

Abstract
Thromboembolic events secondary to rupture or erosion of advanced atherosclerotic plaques represent the leading cause of death worldwide. However, the mechanisms that regulate plaque stability are poorly understood. Human pathology studies show that lesions containing a high ratio of ACTA2+ to CD68+ cells and a thicker extracellular matrix (ECM)-rich fibrous cap are more stable, but there are major ambiguities regarding the origins and functions of ACTA2+ cells within lesions as well as the mechanisms that regulate their investment and/or retention in the protective fibrous cap. The overall hypothesis of our Leducq Fondation PlaqOmics TransAtlantic Network is that detrimental reprogramming of smooth muscle cells (SMC) and other ACTA2+ fibrous cap cells destabilizes atherosclerotic plaques and that there are critical genetic determinants of coronary artery disease (CAD) risk that act in part by impacting phenotypic transitions of fibrous cap cells. Our ultimate goals are to: 1) define distinct SMC phenotypes within human lesions and how these change as a function of lesion severity and vulnerability for rupture or erosion; and 2) to use complementary human and mouse studies to determine mechanisms by which these cells impact lesion pathogenesis and to identify novel therapeutic targets that promote beneficial (plaque stabilizing) changes in the phenotype of SMC and other ECM-producing lesion cells.
Keywords: atherosclerosis, smooth muscle cells, genetic determinants of plaque stability
Atherosclerosis is a progressive disease that is the leading global cause of death(1). Despite decades of research, there remain major ambiguities regarding the mechanisms that control plaque stability and the probability of plaque rupture or erosion leading to possible myocardial infarction (MI) or stroke(2). The general dogma is that: 1) plaques containing a large necrotic core, a thin fibrous cap, and large numbers of CD68+ macrophages (MФ) relative to ACTA2+ smooth muscle cells (SMC) are more prone to rupture; and 2) the primary role of the SMC is beneficial due to its role in formation of a protective fibrous cap (3). However, the members of our PlaqOmics Transatlantic Network https://www.plaqomics.com/members/ have published human pathological, in silico, and animal studies that provide compelling evidence challenging this dogma. Collectively, our data support the view that SMCs play a much more important, diverse, and complex role in lesion pathogenesis than has been generally appreciated.
For example, two recent Nature Medicine studies by the Owens lab, showed that SMCs can have a major protective or detrimental role depending on the nature of their phenotypic transitions (4;5). Transitions mediated by the transcription factor Klf4 led to formation of SMC-derived MФ-marker+ foam cells (4) and exacerbated lesion pathogenesis whereas Oct4-dependent transitions (5) were protective including being critical for investment of SMC into the fibrous cap. Based on data from the CARDIoGRAM consortium, co-led by PlaqOmics members Erdmann/Schunkert, the Klf4 locus was identified to be genome-wide significantly associated with coronary artery disease (CAD) (6). The Pasterkamp and Virmani labs have observed shifts in plaque characteristics underlying clinical manifestations of coronary artery disease and stroke, that mandate a re-evaluation of the classic description of the vulnerable plaque. Notably this includes observing an increased proportion of Acta2+ cell-rich fibrous lesions undergoing plaque erosion rather than plaque rupture contributing to symptomatic cerebrovascular disease (7). The Mayr lab used proteomics to uncover novel extracellular matrix (ECM) proteins upon vascular injury (8) and revealed a plethora of ECM and inflammatory changes that are thought to destabilize the fibrous cap and contribute to lesion instability. Moreover, a recent Science paper(9) by the Björkegren lab, used integrated systems genetic analyses of RNAseq profiles obtained from atherosclerotic and non-atherosclerotic arterial wall samples of CAD patients in the STARNET study with genetic data from CARDIoGRAM. Results showed that downstream effects of genetic determinants in CAD are highly inter-connected in molecular networks acting across cell-types and tissues to cause CAD thus reinforcing the general concept of a more interconnected and multi-functional role of individual plaque cell types. Finally, recent studies by the Leeper lab have identified novel mechanisms that contribute to clonal expansion of SMC within lesions including a potential role for mechanisms that control efferocytotic processes within lesions (10).
Recent joint studies by PlaqOmics members observed a striking overlap between gene loci identified from human genome wide association (GWAS) studies to confer risk for CAD, and those that are expressed in plaque SMCs of atherosclerotic mouse lesions. Not only the Klf4 locus itself (6) but also numerous putative Klf4 or OCT4 target genes in SMC (4;5) display significant signals in CAD GWAS (11). These data provide evidence that our mouse models of advanced atherosclerosis can provide valuable insights into mechanisms that impact the pathogenesis of human atherosclerotic disease. Remarkably, we also found that >80% of SMC-derived cells in advanced lesions lacked detectable SMC markers, and that nearly a third of the MФ-marker+ foam cells within advanced mouse and human coronary artery lesion specimens were of SMC not myeloid origin (4). Consistent with these latter studies, Gordon Francis and co-workers (12) demonstrated that nearly 50% of foam cells within advanced human coronary artery lesions are positive for both Acta2 and CD68 making it uncertain if these are SMCs that have activated MФ markers, MФs that have activated SMC markers, or neither. Moreover, there is recent evidence showing that endothelial cells undergo EndoMT within atherosclerotic lesions and give rise to Acta2+ fibrous cap cells (13). Although enhancement of this process appears to exacerbate lesion development within ApoE−/− WD fed mice, the role of this process in late stage lesion pathogenesis in humans is unknown.
Taken together, the preceding observations show that: 1) one cannot rely on traditional SMC, endothelial cell (EC), and MФ markers to unambiguously distinguish SMC versus other cell types within human atherosclerotic lesion specimens and must rely on use of SMC-, EC-, and macrophage-specific lineage tracing mice and other approaches; 2) previous studies have grossly underestimated the frequency of SMCs within lesions and have misidentified many of lesion SMC as MФs or other cell types; and 3) identification of genomic determinants of SMC phenotypic changes that are important for the stability of human atherosclerotic plaques must include single cell analyses and unambiguous identification of SMCs. Of major interest, ongoing scRNAseq analyses of advanced brachiocephalic lesions from SMC lineage tracing ApoE knockout mice fed a Western diet (WD) for varying periods of time has identified multiple unique clusters of SMC-derived lesion cells of which only a few continue to express classical SMC markers such as Acta2 and Myh11 which have been used in virtually all previous studies in the field to identify lesion SMC.
A major challenge and goal of our Leducq PlaqOmics group is to identify the functions of these SMC subsets, to ascertain which are beneficial or detrimental for late stage lesion pathogenesis in mice, to determine which of these phenotypes are present in human atherosclerotic lesions, and how their frequency or functions vary in stable versus unstable lesions. We aim to identify genetic variants that influence these SMC phenotypic transitions, and ultimately to identify novel therapeutic approaches to enhance plaque stability by promoting beneficial phenotypic changes in SMC and other cells that contribute to formation and maintenance of the protective fibrous cap. Indeed, we hope to identify completely novel therapeutic approaches for doing this that augment the benefits of lipid lowering and antithrombotic therapies, and which are customized based on genetic, genomic, and proteomic profiling to the individual patient.
The overall hypothesis of PlaqOmics is that detrimental reprogramming of SMC destabilizes atherosclerotic plaques and that there are critical genetic determinants of CAD risk that act in part by impacting SMC phenotypic transitions. Our ultimate goals are to: 1) define distinct SMC phenotypes within human lesions and how these change as a function of lesion severity and vulnerability for rupture or erosion; and 2) to use complementary human and mouse studies to determine mechanisms by which these cells impact lesion pathogenesis and to identify novel therapeutic targets that promote beneficial (plaque stabilizing) changes in the phenotype of SMC and other ECM producing lesion cells. We have three specific aims to accomplish our objectives [Figure 1].
Figure 1:
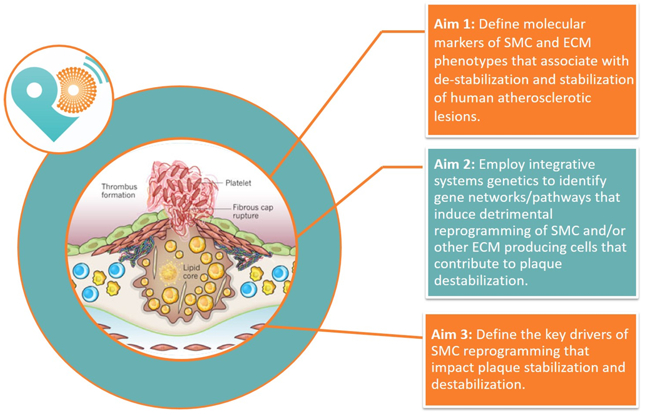
Aims of our PlaqOmics TransAtlantic Leducq Fondation Network. Our PlaqOmics Network is led by G. Owens (University of Virginia) in the United States and G. Pasterkamp (University Medical Center Utrecht) in Europe. The other project leaders are J. Erdmann (University of Lübeck), J. Björkegren (Icahn School of Medicine at Mount Sinai), M. Joner (Technische Universität Münich), M. Mayr (King’s College London), A. Finn (CVPath Institute) and N. Leeper (Stanford University).
Our first aim is to define molecular markers of SMC and ECM phenotypes that associate with de-stabilization and stabilization of human atherosclerotic lesions. This will include identifying SMC subsets in atherosclerotic lesions based on single cell RNAseq analyses on freshly isolated cells from human coronary artery and carotid plaques and cross referencing these data to scRNAseq analyses of mouse lesions from SMC lineage tracing mice to rigorously validate SMC identity.
Our second aim is to employ integrative systems genetics to identify gene networks and within these, pathways that induce detrimental reprogramming of SMC and/or other ECM producing cells that contribute to plaque destabilization. This aim includes: 1) defining candidate genes/pathways that potentially regulate SMC phenotypic transitions critical in regulating lesion stability; 2) identifying co-expression networks that causally affect plaque stability mainly through action of SMCs and other fibrous cap cells; and 3) integrative data analyses using a novel web tool generated by PlaqOmics team member Johan Bjorkegren.
Our third aim is to define the key drivers of SMC reprogramming that impact plaque stabilization and destabilization. We will do this by determining the potential functional roles of these genes in regulating SMC phenotypic transitions critical for plaque stability and elucidate underlying mechanisms and potential therapeutic targets.
In summary, we feel that we have assembled a powerful international team of investigators [Figure 1] with the innovative view that in the current era of preventive medical treatment and risk factor management, the current dogma of the ‘vulnerable plaque” needs to be redefined. Indeed, we are excited and confident that we can achieve unprecedented advances in our understanding of determinants of plaque stability and in particular the role of SMC and other major ECM producing lesion cells.
Acknowledgments
Sources of Research Funding: Leducq Fondation, Paris France
Footnotes
Disclosures: None
Reference List
- 1.Global, regional, and national age-sex specific all-cause and cause-specific mortality for 240 causes of death, 1990–2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet 2015. January 10;385(9963):117–71. PMCID:PMC4340604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pasterkamp G, den Ruijter HM, Libby P. Temporal shifts in clinical presentation and underlying mechanisms of atherosclerotic disease. Nat.Rev.Cardiol 2017. January;14(1):21–9 [DOI] [PubMed] [Google Scholar]
- 3.Bennett MR, Sinha S, Owens GK. Vascular Smooth Muscle Cells in Atherosclerosis. Circ.Res 2016. February 19;118(4):692–702. PMCID:PMC4762053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shankman LS, Gomez D, Cherepanova OA, Salmon M, Alencar GF, Haskins RM, Swiatlowska P, Newman AA, Greene ES, Straub AC, et al. KLF4-dependent phenotypic modulation of smooth muscle cells has a key role in atherosclerotic plaque pathogenesis. Nat.Med 2015. May 18;21(6):628–37. PMCID:PMC4552085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cherepanova OA, Gomez D, Shankman LS, Swiatlowska P, Hess DL, Williams J, Sarmento OF, Alencar GF, Bevard MH, Greene ES, et al. Activation of the pluripotency factor Oct4 in smooth muscle cells is atheroprotective. Nature Medicine Forthcoming. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.van der Harst P, Verweij N. The Identification of 64 Novel Genetic Loci Provides an Expanded View on the Genetic Architecture of Coronary Artery Disease. Circ.Res 2017. December 6; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.van Lammeren GW, den Ruijter HM, Vrijenhoek JE, van der Laan SW, Velema E, De Vries JP, de Kleijn DP, Vink A, de Borst GJ, Moll FL, et al. Time-dependent changes in atherosclerotic plaque composition in patients undergoing carotid surgery. Circulation 2014. June 3;129(22):2269–76 [DOI] [PubMed] [Google Scholar]
- 8.Langley S, Molenaar C, Lu R, Barwari T, Suna G, Yin X, Iglseder B, Paulweber B, Wileit P, Shalhoub J, et al. Extracellular matrix proteomics identifies a molecular signature of symptomatic carotid plaques. J.Clin.Invest Forthcoming. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Franzen O, Ermel R, Cohain A, Akers NK, Di NA, Talukdar HA, Foroughi-Asl H, Giambartolomei C, Fullard JF, Sukhavasi K, et al. Cardiometabolic risk loci share downstream cis- and trans-gene regulation across tissues and diseases. Science 2016. August 19;353(6301):827–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kojima Y, Volkmer JP, McKenna K, Civelek M, Lusis AJ, Miller CL, DiRenzo D, Nanda V, Ye J, Connolly AJ, et al. CD47-blocking antibodies restore phagocytosis and prevent atherosclerosis. Nature 2016. August 4;536(7614):86–90. PMCID:PMC4980260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kessler T, Vilne B, Schunkert H. The impact of genome-wide association studies on the pathophysiology and therapy of cardiovascular disease. EMBO Mol.Med 2016;8(7):688–701. PMCID:PMC4931285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Allahverdian S, Chehroudi AC, McManus BM, Abraham T, Francis GA. Contribution of Intimal Smooth Muscle Cells to Cholesterol Accumulation and Macrophage-Like Cells in Human Atherosclerosis. Circulation 2014. January 30; [DOI] [PubMed] [Google Scholar]
- 13.Chen PY, Qin L, Baeyens N, Li G, Afolabi T, Budatha M, Tellides G, Schwartz MA, Simons M. Endothelial-to-mesenchymal transition drives atherosclerosis progression. J Clin Invest 2015. December;125(12):4514–28. PMCID:PMC4665771 [DOI] [PMC free article] [PubMed] [Google Scholar]