

Abstract
Bacterial cell division is the result of a productive round of the cell cycle to yield two daughter cells. The cell cycle is highly coordinated in Caulobacter crescentus where it is driven by a cell cycle gene-regulatory network that coordinates gene expression with the major cell cycle events such as chromosome replication and cell division. Recent ribosomes profiling data showed that 484 genes undergo changes in translation efficiency during the cell cycle, suggesting a broad role for translational control in cell cycle-regulation. In this chapter, we focus on how to perform ribosome profiling to measure the translation efficiency across cellular mRNAs at key stages in the Caulobacter cell cycle. This methodology relies on the high-yield ludox gradient synchronization of Caulobacter cells followed by ribosome profiling to measure ribosome density and total-RNA-seq to measure mRNA levels.
1. Introduction
In bacteria, cell cycle-regulatory networks control progression of the cell cycle by integrating information about key cell cycle events into their gene expression programs. Caulobacter crescentus has proven to be a powerful model of the bacterial cell cycle due to the high-yield and rapid ability to perform cell synchronization (Evinger & Agabian, 1977; Schrader & Shapiro, 2015). In Caulobacter, the well-defined cell cycle-regulatory network is controlled by a set of master regulators that are arranged in a transcriptional regulatory circuit (Collier, 2016; Lasker, Mann, & Shapiro, 2016; McAdams & Shapiro, 2011). While transcriptional control, DNA methylation, phosphosignaling, protein localization, and regulated proteolysis have well established roles in the cell cycle-regulatory circuit, translational control was recently found to play a significant role where 12.4% its genes have cell cycle-regulated translation (Schrader, Li, Childers, Perez, Weissman, Shapiro et al., 2016). Ribosome profiling now allows genome-wide interrogation of mRNA translation, making it a powerful tool towards understanding the gene-regulatory networks controlling complex cellular events (Brar, Yassour, Friedman, Regev, Ingolia, & Weissman, 2012; Ingolia, Ghaemmaghami, Newman, & Weissman, 2009; Stumpf, Moreno, Olshen, Taylor, & Ruggero, 2013). The methodology described here integrates the high-yield of Caulobacter cell synchronization with the power of ribosome profiling to measure translation efficiency at key stages of the cell cycle. This method allows unbiased interrogation of translational control in bacterial cell cycles with genome-wide coverage.
2. Caulobacter Cell Cycle Ribosome Profiling
Ribosome profiling described here is performed in multiple phases including 2.1- Cell Growth, Synchronization and Harvesting, 2.2- Harvesting Cells, 2.3- Cell Lysis, 2.4- Footprinting and Footprint Extraction, 2.5- Total RNA-Seq Sample Preparation and PAGE Size Selection, and 2.6- Library Generation (Fig. 1). This protocol takes several days to perform (Table I) so it is important to include control samples throughout the process. Typically, ribosome profiling in bacteria uses large volumes of cultures but this protocol is adapted to small samples containing only 30 mL of log-phase cells retrieved upon ludox gradient synchronization. To measure translation efficiency total RNA-seq is compared to ribosome footprints to calculate the translation efficiency, which is the metric measured across the cell cycle.
Figure 1.
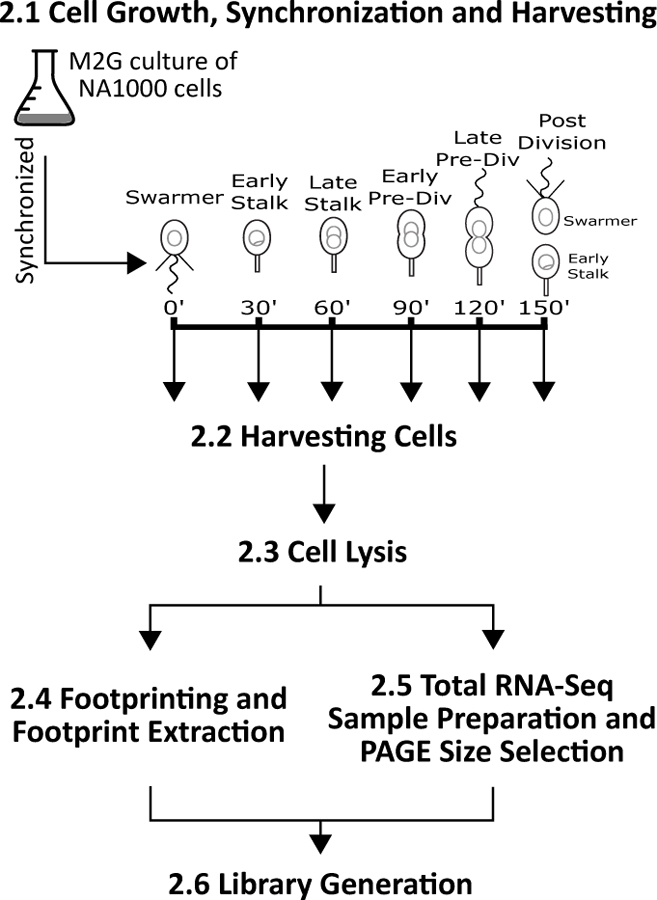
Schematic of Caulobacter ribosome profiling procedure. Section 2.1 - Cell Growth, Synchronization and Harvesting: Grow 1 Liter of NA1000 cells, synchronize, and then grow to one of the cell cycle time points. Section 2.2 - Harvesting Cells: To each cell population, harvest by chloramphenicol pre-treatment (2 min at 28 °C) followed by rapid cell collection on ice. Section 2.3 - Cell Lysis: Lysis is performed under liquid nitrogen and a small portion is saved as a frozen aliquot for total RNA-seq. Section 2.4 - Footprinting and Footprint Extraction: Footprint mRNA fragments, select for size, and recover digested mRNA fragments. Section 2.5 - Total RNA-Seq Sample Preparation and PAGE Size Selection: Use the lysate aliquot from section 2.3 and prepare for total RNA-seq. Section 2.6 - Library Generation: Sequencing libraries are generated in the same way from the products of both ribosome footprinting and total RNA-Seq.
Table I.
Expected completion time for each section.
| Section | Approximate Work Time |
|---|---|
| 2.1- Cell Growth, Synchronization and Harvesting | Three days |
| 2.2- Harvesting Cells | 1 hour |
| 2.3- Cell Lysis | 1.5 hours |
| 2.4- Footprinting and Footprint Extraction | 10 hours |
| 2.5- Total RNA-Seq Sample Preparation and PAGE Size Selection | 13 hours |
| 2.6- Library Generation | Three days |
2.1. Cell Growth, Synchronization and Harvesting
Caulobacter synchronization by ludox gradient synchronization yields a large amount of swarmer cells that can be followed during the cell cycle (Evinger & Agabian, 1977). Briefly, 1 L of mid-log phase NA1000 cells are arrested in growth by carbon starvation, then swarmer cells are isolated by ludox gradient centrifugation. This procedure has been covered in depth recently at both large and small scales (Schrader & Shapiro, 2015). For each ribosome profiling sample, 1 L of cells grown in M2G should be synchronized, and the 30 mL of resulting swarmer cells at OD600=0.5 should be grown to the desired cell cycle stage and rapidly harvested for ribosome profiling and total-RNA-seq.
2.2.1. Equipment
Standard Shaker Incubator
Spectrophotometer
Optical Cuvettes
Corex Tubes
JA-20 Fixed-Angle Aluminum Rotor (Beckman Coulter)
2.2.2. Buffers and Reagents
Caulobacter crescentus Strain NA1000 (Synchronizable Variant).
PYE Media: 2 g of Bactopeptone, 1 g of Yeast Extract, 2 mL of 0.5 M MgSO4, 5 mL of 0.1 M CaCl2, fill up to 1 L with Milli-Q filtered water (autoclaved solution).
20x M2 Salts: 17.4 g of Na2HPO4, 10.6 g of KH2PO4, 10 g of NH4Cl, Resuspend in 1 L of Milli-Q filtered water (autoclaved solution).
M2G Media: 50 mL of 20x M2 Salts, 1 mL of 0.5 M MgSO4, 10 mL of 20% Glucose, 1 mL of Ferrous Sulfate Chelate Solution (Sigma Aldrich), fill to 995 mL with Milli-Q filtered water, and then add 5 mL of 0.1 M CaCl2 (add last after dilution to avoid precipitation). Sterilize with a 0.22 µm filter.
Ludox AS-40 Solution (Sigma).
2.2.3. Procedure
In a shaker incubator at 28 °C and 250 rpm, grow an overnight culture of Caulobacter strain NA1000 in 5 mL of PYE media.
Transfer 200 µL of saturated culture to 25 mL of M2G media and grow overnight in a shaker incubator at 28 °C and 250 rpm.
Read the OD600 of the 25 mL culture using spectrophotometer.
Using a doubling time of 2.33 hours, calculate how much of the 25 mL culture needs to be added to 1 L of culture to give a mid-log (OD600=0.5) 1 L culture in the morning.
Verify the presence of swarmer cells using a phase contrasstrip tubes and perform the following PCR t microscope and a wet mount of the cells.
A detailed video protocol of the synchronization procedure can be found in (Schrader & Shapiro, 2015).
Harvest cells by centrifugation and suspend in 180 mL cold M2 salts.
60 mL of cold ludox solution should be mixed, and the resulting suspension should be pipetted into eight 30 mL corex tubes and spun for 30 min at 6400 g in a JA-20 rotor.
The lower band is the swarmer band. The top of the solution can be aspirated off and discarded. The lower swarmer cell band should be collected from each tube and pooled in a new 50 mL conical tube.
Check again by phase microscopy that the cells are highly enriched for swarmer cells (90–95% motile swarmer cells).
The swarmer cells should be pelleted and washed twice in 30 mL of cold M2 each. Finally, the cells should be suspended in 30 mL of 28 oC M2G media at an OD600=0.5 and placed into a sterile 250 mL flask and shaken at 250 rpm until the desired cell cycle stage.
2.2.4. Notes
After synchronization, an entire 1 L cell synchrony allows for the isolation of one cell cycle ribosome profiling sample.
It is important to examine and confirm that the cells are in the proper phase of the cell cycle by imaging with a wet mount on a phase-contrast microscope. Swarmer cells should be small and motile, stalked cells should be small and immotile, and predivisional cells should be larger with a dumbbell shape morphology.
2.2. Harvesting cells
Caulobacter cells can only be harvested by centrifugation as their cells rapidly clog 0.22 µm filters used for ribosome profiling of B. subtilis or E. coli cells (Oh, Becker, Sandikci, Huber, Chaba, Gloge et al., 2011). Harvesting cells by centrifugation is a process that must be performed in a timely matter to minimize artifacts arising from the chloramphenicol addition. After removal from the shaker incubator, cell temperature needs to be rapidly lowered to 4 °C and maintained there until frozen in liquid nitrogen. A quick harvesting time and rapidly lowering cell temperatures by addition of ice cubes are necessary for the efficient harvesting of ribosomes trapped mRNA fragments.
2.2.1. Equipment
Sorvall Legend X1R Centrifuge (Thermo Scientific)
Fiberlite F15–8 × 50cy Fixed Angle Rotor (Thermo Scientific)
50 mL Conical Tube
Standard Ice Cube Tray
Hammer
Zip-Top Bag
Standard Syringe Needle
2.2.2. Buffers and Reagents
50 mg/mL Chloramphenicol solution in ethanol
Ice cubes of M2G minimal media with 100 µg/ml of Chloramphenicol, 15 mL each, stored at −80 °C.
Resuspension Buffer: 20 mM Tris-HCl pH 8.0, 10 mM MgCl2, 100 mM NH4Cl, 1 mM Chloramphenicol
Lysis Buffer: 20 mM Tris pH 8.0, 10 mM MgCl2, 100 mM NH4Cl, 5 mM CaCl2, 0.4% Triton X100, 0.1% NP-40, 1 mM Chloramphenicol, 100 U/mL RNase free DNase I (Roche)
Liquid Nitrogen
2.2.3. Procedure
Before harvesting the cells, take an ice cube out of the −80o C freezer, place ice cube in zip-top bag, crush using a hammer and place each crushed cube into a 50 mL conical tube and store the tube on ice. Each tube is sufficient for harvesting 30 mL of cells.
For each cell cycle time point, 30 mL of cells at OD600=0.5 are treated with chloramphenicol (addition of 60 µL chloramphenicol to a final concentration of 100 µg/ml) and incubate in 28 oC shaker for 2 minutes at 250 rpm.
Immediately add the cell culture to a 50 mL prechilled conical tube (4 oC) containing 15mL of crushed ice (M2G broth with 100 µg/mL of chloramphenicol), cap the tube and invert twice.
Place tubes in the prechilled, −10 °C F15–8 rotor and spin at 14,000 rpm (22,789 g) for 1.5 min to pellet cells.
Discard supernatant by decanting and suspend cell pellet in 20 mL of prechilled Resuspension Buffer.
Pellet cells again at 14,000 rpm (22,789 g) for 1.5 min to pellet cells in −10 °C F15–8 rotor.
Discard supernatant by decanting and suspend cell pellet in 300 µL of Lysis Buffer.
Slowly drip the cell pellet/lysis buffer suspension into a liquid nitrogen filled 50 mL conical tube.
Poke holes in 50 mL conical lid using a hot needle and pour out remaining liquid nitrogen in the tube. Store the cell pellet in a −80 oC freezer.
2.2.4. Notes
Cell collection should be performed as quickly as possible to avoid artifacts resulting from changes in translation occurring in response to the cell collection procedure.
Cells should be cooled down rapidly by addition to crushed ice (M2G broth with 100 µg/mL of chloramphenicol) and maintained chilled throughout the cell collection procedure.
After the initial spin, the supernatant should still contain small pieces of ice.
Frozen cell pellets can be stored at −80 °C for at least a year.
2.3. Cell Lysis
Cell lysis is performed in a mixer mill with samples frozen in liquid nitrogen to minimize changes in ribosome position occurring in a liquid lysate (Oh et al., 2011). Save a small frozen aliquot for RNA-seq. Most of the lysate is used for ribosome footprinting.
2.3.1. Equipment
Mixer Mill MM 400 (Roche)
Grinding Jar 10 mL (Roche)
12 mm Grinding Ball (Roche)
1.5 mL RNase Free Microcentrifuge Tube
2.3.3. Procedure
Chill assembled jar and grinding ball in liquid nitrogen.
Open jar and add frozen cell pellet/lysis buffer suspension to chilled jar with grinding ball and close lid.
Mill at 15 Hz for 3 min. Chill the sealed jar in liquid nitrogen between each run. Five runs total.
A small scoop of the frozen pulverized cells should be scraped out of the jars using a pre-chilled scoopula and stored in a pre-chilled 1.5 mL microcentrifuge tube at −80 °C for total RNA seq.
To thaw the remaining lysate, place each half, chamber side up, in warm 30 oC water.
2.3.4. Notes
Use cryogenic gloves when working with liquid nitrogen.
2.4. Footprinting and Footprint Extraction
The thawed lysates are then footprinted by addition of micrococcal nuclease (MNase) which generates heterogeneous mRNA footprint sizes unlike the 28 nt footprints generated by eukaryotic ribosomes (Ingolia et al., 2009; O’Connor, Li, Weissman, Atkins, & Baranov, 2013; Oh et al., 2011). MNase reactions can be quenched by addition of EGTA to chelate Ca2+ ions needed for nuclease activity. After footprinting with MNase, the 70S ribosomes are purified on a sucrose gradient. It is important to also run an undigested control on the sucrose gradient to ensure that polysomes were preserved upon cell harvesting and lysis (Figure 2).
Figure 2.
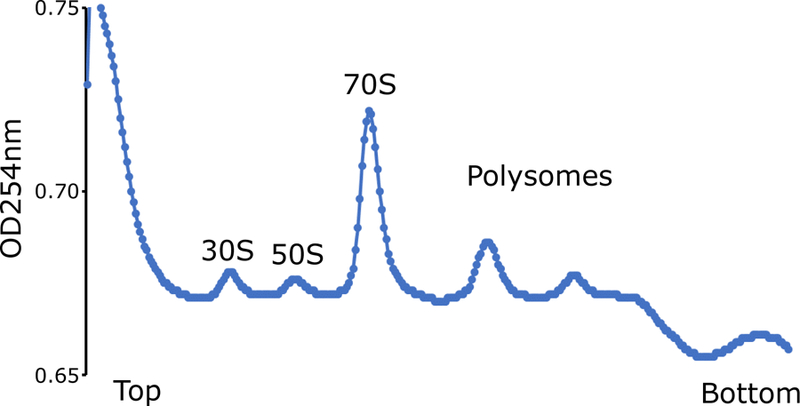
Example of Caulobacter cell cycle polysomes. Undigested cell lysate derived from a single cell cycle timepoint was separated on a 10–55% sucrose gradient.
2.4.1. Equipment
Standard Microcentrifuge
Nanodrop Spectrophotometer (Thermo Fisher)
Open-Top Polyclear Tubes (7030 Seton)
TH-641 Swinging Bucket Rotor Package (Thermo Scientific) (SW 41 Ti equivalent)
Sorvall WX+ Ultracentrifuge (Thermo Scientific)
2 mL Siliconized RNase Free Microcentrifuge Tubes
Standard Thermomixer
Gradient Station (BIO-COMP)
2.4.2. Buffers and Reagents
10 mM Tris-HCl pH 7.0, from 1 M stock (Ambion)
Lysis Buffer: 20 mM Tris-HCl pH 8.0, 10 mM MgCl2, 100 mM NH4Cl, 5 mM CaCl2, 0.4% Triton X100, 0.1% NP-40, 1 mM Chloramphenicol, 100 U/mL RNase free DNase I (Roche)
MNase Enzyme 1500 U/µL (Roche)
SUPERase• In RNase Inhibitor Enzyme 20 U/μL (Ambion)
0.5 M EGTA pH 8.0
10% sucrose solution: 10% sucrose (w/v), 20 mM Tris-HCl pH 8.0, 10 mM MgCl2, 100 mM NH4Cl, 1 mM Chloramphenicol
50% sucrose solution: 50% sucrose (w/v), 20 mM Tris-HCl pH 8.0, 10 mM MgCl2, 100 mM NH4Cl, 1mM Chloramphenicol
Gradient Balance Buffer: 20 mM Tris-HCl pH 8.0, 10 mM MgCl2, 100 mM NH4Cl, 1 mM Chloramphenicol
20% SDS
Acid Phenol Chloroform (Ambion)
Chloroform
3 M NaOAc pH 5.5 (Ambion)
100% isopropanol
80% ethanol from 100% stock
Novex 2x TBE-Urea Sample Buffer (Invitrogen)
2.4.3. Procedure
Continue from section 2.3.3 by recovering the pulverized cells from the thawing chamber and placing them into a pre-chilled 1.5mL microcentrifuge tube for footprinting.
Incubate the lysate for 20 minutes on ice.
Pellet cell debris at 20,000 g for 10 min in a 4 °C microcentrifuge.
Transfer supernatant to a pre-chilled siliconized microcentrifuge tube without disturbing pelleted cells.
Dilute 1 µL of lysate with 99 µL of 10 mM Tris-HCl pH 7.0. Measure A260 by Nanodrop spectrophotometer (blank with 1 µL lysis buffer diluted into 99 µL of 10 mM Tris-HCl pH 7.0).
Digest 500 mg of lysate RNA with 1.3 µL of MNase and 6 µL of SUPERase•In. Add lysis buffer for a 200 µL reaction. Treat undigested polysome control similarly, except without the addition of MNase.
Incubate at 25 °C for 1 h while shaking in a thermomixer at 1400 rpm.
Quench reaction with 2 µL of EGTA and immediately return to ice.
Chill TH-641 swinging rotors and buckets to 4 °C
Fill open-top polyclear tube with 55% sucrose solution until you reach the line on the maker block metal stand (~6 mL).
Carefully layer 10% sucrose solution on top.
Cap open-top polyclear tubes.
From gradient using Gradient Master parameters for a SW 41 Ti rotor, short capped tubes, and 7%−47% weight by volume sucrose gradient.
Remove cap and carefully layer nuclease digested cell lysate and undigested polysome control cell lysate into separate tubes.
Use gradient buffer to balance tubes.
Spin at 35,000 rpm (209,627 g) for 2.5 h at 4 °C.
Fractionate using Gradient Station fractionator, with a piston speed of 0.2 mm/s.
Manually collect fractionated monosomes, cap 2.0 mL siliconized microcentrifuge tube, flash-freeze in liquid nitrogen and stored at −80 °C indefinitely.
To extract total RNA from footprinted monosomes, add 40 µL of 20% SDS to 0.7 mL of thawed monosomes fraction solution. Monosomes may need to be split into two tubes of ~0.7 mL each.
Add 0.7 mL of acid phenol pre-warmed to 65 °C to the tube and mix by vortexing.
Incubate at 65 °C for 5 min while shaking in a thermomixer.
Chill on ice for 5 min.
Spin at 20,000 g for 2 min in a microcentrifuge and transfer supernatant to fresh tube.
Add 0.7 mL of room temperature acid phenol, mix by vortexing, and then incubate at room temperature for 5 min.
Spin at 20,000 g for 2 min in a microcentrifuge and transfer aqueous layer to fresh tube.
Add 0.6 mL of chloroform and mix by vortexing.
Immediately spin at 20,000 g for 1 min in a microcentrifuge and transfer aqueous layer to fresh tube.
Add 75 µL of 3 M NaOAc pH 5.5 and 0.8 mL of 100% isopropanol. Mix by vortexing.
Chill at −80 °C for 30 min.
Pellet RNA at 20,000 g for 1 h in a 4 °C microcentrifuge.
Wash pellet with 0.8 mL of −20 °C 80% ethanol.
Air dry pellet for 5 min and suspend in 20 µL of 10 mM Tris-HCl pH 7.0.
Dilute 1 µL of precipitated RNA with 9 µL of 10 mM Tris pH 7.0 and then quantify with Nanodrop spectrophotometer.
Dilute 50 µg of RNA to 20 µL with 10 mM Tris-HCl pH 7.0 and then add 20 µL of 2x TBE-Urea sample buffer.
Save samples at −20 oC until section 2.5.3 and run size selection for both samples at once on 1x TBE 7M UREA PAGE gel
2.4.4. Notes
When layering 10% sucrose on top of 55% sucrose in an open-top polyclear tube care must be taken, especially in the beginning, to ensure that there is no mixing at the boundary between the two layers.
Carefully transport the sucrose gradient tubes to not disturb the boundary layer. Use smooth, even movements, especially when attaching and removing the buckets from the rotor.
Surplus lysis buffer can be used to balance rotor buckets instead of gradient buffer.
All 6 rotor buckets should be filled with sucrose and balanced for the spin even if all buckets are not loaded with samples.
Fractionated monosomes typically have a final volume of 1.2 mL.
Use siliconized microcentrifuge tubes to avoid transfer loss.
2.5. Total RNA-Seq Sample Preparation and PAGE Size Selection
To measure translation efficiency, mRNA levels are required to normalize the ribosome footprint density. To get high-quality total RNA-seq data, rRNA, which is abundant, should be removed from the sample. rRNA depleted total RNA should then be hydrolyzed under denaturing conditions to yield a size distribution that is similar to ribosome footprint fragment length.
2.5.1. Equipment
Standard Thermomixer
Standard Microcentrifuge
Standard 1-D Vertical Gel Electrophoresis Equipment
2 mL Siliconized Microcentrifuge Tube
Corning Costar Spin-X Centrifuge Tube Filters (Fisher Scentific)
2100 Bioanalyzer (Agilent)
2.5.2. Buffers and Reagents
20% SDS
Acid Phenol Chloroform (Ambion)
Dry Ice
100% ethanol
Chloroform
3 M NaOAc pH 5.5 (Ambion)
0.5 M EDTA
100% isopropanol
80% ethanol from 100% Stock
10 mM Tris-HCl pH 7.0, from 1 M stock (Ambion)
Novex 2x TBE-Urea Sample Buffer (Invitrogen)
10 Base Pair Ladder (Invitrogen)
Novex TBE-Urea Gels, 15%, 10 well (Invitrogen)
1x TBE from 10x stock (Ambion)
SYBR Gold (Invitrogen)
Elution Buffer: 10 mM Tris-HCl pH 7.0, 1 mM EDTA, 0.5 M NaOAC pH 5.5
Glycogen (20 mg/mL)
Bioanalyzer RNA 6000 Nano Kit (Agilent)
Ribo-Zero rRNA Removal Kit: Gram-Negative Bacteria (Illumina)
2x alkaline hydrolysis buffer: 1 µL of 0.5 M EDTA, 30 µL of 0.1 M Na2CO3, 220 µL of 0.1 M NaHCO3
o199p RNA oligo (IDT)
2.5.3. Procedure
To frozen pulverized cell powder aliquot from section 2.3.3, add 0.6 ml of RNA lysis buffer including 40 µL of 20% SDS.
Immediately add 0.7 mL of acid phenol pre-warmed to 65 °C to the tube and mix by vortexing.
Incubate at 65 °C for 5 min while shaking in a thermomixer.
Chill in a dry ice - ethanol bath for 5 minutes (or until completely frozen).
Spin at 20,000 g for 2 min in a microcentrifuge and transfer supernatant to a fresh tube.
Add 0.7 mL of room temperature acid phenol, mix by vortexing, and then incubate at room temperature for 5 min.
Spin at 20,000 g for 2 min in a microcentrifuge and transfer aqueous layer to fresh tube.
Add 0.6 mL of chloroform and mix by vortexing.
Immediately spin at 20,000 g for 1 min in a microcentrifuge and transfer aqueous layer to fresh tube.
Add 65 µL of 3 M NaOAc pH 5.5 (or 1/9 volume) and 0.7 mL of 100% isopropanol (or 1.1 volume), and mix by vortexing.
Chill at −80 °C for 30 minutes.
Pellet RNA at 20,000 g for 1 h in a 4 °C microcentrifuge.
Quickly wash pellet with 0.8 mL of −20 °C 80% ethanol.
Suspend in 50 µL of 10 mM Tris-HCl pH 7.0.
Assess RNA quality before and after ribosomal removal (Figure 3) using Bioanalyzer and RNA 6000 Nano Kit, following manufactures instructions.
5 µg of RNA in a volume of no more than 26 µL can used for the subsequent steps.
Remove 16S and 23S ribosomal RNAs by subtractive hybridization using Ribo-Zero rRNA Removal Kit (Gram-Negative Bacteria). Follow manufactures instructions.
The protocol requires washing the magnetic beads, treating samples with rRNA removal solution, removing RNA, and purifying rRNA depleted samples.
Prepare 2x alkaline hydrolysis buffer and add 12.5 µL of 2x alkaline hydrolysis buffer to 12.5 µL of enriched mRNA.
Incubate at 95 °C for 23 minutes and immediately return to ice.
Add 560 µL of stop buffer to end hydrolysis.
Add 650 µL of 100% isopropanol.Mix by vortexing
Chill at −80 °C for 30 min.
Pellet hydrolyzed RNA at 20,000 g for 1 h in a 4 °C microcentrifuge.
Wash pellet with 0.8 mL of −20 °C 80% ethanol.
Dry and suspend pellet in 10 µL of 10 mM Tris-HCl pH 7.0.
Add 10 µL of 2x TBE-Urea sample buffer.
Prepare 10 base pair ladder in parallel with a solution containing: 1 µL of 10 base pair ladder, 9 µL of 10 mM Tris-HCl pH 7.0, and 10 µL of 2x TBE-Urea sample buffer.
Prepare o199-P control oligo in parallel with a solution containing: 2 µL of 10uM o199-P, 8 µL of 10 mM Tris-HCl pH 7.0, and 10 µL of 2x TBE-Urea sample buffer.
Thaw extracted ribosome footprints sample from section 2.4.3
Denature ladder and RNA samples at 80 °C for 1 min and return to ice.
Set up TBE-Urea Gel 15% in 1x TBE and pre-run for 1 hour at 200 volts.
Wash and then load lanes. Total RNA samples and footprinting samples from section 2.4.3 should be loaded on the same gel. Run for 65 minutes at 200 volts.
Stain gel with 6 µL of SYBR Gold in 60 mL of 1x TBE for 5 min and excise desired bands from 30–40 nt based on ladder and o199p oligo.
RNA recovery from acrylamide gels will be repeated in this procedure so it will be explained in detail here and only referenced subsequently.
Pierce a needle through a 0.5 mL tube and insert gel slice in pierced tube and nest pierced tube into a 2 mL siliconized microcentrifuge tube.
Spin at 20,000 g for 2 min in a microcentrifuge, or until most of the gel has extruded through, and transfer the remaining gel pieces.
Add 0.5 mL of elution buffer and freeze at −80 °C for 20 minutes.
Nutate overnight in a cold room or 4 °C refrigerator.
Incubate at 25 °C for 5 min while shaking in a thermomixer at 1400 rpm.
Transfer gel slurry to a Spin-X centrifuge tube filter using a wide bore pipette or a cut standard tip.
Add 2µL of glycogen and 550µL of 100% isopropanol (1.1 equivalence volume), then mix by vortexing and chill at −20 °C for 1 hour.
Pellet RNA at 20,000 g for 1 h in a 4 °C microcentrifuge and aspirate away the supernatant.
Wash pellet with 0.8 mL of 4 °C 80% ethanol.
Pulse spin to aspirate away remaining ethanol and air dry for 5 min.
Suspend recovered RNA in 15 µL of 10 mM Tris-HCl pH 7.0.
Figure 3.
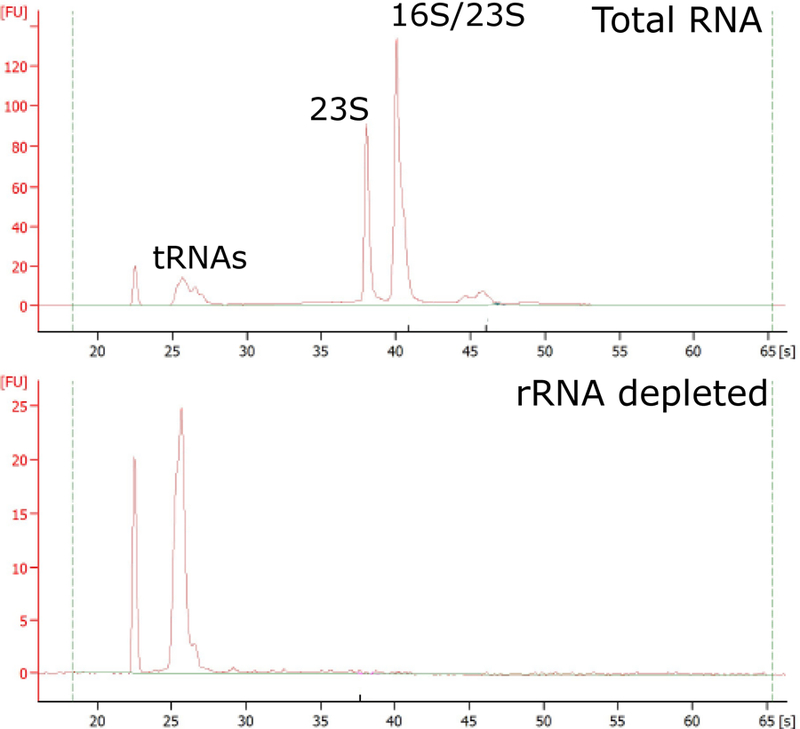
rRNA depletion of total RNA samples for total RNA-seq. Bioanalyzer traces before (top) and after rRNA removal (bottom). Stable RNA peaks are labeled. Caulobacter 23s rRNA is processed into two fragments, one runs at the same size as the 16S rRNA and the other runs at a shorter length. Y-axis scales are different for the two graphs.
2.5.4. Notes
o199p control and ladder help to identify appropriate RNA fragments.
Ribosome footprints may need to be spread across up to 4 lanes of the gel due to high concentration to avoid overloading.
2.6. Library Generation
Library preparation is performed by 3’ end repair, 3’ linker ligation, reverse transcription, single strand cDNA circularization, and finally PCR (Ingolia, Brar, Rouskin, McGeachy, & Weissman, 2012). These steps should be performed on both the footprinted mRNA fragments and the size selected mRNA fragments generated for mRNA-seq. Control o199p RNA oligo can be utilized throughout the library prep to ensure each step has worked efficiently. Quality control steps are noted below to ensure final DNA libraries contain a maximal amount of mRNA fragments. Library generation is performed as described in (Ingolia et al., 2012) but using the T4 RNA ligase KQ mutant (NEB) due to higher efficiency of ligation. DNA oligos utilized here have been adapted to work on the newer illumina sequencers including the Hiseq 4000 and Miniseq, as well as older sequencers (Table II).
Table II.
Oligos for library prep generation
| o199-P | 5’ AUGUACACGGAGUCGACCCGCAACGCGA/3phos/3’ |
| Linker-1 (NEB) | 5’ App/CTGTAGGCACCATCAAT/3ddC 3’ |
| o225-linker-mini-indexed | 5’ /5Phos/AGATCGGAAGAGCGTCGTGTAGGGAAAGAGTGT/iSp18/CACTCA/iSp18/CAAGCAGAAGACGGCATACGAGATATTGATGGTGCCTACAG 3’ |
| JS-ind1 | 5’AATGATACGGCGACCACCGAGATCTACACGATCGGAAGAGCACACGTCTGAACTCCAGTCACATCACGACACTCTTTCCCTACACGACG 3’ |
| JS-ind2 | 5’AATGATACGGCGACCACCGAGATCTACACGATCGGAAGAGCACACGTCTGAACTCCAGTCACCGATGTACACTCTTTCCCTACACGACG 3’ |
| JS-ind3 | 5’AATGATACGGCGACCACCGAGATCTACACGATCGGAAGAGCACACGTCTGAACTCCAGTCACTTAGGCACACTCTTTCCCTACACGACG 3’ |
| JS-ind4 | 5’AATGATACGGCGACCACCGAGATCTACACGATCGGAAGAGCACACGTCTGAACTCCAGTCACTGACCAACACTCTTTCCCTACACGACG 3’ |
| JS-ind5 | 5’AATGATACGGCGACCACCGAGATCTACACGATCGGAAGAGCACACGTCTGAACTCCAGTCACACAGTGACACTCTTTCCCTACACGACG 3’ |
| JS-ind6 | 5’AATGATACGGCGACCACCGAGATCTACACGATCGGAAGAGCACACGTCTGAACTCCAGTCACGCCAATACACTCTTTCCCTACACGACG 3’ |
| JS-ind7 | 5’AATGATACGGCGACCACCGAGATCTACACGATCGGAAGAGCACACGTCTGAACTCCAGTCACCAGATCACACTCTTTCCCTACACGACG 3’ |
| JS-ind8 | 5’AATGATACGGCGACCACCGAGATCTACACGATCGGAAGAGCACACGTCTGAACTCCAGTCACACTTGAACACTCTTTCCCTACACGACG 3’ |
| JS-ind9 | 5’AATGATACGGCGACCACCGAGATCTACACGATCGGAAGAGCACACGTCTGAACTCCAGTCACGATCAGACACTCTTTCCCTACACGACG 3’ |
| JS-ind10 | 5’AATGATACGGCGACCACCGAGATCTACACGATCGGAAGAGCACACGTCTGAACTCCAGTCACTAGCTTACACTCTTTCCCTACACGACG 3’ |
| JS-ind11 | 5’AATGATACGGCGACCACCGAGATCTACACGATCGGAAGAGCACACGTCTGAACTCCAGTCACGGCTACACACTCTTTCCCTACACGACG 3’ |
| JS-ind12 | 5’AATGATACGGCGACCACCGAGATCTACACGATCGGAAGAGCACACGTCTGAACTCCAGTCACCTTGTAACACTCTTTCCCTACACGACG 3’ |
| oJS231 | 5’ CAAGCAGAAGACGGCATACGAGATATTGATGGTGCC 3’ |
2.6.1. Equipment
Standard Thermomixer
Standard Microcentrifuge
Standard 1-D Vertical Gel Electrophoresis Equipment
2 mL Siliconized Microcentrifuge Tubes
0.2 mL PCR Tube
Agilent Bioanalyzer
Qubit Fluorimeter
Corning Costar Spin-X Centrifuge Tube Filters (Fisher Scentific)
Standard Nutator
2.6.2. Buffers and Reagents
10x T4 PNK Buffer supplied with enzyme (New England Biolabs)
T4 PNK Enzyme (New England Biolabs)
SUPERase•In RNase Inhibitor Enzyme 20 U/μL (Ambion)
Dephosphorylation Buffer Master Mix: 2 µL of 10x T4 PNK buffer, 1 µL SUPERase•In (for each reaction).
10 mM Tris-HCl pH 7.0 from 1 M stock (Ambion)
0.5 M EDTA
3 M NaOAc pH 5.5 (Ambion)
Precipitation Master Mix: 10 mM Tris pH 7.0, 3 mM EDTA, 0.375M NaOAc pH 5.5.
Glycogen (20mg/mL)
100% Isopropanol
100% Ethanol
80% Ethanol from 100% stock
50% sterile filtered PEG M.W. 8000
10x T4 RNA Ligase 2 Buffer, supplied with enzyme (New England Biolabs)
100% DMSO
1 µg/µL of Linker-1 (New England Biolabs)
T4 RNA Ligase 2, Truncated K227Q Enzyme (New England Biolabs)
Novex 2x TBE-Urea Sample Buffer (Invitrogen)
Ligation Master Mix: 8 µL of 50% sterile filter PEG M.W., 2 µL of 10x T4 Ligase 2 buffer, 2 µL of 100% DMSO, 1 µL of SUPERase•In, 1 µL of 1 µg/µL of Linker-1 (for each reaction).
10 Base Pair Ladder (Invitrogen)
Novex TBE-Urea Gels, 15%, 10 well (Invitrogen)
1x TBE from 10x stock (Ambion)
SYBR Gold (Invitrogen)
Elution Buffer: 10 mM Tris-HCl pH 7.0, 1 mM EDTA, 0.5 M NaOAC pH 5.5
10 mM dNTP (New England Biolabs)
20 µM o225-linker-mini-indexed (IDT)
5x FSB Buffer supplied with enzyme (Invitrogen)
0.1 M DTT supplied with enzyme (Invitrogen)
Superscript III (Invitrogen)
1 N NaOH
10 mM Tris-HCl pH 8.0 from 1 M stock (Ambion)
Nucleic Acid Master Mix: 1 µL of 10 mM dNTP, 1 µL of 25 µM o225-linker-mini-indexed, 1.5 µL of Milli-Q filtered water (for each reaction)
Reverse Transcription Buffer Master Mix: 4 µL of 5x FSB buffer, 1 µL of 0.1 M DTT, 1 µL of SUPERase•In (for each reaction)
CircLigase Enzyme Kit (Epicentre)
Circularization Master Mix: 2 µL of 10x CircLigase buffer, 1 µL of 1 mM ATP, 1 µL of 50 mM MnCl2 (all components supplied with enzyme)
High fidelity Phusion Enzyme (New England Biolabs)
10 mM dNTP (New England Biolabs)
100 µM DNA oligos (from Table II) (IDT)
Amplification Master Mix: 16.7 µL of 5x High Fidelity buffer (supplied with enzyme), 1.7 µL of dNTP, 0.4 µL of 100 µM JS-ind (use separate oligos #1–12 for each biological sample), 0.4 µL of 100 µM oJS231, 59.2 µL of Milli-Q filtered water, 0.8 µL of High Fidelity Phusion Enzyme (for each sample)
6x DNA loading dye
0.05% Tween 20
EBT: 10 mM Tris-HCl pH 8.0, 0.05% Tween 20
High Sensitivity DNA kit (Agilent)
2.6.3. Procedure
Place the size selected mRNA footprints, size selected total mRNA fragments, and o199-P control sample on ice.
To dephosphorylate the samples, prepare dephosphorylation buffer master mix and add to 15 µL of the sample.
Add 2 µL of T4 PNK and incubate reaction at 37 °C for 1 h.
Prepare precipitation master mix and add 90 µL to each tube.
Heat kill enzyme at 75 °C for 10 min. Acid:Phenol chloroform extraction will work as well.
Add 0.3 mL of 100% ethanol, 2 µL glycogen and mix by vortexing
Chill at −20 °C for 1 h.
Pellet dephosphorylated RNA at 20,000 g for 1 h in a 4 °C microcentrifuge.
Remove supernatant, wash pellet with 0.8 mL of 4 °C 80% ethanol, pellet by spinning at 20,000 g for 15 min in a 4 °C microcentrifuge. Repeat washes two additional times.
Air dry pellet for 5 min.
Suspend pellet in 5 µL of 10 mM Tris pH 7.0.
Prepare the ligation master mix. The PEG will crash out of solution upon addition of DMSO. Warm the tube in your hands to solubilize.
Add 2 µL of T4 RNA Ligase 2, Truncated K227Q recording lot number, mix by vigorously pipetting and incubate reaction at 25 °C for 2.5 h.
Add 20 µL of 2x TBE-Urea sample buffer to the samples.
Prepare 10 bp ladder in parallel: 1 µL of 10 bp ladder, 5 µL of 10 mM Tris-HCl pH 7.0, 6 µL of 2x TBE-Urea sample buffer.
Denature samples at 70 °C for 1 min and return to ice.
Setup 10% TBE-Urea gel in 1x TBE and pre-run for 1 h at 200 volts. It is useful that o199-P negative control and ligated 0199-P positive control are included as sizing controls and to assess efficiency of ligation.
Wash wells and load the gel. Every other lane should be skipped as to not cross contaminate samples.
Run for 50 min at 200 volts.
Stain gel with 6 µL of SYBR gold in 60 mL of 1x TBE for 5 minutes.
Excise the desired band and recover ligated products.
To recover RNA, follow the procedure detailed at the end of section 2.5.3.
Resuspend in 10 µL of 10 mM Tris-HCl pH 7.0.
For reverse transcription, prepare nucleic acid master mix and add 3.5 µL to 10 µL of ligated RNA.
Denature at 60 °C for 5 minutes and return to ice.
Prepare reverse transcription buffer master mix, add 6 µL to the sample, and then add 1 µL of Superscript III.
Incubate at 50 °C for 30 minutes and then add 2.3 µL of 1 N NaOH.
Incubate at 95 °C for 15 min.
To ethanol precipitate add 23 µL of −20 °C 100% ethanol (3 volumes), 2.3 µL of sodium acetate (1/10 volume), and 2 µL of glycogen then mix by vortexing and chill at −20 °C for 1 h.
Pellet cDNA at 20,000 g for 1 h in a 4 °C microcentrifuge and then aspirate away the supernatant. Wash pellet with 1 mL of −20 °C 80% ethanol. Repeat wash 2 more times.
Pulse spin to aspirate away remaining ethanol and air dry for 5 minutes.
After the pellet is dry, suspend the sample in 5 µL of 1x Novex TBE-Urea Sample Buffer.
Prepare 10 bp ladder in parallel: 1 µL of 10 bp ladder, 9 µL of 10 mM Tris-HCl pH 8.0, 10 µL of 2x TBE-Urea sample buffer.
Denature all samples at 80 °C for 2 min and return to ice.
Setup 9% TBE-Urea gel in 1x TBE. Pre-run for 1 h at 200 volts.
Wash lanes and load in samples. Run for 70 min at 200 volts checking predicted size versus dye.
Stain gel with 6 µL of SYBR gold in 60 mL of 1 x TBE for 5 min.
Excise desired bands and recover the reverse transcription products.
To recover DNA pierce a needle through a 0.5 mL tube, insert gel slice in pierced tube, and nest pierced tube into a 2 mL siliconized microcentrifuge tube.
Spin at 20,000 g for 2 minutes in a microcentrifuge or until most of the gel has extruded through. Transfer the remaining gel pieces.
Add 0.5 mL of elution buffer (1/9 volume) and freeze at −80 °C for 20 minutes.
Incubate at 70 °C for 10 min. Shake in thermomixer at 1400 rpm.
Transfer gel slurry to a Spin-X centrifuge tube filter using a wide bore pipette or a cut standard tip.
Add 2 µL of glycogen and 550 µL of 100% isopropanol (1.1 volume), then mix by vortexing and chill at −20 °C for 1 h.
Pellet DNA at 20,000 g for 1 h in a 4 °C microcentrifuge and aspirate away the supernatant.
Wash pellet with 0.8 mL of 4 °C 80% ethanol.
Pulse spin to aspirate away remaining ethanol and air dry for 5 minutes.
After DNA has been recovered, resuspend in 15 µL of 10 mM Tris-HCl pH 8.0.
Add 4 µL of circularization master mix and 1 µL of CircLigase enzyme and incubate at 60 °C for 1 h. Include o225-linker-mini-indexed alone as negative control.
Heat kill enzyme at 80 °C for 10 minutes and then place on ice.
For PCR amplification, prepare amplification master mix and add 79.2 µL to 5µL of circularized ssDNA from proceeding steps. PCR primers containing DNA barcodes to allow for multiplexed sequencing on illumina sequencers should be used (Table II) with a different index oligo for each sample.
- Aliquot 17 µL of PCR mix into four separate PCR strip tubes and perform the following PCR reaction:
Initial denaturing 98°C 30 seconds Denaturing 98°C 10 seconds Annealing 60°C 10 seconds Extension 72°C 5 seconds
Remove PCR tubes after cycles 6, 8, 10 and 12. The sample with the fewest number of cycles with visible PCR product will be gel extracted. As a control, include the o225-linker-mini-indexed to mark the size of no-insert products.
Setup 8% TB polyacrylamide gel in 1x TBE.
Directly load samples and run at 180 volts for 40 min.
Stain gel with 6 µL of SYBR gold in 60 mL of 1x TBE for 5 min.
Excise insert DNA band and recover double stranded DNA products by piercing a needle through a 0.5 mL tube.
Insert gel slice in pierced tube and nest pierced tube into a 2 mL siliconized microcentrifuge tube.
Spin at 20,000 g for 2 minutes in a microcentrifuge, or until most of the gel has extruded through. Transfer the remaining gel pieces.
Add 0.5 mL of elution buffer (1/9 volume) and freeze at −80 °C for 20 min.
Incubate at 70 °C for 10 min in thermomixer at 1400 rpm
Transfer gel slurry to a Spin-X centrifuge tube filter using a wide bore pipette or a cut standard tip.
Add 2 µL of glycogen and 550 µL of 100% isopropanol (1.1 volume), mix by vortexing, and then chill at −20 °C for 1 h.
Pellet DNA at 20,000 g for 1 h in a 4 °C microcentrifuge and aspirate away the supernatant.
Wash pellet with 0.8 mL of 4 °C 80% ethanol.
Pulse spin to aspirate away remaining ethanol and air dry for 5 min.
Suspend amplified DNA in 10 µL of EBT.
For quantification of amplified DNA, dilute 1 µL of amplified DNA with 9 µL of 10mM Tris-HCl pH 8.0.
Assess DNA quality using the bioanalyzer by the steps outlined by Agilent for the High Sensitivity DNA Kit. Quantify DNA concentration using the Qubit fluorimeter.
Samples should then be pooled in equal amounts for Illumina sequencing (50 bp single end) on an Illumina HiSeq instrument.
2.6.4. Notes
Bring the o199 control oligo through each step-in library prep to assess problematic reactions.
Ligation efficiency markedly decreases if PEG is old so it is recommended that PEG be made within a month of use.
T4 RNA Ligase 2, Truncated can be used for ligation instead of T4 RNA Ligase 2, Truncated K227Q mutant, but higher yields have been obtained with the K227Q mutant.
Addition of 1 N NaOH and heating at 95 oC during the reverse transcription steps quenches the reaction by hydrolyzing RNA template.
Loading the gel with small volumes avoids formation of “bunny ears” on gels.
Summary and Conclusion
If followed correctly, this procedure allows the genome-wide measurement of translation at the key stages of the Caulobacter cell cycle. For each ORF, the ratio of ribosome footprint density to total RNA-seq density can be used to calculate translation efficiency, which can be assessed across the cell cycle. Currently, Caulobacter is the only organism with a high-yield drug free method of cell cycle synchronization making it a powerful organism for the study of the bacterial cell cycle. Innovative new methodologies in cell synchronization, such as “baby machines” (Bates, Epstein, Boye, Fahrner, Berg, & Kleckner, 2005), should allow for the expansion of ribosome profiling studies into the cell cycles of other bacterial species.
Acknowledgements
Research reported in this publication was supported by NIGMS of the National Institutes of Health under award number R35GM124733. The authors thank Wayne State University startup funds to JMS.
Footnotes
Competing Interests
The authors declare no competing interests exist.
References
- Bates D, Epstein J, Boye E, Fahrner K, Berg H, & Kleckner N (2005). The Escherichia coli baby cell column: a novel cell synchronization method provides new insight into the bacterial cell cycle. Molecular Microbiology, 57(2), 380–391. doi: 10.1111/j.1365-2958.2005.04693.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brar GA, Yassour M, Friedman N, Regev A, Ingolia NT, & Weissman JS (2012). High-resolution view of the yeast meiotic program revealed by ribosome profiling. Science, 335(6068), 552–557. doi: 10.1126/science.1215110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collier J (2016). Cell cycle control in Alphaproteobacteria. Current Opinion in Microbiology, 30, 107–113. doi: 10.1016/j.mib.2016.01.010 [DOI] [PubMed] [Google Scholar]
- Evinger M, & Agabian N (1977). Envelope-associated nucleoid from Caulobacter crescentus stalked and swarmer cells. Journal of Bacteriology, 132(1), 294–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingolia NT, Brar GA, Rouskin S, McGeachy AM, & Weissman JS (2012). The ribosome profiling strategy for monitoring translation in vivo by deep sequencing of ribosome-protected mRNA fragments. Nature Protocols, 7(8), 1534–1550. doi: 10.1038/nprot.2012.086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingolia NT, Ghaemmaghami S, Newman JRS, & Weissman JS (2009). Genome-Wide Analysis in Vivo of Translation with Nucleotide Resolution Using Ribosome Profiling. Science, 324(5924), 218–223. doi: 10.1126/science.1168978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasker K, Mann TH, & Shapiro L (2016). An intracellular compass spatially coordinates cell cycle modules in Caulobacter crescentus. Current Opinion in Microbiology, 33, 131–139. doi: 10.1016/j.mib.2016.06.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAdams HH, & Shapiro L (2011). The architecture and conservation pattern of whole-cell control circuitry. Journal of Molecular Biology, 409(1), 28–35. doi: 10.1016/j.jmb.2011.02.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connor PB, Li GW, Weissman JS, Atkins JF, & Baranov PV (2013). rRNA:mRNA pairing alters the length and the symmetry of mRNA-protected fragments in ribosome profiling experiments. Bioinformatics, 29(12), 1488–1491. doi: 10.1093/bioinformatics/btt184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh E, Becker AH, Sandikci A, Huber D, Chaba R, Gloge F, . . . Bukau B (2011). Selective ribosome profiling reveals the cotranslational chaperone action of trigger factor in vivo. Cell, 147(6), 1295–1308. doi: 10.1016/j.cell.2011.10.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrader JM, Li GW, Childers WS, Perez AM, Weissman JS, Shapiro L, & McAdams HH (2016). Dynamic translation regulation in Caulobacter cell cycle control. Proceedings of the National Academy of Sciences of the United States of America, 113(44), E6859–E6867. doi: 10.1073/pnas.1614795113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrader JM, & Shapiro L (2015). Synchronization of Caulobacter crescentus for investigation of the bacterial cell cycle. Journal of Visualized Experiments(98), e52633. doi: 10.3791/52633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stumpf CR, Moreno MV, Olshen AB, Taylor BS, & Ruggero D (2013). The translational landscape of the mammalian cell cycle. Molecular Cell, 52(4), 574–582. doi: 10.1016/j.molcel.2013.09.018 [DOI] [PMC free article] [PubMed] [Google Scholar]