

ABSTRACT
Typical crystallizable fragment (Fc) glycans attached to the CH2 domain in therapeutic monoclonal antibodies (mAbs) are core-fucosylated and asialo-biantennary complex-type glycans, e.g., G2F (full galactosylation), G1aF (terminal galactosylation on the Man α1-6 arm), G1bF (terminal galactosylation on the Man α1-3 arm), and G0F (non-galactosylation). Terminal galactose (Gal) residues of Fc-glycans are known to influence effector functions such as antibody-dependent cell-mediated cytotoxicity and complement-dependent cytotoxicity (CDC), but the impact of the G1F isomers (G1aF and G1bF) on the effector functions has not been reported. Here, we prepared four types of glycoengineered anti-CD20 mAbs bearing homogeneous G2F, G1aF, G1bF, or G0F (G2F mAb, G1aF mAb, G1bF mAb, or G0F mAb, respectively), and evaluated their biological activities. Interestingly, G1aF mAb showed higher C1q- and FcγR-binding activities, CDC activity, and FcγR-activation property than G1bF mAb. The activities of G1aF mAb and G1bF mAb were at the same level as G2F mAb and G0F mAb, respectively. Hydrogen–deuterium exchange/mass spectrometry analysis of dynamic structures of mAbs revealed the greater involvement of the terminal Gal residue on the Man α1-6 arm in the structural stability of the CH2 domain. Considering that mAbs interact with FcγR and C1q via their hinge proximal region in the CH2 domain, the structural stabilization of the CH2 domain by the terminal Gal residue on the Man α1-6 arm of Fc-glycans may be important for the effector functions of mAbs. To our knowledge, this is the first report showing the impact of G1F isomers on the effector functions and dynamic structure of mAbs.
Abbreviations: ABC, ammonium bicarbonate solution; ACN, acetonitrile; ADCC, antibody-dependent cell-mediated cytotoxicity; C1q, complement component 1q; CDC, complement-dependent cytotoxicity; CQA, critical quality attribute; Endo, endo-β-N-acetylglucosaminidase; FA, formic acid; Fc, crystallizable fragment; FcγR, Fcγ receptors; Fuc, fucose; Gal, galactose; GlcNAc, N-acetylglucosamine; GST, glutathione S-transferase; HER2, human epidermal growth factor receptor 2; HDX, hydrogen–deuterium exchange; HILIC, hydrophilic interaction liquid chromatography; HLB-SPE, hydrophilic-lipophilic balance–solid-phase extraction; HPLC, high-performance liquid chromatography; mAb, monoclonal antibody; Man, mannose; MS, mass spectrometry; PBS, phosphate-buffered saline; SGP, hen egg yolk sialylglycopeptides.
KEYWORDS: Therapeutic monoclonal antibody, glycoengineering, galactose, complement-dependent cytotoxicity, antibody-dependent cell-mediated cytotoxicity
Introduction
Monoclonal antibodies (mAbs) are one of the most successful biological drug classes, and more than 40 mAb products have been approved for use in the United States, Japan, and the European Union.1,2 Currently, therapeutic mAbs are being used for treatments of various diseases including cancers and inflammatory diseases.3 Certain types of mAbs exhibit their efficacy via immune effector functions, such as antibody-dependent cell-mediated cytotoxicity (ADCC) and complement-dependent cytotoxicity (CDC).4 ADCC and CDC are important as major pharmacological functions of some cancer therapeutics, such as rituximab, trastuzumab, and alemtuzumab.5 ADCC is caused by activation of immune effector cells via the interactions of the crystallizable fragment (Fc) with Fcγ receptors (FcγRs).6 CDC is triggered by an interaction between the Fc region and complement component 1q (C1q), resulting in complement cascade activation and subsequent lysis of target cells. Thus, these Fc-mediated effector functions of mAbs could be controlled by modulating Fc–FcγRs and Fc–C1q interactions. It is known that Fc glycosylation is one of the important factors that affect these interactions and effector functions, and the glycan heterogeneity is considered as one of the critical attributes associated with the efficacy of therapeutic mAb products.
Mammalian cell-produced therapeutic mAbs are N-glycosylated at the Asn297 residue (EU numbering) in the CH2 domain of the Fc region.7 The major glycans in the Fc region are core-fucosylated biantennary complex-type glycans bearing 0–2 moles of non-reducing terminal galactose (Gal) residues (G0F, G1F, and G2F). G1F has two isomers, G1aF and G1bF. The Gal residue of G1aF and G1bF are located on the non-reducing terminal of mannose (Man) α1-6 and Man α1-3 arms of G1F glycan, respectively (Figure 1).8,9 Previous studies have reported that certain components of Fc-glycans dramatically affect effector functions.4,10 For example, the removal of the core-fucose (Fuc) residue enhances ADCC by increasing the binding affinity of mAb to FcγRIII.11,12 Although it has been reported that Fc-galactosylation does not affect ADCC, recent studies have shown that mAbs with galactosylated Fc-glycan exhibit higher ADCC than those with agalactosylated Fc-glycan.13,14 Fc-glycan galactosylation also enhances C1q-binding and CDC activities of IgG antibodies.15 However, it is unclear whether the effector functions differ between mAbs bearing major isomeric glycans, such as mAb bearing G1aF glycan (G1aF mAb) and mAb bearing G1bF glycan (G1bF mAb).
Figure 1.
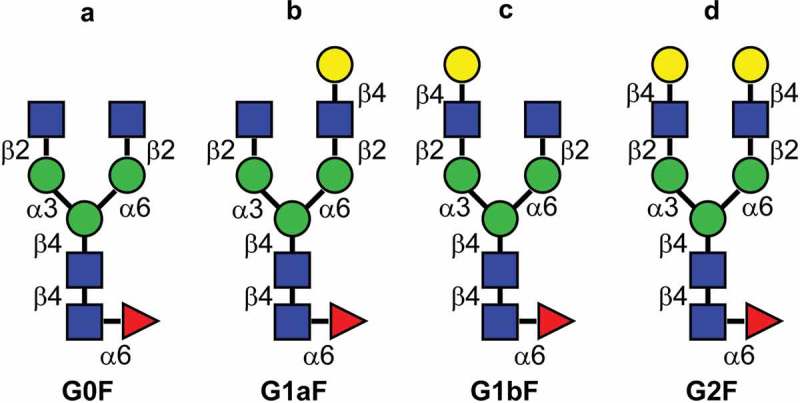
Major glycan structures of therapeutic mAbs. (a) Core-fucosylated agalacto-biantennary complex-type glycan (G0F); (b) core-fucosylated biantennary complex-type glycan with galactosylation on the Man α1-6 arm (G1aF); (c) core-fucosylated biantennary complex-type glycan with galactosylation on the Man α1-3 arm (G1bF); (d) core-fucosylated and fully galactosylated biantennary complex-type glycan (G2F). Yellow circle, Gal; green circle, Man; blue square, N-acetylglucosamine; red triangle, fucose.
Chemoenzymatic transglycosylation technology for preparation of mAbs with homogeneous Fc-glycan has made remarkable progress recently. Using glycoengineered mAbs with homogeneous Fc-glycan, the effects of each glycan on biological functions and physicochemical properties have been actively investigated.16–20 For example, Huang et al. reported that an anti-CD20 mAb with G2 glycan (non-fucosylated and fully galactosylated complex type biantennary) showed higher FcγRIIIa binding activity compared with commercially available rituximab.18 Kurogochi et al. also prepared anti-human epidermal growth factor receptor 2 (HER2) mAbs with homogeneous pauchi-mannose type glycan and non-fucosylated glycans (G0, G2 and fully sialylated G2 glycans), and demonstrated that the anti-HER2 mAb with G2 glycan exhibited higher FcγRIIIa binding activity than the mAbs with other glycans.19 Recently, Wada et al. produced recombinant human mAbs with G0, G2, fully sialylated G2, G0F, G2F, and fully sialylated G2F glycans, and showed impacts of each of the Fc-glycans on biological activities, thermal stabilities, and aggregation propensities in detail.17 As shown in these previous reports, the effects of agalactosylated, fully galactosylated glycans, or fully sialylated Fc-glycans on biological activities and physicochemical properties of mAbs are well documented; however, the effects of structural galactosylated isomers in Fc-glycans on biological and physicochemical properties of mAbs have not been reported previously.
In this study, using the original chemoenzymatic transglycosylation approach, we prepared four anti-CD20 mAbs bearing homogeneous complex-type biantennary glycans: G2F (full galactosylation of both Man α1-3 and Man α1-6 arms), G1aF, G1bF, and G0F (non-galactosylation). We evaluated their biological activities (antigen-binding, FcγR- and C1q-binding, CDC, and FcγR-activation), and analyzed their structural stability. To the best of our knowledge, this study is the first to demonstrate the different impacts of G1F isomers on effector functions and the dynamic structure of a mAb.
Results
Preparation of anti-CD20 mAbs with homogeneous glycans
Starting with commercially available anti-CD20 rituximab (Rituxan®), we prepared four types of glycoengineered anti-CD20 mAbs (G0F mAb, G1aF mAb, G1bF mAb, and G2F mAb) with core-fucosylated homogeneous glycans by a chemoenzymatic approach using some endo-β-N-acetylglucosaminidases (Endo) and oxazolinated glycans. Figure 2 shows the schematic of the workflow for sample preparation. All oxazolinated glycans (G2-Oxa, G1a-Oxa, G1b-Oxa, and G0-Oxa) were prepared using hen egg yolk sialylglycopeptides (SGP). G2-Oxa and G0-Oxa were prepared as previously reported.19,21 G1a-OH and G1b-OH were prepared by treating SGP with mild acid, β1-4 galactosidase, α2-3, 6, 8 neuraminidase, and wild-type Endo S,20,22,23 and finally converted into oxazolinated glycans (G1a-Oxa and G1b-Oxa) as shown in Supplementary Figure S1.19–21 Their structures were confirmed by multiple-stage mass spectrometry (MS) and 1H-NMR analysis (see “Preparation of oxazolinated glycans” in Materials and Methods).24 For truncating Fc-glycans, anti-CD20 mAbs were digested with wild-type Endo S and Endo D. The mAbs with truncated Fc-glycans were transglycosylated with Endo F3 mutant (D165Q), which shows high substrate specificity toward (Fucα1-6)GlcNAcβ1-Asn group in transglycosylation,25 in the presence of each oxazolinated glycan. The anti-CD20 mAbs with two core-fucosylated homogeneous N-glycan chains were separated from those with (±Fucα1-6)GlcNAcβ1-Asn group(s) using cation-exchange column chromatography.
Figure 2.

Preparation of anti-CD20 mAbs with core-fucosylated homogeneous N-glycans. This illustration details the preparation of anti-CD20 mAbs having two N-glycan chains with G2F structure. (1) digestion with wild-type Endo S and Endo D for truncating N-glycans, (2) transglycosylation with Endo F3 mutant (D165Q) in the presence of oxazolinated glycans as a glycan donor, (3) the separation and purification of anti-CD20 mAbs with two core-fucosylated N-glycan chains using cation-exchange column chromatography. Yellow circle, Gal; green circle, Man; blue square, N-acetylglucosamine; red triangle, fucose.
Glycan homogeneities of glycoengineered anti-CD20 mAbs
To confirm the glycan homogeneities of glycoengineered anti-CD20 mAbs, Fc-glycans were enzymatically released, followed by labeling with a 2-AB fluorescent dye and analysis using high-performance liquid chromatography (HPLC). Glycan profiling of the commercially available anti-CD20 therapeutic mAb product resulted in the detection of 2AB-labeled G0F, G1aF, G1bF, and G2F, in this order, during retention times of 20–30 min (Figure 3(a)).26 Figure. 3(b–e) shows the glycan profiles of G0F mAb, G1aF mAb, G1bF mAb, and G2F mAb, respectively. The glycan purity (%) of each glycoengineered anti-CD20 mAb was 95.0–96.9% (Supplementary Table S1). To further clarify the homogeneity of the glycoengineered mAb, we then evaluated the transglycosylation level of each mAb by analyzing site-specific N-glycosylation of tryptic digests from mAbs. Fc-glycopeptides with a GlcNAc and non-glycosylated peptides were not detected in all mAbs (Table 1). Although Fc-glycopeptides with a (Fucα1-6)GlcNAc were detected in G0F mAb and G2F mAb, the content ratios were sufficiently low (≤1.2%). To confirm the effects of terminal Gal residues of N-glycans on the antigen-binding properties of glycoengineered anti-CD20 mAbs, we performed a cell-based competitive binding assay (Supplementary Figure S2). This revealed that all glycoengineered mAbs we used in this study have similar CD20-binding properties, suggesting that the galactosylation level does not affect the antigen-binding properties of anti-CD20 mAbs. These data confirmed the high Fc-glycan homogeneities and similar antigen binding properties of each glycoengineered anti-CD20 mAb used in this study.
Figure 3.
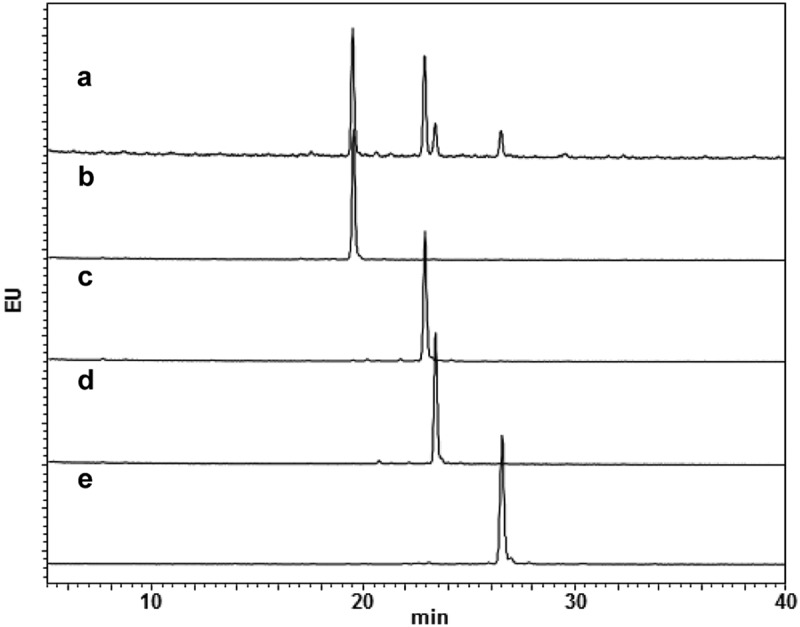
Glycan profiles of mAbs with homogeneous glycans. Glycan profiles of commercially available anti-CD20 mAb (a) and glycoengineered G0F mAb (b), G1aF mAb (c), G1bF mAb (d), and G2F mAb (e). The glycan profiles were obtained using HPLC analysis of 2-AB-labeled glycans.
Table 1.
Fc-glycopeptides analysis of mAbs with homogenous glycan.
| Deduced glycan | Relative peak area (%) of each glycopeptide against total peak area of EEQYNSTYR peptides |
|||
|---|---|---|---|---|
| G0F-mAb | G1aF-mAb | G1bF-mAb | G2F-mAb | |
| G0F lacking a GlcNAc | –a | 0.7 | 0.6 | – |
| G0F | 98.4 | 1.1 | 0.2 | – |
| G1F (G1aF + G1bF) | 0.8 | 97.9 | 98.4 | 0.9 |
| G2F | 0.3 | 0.3 | 0.9 | 97.9 |
| (Fucα1-6)GlcNAcb | 0.5 | – | – | 1.2 |
| GlcNAcb | – | – | – | – |
| non-glycosylated peptide | – | – | – | – |
EEQYNSTYR peptide was obtained by tryptic digestion of mAbs. a, not detected; b, reducing terminal N-acetylglucosamine from Fc-glycan; underline, N-glycosylation site at Asn 297(EU numbering). Structures of G0F, G1aF, G1bF, and G2F: see Figure 1. GlcNAc, N-acetylglucosamine; Fuc, fucose.
Evaluation of biological activities of glycoengineered anti-CD20 mAbs: CDC activities
We previously reported that, using Fc receptor affinity column chromatography, mAbs can be separated into three fractions that show different galactosylation levels, and that their CDC activity is correlated with the abundance of terminal Gal residues of N-glycans.13 To examine the effects of terminal Gal residues on the Man α1-3 and Man α1-6 arms of Fc-glycans on the effector functions of mAbs, we evaluated the CDC activities of G0F mAb, G1aF mAb, G1bF mAb, and G2F mAb (Figure 4(a)). As expected, G2F mAb showed higher CDC activities than G0F mAb. Interestingly, a difference in the biological activities of G1aF mAb and G1bF mAb was clearly detected: G1aF mAb showed higher activity than G1bF mAb. The activities of G1aF mAb and G1bF mAb were at the same level as G2F mAb and G0F mAb, respectively. In addition, we assessed the CDC activities of G1aF mAb and G1bF mAb mixtures in different ratios (Figure 4(b)). The results showed that the CDC activity of the mixture increased with an increasing proportion of G1aF mAb. Reportedly, C1q-binding activities of mAbs are associated with their CDC activities.18 We then assessed the binding activities of the glycoengineered mAbs to human C1q. As shown in Figure 4(c), similar to the CDC activity, the C1q-binding activity was increased as follows: G2F = G1aF > G1bF = G0F. These results suggested that the terminal Gal residue on the Man α1-6 arm of Fc-glycans plays an important role in the CDC activity.
Figure 4.

CDC and C1q-binding activities of glycoengineered anti-CD20 mAbs. (a, b) CDC activities of glycoengineered anti-CD20 mAbs. Raji cells were incubated with 16% human serum and were serially diluted with glycoengineered anti-CD20 mAbs. (a) Percentages of cell lysis plotted against mAb concentrations. (b) CDC activity of 1 µg/ml of G1aF mAb and G1bF mAb mixtures in different ratios. (c) C1q binding of glycoengineered anti-CD20 mAbs. Raji cells were opsonized with anti-CD20 mAbs and incubated with human serum. The cells were stained with FITC-conjugated anti-C1q antibody and the C1q-binding level was analyzed by flow cytometer. Data are presented as mean ± SD (n = 3).
FcγR-binding and activation properties of glycoengineered anti-CD20 mAbs
We performed surface plasmon resonance (SPR) analysis using the extracellular domains of human FcγRI, FcγRIIa, FcγRIIb, FcγRIIIa, and FcγRIIIb in order to assess the effect of terminal Gal residues on FcγR-binding properties. Some previous studies suggested that the galactosylation level of N-glycans affects the FcγRIIIa-binding affinity.13,14 In agreement with previous studies, G2F mAb showed higher FcγRIIIa binding activity than G0F mAb. Interestingly, G1aF mAb showed higher FcγRIIIa binding activity than G1bF mAb. The FcγRIIIa binding activity of G1aF mAb and G1bF mAb were at the same level as G2F mAb and G0F mAb, respectively (Figure 5). We also found that the FcγRIIa, FcγRIIb, and FcγRIIIb binding activities of G1aF mAb and G2F mAb were higher than those of G1bF mAb and G0F mAb, which was similar to FcγRIIIa binding activity; whereas the FcγRI binding was not affected by the galactosylation level of N-glycans (Supplementary Figure S3).
Figure 5.

Binding of glycoengineered anti-CD20 mAbs to human FcγRs. SPR analysis was used to measure the binding of anti-CD20 mAbs to human FcγRI, FcγRIIa, and FcγRIIIa. Binding sensorgrams corrected by both the surface blank and buffer injection control are represented.
FcγR-binding activity is highly associated with the effector functions of mAbs induced by FcγR activation. FcγRIIa and FcγRIIIa activations contribute to antibody-dependent cellular phagocytosis mediated by immune cells such as macrophages and dendritic cells, and the FcγRIIIa activation in natural killer cells is the trigger of ADCC.27,28 To evaluate the FcγR activation properties of glycoengineered anti-CD20 mAbs, we performed a reporter assay using CD20-positive Raji cells (target cells) and human FcγR-expressing reporter cells (Jurkat/FcγR/NFAT-Luc cells; effector cells). We previously revealed that this reporter assay reflected FcγRs activation properties and could be used as a surrogate of ADCC.29 As shown in Figure 6(a,b), the order of FcγRIIa and FcγRIIIa activation properties of glycoengineered mAbs were the same as the FcγR-binding activities (G2F = G1aF > G1bF = G0F). These findings indicate that the terminal Gal residue on the Man α1-6 arm of Fc-glycans affects the FcγRIIa- and FcγRIIIa-binding and activation properties of mAbs compared with that on the Man α1-3 arm.
Figure 6.

FcγR activation properties of glycoengineered anti-CD20 mAbs. FcγRIIa (a) and FcγRIIIa (b) activation properties of glycoengineered mAbs. Jurkat/FcγRIIa/NFAT-Luc or Jurkat/FcγRIIIa/NFAT-Luc reporter cells were incubated with serially diluted anti-CD20 mAbs in the presence of Raji cells. FcγR activation was evaluated by assessing the luminescence intensity.
Effect of Fc-glycans on the structural stability of CH2 domain
Several studies have reported that Fc-glycans affect the conformational stability of the CH2 domain.13,30,31 However, the effect of terminal Gal residues on the Man α1-3 or Man α1-6 arm of Fc-glycans on the stability of the CH2 domain is still not well understood. Therefore, we subjected the glycoengineered anti-CD20 mAbs to hydrogen–deuterium exchange (HDX)/MS to compare the structural stability of their CH2 domains. After deuterium labeling at pH 7.4 from 10 s to 960 min, the reaction was quenched and the resultant mAbs were pepsin-digested. A comparison of deuterium uptake plots of the peptides at Phe245–Met256 residues are shown in Figure 7. Surprisingly, some peptides derived from the CH2 domain of G1aF mAb incorporated fewer deuterium atoms than those incorporated by G1bF mAb. In addition, the deuterium uptake plots of G1aF mAb and G1bF mAb were similar to those of G2F mAb and G0F mAb, respectively. These findings suggest that the CH2 domains of mAbs bearing the terminal Gal residue on the Man α1-6 arm are more stable than those of the mAbs bearing that on the Man α1-3 arm. Considering the higher FcγR activation properties and CDC activities in G1aF mAb and G2F mAb compared with G1bF mAb and G0F mAb, the stabilization of the CH2 domain modulated by the terminal Gal residue on the Man α1-6 arm of Fc-glycans could play an important role in the regulation of FcγR- and C1q-binding activities.
Figure 7.
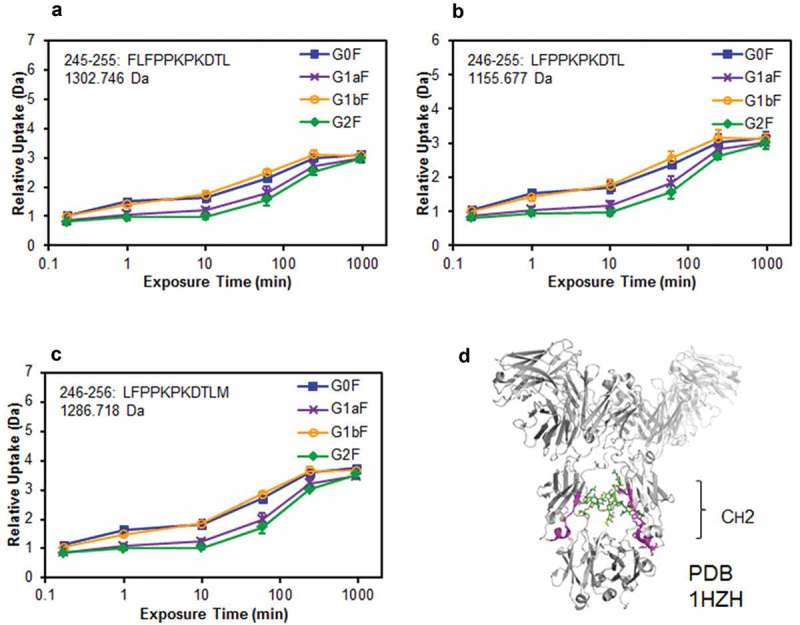
Comparison of structural stabilities of the CH2 domain of glycoengineered anti-CD20 mAbs using HDX/MS. Deuterium uptake plots of peptides Phe245–Leu255 (FLFPPKPKDTL) (a), Leu246–Leu255 (LFPPKPKDTL) (b), and Leu246–Met256 (LFPPKPKDTLM) (c) in the H-chain. (d) Physical representations of the crystal structures (PDB 1HZH) of peptides at Phe245–Met256 (magenta ribbons).
Discussion
In this study, we prepared four anti-CD20 mAbs bearing homogeneous glycan including two G1F isomers by original transglycosylation technology (Figure 2), and evaluated their biological activities (antigen-binding, FcγR- and C1q-binding, CDC, and FcγR-activation) and structural stability. The G1aF and G1bF glycans are positional isomers of G1F glycan, and their terminal Gal residues are located on the non-reducing terminus of Man α1-6 and Man α1-3 arms of G1F glycan, respectively. It is known that both isomers are attached to the mAbs produced by Chinese hamster ovary cells. It has been suggested that Fc-glycans are associated with the structural stability of Fc region of mAbs. Structurally, the Man α1-6 arm extends toward the CH2-CH3 elbow region, and the Man α1-3 arm extends into the inner space of the CH2 domain in the Fc region.32 Glycan components of the Man α1-6 arm interact with the inner surface of the CH2-CH3 elbow region, and the Man residues on the Man α1-3 arm interact with one another within the inner space of the CH2 domain.13,33 Reportedly, multiple interactions between the glycan components of the Man α1-6 arm and protein portion of a mAb affect the conformational stability of the CH2 domain.10,13,15,34 However, the roles of terminal Gal residues in effector functions of mAbs have not been completely understood. Particularly, it is unclear whether the effector functions differ between mAbs with a terminal galactose on Man α1-6 arm and Man α1-3 arm (G1aF mAb and G1bF mAb). Therefore, our study focused on the impact of G1F isomers in Fc-glycan on the biological activities and dynamic structure of mAbs.
Activation of the classical complement pathway, which is the mechanism of CDC, is triggered by the binding of the C1 complex, formed by the recognition molecule C1q and serine proteases C1r and C1s, to the antigen–antibody complex.35 In this study, we demonstrated that the terminal Gal residue on the Man α1-6 arm of Fc-glycans is more critical for modulating C1q-binding and CDC activities than those on the Man α1-3 arm. Although it was reported that the galactosylation level of N-glycan affects the CDC activity of mAbs,13,15 we demonstrated that the mono-galactosylated glycan isomers (G1aF and G1bF) have different CDC activities. In addition, we evaluated the FcγR-binding activities of four glycoengineered mAbs. Although previous studies showed inconsistent results about the effect of Fc-galactosylation on FcγRIIIa binding affinity,13,14,36,37 our results clearly demonstrated that the galactosylation on the Man α1-6 arm of Fc-glycans increased the FcγRIIIa binding and activation property. One reason for the previous inconsistent results could be that mixtures with differences in the ratio of G1aF and G1bF mAbs were used as typical G1F mAb samples in each study. We also revealed that the FcγRIIa, FcγRIIb, and FcγRIIIb binding activities were increased depending on the galactosylation of the Man α1-6 arm of Fc-glycans (Supplementary Figure S3). These findings were supported by several reports.37–39 In light of these data, enhancements of FcγRs binding activities could be commonly dependent on the galactosylation of the Man α1-6 arm of Fc-glycans.
The C1q- and FcγRs-binding sites of human IgG1 are located in the CH2 domain that contains the N-glycosylation site, and Fc-glycans are known to play a critical role in C1q- and FcγRs-binding.4,40 HDX/MS revealed that galactosylation on the Man α1-6 arm modulates the structural stability of the neighboring region (Phe245–Met256) of the N-glycosylation site (Asn297) in the CH2 domain. Previous studies have reported that the Gal residue on the Man α1-6 arm non-covalently interacts with the Lys250 residue (EU numbering 246) in the CH2 domain and that the confirmation of the CH2 domain is stabilized through CH–π interactions between the Man α1-6 arm and two Phe residues (Phe245 and Phe247; EU numbering 241 and 243, respectively).10,33,41 We previously reported that Gal residue interactions with Lys250, decrease the entropic disadvantage of the binding with FcγRIIIa. On the other hand, in the crystal structure, the Gal residue on the Man α1-3 arm is exposed to the solvent (PDB ID; 4CDH). Thus, we hypothesized that the entropic effect of the Gal residue on the Man α1-3 arm is quite small.13 Our finding that the Gal residue on Man α1-3 showed little impact on effector function and structural stability supported this hypothesis. Considering the aforementioned findings from previous studies and our results, we conclude that structural stabilization in the CH2 domain, resulting from interactions between the terminal Gal residue on the Man α1-6 arm of Fc-glycans and the peptide region containing Lys250, is important for enhancing the multiple effector functions of mAbs (CDC and FcγRs activation).
In addition to demonstrating the novel finding in the structure-activity relationships of mAbs bearing different Fc-glycan, our results impact the quality control of therapeutic mAbs. Therapeutic mAbs have various critical quality attributes (CQAs) that affect safety and efficacy of the product, and the galactosylation level of Fc-glycan can be a CQA for therapeutic mAbs exhibiting immune effector functions containing ADCC and CDC.8 In quality control of that kind of therapeutic mAbs, the evaluation of glycan structure to ensure the consistency of galactosylation level could be required. Generally, the galactosylation level has been evaluated without distinguishing G1F glycan isomers;42 however, considering our findings, the Gal residues on Man α1-6 arm and Man α1-3 arm of Fc-glycans should be evaluated distinctly in the galactosylation analysis related to effector functions.
In conclusion, this study demonstrated that the terminal Gal residue of the Man α1-6 arm of Fc-glycans affects CDC activity, C1q and FcγRs binding activities, and FcγRs activation properties. Furthermore, we suggest that the terminal Gal residue of Man α1-6 arm of Fc-glycans confers structural stability to the CH2 domain, enhancing the Fc-mediated effector functions of therapeutic mAbs. This is the first report demonstrating the impact of G1F glycan isomers on the effector functions and dynamic structure of mAbs. Our finding can contribute to the appropriate quality control of therapeutic mAbs and the development of novel glycoengineered mAbs.
Materials and methods
Materials
Anti-CD20 mAb (rituximab, Rituxan®) was purchased from Zenyaku Kogyo Co. Ltd. (Tokyo, Japan). SGP and α2-3, 6, 8 neuraminidase were purchased from Fushimi Pharmaceutical (Cat. No. 171801, Kagawa, Japan) and New England Biolabs (Cat. No. P0720S, Ipswich, MA, USA), respectively. Sephadex G-25 was purchased from GE Healthcare Life Sciences (Chicago, IL, USA). Iatrobeads 6RS-8060 was purchased from LSI Medience Corporation (Tokyo, Japan). Raji cells (JCRB9012) were obtained from the Japanese Collection of Research Bioresources Cell Bank (Osaka, Japan). The development of Jurkat/FcγR/NFAT-Luc cells, which express human FcγRIIa or FcγRIIIa and NFAT-driven luciferase reporters, was previously reported.29 All cells were maintained at 37°C in 5% CO2 with RPMI 1640 medium (Life Technologies, Carlsbad, CA, USA) supplemented with 10% heat-inactivated fetal bovine serum. Recombinant extracellular domains of human FcγRs were purchased from Sino Biological (Beijing, China).
Preparation of enzymes
To construct the expression plasmid for β1-4 galactosidase, the DNA fragment encoding it was amplified from Bacteroides thetaiotaomicron genomic DNA by PCR and cloned into pGEX-6P-1 vector (GE Healthcare). For construction of the expression plasmid for Endo F3 mutant (D165Q),25 firstly the artificial gene of Endo F3 from Elizabethkingia meningoseptica was synthesized and cloned into pTAC-2 vector (BioDynamics Laboratory Inc., Tokyo, Japan). Secondly, site-directed mutation for D165Q was introduced into the above-cloned DNA using KOD-plus-Mutagenesis Kit (Toyobo, Osaka, Japan). Then, the DNA fragment lacking signal peptide sequence (1–39) was amplified by PCR and cloned into pGEX-6P-1 vector. To construct the expression plasmid for wild-type Endo S, the DNA fragment encoding it lacking signal peptide sequence (1–36) was amplified from Streptococcus pyogenes genomic DNA by PCR and cloned into pFN18A Halo Tag T7 Flexi Vector (Promega, Madison, WI, USA). These three expression plasmids were separately transformed into Escherichia coli BL21 (DE3), and then corresponding proteins were expressed with standard procedures. Glutathione S-transferase (GST)-tagged β1-4 galactosidase and Endo F3 (D165Q) were purified by affinity chromatography with Glutathione sepharose, and then GST-tags of these purified proteins were cleaved with protease recognizing the cleavage site. Halo-tagged wild-type Endo S was immobilized on HaloLinkTM Resin (Promega) according to the manufacturer’s instructions.
Preparation of oxazolinated glycans (G1a- and G1b-Oxa)
G1a- and G1b-Oxa were prepared from SGP. Partial desialylation of SGP (356.4 mg, 124 μmol) was achieved by 20 mM HCl at 60°C for 4 h followed by neutralization with 1 M Na2CO3. The products were then injected into a φ10 × 250 mm Mightysil RP-18GP Aqua column (Kanto Chemical, Tokyo, Japan) in a reverse-phase HPLC system (GL Science, Tokyo, Japan), and the monosialyl glycopeptides (S1a- and S1b-peptide) were separated using a gradient of buffers A (100 mM AcOH-Et3N, pH 4.0) and B (0.05% 1-butanol in A buffer): a flow rate of 2.5 mL/min with a linear gradient of 10–100% of B buffer over 25 min (the monitoring absorbance, 220 nm) (Supplementary Figure S4). The peak eluting at 8.5 min was lyophilized and desalted by gel filtration (Sephadex G-25, φ20 × 600 mm, 0.05 M NH4OH) to yield 59.6 mg of S1b-peptide (19%). The peak eluting at 11.7 min was lyophilized and desalted by gel filtration (Sephadex G-25) to yield 62.9 mg of S1a-peptide (20%). S1a-peptide (48.2 mg, 18.7 μmol) and S1b-peptide (27.9 mg, 10.5 μmol) were separately incubated at 37°C for 6 h with β1-4 galactosidase. The reactions were heated at 70°C for 1 h to inactivate the enzyme, and incubated with α2-3, 6, 8 neuraminidase at 37°C for 24 h. The resulting products were subjected to a Sephadex G-25 gel filtration column equilibrated with 0.05 M NH4OH to obtain G1a-peptide (38.5 mg; 97%) and G1b-peptide (22.3 mg; 97%). The structural assignments of G1a- and G1b-peptide were based on diagnostic fragmentation in the negative ion collision-induced dissociation spectra (Supplementary Figure S5).24 The G1a-peptide (21.1 mg, 10.0 μmol) and G1b-peptide (22.3 mg, 10.5 μmol) were then subjected to hydrolysis with wild-type Endo S at 37°C for 24 h. G1a-OH (12.6 mg, 99%) and G1b-OH (13.4 mg; 99%) were obtained after purification using Iatrobeads column chromatography (ethly acetate/methanol/water = 2:1:1 (v/v/v) as eluent) followed by Sephadex G-25 gel filtration (0.05 M NH4OH). G1a- and G1b-OH were finally converted into oxazolines according to previously described method.19,21 G1a- and G1b-Oxa were identified using 1H-NMR analysis, which showed the characteristic signal of the reducing end anomeric proton of oxazolines form as a doublet peak with a specific coupling constant (G1a-Oxa; 6.09 ppm, 6.87 Hz, G1b-Oxa; 6.09 ppm, 6.87 Hz) (Supplementary Figure S6).
Preparation of anti-CD20 mAbs with homogeneous glycans
The preparation of glycoengineered anti-CD20 mAbs with homogeneous glycans was performed as previously described.19,20 Figure 2 shows the schematic for the workflow of sample preparation. For truncating Fc-N-glycans, anti-CD20 mAb was digested with immobilized Halo-tagged wild-type Endo S and Remove-iT Endo D (Cat. No. P0742S, New England Biolabs) at 37°C for 4 h. Immobilized Endo S was removed from the reaction mixture by centrifugation; subsequently, Endo D was removed using affinity chromatography using chitin resin (New England Biolabs). mAbs containing (±Fucα1-6)GlcNAcβ1-Asn groups were isolated on Ab-Capcher Mag beads (ProteNova, Kagawa, Japan). The isolated mAbs, as glycan acceptors, were mixed with oxazolinated glycans (G2-Oxa, G1a-Oxa, G1b-Oxa, or G0-Oxa), as glycan donors (donor/acceptor molar ratio, 100:1), and glycosynthase Endo F3 (D165Q) in 50 mM Tris-HCl buffer (pH 7.0) at 37°C for 1 h. The mAbs in the reaction mixture were isolated on Ab-Capcher ExTra beads (ProteNova). In addition, to eliminate the anti-CD20 mAbs with the GlcNAc group(s) and (Fucα1-6)GlcNAc group(s), glycoengineered anti-CD20 mAbs were loaded on to a cation-exchange column, ProPacTM WCX-10 column (9 × 250 nm, Thermo Fisher Scientific, Waltham, MA, USA) that was equipped with ÄKTAexplorer 10S system (GE Healthcare) at 4°C. A linear gradient was used [phase A: 10 mM sodium acetate (pH 4.3); phase B: 10 mM sodium acetate (pH 4.3) + 1.0 M NaCl] at a constant flow of 5.0 ml/min.19,20,43 The anti-CD20 mAbs with two core-fucosylated homogeneous N-glycan chains were collected and concentrated using an Amicon Ultra-4 (MerckMillipore, Burlington, MA, USA) in phosphate-buffered saline (PBS).
Glycan profiling: enzymatic N-glycan release by PNGase F
Each mAb sample (5 μg) was desalted using acetone precipitation. The precipitant was dissolved in 44 μL of 20 mM trimethylamine acetate (pH 6.0) containing 0.5% sodium dodecyl sulfate and 25 mmol/mL dithiothreitol. This mixture was heated at 65°C for 10 min. Next, it was cooled on ice, and 5 μL of 10% Triton X and 0.5 μL of PNGase F (1 unit/μL, Cat. No. 11365193001, Roche Diagnostics, Mannheim, Germany) were added to the sample. Enzymatic release was performed at 37°C for 15 min. A digesting agent was loaded onto a hydrophilic-lipophilic balance-solid-phase extraction (HLB-SPE) column (1 mL, 30 mg; Waters, Milford, MA, USA) conditioned using 1 mL of methanol followed by 1 mL of water. Subsequently, the column was washed twice with 0.3 mL of 10% methanol containing 100 μmol/mL of acetic acid. Both flow through and washing solvents were combined and dried using a centrifugal evaporator at 45°C.
Fluorescence labeling and HPLC analysis of released N-glycan
Here, 2-aminobenzamide and sodium cyanoborohydride were dissolved in 50 mg/mL and 63 mg/mL in dimethylsulfoxide/acetic acid mixture (70:30).44 A labeling solution (5 μL) was added to the dried glycan sample, and labeling was performed at 65°C for 3 h. Excess labeling solution was removed by hydrophilic interaction liquid chromatography (HILIC)-SPE.45 Briefly, 300 mL of 95% acetonitrile (ACN) was added to the sample, which was then applied to the HLB-SPE column (Waters) conditioned using 1 mL of 95% ACN. After rinsing the column with 1 mL of 95% ACN, labeled N-glycans were eluted using 1 mL of 20% ACN. The eluate was dried using a centrifugal evaporator and then dissolved with 8 μL of water. Of this diluted solution, 1 μL was analyzed using HILIC using a Waters H-class bio chromatograph system (Waters) equipped with an Acquity UPLC BEH Amide column (dimension, 2.1 × 150 mm; particle size, 1.7 μm; Waters). The mobile phase used was 100 mmol/mL of ammonium formate (pH 4.5) (A) and ACN (B). The column temperature was maintained at 45°C with a flow rate of 0.25 mL/min. The column was equilibrated with 75% of B. After injection, the mobile phase was held at 75% of B for 2 min; then, proportion of B was increased to 50% over 50 min. Fluorescence detection was performed at an excitation wavelength of 330 nm and an emission wavelength of 420 nm.
Fc-glycosylation analysis
A protein sample (5 µg) was dissolved in 350 µL of 25 mM ammonium bicarbonate solution (ABC) at pH 9.0. After adding 4 µL of 25 mM ABC containing 250 mM tris(2-carboxyethyl)phosphine hydrochloride, the solution was incubated at 60°C for 30 min. Subsequently, 25 mM ABC containing 5 µL of 500 mM iodoacetamide was added to the solution, and the mixture was incubated at 37°C for 30 min under dark conditions. Carboxymethylated proteins were incubated with 2 µg of trypsin and lysyl-endopeptidase C mixture (Mass Spec Grade Trypsin/Lys-C Mix; Mass Spec Grade, Promega) at 37°C overnight, and the reaction was quenched by adding an equal volume of 0.1% formic acid (FA) solution. The digest was desalted using an Oasis HLB microplate SPE cartridge (Waters) and then reconstituted in 0.1% FA. The sample solution was analyzed using liquid chromatography (LC)/MS using an Orbitrap Elite Mass Spectrometer (Thermo Fisher Scientific) connected to an UltiMate 3000 Nano LC system (Thermo Fisher Scientific). The analytical column used was the reverse-phase column, MonoCap C18 HighResolution 2000 (dimension, 0.1 × 2000 mm; GL Sciences, Tokyo, Japan). The mobile phase used was 0.1% FA (A buffer) and ACN containing 0.1% FA (B buffer). The peptides were eluted at a flow rate of 500 nL/min with a gradient of 2–50% of B buffer over 180 min. MS conditions were as follows: electrospray voltage = 2.0 kV in positive-ion mode; full mass range (m/z) = 700–2000; full mass resolution = 60,000; and collision energy = 25% for collision-induced dissociation–MS/MS. Full mass spectra and MS/MS spectra were acquired using the Orbitrap Elite Mass Spectrometer and a linear ion trap, respectively. Spectral data obtained using MS/MS were subjected to peptide-mapping analysis using the BioPharma Finder 3.0 software (Thermo Fisher Scientific). Searches for the amino acid sequence of rituximab with carbamidomethylation (57.021 Da) of Cys residues as a static modification and glycosylations with G0F (1444.534 Da), G1F (1606.587 Da), G2F (1768.640 Da), and G0F–HexNAc (1241.454 Da) as variable modifications were performed using the FASTA database.
Cell-based assays (competitive CD20-binding, CDC, C1q binding, FcγR activation)
Cell-based assays of glycoengineered anti-CD20 mAbs with homogeneous glycans were performed as previously described.29,46
Competitive CD20 binding assay was performed by cell-based electrochemiluminescence assay. Briefly, Raji cells were seeded onto an MSD high-bind plate (Meso Scale discovery, Rockville, MD). After blocking with 3% blocker A (Meso Scale discovery), the cells were incubated with a pre-mixed sample (1% blocker A, 0.5 µg/ml biotin-labelled rituximab, 0.5 µg/ml SULFO-TAG streptavidin, and serially diluted anti-CD20 mAbs) for 1 h at room temperature. The binding level of biotin-labelled rituximab was measured using MESO QuickPlex SQ 120 (Meso Scale Discovery).
In the CDC assay, Raji cells were incubated with Opti-MEM (Life Technologies) containing 16% human AB serum (Cat. No. H4522, Sigma-Aldrich, St. Louis, MO) and serially diluted anti-CD20 mAbs for 4 h. The cell lysis was measured using a CytoTox-Glo Cytotoxicity Assay kit (Promega).
In C1q binding assay, Raji cells were opsonized with 10 µg/mL of anti-CD20 mAbs and then cultured with 5% human AB serum at 37°C for 30 min. The cells were stained with FITC-conjugated anti-human C1q antibody (ab4223; Abcam, Cambridge, UK) in room temperature. The fluorescence intensity was measured using BD FACSCanto II flow cytometer (BD Biosciences, San Jose, CA, USA) and corrected by the background signal from the sample without mAbs.
In the FcγR reporter assay, Raji cells (target cells) and Jurkat/FcγRIIa/NFAT-Luc or Jurkat/FcγRIIIa/NFAT-Luc cells (effector cells) were seeded at an effector:target ratio of 10:1, and then cultured with serially diluted glycoengineered anti-CD20 mAbs. After incubation at 37°C for 4 h in 5% CO2, luciferase activity was assessed using the ONE-Glo Luciferase Assay System (Promega).
Surface plasmon resonance analysis
A Biacore T200 SPR biosensor (GE Healthcare) and CM5 sensor chip were used to evaluate the binding properties of glycoengineered anti-CD20 mAbs. The binding of glycoengineered anti-CD20 mAbs to recombinant human FcγRs with C-terminal polyhistidine tag (Cat. No. 10256-H08H, 10374-H08H1, 10259-H08H, 10389-H08H1, and 11046-H08H, Sino Biological) was assessed as described previously.47 In all experiments, each sensorgram from the sample flow cell was corrected by both surface blank and buffer injection control (double reference).
HDX/MS
HDX/MS was performed as previously described.13 Anti-CD20 mAbs (2 mg/mL) were diluted 20-fold with PBS in D2O (pH 7.0). The diluted solutions were separately incubated at 10°C for 0, 0.17, 1, 10, 60, 240, and 960 min. Next, deuterium-labeled samples were quenched by adjusting the pH to 2.25 using equal volumes of prechilled quenching buffer (1°C) containing 300 mM Tris (2-carboxyethyl) phosphine hydrochloride (Sigma–Aldrich) and 4 M guanidine-HCl (Thermo Fisher Scientific) using the CTC PAL sample manager (LEAP Technologies, NC, USA). All time points were determined using three independent labeling experiments. After quenching, the solutions were subjected to online pepsin digestion and were analyzed using LC/MS using the nanoACQUITY UPLC system (Waters) connected to the SynaptG2-S quad–time-of-flight mass spectrometer (Waters). Online pepsin digestion was performed using a Poroszyme Immobilized Pepsin Cartridge (dimension, 2.1 × 30 mm; Applied Biosystems, CA, USA) in a solution of 0.1% FA and 10 mM guanidine-HCl (pH 2.25) at 10°C for 2 min and at a flow rate of 300 μL/min. A reversed-phase ACQUITY UPLC BEH C18 column (dimension, 1.0 × 100 mm; particle size, 1.7 μm; Waters) was used as the analytical column with a mobile phase comprising 0.1% FA (pH 2.25; A buffer) and 0.1% FA containing 90% ACN (pH 2.25; B buffer). The deuterated peptides were eluted at a flow rate of 30 μL/min with a gradient of 13–85% of B buffer over 8 min. MS conditions were as follows: electrospray voltage = 2.8 kV in positive-ion mode; sampling cone at 40 V; source temperature = 80°C; desolation temperature = 100°C; and mass range (m/z) = 100–2000. MSE was performed by a series of low and high collision energies ramping from 10 to 40 V. MSE spectra were subjected to a database search using the ProteinLynx Global Server (PLGS) version 2.5.3 against an in-house database containing the amino acid sequence of rituximab. Search results and MS raw files were used to analyze deuteration levels of the peptic fragments using the DynamX 2.0 software (Waters). The measurement was performed in triplicate.
Funding Statement
This work was supported in part by Japan Agency for Medical Research and Development (AMED) under Grant Number [JP18mk0101081].
Acknowledgments
We thank Ms. M. Mori, Dr. S. Takashima, and Dr. M. Kurogochi from The Noguchi Institute for preparing glycoengineered mAbs.
Declaration of interest
The authors have no conflict of interest to declare.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s websites.
References
- 1.Reichert JM. Marketed Therapeutic Antibodies Compendium. MAbs. 2012;4:413–15. doi: 10.4161/mabs.19931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Singh S, Kumar NK, Dwiwedi P, Charan J, Kaur R, Sidhu P, Chugh VK. Monoclonal Antibodies: A Review. Curr Clin Pharmacol. 2018;13:85–99. doi: 10.2174/1574884712666170809124728. [DOI] [PubMed] [Google Scholar]
- 3.Kaplon H, Reichert JM. Antibodies to Watch in 2018. MAbs. 2018;10:183–203. doi: 10.1080/19420862.2018.1415671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang X, Mathieu M, Brezski RJ. IgG Fc engineering to modulate antibody effector functions. Protein Cell. 2018;9:63–73. doi: 10.1007/s13238-017-0473-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Suzuki M, Kato C, Kato A. Therapeutic antibodies: their mechanisms of action and the pathological findings they induce in toxicity studies. J Toxicol Pathol. 2015;28:133–39. doi: 10.1293/tox.2015-0031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zafir-Lavie I, Michaeli Y, Reiter Y. Novel antibodies as anticancer agents. Oncogene. 2007;26:3714–33. doi: 10.1038/sj.onc.1210372. [DOI] [PubMed] [Google Scholar]
- 7.Edelman GM, Cunningham BA, Gall WE, Gottlieb PD, Rutishauser U, Waxdal MJ. The covalent structure of an entire gammag immunoglobulin molecule. Proc Natl Acad Sci U S A. 1969;63:78–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Reusch D, Tejada ML. Fc glycans of therapeutic antibodies as critical quality attributes. Glycobiology. 2015;25:1325–34. doi: 10.1093/glycob/cwv065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Visser J, Feuerstein I, Stangler T, Schmiederer T, Fritsch C, Schiestl M. Physicochemical and functional comparability between the proposed biosimilar rituximab Gp2013 and originator rituximab. BioDrugs. 2013;27:495–507. doi: 10.1007/s40259-013-0036-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li W, Zhu Z, Chen W, Feng Y, Dimitrov DS. Crystallizable fragment glycoengineering for therapeutic antibodies development. Front Immunol. 2017;8:1554. doi: 10.3389/fimmu.2017.01554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ishida T, Joh T, Uike N, Yamamoto K, Utsunomiya A, Yoshida S, Saburi Y, Miyamoto T, Takemoto S, Suzushima H, et al. Defucosylated Anti-CCR4 monoclonal antibody (KW-0761) for relapsed adult T-cell leukemia-lymphoma: a multicenter phase II study. J Clin Oncol. 2012;30:837–42. doi: 10.1200/JCO.2011.37.3472. [DOI] [PubMed] [Google Scholar]
- 12.Shields RL, Lai J, Keck R, O‘Connell LY, Hong K, Meng YG, Weikert SH, Presta LG. Lack of fucose on human IgG1 N-linked oligosaccharide improves binding to human FcγRIII and antibody-dependent cellular toxicity. J Biol Chem. 2002;277:26733–40. doi: 10.1074/jbc.M202069200. [DOI] [PubMed] [Google Scholar]
- 13.Kiyoshi M, Caaveiro JMM, Tada M, Tamura H, Tanaka T, Terao Y, Morante K, Harazono A, Hashii N, Shibata H, et al. Assessing the heterogeneity of the Fc-Glycan of a therapeutic antibody using an engineered FcγReceptor IIIa-immobilized column. Sci Rep. 2018;8:3955. doi: 10.1038/s41598-018-22199-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thomann M, Reckermann K, Reusch D, Prasser J, Tejada ML. Fc-galactosylation modulates antibody-dependent cellular cytotoxicity of therapeutic antibodies. Mol Immunol. 2016;73:69–75. doi: 10.1016/j.molimm.2016.03.002. [DOI] [PubMed] [Google Scholar]
- 15.Peschke B, Keller CW, Weber P, Quast I, Lunemann JD. Fc-galactosylation of human immunoglobulin gamma isotypes improves C1q binding and enhances complement-dependent cytotoxicity. Front Immunol. 2017;8:646. doi: 10.3389/fimmu.2017.00646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu CP, Tsai TI, Cheng T, Shivatare VS, Wu CY, Wong CH. Glycoengineering of antibody (Herceptin) through yeast expression and in vitro enzymatic glycosylation. Proc Natl Acad Sci U S A. 2018;115:720–25. doi: 10.1073/pnas.1718172115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wada R, Matsui M, Kawasaki N. Influence of N-glycosylation on effector functions and thermal stability of glycoengineered IgG1 monoclonal antibody with homogeneous glycoforms. MAbs. 2019;11:350–72. doi: 10.1080/19420862.2018.1551044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huang W, Giddens J, Fan SQ, Toonstra C, Wang LX. Chemoenzymatic glycoengineering of intact IgG antibodies for gain of functions. J Am Chem Soc. 2012;134:12308–18. doi: 10.1021/ja3051266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kurogochi M, Mori M, Osumi K, Tojino M, Sugawara S, Takashima S, Hirose Y, Tsukimura W, Mizuno M, Amano J, et al. Glycoengineered monoclonal antibodies with homogeneous glycan (M3, G0, G2, and A2) using a chemoenzymatic approach have different affinities for FcγRIIIa and variable antibody-dependent cellular cytotoxicity activities. PLoS One. 2015;10:e0132848. doi: 10.1371/journal.pone.0132848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tsukimura W, Kurogochi M, Mori M, Osumi K, Matsuda A, Takegawa K, Furukawa K, Shirai T. Preparation and biological activities of anti-HER2 monoclonal antibodies with fully core-fucosylated homogeneous bi-antennary complex-type glycans. Biosci Biotechnol Biochem. 2017;81:2353–59. doi: 10.1080/09168451.2017.1394813. [DOI] [PubMed] [Google Scholar]
- 21.Noguchi M, Tanaka T, Gyakushi H, Kobayashi A, Shoda S. Efficient synthesis of sugar oxazolines from unprotected N-acetyl-2-amino sugars by using chloroformamidinium reagent in water. J Org Chem. 2009;74:2210–12. doi: 10.1021/jo8024708. [DOI] [PubMed] [Google Scholar]
- 22.Collin M, Olsen A. Endo S, a novel secreted protein from streptococcus pyogenes with endoglycosidase activity on human IgG. Embo J. 2001;20:3046–55. doi: 10.1093/emboj/20.12.3046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Collin M, Olsen A. Effect of SpeB and EndoS from streptococcus pyogenes on human immunoglobulins. Infect Immun. 2001;69:7187–89. doi: 10.1128/IAI.69.11.7187-7189.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Harvey DJ. Fragmentation of negative ions from carbohydrates: part 2. fragmentation of high-mannose N-Linked glycans. J Am Soc Mass Spectrom. 2005;16:631–46. doi: 10.1016/j.jasms.2005.01.005. [DOI] [PubMed] [Google Scholar]
- 25.Giddens JP, Lomino JV, Amin MN, Wang LX. Endo-F3 glycosynthase mutants enable chemoenzymatic synthesis of core-fucosylated triantennary complex type glycopeptides and glycoproteins. J Biol Chem. 2016;291:9356–70. doi: 10.1074/jbc.M116.721597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Reusch D, Haberger M, Falck D, Peter B, Maier B, Gassner J, Hook M, Wagner K, Bonnington L, Bulau P, et al. Comparison of methods for the analysis of therapeutic immunoglobulin G Fc-glycosylation profiles-part 2: mass spectrometric methods. MAbs. 2015;7:732–42. doi: 10.1080/19420862.2015.1045173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ibuki C, Seino Y, Otsuka T, Kimata N, Inami T, Munakata R, Mizuno K. Switching to pitavastatin in statin-treated low Hdl-C patients further improves the lipid profile and attenuates minute myocardial damage. J Clin Med Res. 2012;4:385–92. doi: 10.4021/jocmr1108w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fritz-Rdzanek A, Grzybowski W, Beta J, Durczynski A, Jakimiuk A. He4 protein and smrp: potential novel biomarkers in ovarian cancer detection. Oncol Lett. 2012;4:385–89. doi: 10.3892/ol.2012.757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tada M, Ishii-Watabe A, Suzuki T, Kawasaki N. Development of a cell-based assay measuring the activation of fcgammariia for the characterization of therapeutic monoclonal antibodies. PLoS One. 2014;9:e95787. doi: 10.1371/journal.pone.0095787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu H, Nowak C, Andrien B, Shao M, Ponniah G, Neill A. Impact of IgG Fc-oligosaccharides on recombinant monoclonal antibody structure, stability, safety, and efficacy. Biotechnol Prog. 2017;33:1173–81. doi: 10.1002/btpr.2498. [DOI] [PubMed] [Google Scholar]
- 31.Liu H, Bulseco GG, Sun J. Effect of posttranslational modifications on the thermal stability of a recombinant monoclonal antibody. Immunol Lett. 2006;106:144–53. doi: 10.1016/j.imlet.2006.05.011. [DOI] [PubMed] [Google Scholar]
- 32.Krapp S, Mimura Y, Jefferis R, Huber R, Sondermann P. Structural analysis of human IgG-Fc glycoforms reveals a correlation between glycosylation and structural integrity. J Mol Biol. 2003;325:979–89. [DOI] [PubMed] [Google Scholar]
- 33.Raju TS. terminal sugars of Fc glycans influence antibody effector functions of IgGs. Curr Opin Immunol. 2008;20:471–78. doi: 10.1016/j.coi.2008.06.007. [DOI] [PubMed] [Google Scholar]
- 34.Harbison AM, Brosnan LP, Fenlon K, Fadda E. Sequence-to-structure dependence of isolated IgG Fc complex biantennary N-glycans: a molecular dynamics study. Glycobiology. 2019;29:94–103. doi: 10.1093/glycob/cwy097. [DOI] [PubMed] [Google Scholar]
- 35.Mortensen SA, Sander B, Jensen RK, Pedersen JS, Golas MM, Jensenius JC, Hansen AG, Thiel S, Andersen GR. Structure and Activation of C1, the Complex Initiating the Classical Pathway of the Complement Cascade. Proc Natl Acad Sci U S A. 2017;114:986–91. doi: 10.1073/pnas.1616998114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shinkawa T, Nakamura K, Yamane N, Shoji-Hosaka E, Kanda Y, Sakurada M, Uchida K, Anazawa H, Satoh M, Yamasaki M, et al. The absence of fucose but not the presence of galactose or bisecting N-acetylglucosamine of human IgG1 complex-type oligosaccharides shows the critical role of enhancing antibody-dependent cellular cytotoxicity. J Biol Chem. 2003;278:3466–73. doi: 10.1074/jbc.M210665200. [DOI] [PubMed] [Google Scholar]
- 37.Dashivets T, Thomann M, Rueger P, Knaupp A, Buchner J, Schlothauer T. Multi-angle effector function analysis of human monoclonal IgG glycovariants. PLoS One. 2015;10:e0143520. doi: 10.1371/journal.pone.0143520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Thomann M, Schlothauer T, Dashivets T, Malik S, Avenal C, Bulau P, Ruger P, Reusch D. In vitro glycoengineering of IgG1 and its effect on Fc receptor binding and Adcc activity. PLoS One. 2015;10:e0134949. doi: 10.1371/journal.pone.0134949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Roberts JT, Barb AW. A single amino acid distorts the fc gamma receptor IIIb/CD16b structure upon binding immunoglobulin G1 and reduces affinity relative to CD16a. J Biol Chem. 2018. doi: 10.1074/jbc.RA118.005273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Subedi GP, Barb AW. The structural role of antibody N-glycosylation in receptor interactions. Structure. 2015;23:1573–83. doi: 10.1016/j.str.2015.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Houde D, Peng Y, Berkowitz SA, Engen JR. Post-translational modifications differentially affect IgG1 conformation and receptor binding. Mol Cell Proteomics. 2010;9:1716–28. doi: 10.1074/mcp.M900540-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Raju TS, Jordan RE. Galactosylation variations in marketed therapeutic antibodies. MAbs. 2012;4:385–91. doi: 10.4161/mabs.19868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang S, Ionescu R, Peekhaus N, Leung JY, Ha S, Vlasak J. Separation of post-translational modifications in monoclonal antibodies by exploiting subtle conformational changes under mildly acidic conditions. J Chromatogr A. 2010;1217:6496–502. doi: 10.1016/j.chroma.2010.08.044. [DOI] [PubMed] [Google Scholar]
- 44.Bigge JC, Patel TP, Bruce JA, Goulding PN, Charles SM, Parekh RB. Nonselective and efficient fluorescent labeling of glycans using 2-Amino benzamide and anthranilic acid. Anal Biochem. 1995;230:229–38. doi: 10.1006/abio.1995.1468. [DOI] [PubMed] [Google Scholar]
- 45.Anumula KR, Dhume ST. High resolution and high sensitivity methods for oligosaccharide mapping and characterization by normal phase high performance liquid chromatography following derivatization with highly fluorescent anthranilic acid. Glycobiology. 1998;8:685–94. [DOI] [PubMed] [Google Scholar]
- 46.Aoyama M, Tada M, Tatematsu KI, Hashii N, Sezutsu H, Ishii-Watabe A. Effects of amino acid substitutions on the biological activity of Anti-CD20 monoclonal antibody produced by transgenic silkworms (Bombyx Mori). Biochem Biophys Res Commun. 2018;503:2633–38. doi: 10.1016/j.bbrc.2018.08.015. [DOI] [PubMed] [Google Scholar]
- 47.Tada M, Tatematsu K, Ishii-Watabe A, Harazono A, Takakura D, Hashii N, Sezutsu H, Kawasaki N. Characterization of Anti-CD20 monoclonal antibody produced by transgenic silkworms (Bombyx Mori). MAbs. 2015;7:1138–50. doi: 10.1080/19420862.2015.1078054. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.