

Abstract
It is well established that selective activation of group I metabotropic glutamate (mGlu) receptors induces LTD of synaptic transmission at Schaffer collateral–CA1 synapses. In contrast, application of 1S,3R-ACPD, a mixed agonist at group I and group II mGlu receptors, induces LTP. Using whole-cell recordings from CA1 pyramidal cells and field recordings in the hippocampal CA1 region, we investigated the specific contribution of group II mGlu receptors to synaptic plasticity at Schaffer collateral–CA1 synapses in acute slices of adult mice. Pharmacological activation of group II mGlu receptors (mGlu2 and mGlu3 receptors) with the specific agonist LY354740 in conjunction with electrical stimulation induced postsynaptic LTP. This form of plasticity requires coactivation of NMDA receptors (NMDARs). Group II mGlu receptor activation led to PKC-dependent phosphorylation of the GluN1 subunit. We found that both synaptic and extrasynaptic NMDARs, which are differentially modulated by mGlu2 and mGlu3 receptors, contribute to LTP induction. Furthermore, LTP initiated by activation of group II mGlu receptors was not occluded by LTP induced with high-frequency trains of stimuli. However, the phosphorylation of NMDARs mediated by group II mGlu receptor activation led to a priming effect that enhanced subsequent high-frequency stimulation-induced LTP. These findings reveal a novel metaplastic mechanism through which group II mGlu receptors modulate synaptic function at the Schaffer collateral input to CA1 pyramidal cells, thereby lowering the threshold to induce plasticity.
SIGNIFICANCE STATEMENT The group II metabotropic glutamate (mGlu II) receptors exert a well characterized action on presynaptic neuron terminals to modulate neurotransmitter release. Here, we show that these receptors also have postsynaptic effects in promoting the induction of synaptic plasticity. Using an electrophysiological approach including field and whole-cell patch recording in hippocampi from wild-type and transgenic mice, we show that activation of group II mGlu receptors enhances NMDA receptor (NMDAR)-mediated currents through PKC-dependent phosphorylation. This priming of NMDARs lowers the threshold for the induction of LTP of synaptic transmission. These findings may also provide new insights into the mechanisms through which drugs targeting mGlu II receptors alleviate hypoglutamatergic conditions such as those occurring in certain brain disorders such as schizophrenia.
Keywords: CA1, hippocampus, LTP, metabotropic glutamate receptor, metaplasticity, NMDA receptor
Introduction
Group II metabotropic glutamate receptors (mGlu II) comprising the mGlu2 and the mGlu3 receptor subtypes are present throughout the brain with high expression in the hippocampus (Tamaru et al., 2001). Their role as presynaptic autoreceptors in the negative feedback regulation of glutamate release is well established at the mossy fiber–CA3 synapse (Kamiya et al., 1996; Scanziani et al., 1997), at CA1 synapses onto subicular neurons (Kintscher et al., 2012), and at the lateral perforant path (Aksoy-Aksel and Manahan-Vaughan, 2015) and medial perforant path between the entorhinal cortex and CA1 pyramidal cells (Macek et al., 1996; Capogna, 2004). More recently, postsynaptic actions have been characterized including the initiation by somatodendritic mGlu3 receptors of theta oscillations in the hippocampal CA3 area (Ster et al., 2011) and the activation by mGlu2 receptors of a dendritic shunt in dentate granule cells involving a G-protein-coupled inwardly rectifying potassium conductance (Brunner et al., 2013). mGlu II receptors are also implicated in learning and synaptic plasticity. Spatial memory encoding in rats is deficient after blockade of mGlu II receptors (Altinbilek and Manahan-Vaughan, 2009). In addition, working memory and performance on a spatial novelty task relying on the explorative drive of the animal are impaired in double knock-out (KO) (GRM2/3) mice (Lyon et al., 2011).
At the mossy fiber–CA3 synapse, activation of mGlu2 receptors at presynaptic terminals induces LTD (Yokoi et al., 1996), presumably by inhibition of adenylyl cyclase (Conn and Pin, 1997). However, at Schaffer collateral synapses onto CA1 pyramidal cells, the roles of mGlu II receptors in synaptic plasticity remain unclear. Earlier studies reported that application of the metabotropic agonist 1S,3R-ACPD induces LTP at these synapses (Bashir et al., 1993; Bortolotto et al., 1994), but 1S,3R-ACPD was later shown to be an agonist at both mGlu I and mGlu II receptors. Selective activation of mGlu I receptors induces LTD at these synapses (Palmer et al., 1997; Fitzjohn et al., 1999; Huber et al., 2000). We therefore hypothesized that the LTP observed in response to a mixed agonist is mediated primarily by mGlu II receptors. Indeed, our experimental results indicate that activation of mGlu II receptors leads to the phosphorylation of NMDA receptors, which facilitates the induction of LTP.
Materials and Methods
Tissue preparation.
Acute slices were prepared from juvenile male mice at postnatal day 21 (P21) to P27 following a protocol approved by the Veterinary Department of the Canton of Zurich (approval ID: 81–2014). Transgenic animals used in our experiments were mGlu2−/− (RRID:MGI:3629946) (Morishima et al., 2005) and mGlu3−/− (Corti et al., 2007) mice. Mice were decapitated and brains quickly removed in ice-cold artificial CSF (ACSF) solution containing the following (in mm): 125 NaCl, 2.5 KCl, 1.25 NaH2PO4, 25 NaCHCO3, 1 MgCl2, 2 CaCl2, and 10 glucose, pH 7.4, equilibrated with 95% O2 and 5% CO2. Next, 350 μm (for field) or 300 μm (for patch) thick sagittal acute slices were prepared with a vibratome (HM 650V; Microm International) in the same ice-cold ACSF. Sections were incubated in ASCF for 20 min at 34°C and then kept at room temperature for at least 1 h before recording.
Drugs.
d-(−)-2-amino-5-phosphonopentanoic acid (D-AP5), NBQX disodium salt, LY341495, Ro 25–6981 maleate salt, MK-801, genistein, and cyclopiazonic acid (CPA) were from Abcam. LY354740, bisindolylmaleide II, chelerytherine chloride, and H89 dihydrochloride were from Tocris Bioscience. N-ethyl-maleimide (NEM) was from Sigma-Aldrich and NVP-AAM077-NX-6 was a generous gift from Novartis Pharma.
Electrophysiology.
Recordings were obtained using an Axopatch 200B amplifier (Molecular Devices). Acute slices were transferred to a recording chamber mounted on an upright microscope (Axioscope FS1; Zeiss). Slices were superfused continuously at 2 ml/min with ACSF at 28°C. Single test pulses were delivered to the Schaffer collaterals (stratum radiatum of CA1) at 0.05 Hz via a monopolar electrode. Input–output curves were generated for each slice and the stimulation intensity was adjusted to 40–50% of the maximum response. After at least 10 min of stable baseline fEPSPs, LTP was induced either by bath application of LY354740 (1 μm for 10 min), a specific group II mGlu receptor agonist, or by 3 trains of 100 Hz stimulation of 1 s duration separated by 20 s of high-frequency stimulation (HFS). Alternatively, after a 5 min baseline, slices were pretreated with a group II mGlu receptor antagonist LY341495 (4 μm), a membrane permeable blocker of G-proteins, NEM (100 μm), or an NMDAR antagonist, D-AP5 (40 μm) for 10 min before coapplication with LY354740. Slices were pretreated with a PKA antagonist, H89 (10 μm), a GluN2A antagonist, NVP-AAM077 (0.1 μm), or a GluN2B antagonist, Ro25–69813 (3 μm), for 20 min before coapplication with LY354740 and were pretreated with PKC antagonists, either bisindolylmaleide II (1 μm) or chelerytherine (10 μm), or a protein tyrosine kinase inhibitor, genistein (30 μm), for 25 min before coapplication with LY354740. The use-dependent NMDAR blocker MK-801 (40 μm) was bath applied for 20 min while stimulating at 0.05 Hz and then washed out for 10 min before applying LY354740 for 10 min. Slices were incubated for 2 h in the Ca2+-ATPase inhibitor CPA (20 μm) before coapplication with KCl (100 mm) for 5 min. CPA was then washed out for 10 min before bath application of LY354740 (1 μm for 10 min). Evoked responses were analyzed by measuring the slope of individual fEPSPs. The slopes from three sequential sweeps were averaged. All slopes were normalized to the average slope calculated during the predrug period (percentage of baseline).
Whole-cell recordings were performed by using the blind patch-clamp method (Blanton et al., 1989). Recordings from CA1 pyramidal neurons were obtained with patch pipettes (5–6 MΩ) filled with intracellular solution containing the following (in mm): 135 K-gluconate, 5 KCl, 10 HEPES, 1 EGTA, 5 phosphocreatine (CrP), 2 MgATP, and 0.4 NaGTP, pH 7.2. Membrane potentials were corrected for junction potentials. EPSCs were recorded at −70 mV and EPSPs were recorded in current-clamp mode at resting potential. After at least 5 min of stable baseline recording, LTP was induced by bath application of LY354740 for 10 min and then washed out for 30 min.
NMDA-mediated EPSCs were recorded at −70 mV with ACSF containing a low magnesium concentration (0.1 mm) and in presence of an AMPA receptor antagonist (NBQX, 25 μm). The amplitudes from three sequential sweeps were averaged. Input resistance was either monitored continuously or calculated by measuring the amplitude of the steady-state current evoked during a −5 mV voltage step delivered 50 ms before the test stimulation. Cells were excluded if a change of > 20% occurred during the recording.
All data were analyzed offline using pClamp 9 software (Molecular Devices) and are reported as the mean ± SEM. Statistical comparisons were made using two-tailed unpaired or paired Student's t tests. Differences were considered significant at p < 0.05.
Western blots.
Acute slices were prepared as described in the tissue preparation section. Slices were removed from the recording chamber 10 min after the application of LY354740 in the bath or 5 min after HFS. The CA1 region was isolated under a binocular dissection microscope. CA1 tissue was flash frozen in liquid nitrogen and stored at −80°C before extraction in 50 μl of RIPA buffer (50 mm Tris-HCl, pH 8.0, 150 mm NaCl, 0.1% SDS, 1% NP-40, and 0.5% sodium deoxycholate) containing protease inhibitor mixture (1:200), PMSF (1:500), and phosphatase inhibitor 2 and 3 (1:200). After homogenization of samples, sonication for 10 min at 4°C, and centrifugation for 10 min at 13,000 × g at 4°C, the supernatant was collected in Eppendorf tubes. Hippocampal proteins (20 μg) were resolved by electrophoresis on an 8% polyacrylamide gel and transferred to nitrocellulose membranes. Membranes were then incubated in a blocking solution containing 5% Western blocking solution (Roche) in TBST 0.05% (Tris-base 0.1 m, NaCl 1.5 m and 0.05% Tween 20, pH 8.0) for 2 h at room temperature and incubated in primary antibodies overnight at 4°C. The following antibodies were used; rabbit anti-GluN1 phospho S896 (1:1000, RRID:AB_1310496; Abcam) and rabbit anti-GAPDH (1:1000, RRID:AB_561053; Cell Signaling Technology). An antibody-blocking peptide against the rabbit anti-GluN1 phospho S896 antibody was used to confirm its specificity (1:100, ab203399; Abcam). After 4 washes of 20 min in TBST, membranes were incubated for 1 h at room temperature with a secondary antibody (goat anti-rabbit, IRDye 680 nm, 1:10000, Li-Cor Biosciences). Protein signal and intensity were detected by infrared fluorescence using an Odyssey IR scanner (Li-Cor Biosciences). Bands were quantified with ImageJ software and normalized to GAPDH values.
Results
Group II mGlu-receptor-dependent LTP at Schaffer collateral–CA1 synapses
Because selective activation of mGlu I receptors in the CA1 area of hippocampus induces LTD (Palmer et al., 1997; Fitzjohn et al., 1999), but coactivation of mGlu I and II receptors with the nonselective agonist ACPD induces LTP (Bashir et al., 1993; Bortolotto et al., 1994), we hypothesized that selective activation of group II mGlu receptors may be sufficient to induce LTP. Indeed, bath application of LY354740 (LY35, 1 μm, 10 min), a highly selective agonist of mGlu II receptors, induced a significant potentiation of the CA1 fEPSP (to 137.1 ± 6.1%, p = 0.0004, n = 12; Fig. 1A,C) in acute slices from adult mouse. Application of the mGlu II receptor antagonist LY341495 (4 μm) (Schoepp et al., 1999) bath applied for 20 min before and during LY354740 application prevented LTP (94.2 ± 9.2%, p = 0.55, n = 6; Fig. 1A,C), confirming that this form of LTP is initiated by mGlu II receptor activation.
Figure 1.

Characterization of transduction pathways mediating group II mGlu-receptor-induced LTP at the Schaffer collateral–CA1 pyramidal cell synapse. A, fEPSPs evoked in CA1 pyramidal cells by stimulation of Schaffer collaterals at low freuency (0.05 Hz) undergo LTP after activation of group II mGlu receptors with bath-applied LY354740 (1 μm, 10 min). LTP is prevented by LY341495 (4 μm, 10 min) a selective group II mGlu receptor antagonist. The inset shows representative traces (averages of six sweeps) before (1) and after (2) drug application. B, Induction of LTP is blocked by NEM (100 μm, 10 min), a membrane-permeable blocker of G-proteins, and by bisindolylmaleimide (1 μm, 20 min), a PKC antagonist. Inset, Representative traces (averages of six sweeps) before (1) and after (2) drug application. Scale bars, 0.5 mV and 10 ms. C, Quantification of fEPSP slope changes under a variety of pharmacological conditions (LY354740 vs LY354740 + LY341495: **p < 0.01, 2-tailed unpaired Student's t test). The antagonists NEM (100 μm, 10 min), bisindolylmaleimide (1 μm, 20 min), chelerytherine (10 μm, 20 min), CPA (20 μm), H89 (10 μm, 20 min), and genistein (30 μm, 30 min) were all applied in combination with LY354740 (1 μm, 10 min). ****p < 0.0001, *p < 0.05 indicates significantly different from slices treated only with LY354740 (ANOVA followed by Tukey's post hoc test; F(6, 42) = 8.642). D, Paired-pulse ratio (PPR) of evoked fEPSPs (interstimulus interval = 50 ms, averages of six sweeps) after LTP induction. Scale bars, 0.5 mV and 10 ms. E, PPR is not modified significantly (two-tailed paired Student's t test) after application of LY354740, consistent with a postsynaptic mechanism of LTP.
mGlu II receptors can couple to Gi/o-proteins to decrease activity of adenylyl cyclase (Niswender and Conn, 2010). We investigated whether perturbing this classical pathway would inhibit LTP. Blocking G-proteins with bath-applied NEM (100 μm, 10 min) prevented LTP (96.5 ± 10%, p = 0.0005, n = 4; Fig. 1B), but the PKA inhibitor H89 (10 μm, 20 min) only partially reduced LTP (112.5 ± 4.6%, p = 0.0151, n = 8; Fig. 1C), suggesting that an additional signaling pathway contributes to mGlu II-receptor-induced LTP. Previous work has shown that stimulation of mGlu II receptors can also activate tyrosine kinases (Trepanier et al., 2013) and PKC (Otani et al., 2002; Tyszkiewicz et al., 2004). We found that mGlu II-receptor-induced LTP was not affected by inhibiting tyrosine kinase with genistein (30 μm, 30 min; 121.8 ± 3.4%, p = 0.29, n = 8; Fig. 1C), but was prevented in the presence of either of two antagonists for PKC, bisindolylmaleimide (1 μm, 20 min, 101.8 ± 6.1%, p = 0.0003, n = 6) and chelerythrine (10 μm, 20 min, 89.5 ± 4.5%, p < 0.0001, n = 6) (Fig. 1B,C). Activation of PKC isoforms (Toullec et al., 1991) is calcium dependent. Because the turnover of phosphoinositides leads to activation of PKC and to the production of IP3, which subsequently induces release calcium stores (Nishizuka, 1986), we tested whether intracellular release of calcium stores was involved in this signaling pathway. mGlu II-dependent LTP was reduced significantly when calcium stores were depleted after treatment with CPA, a Ca2+-ATPase inhibitor (20 μm, 1 h: 109.2 ± 4.7%, p = 0.0041, n = 7; Fig. 1C). The inhibitors alone had no effect on baseline transmission. These data suggest that mGlu II-receptor-dependent LTP involves a noncanonical signaling pathway involving the release of intracellular calcium stores and PKC activation.
In the hippocampus, mGlu II receptors exhibit both presynaptic and postsynaptic expression (Petralia et al., 1996; Shigemoto et al., 1997; Tamaru et al., 2001). We observed no difference in the paired-pulse ratio recorded under baseline conditions (2 ± 0.2; Fig. 1D,E) and during stable LTP at 60 min after wash-out of LY354740 (1.9 ± 0.2, p = 0.53, n = 6; Fig. 1D,E). We thus conclude that the mGlu II receptors mediating LTP in CA1 pyramidal cells are localized postsynaptically.
mGlu II-receptor-dependent LTP requires activation of NMDARs
We noted that pharmacological activation of mGlu II receptors in the absence of Schaffer collateral stimulation at 0.05 Hz failed to induce LTP (with synaptic stimulation: 134.1 ± 6.3%; no stimulation: 96.9 ± 9.2%, p = 0.001, n = 6; Fig. 2A,B). Furthermore, bath application of the specific NMDAR antagonist D-AP5 (40 μm) 10 min before and during LY354740 application prevented LTP (98 ± 5.3%, p = 0.0004, n = 6; Fig. 2A,B), demonstrating that this form of metabotropic plasticity was dependent on the activation of NMDARs.
Figure 2.

Induction of LTP by group II mGlu receptor activation requires NMDAR activation. A, fEPSPs recorded in the stratum radiatum express LTP only if group II mGlu receptor activation (LY354740, 1 μm, 10 min) occurs concomitantly with repetitive Schaffer collateral stimulation (0.05 Hz). LTP is prevented by blocking NMDARs (D-AP5, 40 μm, 10 min). Insets, Representative traces (averages of six sweeps) before (1) and after (2) drug application. Scale bars, 0.5 mV and 10 ms. B, Quantification of fEPSP slope changes under the conditions presented in A (no stimulation, D-AP5). ****p < 0.0001, *p < 0.05 indicates significantly different from slices treated only with LY354740 (ANOVA followed by Tukey's post hoc test; F(2, 18) = 14.23). C, Whole-cell patch-clamp recordings of responses evoked by Schaffer collateral stimulation show potentiation by activation of group II mGlu receptors during experiments in current-clamp mode, but not in voltage-clamp mode at −70 mV, a potential at which NMDARs are blocked significantly by magnesium. Insets, Representative averaged traces. Scale bars, 10 mV/20 pA and 5 ms. D, Quantification of EPSPs and EPSCs showing LY354740-induced LTP only in current-clamp mode, two-tailed paired Student's t test. E, Pharmacologically isolated NMDA currents (NBQX, 25 μm, 10 min) evoked by Schaffer collateral stimulation at a holding potential of −70 mV with low extracellular magnesium (0.1 mm) are potentiated in response to activation of group II mGlu receptors (LY354740, 1 μm, 10 min). NMDAR-mediated currents are not potentiated if PKC is blocked (bisindolylmaleimide, 1 μm, 20 min). Insets, Representative traces (averages of six sweeps) before (1) and after (2) drug application and after 40 μm D-AP5 (3) to confirm the selective activation of NMDARs. Scale bars, 20 ms and 20 pA. F, Quantification of EPSCs before, during, and after application of LY354740 in control conditions and when PKC is blocked. ****p < 0.0001, *p < 0.05 indicates significantly different from the baseline (ANOVA followed by Tukey's post hoc test; LY35: F(2, 17) = 100.3; LY35 + bisindolylmaleimide; F(2, 15) = 225.7). G, Western blots showing phosphorylation at serine 896 of the GluN1 subunit (GluN1 p-S896) in control conditions after application of 1 μm LY354740 alone or in combination with 1 μm bisindolylmaleimide. The specificity of the primary antibody against GluN1 p-S896 was confirmed by addition of an antibody-blocking peptide. The protein ladder indicates molecular weight markers (M) in kDa. H, Protein quantification reveals increased phosphorylation at GluN1 p-S896 after activation of group II mGlu receptors. This effect is blocked by the PKC antagonist. ****p < 0.0001, *p < 0.05 indicates significantly different from the control (ANOVA followed by Tukey's post hoc test; F(2, 16) = 4.119).
The requirement of NMDAR activation was also apparent in experiments in which the effect of mGlu II receptor activation on LTP induction was compared in CA1 pyramidal cells recorded either in current-clamp mode at resting potential or in voltage-clamp mode at −70 mV. In current-clamp mode, Schaffer collateral stimulation at 0.05 Hz evoked sufficient depolarization to alleviate magnesium block from NMDAR channels, allowing LTP (139.3 ± 13%, p = 0.019, n = 8; Fig. 2C,D). In contrast, voltage clamping the soma of CA1 pyramidal cells at −70 mV did not permit an NMDAR-mediated response sufficient to induce LTP (99.6 ± 6.8%, p = 0.94, n = 5; Fig. 2C,D).
Next we investigated whether mGlu II receptor activation modulates NMDAR function. NMDAR-mediated EPSCs were evoked in CA1 pyramidal cells voltage clamped at −70 mV in the presence of low magnesium (0.1 mm) and NBQX (25 μm, 10 min) to block AMPA/kainate receptors. Under these conditions, application of LY354740 (1 μm, 10 min) resulted in a significant increase in amplitude of NMDA EPSCs within 2 min of addition to the bath (126.9 ± 7%, p = 0.0018, n = 8; Fig. 2E,F), an effect that was, however, reversed within 2 min of agonist wash-out. This enhancement of the NMDAR-mediated EPSC was no longer observed after blocking PKC (bisindolylmaleimide, 1 μm, 20 min; 96.3 ± 5.2%, p = 0.7, n = 6; Fig. 2E,F). At the end of the experiments, application of D-AP5 (40 μm, >5 min) abolished EPSCs, confirming their mediation by NMDARs. A study in prefrontal cortical neurons has shown that activation of mGlu II receptors increases NMDAR-mediated currents through a process involving phosphorylation of the GluN1 NMDAR subunit at Ser896 (Tyszkiewicz et al., 2004). We therefore tested whether a similar phosphorylation of the GluN1 NMDAR subunit in response to mGlu II receptor activation occurs in CA1 pyramidal cells. Indeed, application of LY354740 (1 μm, 10 min) increased phosphorylation at GluN1 p-S896 in the CA1 region by ∼70% (LY354740, 1 μm, 10 min: 173.3 ± 18.5%, p = 0.03, n = 5; Fig. 2G,H) compared with control slices (no LY354740: n = 7). This increase in phosphorylation, observed in the Western blots, was no longer significant when mGlu II receptors were activated in presence of a PKC inhibitor (bisindolylmaleimide, 1 μm, 20 min; 143 ± 15.3%, p = 0.2, n = 7; Fig. 2G,H). We used a blocking peptide to confirm the specificity of the anti-GluN1 phospho S896 antibody. Therefore, activation of PKC by mGlu II receptors enhances NMDAR function by increasing GluN1 phosphorylation.
mGlu2 and mGlu3 receptors activate synaptic and extrasynaptic NMDARs differentially to promote LTP induction
Depending on their subtype, mGlu receptors can be localized perisynaptically and/or extrasynaptically, whereas NMDARs are found both within and outside of the synapse. We performed experiments to determine the contribution to mGlu II-receptor-dependent LTP of NMDARs containing the GluN2B subunit, which are primarily expressed outside of the synapse, and of receptors containing the GluN2A subunits, which represent the majority of NMDARs within the synapse; the remainder correspond to heterotrimeric receptors in adult rat hippocampus (Papouin et al., 2012). Our results indicate a similar distribution in adult mouse as blocking NR2B-containing receptors with Ro25–6981 (3 μm) reduced EPSC amplitude by only 13.8 ± 1.4% (p = 0.02; n = 5), whereas subsequent addition of NVP-AAM077 at 0.1 μm, the highest concentration that is still specific for NR2A-containing receptors (Bartlett et al., 2007), reduced EPSC amplitude by 63 ± 4.9% (p < 0.0001, n = 5; Fig. 3A). The remaining response was blocked by D-AP5 (91.6 ± 2.1%, p < 0.0001, n = 5). The incomplete block with combined application of NR2A and NR2B antagonists is likely to reflect the presence of heterotrimeric receptors, which appear to be insensitive to subunit-specific antagonists (Delaney et al., 2013). Under control conditions, a similar reduction in mGlu II-dependent LTP was observed with blockade of GluN2A subunit-containing NMDARs (NVP-AAM077, 0.1 μm, 20 min; 118.9 ± 5.7%, p = 0.028, n = 7) as with blockade of GluN2B subunit-containing NMDARs (Ro25–6981, 3 μm, 20 min; 118.3 ± 3.6%, p = 0.024, n = 7; Fig. 3B,C). Selective inhibition of synaptic NMDARs with the use-dependent antagonist MK-801 (40 μm, 20 min) reduced mGlu II-receptor-dependent LTP by more than half (118.4 ± 2.7%, p = 0.01, n = 7). Additional blockade of GluN2B subunit-containing NMDARs by Ro25–6981 completely prevented LTP (Ro25–6981, 3 μm, 20 min; 106.4 ± 2%, p = 0.04, n = 6; Fig. 3D,E). Conversely, blocking GluN2A-subunit-containing NMDARs with NVP-AAM077 after MK-801 treatment did not lead to a further reduction in LTP (NVP-AAM077, 0.1 μm, 20 min; 116.5 ± 4.6%, p = 0.9, n = 6; Fig. 3D,E). Therefore, mGlu II receptors target both synaptic and extrasynaptic NMDARs to trigger LTP (Fig. 3F).
Figure 3.
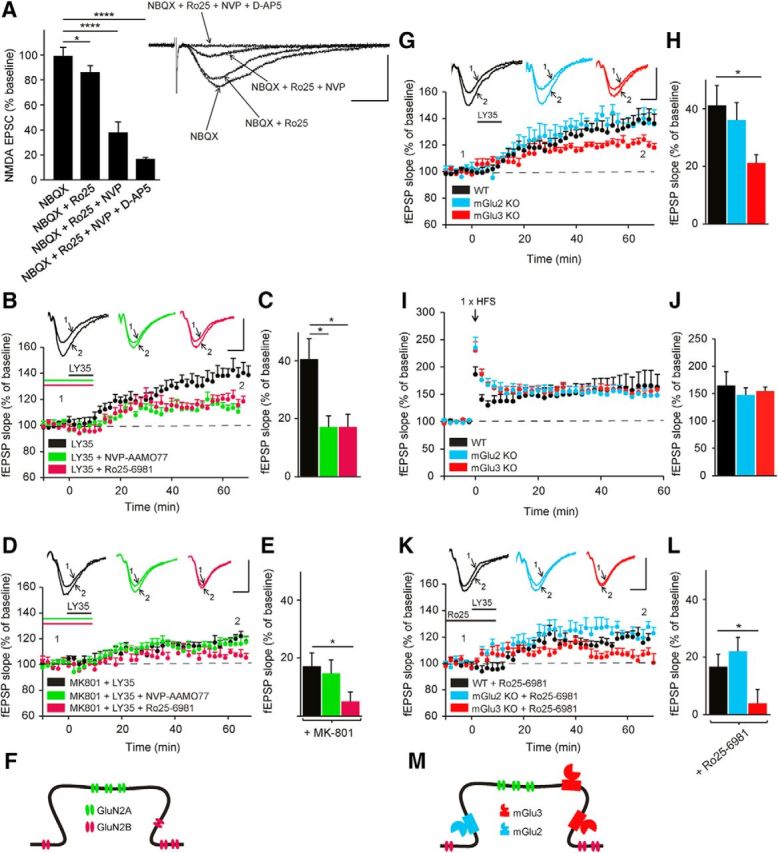
Characterization of NMDAR subunits that are targeted by group II mGlu receptor subtypes to induce LTP in CA1 pyramidal cells. A, Pharmacologically isolated NMDA currents (NBQX, 25 μm) evoked by Schaffer collateral stimulation at a holding potential of −70 mV with low extracellular magnesium (0.1 mm) were slightly diminished in the presence of a GluN2B NMDAR subunit antagonist (Ro25–69813, 3 μm, 20 min), but were strongly reduced after applying a GluN2A NMDAR subunit antagonist (NVP-AAM077, 0.1 μm, 20 min). Inset, Sample traces of NMDA-EPSCs showing effects of antagonists (****p < 0.0001, *p < 0.05; ANOVA followed by Tukey's post hoc test; F(3, 15) = 179.1). Scale bars, 25 ms and 40 pA. B, LTP is significantly reduced in the presence of antagonists for either the GluN2A (NVP-AAM077, 0.1 μm, 20 min) or the GluN2B NMDAR subunit (Ro25–69813, 3 μm, 20 min). C, Quantification of fEPSP slope changes under the conditions presented in B. *p < 0.05 indicates significantly different from slices treated with LY354740 alone (ANOVA followed by Tukey's post hoc test; F(2, 19) = 5.565). D, After bath application of the use-dependent blocker MK-801 (40 μm, 20 min) to block the synaptic NMDARs selectively, subsequent blocking of GluN2A subunits has no further effect, whereas blocking of GluN2B subunits leads to an additional pronounced reduction in LTP. E, Quantification of fEPSP slope changes under the conditions presented in D. *p < 0.05 indicates significantly different from slices treated with LY354740 alone and MK801 (ANOVA followed by Tukey's post hoc test; F(2, 16) = 3.799). F, Proposed scheme showing the two different pools of NMDARs involved in the induction of LTP, one synaptic including mainly GluN2A subunits and the other extrasynaptic including mainly GluN2B subunits. G, Comparison of group II mGlu-receptor-induced LTP in wild-type, mGlu2 KO, and mGlu3 KO mice shows that only the absence of the mGlu3 receptor reduces LTP. When mGlu2 and mGlu3 receptors are both blocked (LY341495, 4 μm, 10 min), LTP is prevented. H, Quantification of fEPSP slope changes under the conditions presented in G. *p < 0.05 indicates significantly different from WT mice (ANOVA followed by Tukey's post hoc test; F(2, 17) = 4.09). I, Single train of HFS induces LTP that was not significantly different in WT, mGlu2, and mGlu3 KO slices. J, Quantification of fEPSP slope changes under the conditions presented in I (ANOVA followed by Tukey's post hoc test; F(2, 13) = 0.2183). K, After blockade of the GluN2B NMDAR subunit, LTP was prevented in mGlu3 KO mice and reduced in wild-type and mGlu2 KO mice. L, Quantification of fEPSP slope changes under the conditions presented in K. *p < 0.05 indicates significantly different from WT + Ro25–69813 mice (ANOVA followed by Tukey's post hoc test; F(2, 18) = 7.927). M, Working model illustrating how distinct mGlu2 and mGlu3 receptor localization may lead to preferential modulation of different pools of NMDARs, with only the mGlu3 receptor able to modulate both synaptic and extrasynaptic NMDARs. Scale bars for the field traces: 0.5 mV and 10 ms.
We then used KO mice to assess the contribution of mGlu2 and mGlu3 receptors to LTP. mGlu II-receptor-dependent LTP was reduced significantly in mGlu3 KO mice (121.0 ± 1.7%, p = 0.03, n = 6), but not in mGlu2 KO mice (136.1 ± 3.7%, p = 0.7, n = 7), compared with WT mice (141.1 ± 6.7%, n = 8; Fig. 3G,H). These findings indicate that activation of the mGlu2 receptor alone is not sufficient to induce maximal LTP compared with mGlu3. Control experiments suggest that this effect is not the consequence of mGlu II receptor KO causing a generalized impairment in synaptic plasticity (Fig. 3I,J). LTP induced by a standard HFS protocol resulted in LTP of similar magnitude in wild-type slices (167.1 ± 26%, p = 0.04, n = 6), as in mGlu2 KO (147.7 ± 12.8%, p = 0.01, n = 5) and in mGlu3 KO slices (154.5 ± 6.2%, p = 0.001, n = 5).
It has been shown in several brain regions that postsynaptic mGlu2 receptors are located at extrasynaptic sites remote from the postsynaptic density, whereas postsynaptic mGlu3 receptors are expressed both synaptically and extrasynaptically (Tamaru et al., 2001). This distinct distribution pattern suggests that mGlu2 and mGlu3 receptors may differ in their ability to modulate synaptic versus extrasynaptic pools of NMDARs to induce LTP. We tested this hypothesis by examining the effects of blocking either GluN2A (mostly synaptic NMDARs) or GluN2B (mostly extrasynaptic NMDARs) subunit-containing NMDARs in mGlu2 and mGlu3 KO mice. When the GluN2B subunit was blocked, mGlu II-receptor-dependent LTP was reduced but still present in WT (118.3 ± 3.6%, n = 7) and mGlu2 receptor KO slices (122.2 ± 2.8%, p = 0.64, n = 8), but was completely blocked in mGlu3 receptor KO slices (104.6 ± 2.7%, p =0.023, n = 6; Fig. 3K,L). When the GluN2A subunit was blocked, mGlu II-receptor-dependent LTP was reduced to a similar extent in WT (118.9 ± 5.7%, p = 0.027, n = 7), mGlu2 KO (121.1 ± 3.3%, p = 0.018, n = 8), and mGlu3 KO mice (120.9 ± 1.1%, p = 0.024, n = 6). These results are consistent with a scenario (Fig. 3M) in which mGlu3 receptors can access both synaptic and extrasynaptic NMDARs, whereas mGlu2 receptors modulate only extrasynaptic NMDARs.
Comparing mGlu II-receptor-dependent LTP with HFS-LTP
Because mGlu II-receptor-dependent LTP and classical HFS-LTP in the hippocampus are both NMDAR dependent, we investigated whether they share similar mechanisms by performing occlusion experiments. After stable pharmacological mGlu II-induced LTP (138.4 ± 6.6%, p = 0.01, n = 4; LY354740, 1 μm, 10 min; Fig. 4A,B), additional potentiation was induced by saturating HFS-LTP (3 trains at 100 Hz for 1 s with an interstimulus interval of 20 s). The HFS-LTP protocol was repeated 3 times with a 15 min interval to obtain saturating LTP (274.8 ± 38.1%, p = 0.037, n = 4; Fig. 4A,B). When the order was reversed, with HFS-LTP (251.2 ± 28.9%, n = 4; Fig. 4C,D) preceding mGlu II-receptor-dependent LTP, a further significant increase in LTP was again observed (284.6 ± 8.2%, p = 0.027, n = 4; Fig. 4C,D). The mGlu II-receptor-dependent LTP and HFS-LTP do not occlude each other. Moreover, as shown in Figure 4, E and G, inhibiting mGlu II receptors did not impair HFS-induced LTP (control: 165.4 ± 26.4%, n = 6; LY341495: 164.2 ± 22.0%, n = 4, p =0.97), indicating that the HFS protocol does not activate mGlu II receptors sufficiently to induce LTP. Together, these results show that the mGlu II-receptor-dependent LTP and HFS-LTP do not share common induction mechanisms.
Figure 4.
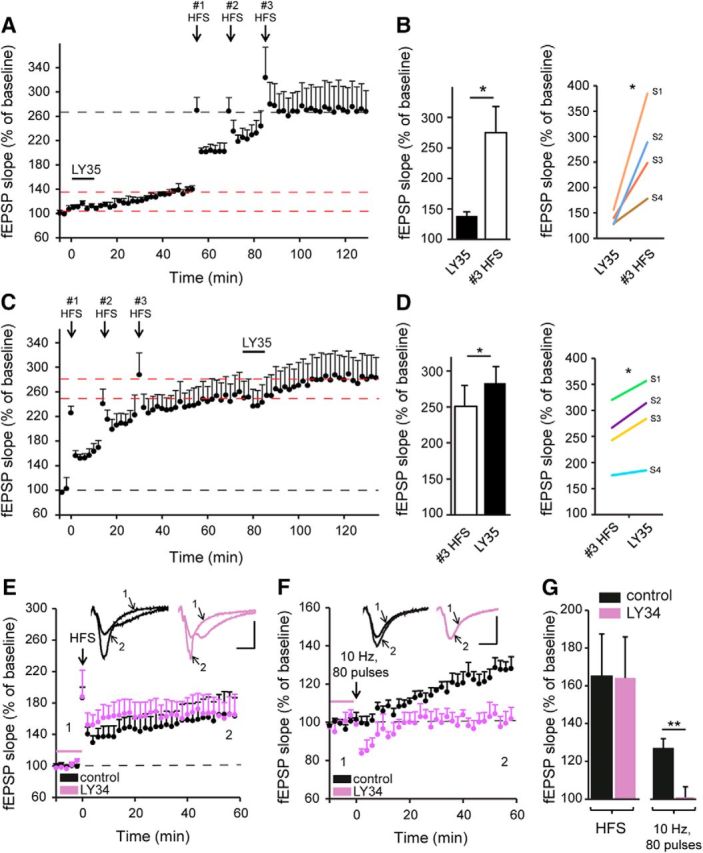
Two modes of LTP induction, either by HFS or by group II mGlu receptor activation, do not mutually occlude each other. A, LTP induced by group II mGlu receptor activation (LY354740, 1 μm, 10 min) reached a peak steady-state level after 45 min. At this point, HFS (3 trains at 100 Hz for 1 s separated by 20 s) repeated 3 times leads to a significant further increase in LTP. B, Quantification of fEPSP slope changes under the conditions presented in A. Two-tailed paired Student's t test. Left, Means for all slices. Right, Responses for each slice. C, In reverse order experiments, HFS was applied three times to achieve saturating LTP. Subsequent pharmacological activation of group II mGlu receptor activation induced a further increase in LTP. D, Quantification of fEPSP slope changes under the conditions presented in C. Two-tailed paired Student's t test. Left, Means for all slices. Right, responses for each slice. E, LTP induction by HFS (3 × 100 Hz, ISI: 20 s) is not impaired by group II mGlu receptor antagonist (LY341495, 4 μm, 10 min). F, LTP induced by 80 pulses at 10 Hz is blocked by a group II mGlu receptor antagonist (LY341495, 4 μm, 10 min). G, Quantification of fEPSP slope changes under the conditions presented in E and F. *p < 0.05, **p < 0.01, two-tailed unpaired Student's t test. Scale bars, 0.5 mV and 10 ms.
In the hippocampus, theta rhythm (5–14 Hz) is the dominant neuronal oscillation and plays a crucial role in neuronal plasticity to promote learning and memory. mGlu II receptor activation contribute to the generation of hippocampal theta oscillations (Ster et al., 2011). We investigated whether mGlu II-LTP could be induced by a protocol of LFS in a theta range (80 pulses at 10 Hz). This protocol induced LTP (127.4 ± 5.2%, p = 0.0066, n = 5; Fig. 4F,G) that was inhibited by the mGlu II receptor antagonist LY341495 (101.3 ± 4.6%, p = 0.0098, n = 4; Fig. 4F,G). These results indicate that mGlu II–LTP can be induced by theta-frequency oscillations.
Priming of NMDARs by mGlu II receptor activation mediates metaplasticity
Our observation that activation of mGlu II receptors results in a rapid increase within 5 min in the amplitude of NMDAR-mediated EPSCs (Fig. 2E), but a much slower induction of LTP (20 min) (Fig. 1A), suggests a metaplastic response (Abraham, 2008) whereby mGlu II-receptor-mediated modulation of NMDARs enhances the generation of LTP. We further characterized this phenomenon by inducing subsaturating LTP with 1 train of HFS (100 Hz, 1 s) and comparing the response with or without prior activation of mGlu II receptors (LY354740,1 μm, 10 min). For these experiments, we did not stimulate the Schaffer collateral pathway during pharmacological activation of mGlu II receptors. Pharmacological priming resulted in a significant increase in LTP (control: 32.8 ± 5.1%, n = 6; priming: 65.4 ± 5.5%, n = 7, p = 0.0014; Fig. 5A,B). We then investigated whether priming differs in WT and mGlu II receptor KO mice. Priming, expressed as the value of HFS-LTP with LY354740 normalized to the value of HFS-LTP without LY354740, had the greatest effect in WT mice (111.5 ± 9.8%, n = 6; Fig. 5C) and in mGlu2 KO mice (86.3 ± 10.1%, p =0.35, n = 6; Fig. 5C), but was significantly reduced in mGlu3 KO mice (53.2 ± 15.5%, p = 0.013, n = 6; Fig. 5C). This result is consistent with the decrease in LTP observed in the absence of the mGlu3 receptor (Fig. 3D,F). To gain insight into the priming mechanism, we examined the phosphorylation status of Ser-896 of the GluN1 subunit of NMDARs during these experiments. Activation of mGlu II receptors alone (LY354740, 1 μm, 10 min) led to almost a doubling of the phosphorylation level of GluN1 Ser896 (no stimulation, no LY354740: n = 7; no stimulation + LY354740: n = 4, 198.7 ± 36.3%, p = 0.026; Fig. 5D,E). Therefore, mGlu II-receptor-mediated NMDAR phosphorylation does not require synaptic stimulation. Therefore, NMDAR phosphorylation without synaptic stimulation is not sufficient to induce LTP (Fig. 2A,B). These findings are consistent with a priming role for mGlu II receptors in which the phosphorylation of NMDARs renders synapses more competent to undergo LTP.
Figure 5.
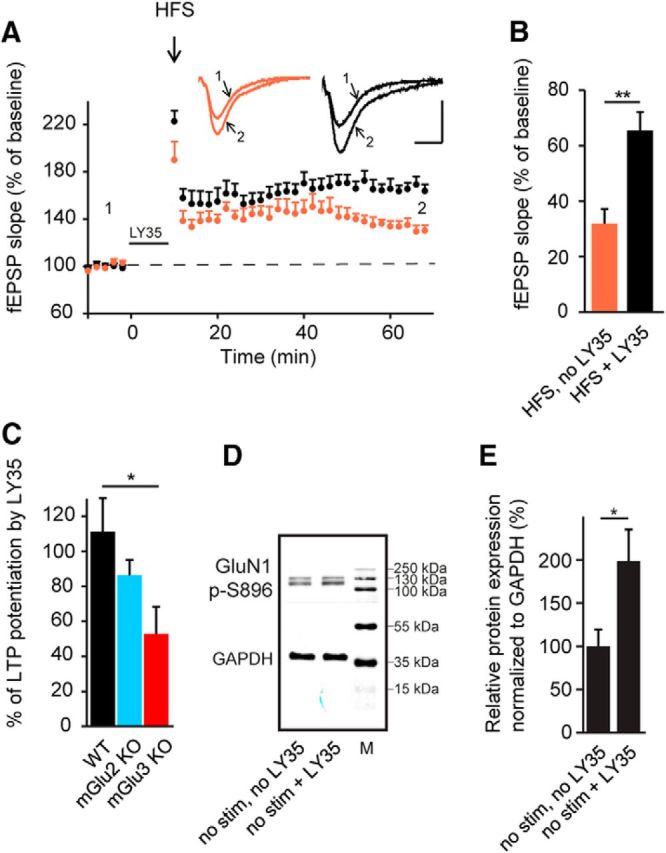
Pharmacological priming of group II mGlu receptors enhances HFS-induced LTP. A, LTP induced by HFS (3 trains at 100 Hz for 1 s separated by 20 s) was potentiated by prior priming of group II mGlu receptors with LY354740 (1 μm, 10 min). Insets, Representative traces (averages of six sweeps) before (1) and after (2) LTP induction. Scale bars, 0.5 mV and 10 ms. B, Quantification of fEPSP slope changes under the conditions presented in A. Two-tailed unpaired Student's t test. C, Net increase in LTP after priming with LY354740 in WT, mGlu2 KO, and mGlu3 KO mice. LTP is significantly reduced in mGlu3 KO mice compared with WT and mGlu2 KO mice. ****p < 0.0001, *p < 0.05 indicates significantly different from WT mice (ANOVA followed by Tukey's post hoc test; F(2, 15) = 5.411). D, Western blots showing phosphorylation at serine 896 of the GluN1 subunit (GluN1 p-S896) under different conditions. The protein ladder shows molecular weight markers (M) in kDa. E, Protein quantification indicates greater levels of phosphorylation at GluN1 p-S896 when group II mGlu receptors are activated even in the absence of stimulation, indicating that both phosphorylation and activation of NMDARs are required to initiate plasticity. *p < 0.05, **p < 0.01, two-tailed unpaired Student's t test.
Discussion
Our data show that selective activation of mGlu II receptors at Schaffer collateral synapses onto CA1 pyramidal neurons renders synapses more competent for the induction of LTP through a mechanism involving the phosphorylation of synaptic and extrasynaptic NMDARs. Accumulating evidence suggests that group II mGlu receptors are positively coupled to PLC/PKC pathways, in addition to the canonical adenylyl cyclase–PKA pathway. Previous studies have demonstrated that activation of mGlu II receptors can increase PLC activity in hippocampus (Klein et al., 1997) and prefrontal cortex (Otani et al., 2002) and PKC activity in the prefrontal cortex (Otani et al., 2002; Tyszkiewicz et al., 2004) probably via the βγ complex of the Gi/o protein (Rebecchi and Pentyala, 2000). Consistent with these data, we found that inhibition of PKC completely blocked mGlu II-receptor-dependent LTP. A similar PLC–PKC-dependent transduction mechanism has been described at the parallel fiber–Purkinje cell synapse, where activation of mGlu4 receptors inhibits transmission (Abitbol et al., 2012), and in cultured cerebellar granule cells, where mGlu7 receptor activation blocks P/Q-type calcium channels (Perroy et al., 2000). Furthermore, PKC activation is typically a calcium-dependent process, which is consistent with our observation that LTP initiated by mGlu II receptors was strongly reduced after depletion of intracellular calcium stores.
In our experiments, blockade of the adenylyl cyclase–PKA pathway also reduced LTP, although to a lesser extent. Recent studies suggest that the main role of the adenylyl cyclase–PKA pathway in neurons is to modulate the functional expression of mGlu II receptors. PKA can inhibit the dephosphorylation by protein phosphatase 2C of Ser-845 in the C-terminal tail of the mGlu3 receptor (Flajolet et al., 2003). Therefore, the interaction of the mGlu3 receptor with the endocytic machinery may be reduced, thereby stabilizing membrane expression as described for other receptors (Chen and Roche, 2007). Alternatively, PKA-mediated phosphorylation may enhance mGlu3-receptor-initiated signal transduction (Uematsu et al., 2015). Therefore, mGlu II receptors appear to signal both through inhibition of the adenylyl cyclase–PKA pathway and through activation of a PLC–PKC-dependent pathway. The enhancement of NMDAR-evoked currents in CA1 pyramidal neurons in response to mGlu II receptor activation has also been reported to involve Src tyrosine kinase signaling (Trepanier et al., 2013). However, in a previous study in pyramidal neurons of the prefrontal cortex (Tyszkiewicz et al., 2004), as well as in our recordings from CA1 pyramidal neurons, this effect could not be confirmed, possibly reflecting a difference in experimental conditions. We found that mGlu II receptor activation led to PKC-dependent phosphorylation of the GluN1 subunit at Ser896 as described previously (Tyszkiewicz et al., 2004), which may account for the observed enhancement in synaptic plasticity. We cannot, however, rule out an involvement of PKC phosphorylation of GluN2A and GluN2B subunits that could contribute to this effect.
Unlike classical NMDAR-dependent LTP, mGlu II-receptor-dependent LTP exhibited a slow time course of potentiation. Because activation of mGlu II receptors mobilizes intracellular Ca2+ stores via IP3 transduction (Otani et al., 2002), the slow kinetics of LTP may reflect a gradual rise in calcium. mGlu II receptors may also act indirectly to increase intracellular calcium by phosphorylating the GluN1 subunit, which can enhance calcium influx through NMDARs by increasing the open probability of the channel (Chen and Huang, 1992) and by promoting receptor trafficking to the synapse (Lan et al., 2001; Groc et al., 2004). In our experiments, the activation of mGlu II receptors alone was insufficient to induce LTP; additional stimulation of the Schaffer collateral pathway was required. This stimulation depolarizes the targeted CA1 pyramidal cells, which, again, can increase intracellular calcium influx by relieving magnesium block from NMDAR channels (Mayer et al., 1984; Nowak et al., 1984). Therefore, mGlu II receptor activation and CA1 pyramidal cell depolarization can act synergistically to raise intracellular calcium concentration to facilitate the induction of LTP (Raymond and Redman, 2002; Cavazzini et al., 2005). Interestingly, mGlu1 receptors also operate synergistically with NMDARs to induce LTP at Schaffer collateral synapses by reducing the negative feedback regulation of SK channels on NMDA-receptor mediated currents (Tigaret et al., 2016).
Our results show that mGlu II-receptor-dependent LTP was decreased significantly in mGlu3 receptor KO mice, but not in mGlu2 receptor KO mice. These findings are likely to reflect the differential somatodendritic localization of these receptors. Expression of mGlu2 receptors is not observed in the vicinity of the synapse (Luján et al., 1997), suggesting that they modulate an extrasynaptic pool of NMDARs preferentially. In contrast, mGlu3 receptors are present both extrasynaptically and in the proximity of the postsynaptic density (Tamaru et al., 2001), which would allow them to act also on synaptic NMDARs. Our experiments validate this scenario because LTP mediated by synaptic GluN2A subunit-containing NMDARs was induced only after activation of mGlu3 receptors, whereas LTP mediated by extrasynaptic GluN2B subunit-containing NMDARs was observed after activation of both mGlu2 and mGlu3 receptors. The distinct spatial organization of mGlu II receptor subtypes may also reflect specific interactions with scaffolding proteins (Pawson and Scott, 1997; Kim and Sheng, 2004). At present, however, little information is available about these signal transduction mechanisms except for the interaction of the mGlu3 receptor with protein phosphatase 2C and PICK1 (Chung et al., 2000; Hirbec et al., 2002).
Our experiments provide evidence for at least two distinct mechanisms that contribute to mGlu II-receptor-dependent LTP, one that operates through the priming of NMDARs and the other through the release of intracellular calcium stores. Therefore, mGlu II receptor activation in isolation does not induce LTP, but rather exerts a metaplastic effect on NMDAR-mediated synaptic plasticity, thereby altering the threshold for LTP induction (Abraham, 2008). The HFS protocol did not occlude mGlu II-dependent LTP, which suggests that, in addition to NMDARs, mGlu II receptors modulate further targets affecting synaptic plasticity. An attractive candidate mechanism may involve an action on inhibitory circuits because activation of hippocampal mGlu II receptors has been shown to depress inhibitory transmission (Poncer et al., 1995) and to induce LTD at inhibitory synapses (Liu et al., 1993), both of which could contribute to the enhanced LTP that we observed.
Our experiments indicate that metaplasticity involving the priming of NMDARs is mediated by the mGlu3 receptor. Moreover, consistent with our previous work showing the involvement of mGlu3 receptors in the generation of theta oscillations (Ster et al., 2011), we demonstrate that mGlu II-dependent LTP can be induced by stimulation of Schaffer collaterals at theta frequency. Theta oscillations are also implicated in the facilitation of LTP generation in response to exposure to a novel environment (Li et al., 2003). It is thus of interest that coactivation of mGlu II receptors and NMDARs allows the induction of LTP in response to patterns of stimulation that are more physiological than the high-frequency trains used in classical LTP protocols.
Genetic and clinical studies have provided evidence for the involvement of mGlu II receptors in schizophrenia (Patil et al., 2007; Harrison et al., 2008), in which abnormal theta oscillations have been reported (Ford et al., 2007). Given that schizophrenia is characterized by a hypoglutamatergic state (Gao et al., 2000), our finding that mGlu II receptors can act postsynaptically to increase NMDAR function suggests that their activation may represent one of the mechanisms for the antipsychotic effects of treatment with an mGlu2/3 agonist (Patil et al., 2007).
Footnotes
This work was supported by the Swiss National Science Foundation (Grant 31003A_162558). We thank D. Göckeritz-Dujmovic, S. Giger, H. Kasper, F. David, and P. Morciano for technical assistance; Beat Gähwiler for valuable discussions; Shigetada Nakanishi for permission to use mGlu2−/− mice; and Corrado Corti, Mauro Corsi, and GlaxoSmithKline for permission to use mGlu3−/− mice.
The authors declare no competing financial interests.
References
- Abitbol K, McLean H, Bessiron T, Daniel H. A new signalling pathway for parallel fibre presynaptic type 4 metabotropic glutamate receptors (mGluR4) in the rat cerebellar cortex. J Physiol. 2012;590:2977–2994. doi: 10.1113/jphysiol.2012.232074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abraham WC. Metaplasticity: tuning synapses and networks for plasticity. Nat Rev Neurosci. 2008;9:387–399. doi: 10.1038/nrn2356. [DOI] [PubMed] [Google Scholar]
- Aksoy-Aksel A, Manahan-Vaughan D. Synaptic strength at the temporoammonic input to the hippocampal CA1 region in vivo is regulated by NMDA receptors, metabotropic glutamate receptors and voltage-gated calcium channels. Neuroscience. 2015;309:191–199. doi: 10.1016/j.neuroscience.2015.03.014. [DOI] [PubMed] [Google Scholar]
- Altinbilek B, Manahan-Vaughan D. A specific role for group II metabotropic glutamate receptors in hippocampal long-term depression and spatial memory. Neuroscience. 2009;158:149–158. doi: 10.1016/j.neuroscience.2008.07.045. [DOI] [PubMed] [Google Scholar]
- Bartlett TE, Bannister NJ, Collett VJ, Dargan SL, Massey PV, Bortolotto ZA, Fitzjohn SM, Bashir ZI, Collingridge GL, Lodge D. Differential roles of NR2A and NR2B-containing NMDA receptors in LTP and LTD in the CA1 region of two-week old rat hippocampus. Neuropharmacology. 2007;52:60–70. doi: 10.1016/j.neuropharm.2006.07.013. [DOI] [PubMed] [Google Scholar]
- Bashir ZI, Bortolotto ZA, Davies CH, Berretta N, Irving AJ, Seal AJ, Henley JM, Jane DE, Watkins JC, Collingridge GL. Induction of LTP in the hippocampus needs synaptic activation of glutamate metabotropic receptors. Nature. 1993;363:347–350. doi: 10.1038/363347a0. [DOI] [PubMed] [Google Scholar]
- Blanton MG, Lo Turco JJ, Kriegstein AR. Whole cell recording from neurons in slices of reptilian and mammalian cerebral cortex. J Neurosci Methods. 1989;30:203–210. doi: 10.1016/0165-0270(89)90131-3. [DOI] [PubMed] [Google Scholar]
- Bortolotto ZA, Bashir ZI, Davies CH, Collingridge GL. A molecular switch activated by metabotropic glutamate receptors regulates induction of long-term potentiation. Nature. 1994;368:740–743. doi: 10.1038/368740a0. [DOI] [PubMed] [Google Scholar]
- Brunner J, Ster J, Van-Weert S, Andrási T, Neubrandt M, Corti C, Corsi M, Ferraguti F, Gerber U, Szabadics J. Selective silencing of individual dendritic branches by an mGlu2-activated potassium conductance in dentate gyrus granule cells. J Neurosci. 2013;33:7285–7298. doi: 10.1523/JNEUROSCI.4537-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capogna M. Distinct properties of presynaptic group II and III metabotropic glutamate-receptor-mediated inhibition of perforant pathway-CA1 EPSCs. Eur J Neurosci. 2004;19:2847–2858. doi: 10.1111/j.1460-9568.2004.03378.x. [DOI] [PubMed] [Google Scholar]
- Cavazzini M, Bliss T, Emptage N. Ca2+ and synaptic plasticity. Cell Calcium. 2005;38:355–367. doi: 10.1016/j.ceca.2005.06.013. [DOI] [PubMed] [Google Scholar]
- Chen BS, Roche KW. Regulation of NMDA receptors by phosphorylation. Neuropharmacology. 2007;53:362–368. doi: 10.1016/j.neuropharm.2007.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Huang LY. Protein kinase C reduces Mg2+ block of NMDA-receptor channels as a mechanism of modulation. Nature. 1992;356:521–523. doi: 10.1038/356521a0. [DOI] [PubMed] [Google Scholar]
- Chung HJ, Xia J, Scannevin RH, Zhang X, Huganir RL. Phosphorylation of the AMPA receptor subunit GluR2 differentially regulates its interaction with PDZ domain-containing proteins. J Neurosci. 2000;20:7258–7267. doi: 10.1523/JNEUROSCI.20-19-07258.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conn PJ, Pin JP. Pharmacology and functions of metabotropic glutamate receptors. Annu Rev Pharmacol Toxicol. 1997;37:205–237. doi: 10.1146/annurev.pharmtox.37.1.205. [DOI] [PubMed] [Google Scholar]
- Corti C, Battaglia G, Molinaro G, Riozzi B, Pittaluga A, Corsi M, Mugnaini M, Nicoletti F, Bruno V. The use of knock-out mice unravels distinct roles for mGlu2 and mGlu3 metabotropic glutamate receptors in mechanisms of neurodegeneration/neuroprotection. J Neurosci. 2007;27:8297–8308. doi: 10.1523/JNEUROSCI.1889-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaney AJ, Sedlak PL, Autuori E, Power JM, Sah P. Synaptic NMDA receptors in basolateral amygdala principal neurons are triheteromeric proteins: physiological role of GluN2B subunits. J Neurophysiol. 2013;109:1391–1402. doi: 10.1152/jn.00176.2012. [DOI] [PubMed] [Google Scholar]
- Flajolet M, Rakhilin S, Wang H, Starkova N, Nuangchamnong N, Nairn AC, Greengard P. Protein phosphatase 2C binds selectively to and dephosphorylates metabotropic glutamate receptor 3. Proc Natl Acad Sci U S A. 2003;100:16006–16011. doi: 10.1073/pnas.2136600100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford JM, Krystal JH, Mathalon DH. Neural synchrony in schizophrenia: from networks to new treatments. Schizophr Bull. 2007;33:848–852. doi: 10.1093/schbul/sbm062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzjohn SM, Kingston AE, Lodge D, Collingridge GL. DHPG-induced LTD in area CA1 of juvenile rat hippocampus; characterisation and sensitivity to novel mGlu receptor antagonists. Neuropharmacology. 1999;38:1577–1583. doi: 10.1016/S0028-3908(99)00123-9. [DOI] [PubMed] [Google Scholar]
- Gao XM, Sakai K, Roberts RC, Conley RR, Dean B, Tamminga CA. Ionotropic glutamate receptors and expression of N-methyl-D-aspartate receptor subunits in subregions of human hippocampus: effects of schizophrenia. Am J Psychiatry. 2000;157:1141–1149. doi: 10.1176/appi.ajp.157.7.1141. [DOI] [PubMed] [Google Scholar]
- Groc L, Heine M, Cognet L, Brickley K, Stephenson FA, Lounis B, Choquet D. Differential activity-dependent regulation of the lateral mobilities of AMPA and NMDA receptors. Nat Neurosci. 2004;7:695–696. doi: 10.1038/nn1270. [DOI] [PubMed] [Google Scholar]
- Harrison PJ, Lyon L, Sartorius LJ, Burnet PW, Lane TA. The group II metabotropic glutamate receptor 3 (mGluR3, mGlu3, GRM3): expression, function and involvement in schizophrenia. J Psychopharmacol. 2008;22:308–322. doi: 10.1177/0269881108089818. [DOI] [PubMed] [Google Scholar]
- Hirbec H, Perestenko O, Nishimune A, Meyer G, Nakanishi S, Henley JM, Dev KK. The PDZ proteins PICK1, GRIP, and syntenin bind multiple glutamate receptor subtypes: analysis of PDZ binding motifs. J Biol Chem. 2002;277:15221–15224. doi: 10.1074/jbc.C200112200. [DOI] [PubMed] [Google Scholar]
- Huber KM, Kayser MS, Bear MF. Role for rapid dendritic protein synthesis in hippocampal mGluR-dependent long-term depression. Science. 2000;288 doi: 10.1126/science.288.5469.1254. 1254–1256-1257. [DOI] [PubMed] [Google Scholar]
- Kamiya H, Shinozaki H, Yamamoto C. Activaton of metabotropic glutamate receptor type 2/3 suppresses transmission at rat hippocampal mossy fibre synapses. J Physiol. 1996;493:447–455. doi: 10.1113/jphysiol.1996.sp021395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim E, Sheng M. PDZ domain proteins of synapses. Nat Rev Neurosci. 2004;5:771–781. doi: 10.1038/nrn1517. [DOI] [PubMed] [Google Scholar]
- Kintscher M, Breustedt J, Miceli S, Schmitz D, Wozny C. Group II metabotropic glutamate receptors depress synaptic transmission onto subicular burst firing neurons. PLoS One. 2012;7:e45039. doi: 10.1371/journal.pone.0045039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein J, Iovino M, Vakil M, Shinozaki H, Löffelholz K. Ontogenetic and pharmacological studies on metabotropic glutamate receptors coupled to phospholipase D activation. Neuropharmacology. 1997;36:305–311. doi: 10.1016/S0028-3908(97)00024-5. [DOI] [PubMed] [Google Scholar]
- Lan JY, Skeberdis VA, Jover T, Grooms SY, Lin Y, Araneda RC, Zheng X, Bennett MV, Zukin RS. Protein kinase C modulates NMDA receptor trafficking and gating. Nat Neurosci. 2001;4:382–390. doi: 10.1038/86028. [DOI] [PubMed] [Google Scholar]
- Li S, Cullen WK, Anwyl R, Rowan MJ. Dopamine-dependent facilitation of LTP induction in hippocampal CA1 by exposure to spatial novelty. Nat Neurosci. 2003;6:526–531. doi: 10.1038/nn1049. [DOI] [PubMed] [Google Scholar]
- Liu YB, Disterhoft JF, Slater NT. Activation of metabotropic glutamate receptors induces long-term depression of GABAergic inhibition in hippocampus. J Neurophysiol. 1993;69:1000–10004. doi: 10.1152/jn.1993.69.3.1000. [DOI] [PubMed] [Google Scholar]
- Luján R, Roberts JD, Shigemoto R, Ohishi H, Somogyi P. Differential plasma membrane distribution of metabotropic glutamate receptors mGluR1 alpha, mGluR2 and mGluR5, relative to neurotransmitter release sites. J Chem Neuroanat. 1997;13:219–241. doi: 10.1016/S0891-0618(97)00051-3. [DOI] [PubMed] [Google Scholar]
- Lyon L, Burnet PW, Kew JN, Corti C, Rawlins JN, Lane T, De Filippis B, Harrison PJ, Bannerman DM. Fractionation of spatial memory in GRM2/3 (mGlu2/mGlu3) double knockout mice reveals a role for group II metabotropic glutamate receptors at the interface between arousal and cognition. Neuropsychopharmacology. 2011;36:2616–2628. doi: 10.1038/npp.2011.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macek T, Winder D, Gereau R, Ladd C, Conn P. Differential involvment of group II and group III mGluRs as autoreceptors at lateral and medial perforant path synapses. J Neurophysiol. 1996;76:3798–3806. doi: 10.1152/jn.1996.76.6.3798. [DOI] [PubMed] [Google Scholar]
- Mayer ML, Westbrook GL, Guthrie PB. Voltage-dependent block by Mg2+ of NMDA responses in spinal cord neurones. Nature. 1984;309:261–263. doi: 10.1038/309261a0. [DOI] [PubMed] [Google Scholar]
- Morishima Y, Miyakawa T, Furuyashiki T, Tanaka Y, Mizuma H, Nakanishi S. Enhanced cocaine responsiveness and impaired motor coordination in metabotropic glutamate receptor subtype 2 knockout mice. Proc Natl Acad Sci U S A. 2005;102:4170–4175. doi: 10.1073/pnas.0500914102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishizuka Y. Studies and perspectives of protein kinase C. Science. 1986;233:305–3012. doi: 10.1126/science.3014651. [DOI] [PubMed] [Google Scholar]
- Niswender CM, Conn PJ. Metabotropic glutamate receptors: physiology, pharmacology, and disease. Annu Rev Pharmacol Toxicol. 2010;50:295–322. doi: 10.1146/annurev.pharmtox.011008.145533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowak L, Bregestovski P, Ascher P, Herbet A, Prochiantz A. Magnesium gates glutamate-activated channels in mouse central neurones. Nature. 1984;307:462–465. doi: 10.1038/307462a0. [DOI] [PubMed] [Google Scholar]
- Otani S, Daniel H, Takita M, Crépel F. Long-term depression induced by postsynaptic group II metabotropic glutamate receptors linked to phospholipase C and intracellular calcium rises in rat prefrontal cortex. J Neurosci. 2002;22:3434–3444. doi: 10.1523/JNEUROSCI.22-09-03434.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer MJ, Irving AJ, Seabrook GR, Jane DE, Collingridge GL. the group I mGlu receptor agonist DHPG induces a novel form of LTD in the CA1 region of the hippocampus. Neuropharmacology. 1997;36:1517–1532. doi: 10.1016/S0028-3908(97)00181-0. [DOI] [PubMed] [Google Scholar]
- Papouin T, Ladépêche L, Ruel J, Sacchi S, Labasque M, Hanini M, Groc L, Pollegioni L, Mothet JP, Oliet SH. Synaptic and extrasynaptic NMDA receptors are gated by different endogenous coagonists. Cell. 2012;150:633–646. doi: 10.1016/j.cell.2012.06.029. [DOI] [PubMed] [Google Scholar]
- Patil ST, Zhang L, Martenyi F, Lowe SL, Jackson KA, Andreev BV, Avedisova AS, Bardenstein LM, Gurovich IY, Morozova MA, Mosolov SN, Neznanov NG, Reznik AM, Smulevich AB, Tochilov VA, Johnson BG, Monn JA, Schoepp DD. Activation of mGlu2/3 receptors as a new approach to treat schizophrenia: a randomized Phase 2 clinical trial. Nat Med. 2007;13:1102–1107. doi: 10.1038/nm1632. [DOI] [PubMed] [Google Scholar]
- Pawson T, Scott JD. Signaling through scaffold, anchoring, and adaptor proteins. Science. 1997;278:2075–2080. doi: 10.1126/science.278.5346.2075. [DOI] [PubMed] [Google Scholar]
- Perroy J, Prezeau L, De Waard M, Shigemoto R, Bockaert J, Fagni L. Selective blockade of P/Q-type calcium channels by the metabotropic glutamate receptor type 7 involves a phospholipase C pathway in neurons. J Neurosci. 2000;20:7896–7904. doi: 10.1523/JNEUROSCI.20-21-07896.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petralia RS, Wang YX, Niedzielski AS, Wenthold RJ. The metabotropic glutamate receptors, mGluR2 and mGluR3, show unique postisynaptic, presynaptic and glial localizations. Neuroscience. 1996;71:949–976. doi: 10.1016/0306-4522(95)00533-1. [DOI] [PubMed] [Google Scholar]
- Poncer JC, Shinozaki H, Miles R. Dual modulation of synaptic inhibition by distinct metabotropic glutamate receptors in the rat hippocampus. J Physiol. 1995;485:121–134. doi: 10.1113/jphysiol.1995.sp020717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raymond CR, Redman SJ. Different calcium sources are narrowly tuned to the induction of different forms of LTP. J Neurophysiol. 2002;88:249–255. doi: 10.1152/jn.2002.88.1.249. [DOI] [PubMed] [Google Scholar]
- Rebecchi MJ, Pentyala SN. Structure, function, and control of phosphoinositide-specific phospholipase C. Physiol Rev. 2000;80:1291–1335. doi: 10.1152/physrev.2000.80.4.1291. [DOI] [PubMed] [Google Scholar]
- Scanziani M, Salin PA, Vogt KE, Malenka RC, Nicoll RA. Use-dependent increases in glutamate concentration activate presynaptic metabotropic glutamate receptors. Nature. 1997;385:630–634. doi: 10.1038/385630a0. [DOI] [PubMed] [Google Scholar]
- Schoepp D, Jane D, Monn J. Pharmacological agents acting at subtypes of metabotropic glutamate receptors. Neuropharmacology. 1999;38:1431–1476. doi: 10.1016/S0028-3908(99)00092-1. [DOI] [PubMed] [Google Scholar]
- Shigemoto R, Kinoshita A, Wada E, Nomura S, Ohishi H, Takada M, Flor P, Neki A, Abe T, Nakanishi S, Mizuno N. Differential presynaptic localization of metabotropic glutamate receptor subtypes in the rat hippocampus. J Neurosci. 1997;17:7503–7522. doi: 10.1523/JNEUROSCI.17-19-07503.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ster J, Mateos JM, Grewe BF, Coiret G, Corti C, Corsi M, Helmchen F, Gerber U. Enhancement of CA3 hippocampal network activity by activation of group II metabotropic glutamate receptors. Proc Natl Acad Sci U S A. 2011;108:9993–9997. doi: 10.1073/pnas.1100548108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamaru Y, Nomura S, Mizuno N, Shigemoto R. Distribution of metabotropic glutamate receptor mGluR3 in the mouse CNS: differential locationrelative to pre- and postsynaptic sites. Neuroscience. 2001;106:481–503. doi: 10.1016/S0306-4522(01)00305-0. [DOI] [PubMed] [Google Scholar]
- Tigaret CM, Olivo V, Sadowski JH, Ashby MC, Mellor JR. Coordinated activation of distinct Ca++ sources and metabotropic glutamate receptors encodes Hebbian synaptic plasticity. Nat Commun. 2016;7:10289. doi: 10.1038/ncomms10289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toullec D, Pianetti P, Coste H, Bellevergue P, Grand-Perret T, Ajakane M, Baudet V, Boissin P, Boursier E, Loriolle F, Duhamell L, Charon D, Kirilovsky J. The bisindolylmaleimide GF 109203X is a potent and selective inhibitor of protein kinase C. J Biol Chem. 1991;266:15771–15781. [PubMed] [Google Scholar]
- Trepanier C, Lei G, Xie YF, MacDonald JF. Group II metabotropic glutamate receptors modify N-methyl-D-aspartate receptors via Src kinase. Sci Rep. 2013;3:926. doi: 10.1038/srep00926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyszkiewicz JP, Gu Z, Wang X, Cai X, Yan Z. Group II metabotropic glutamate receptors enhance NMDA receptor currents via a protein kinase C-dependent mechanism in pyramidal neurones of rat prefrontal cortex. J Physiol. 2004;554:765–777. doi: 10.1113/jphysiol.2003.056812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uematsu K, Heiman M, Zelenina M, Padovan J, Chait BT, Aperia A, Nishi A, Greengard P. Protein kinase A directly phosphorylates metabotropic glutamate receptor 5 to modulate its function. J Neurochem. 2015;132:677–686. doi: 10.1111/jnc.13038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoi M, Kobayashi K, Manabe T, Takahashi T, Sakagushi I, Kasuura G, Shigemoto R, Ohishi H, Nomura S, Nakamura K, Nakao K, Katsuki M, Nakanishi S. Impairment of hippocampal mossy fiber LTD in mice lacking mGluR2. Science. 1996;273:645–647. doi: 10.1126/science.273.5275.645. [DOI] [PubMed] [Google Scholar]