variable fragment
SPR Surface plasmon resonance
TALE Transcription activator-like effector
VH Variable heavy
VL Variable light
Supplemental material
Supplementary data for this article can be accessed here.
References
- 1.Bradbury ARM, Sidhu S, Dubel S, McCafferty J.. Beyond natural antibodies: the power of in vitro display technologies. Nat Biotechnol. 2011;29:245–54. doi: 10.1038/nbt.1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Binz HK, Amstutz P, Pluckthun A. Engineering novel binding proteins from nonimmunoglobulin domains. Nat Biotechnol. 2005;23:1257–68. doi: 10.1038/nbt1127. [DOI] [PubMed] [Google Scholar]
- 3.Chao G, Lau WL, Hackel BJ, Sazinsky SL, Lippow SM, Wittrup KD. Isolating and engineering human antibodies using yeast surface display. Nat Protoc. 2006;1:755–68. doi: 10.1038/nprot.2006.94. [DOI] [PubMed] [Google Scholar]
- 4.Melidoni AN, Dyson MR, Wormald S, McCafferty J. Selecting antagonistic antibodies that control differentiation through inducible expression in embryonic stem cells. Proc Natl Acad Sci. 2013;110:17802–07. doi: 10.1073/pnas.1312062110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang H, Wilson IA, Lerner RA. Selection of antibodies that regulate phenotype from intracellular combinatorial antibody libraries. Proc Natl Acad Sci. 2012;109:15728–33. doi: 10.1073/pnas.1214275109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Beerli RR, Bauer M, Buser RB, Gwerder M, Muntwiler S, Maurer P, Saudan P, Bachmann MF. Isolation of human monoclonal antibodies by mammalian cell display. Proc Natl Acad Sci. 2008;105:14336–41. doi: 10.1073/pnas.0805942105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Breous-Nystrom E, Schultze K, Meier M, Flueck L, Holzer C, Boll M, Seibert V, Schuster A, Blanusa M, Schaefer V, et al. Retrocyte Display® technology: generation and screening of a high diversity cellular antibody library. Methods. 2013;65:1–11. [DOI] [PubMed] [Google Scholar]
- 8.Waldmeier L, Hellmann I, Gutknecht CK, Wolter FI, Cook SC, Reddy ST, Grawunder U, Beerli RR. Transpo-mAb display: transposition-mediated B cell display and functional screening of full-length IgG antibody libraries. Mabs. 2016;8:726–40. doi: 10.1080/19420862.2016.1196521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sadelain M, Papapetrou EP, Bushman FD. Safe harbours for the integration of new DNA in the human genome. Nat Rev Cancer. 2012;12:51–58. doi: 10.1038/nrc3179. [DOI] [PubMed] [Google Scholar]
- 10.Zhou C, Jacobsen FW, Cai L, Chen Q, Shen WD. Development of a novel mammalian cell surface antibody display platform. Mabs. 2010;2:508–18. doi: 10.4161/mabs.2.5.13089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li CZ, Liang ZK, Chen ZR, Lou HB, Zhou Y, Zhang ZH, Yu F, Liu S, Zhou Y, Wu S, et al. Identification of HBsAg-specific antibodies from a mammalian cell displayed full-length human antibody library of healthy immunized donor. Cell Mol Immunol. 2012;9:184–90. doi: 10.1038/cmi.2011.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Matreyek KA, Stephany JJ, Fowler DM. A platform for functional assessment of large variant libraries in mammalian cells. Nucleic Acids Res. 2017;45:e102. doi: 10.1093/nar/gkx183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rouet P, Smih F, Jasin M. Introduction of double-strand breaks into the genome of mouse cells by expression of a rare-cutting endonuclease. Mol Cell Biol. 1994;14:8096–106. doi: 10.1128/mcb.14.4.2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Porteus MH, Baltimore D. Chimeric nucleases stimulate gene targeting in human cells. Science. 2003;300:763. doi: 10.1126/science.1078395. [DOI] [PubMed] [Google Scholar]
- 15.Bogdanove AJ, Voytas DF. TAL effectors: customizable proteins for DNA targeting. Science. 2011;333:1843–46. doi: 10.1126/science.1204094. [DOI] [PubMed] [Google Scholar]
- 16.Sampson TR, Weiss DS. Exploiting CRISPR/Cas systems for biotechnology. Bioessays. 2014;36:34–38. doi: 10.1002/bies.v36.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mason DM, Weber CR, Parola C, Meng SM, Greiff V, Kelton WJ, Reddy ST. High-throughput antibody engineering in mammalian cells by CRISPR/Cas9-mediated homology-directed mutagenesis. Nucleic Acids Res. 2018. doi: 10.1093/nar/gky550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Luo Y, Liu C, Cerbini T, San H, Lin Y, Chen G, Rao MS, Zou J. Stable enhanced green fluorescent protein expression after differentiation and transplantation of reporter human induced pluripotent stem cells generated by AAVS1 transcription activator-like effector nucleases. Stem Cells Transl Med. 2014;3:821–35. doi: 10.5966/sctm.2013-0212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gronwald RGK, Grant FJ, Haldeman BA, Hart CE, O’Hara PJ, Hagen FS, Ross R, Bowen-Pope D, Murray MJ. Cloning and expression of a cDNA coding for the human platelet-derived growth factor receptor: evidence for more than one receptor class. Proc Natl Acad Sci. 1988;85:3435-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Foote J, Winter G. Antibody framework residues affecting the conformation of the hypervariable loops. J Mol Biol. 1992;224:487–99. doi: 10.1016/0022-2836(92)91010-M. [DOI] [PubMed] [Google Scholar]
- 21.Dyson MR, Zheng Y, Zhang C, Colwill K, Pershad K, Kay BK, Pawson T, McCafferty J. Mapping protein interactions by combining antibody affinity maturation and mass spectrometry. Anal Biochem. 2011;417:25–35. doi: 10.1016/j.ab.2011.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schofield DJ, Pope AR, Clementel V, Buckell J, Chapple S, Clarke KF, Conquer JS, Crofts AM, Crowther SR, Dyson MR, et al. Application of phage display to high throughput antibody generation and characterization. Genome Biol. 2007;8:R254. doi: 10.1186/gb-2007-8-5-r81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.DiCara DM, Chirgadze DY, Pope AR, Karatt-Vellatt A, Winter A, Slavny P, van Den Heuvel J, Parthiban K, Holland J, Packman LC, et al. Characterization and structural determination of a new anti-MET function-blocking antibody with binding epitope distinct from the ligand binding domain. Sci Rep. 2017;7:9000. doi: 10.1038/s41598-017-09460-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelsen TS, Heckl D, Ebert BL, Root DE, Doench JG, et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science. 2014;343:84–87. doi: 10.1126/science.1247005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang T, Wei JJ, Sabatini DM, Lander ES. Genetic screens in human cells using the CRISPR-Cas9 system. Science. 2014;343:80–84. doi: 10.1126/science.1246981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cristea S, Freyvert Y, Santiago Y, Holmes MC, Urnov FD, Gregory PD, Cost GJ. In vivo cleavage of transgene donors promotes nuclease-mediated targeted integration. Biotechnol Bioeng. 2013;110:871–80. doi: 10.1002/bit.24733. [DOI] [PubMed] [Google Scholar]
- 27.Sakuma T, Takenaga M, Kawabe Y, Nakamura T, Kamihira M, Yamamoto T. Homologous Recombination-Independent Large Gene Cassette Knock-in in CHO Cells Using TALEN and MMEJ-Directed Donor Plasmids. Ijms. 2015;16:23849–66. doi: 10.3390/ijms161023849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pogson M, Parola C, Kelton WJ, Heuberger P, Reddy ST. Immunogenomic engineering of a plug-and-(dis)play hybridoma platform. Nat Commun. 2016;7:12535. doi: 10.1038/ncomms12535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McVey M, Lee SE. MMEJ repair of double-strand breaks (director’s cut): deleted sequences and alternative endings. Trends Genet. 2008;24:529–38. doi: 10.1016/j.tig.2008.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chapple SDJ, Crofts AM, Shadbolt SP, McCafferty J, Dyson MR. Multiplexed expression and screening for recombinant protein production in mammalian cells. BMC Biotechnol. 2006;6. doi: 10.1186/1472-6750-6-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Geissmann Q. OpenCFU, a new free and open-source software to count cell colonies and other circular objects. PLoS One. 2013;8:e54072. doi: 10.1371/journal.pone.0054072. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.