

Abstract
Neurotrophin-3 (NT-3) is a secreted neurotrophic factor that binds neurotrophin receptor tyrosine kinase C (TrkC), which in turn binds to presynaptic protein tyrosine phosphatase σ (PTPσ) to govern excitatory synapse development. However, whether and how NT-3 cooperates with the TrkC-PTPσ synaptic adhesion pathway and TrkC-mediated intracellular signaling pathways in rat cultured neurons has remained unclear. Here, we report that NT-3 enhances TrkC binding affinity for PTPσ. Strikingly, NT-3 treatment bidirectionally regulates the synaptogenic activity of TrkC: at concentrations of 10–25 ng/ml, NT-3 further enhanced the increase in synapse density induced by TrkC overexpression, whereas at higher concentrations, NT-3 abrogated TrkC-induced increases in synapse density. Semiquantitative immunoblotting and optogenetics-based imaging showed that 25 ng/ml NT-3 or light stimulation at a power that produced a comparable level of NT-3 (6.25 μW) activated only extracellular signal-regulated kinase (ERK) and Akt, whereas 100 ng/ml NT-3 (light intensity, 25 μW) further triggered the activation of phospholipase C-γ1 and CREB independently of PTPσ. Notably, disruption of TrkC intracellular signaling pathways, extracellular ligand binding, or kinase activity by point mutations compromised TrkC-induced increases in synapse density. Furthermore, only sparse, but not global, TrkC knock-down in cultured rat neurons significantly decreased synapse density, suggesting that intercellular differences in TrkC expression level are critical for its synapse-promoting action. Together, our data demonstrate that NT-3 is a key factor in excitatory synapse development that may direct higher-order assembly of the TrkC/PTPσ complex and activate distinct intracellular signaling cascades in a concentration-dependent manner to promote competition-based synapse development processes.
SIGNIFICANCE STATEMENT In this study, we present several lines of experimental evidences to support the conclusion that neurotrophin-3 (NT-3) modulates the synaptic adhesion pathway involving neurotrophin receptor tyrosine kinase C (TrkC) and presynaptic protein tyrosine phosphatase σ (PTPσ) in a bidirectional manner at excitatory synapses. NT-3 acts in concentration-independent manner to facilitate TrkC-mediated presynaptic differentiation, whereas it acts in a concentration-dependent manner to exert differential effects on TrkC-mediated organization of postsynaptic development. We further investigated TrkC extracellular ligand binding, intracellular signaling pathways, and kinase activity in NT-3-induced synapse development. Last, we found that interneuronal differences in TrkC levels regulate the synapse number. Overall, these results suggest that NT-3 functions as a positive modulator of synaptogenesis involving TrkC and PTPσ.
Keywords: excitatory synapse, neurotrophin-3, PTPσ, synaptic cell adhesion, TrkC
Introduction
Synapse development requires precise coordination between presynaptic and postsynaptic neurons to promote properties characteristic of a particular type of synapse. This coordination is achieved through the action of a variety of molecular mechanisms organized by trans-synaptic adhesion molecules, some of which have been referred to as “synapse organizers” because of their ability to induce presynaptic or postsynaptic differentiation in simplified experimental systems (Biederer and Scheiffele, 2007; Südhof, 2008; Ko, 2012; Um and Ko, 2013; Ko et al., 2015a). In addition to membrane-anchored synapse organizers, secreted soluble factors are also critical for various synapse assembly processes at neuromuscular junctions and in the CNS (Johnson-Venkatesh and Umemori, 2010). Notably, classical morphogens (e.g., sonic hedgehog, TGFβ/BMP, and Wnts), fibroblast growth factors (e.g., FGF7 and FGF22), neurotrophic factors (e.g., BDNF or GDNF), and glial factors (e.g., thrombospondins) have been implicated in organizing presynaptic and postsynaptic differentiation of mammalian neurons (Johnson-Venkatesh and Umemori, 2010). However, how diverse classes of synapse organizers mediate distinct phases of synapse development has not been investigated exclusively.
Among emerging synapse organizers, the neurotrophin receptor tyrosine kinase C (TrkC) has been reported to act as both a structural lynchpin and functional coordinator because it interacts with protein tyrosine phosphatase σ (PTPσ) extracellularly and activates a host of signaling cascades intracellularly (for review, see Um and Ko, 2013). TrkC, together with TrkA and TrkB, constitute the Trk superfamily, the members of which exhibit distinct patterns of binding to four neurotrophins: nerve growth factor (NGF), brain-derived neurotrophic factor (BDNF), neurotrophin-3 (NT-3), and neurotrophin-4 (NT-4). NGF binds to TrkA; BDNF and NT-4 bind to TrkB; and NT-3 binds to TrkC. These ligand–Trk interactions regulate activity-dependent synapse development in the CNS (Park and Poo, 2013). BDNF signaling through TrkB, in particular, has been studied extensively in various synaptogenesis contexts (for review, see Park and Poo, 2013). It was shown recently that BDNF-dependent TrkB receptor trafficking and signaling are dictated by cis interactions of TrkB with Slitrk5, another synapse organizer that also binds to PTPσ (Takahashi et al., 2012; Yim et al., 2013; Song et al., 2015). BDNF stimulation switches the binding partner of Slitrk5 from PTPσ to TrkB (Song et al., 2015), suggesting that BDNF modulates a specific synaptic adhesion pathway selectively. Although this hypothesis is conceptually compelling, it is not clear whether other neurotrophins similarly modulate synapse development.
In the present study, we found that NT-3 promotes and is required for TrkC-dependent presynaptic differentiation through increased binding of dimerized NT-3 with PTPσ. Intriguingly, NT-3 boosts TrkC-induced synapse density in a concentration-dependent manner. In a concentration range of 10–25 ng/ml, NT-3 increases synapse density, whereas, at 100 ng/ml, NT-3 dampens TrkC-triggered synapse density, possibly through downregulation of total TrkC levels. Moreover, quantitative immunoblotting and optogenetics-based imaging experiments revealed that NT-3 activates ERK and Akt, but not phospholipase C (PLC)-γ1 or cAMP response-element-binding protein (CREB), at a concentration of 25 ng/ml. In contrast, it activates PLC-γ1 and CREB, in addition to ERK and Akt, at a concentration of 100 ng/ml. This concentration-dependent alteration of intracellular signaling cascades does not require the presence of PTPσ. However, gain-of-function assays and molecular replacement approaches using various TrkC point mutants showed that the synapse-density-promoting activity of TrkC requires extracellular binding to NT-3 and PTPσ; signaling through the primary intracellular signaling pathways ERK, Akt, PLC-γ1, and CREB; and TrkC kinase activity. These results suggest that simply activating PLC-γ1 and CREB does not trigger the synaptotoxic effects of high concentrations of NT-3. Last, we found that relative differences in TrkC levels between neurons dictate TrkC-mediated synapse density in hippocampal neurons, reminiscent of the mechanism that operates in regulating cortical and cerebellar synapse number (Kwon et al., 2012; Joo et al., 2014). Collectively, our data suggest that NT-3 acts via modulation of the TrkC/PTPσ synaptic adhesion pathway to regulate synapse development in a local-concentration-dependent manner.
Materials and Methods
Construction of expression vectors.
pDisplay-mTrkC encoding full-length mouse TrkC (GenBank No. AY336094), excluding its signal peptide (aa 1-31), was cloned into the pDisplay vector via XmaI and SacII sites. pcDNA3.1 HA-rTrkC (a gift from Dr. Patrick Mehlen, UMR Inserm, France), encodes full-length rat TrkC (GenBank No. NM_001270656). The following TrkC constructs were generated by site-directed mutagenesis using pcDNA3.1 HA-rTrkC as a backbone: TrkC D240A/D242A (a PTPσ-binding-defective mutant); TrkC N366A/T369A (an NT-3-binding-defective mutant); TrkC Y516F (a mutant defective for tyrosine phosphorylation at Y516); and TrkC Y820F (a mutant defective for tyrosine phosphorylation at Y820); TrkC D679N (a kinase-activity-defective mutant); and TrkC E543D. Small hairpin (interfering) RNA (shRNA) lentiviral vectors against TrkC were constructed by annealing, phosphorylating, and subcloning the following oligonucleotides targeting rat TrkC sequences into XhoI and XbaI sites of a single KD vector (l-309) immediately downstream of the human H1 promoter: T1-CDS, 5′-TGC AGC AAG ACT GAG ATC A-3′; T2-CDS, 5′-CGG TCC AAA TTT GGA ATG A-3′; T3-CDS, 5′-CGG CTG AAT ATT AAG GAG A-3′; T4-CDS, 5′-GAA TAT CAC TTC CAT ACA C-3′; T5-CDS, 5′-ACG CTG AGC CTT CGG GAA T-3′; T6-untranslated region (UTR), 5′-GCG TTT ATA CTC TGT TGC CTC CTC T-3′; T7-UTR, 5′-CTT CAA CTC CTT TCG TCC ATC TTT G-3′; T8-UTR, 5′-CAT CTT TGA CCG AAA CGA ACA TCT T-3′; and T9-CDS, 5′-GCA GCA AGA CTG AGA TCA A-3′. Only T4-CDS and T7-UTR were effective in knocking down TrkC mRNA levels. T9-CDS is identical to the oligonucleotide used in the study of Takahashi et al. (2011). For construction of the Opto-TrkC construct (Lyn-CytoTrkC-PHR-P2A-FP; see Fig. 5A), the PHR domain from the Opto-TrkB vector (Chang et al., 2014) was amplified by PCR. The myristoylation sequence (lyn-tagged for membrane insertion) was added to the N terminus of CRY2PHR and the cytoplasmic region of mouse TrkC (aa 454–825) was inserted between the myristoylation sequence and CRY2PHR using the In-Fusion cloning system (Clontech), following the manufacturer's instructions. P2A (2A peptide sequence; 57 bp) was inserted into iRFP-N1 vector (P2A-iRFP682-N1; Clontech) and the Opto-TrkC vector was inserted at the N terminus of the P2A-iRFP682-N1 vector for visualization. pmCitrine-N1-rTrkC, encoding full-length rat TrkC, was cloned into the pmCitrine-N1 vector (Clontech) via NheI and NotI sites.
Figure 5.
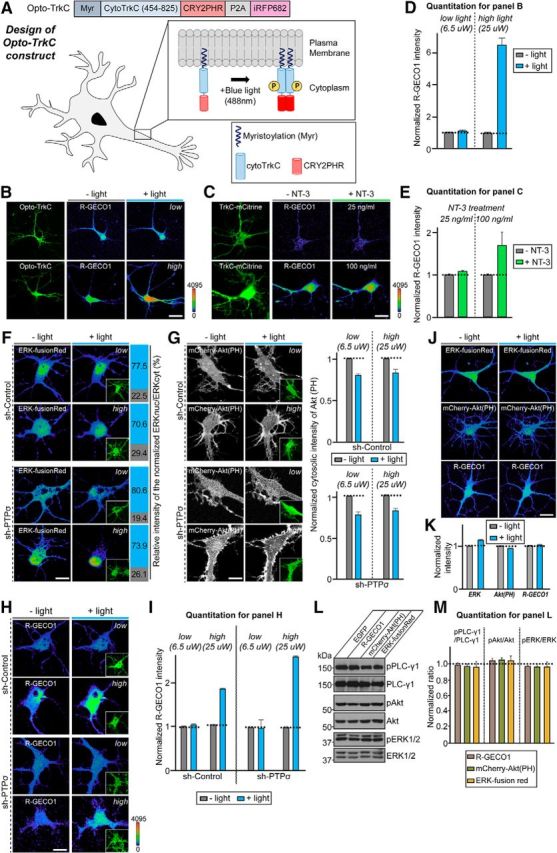
Light illumination elicits distinct Opto-TrkC activation in an intensity-dependent, but PTPσ-independent, fashion. A, Schematic illustration of the Opto-TrkC system in cultured neurons. The Opto-TrkC vector consists of a myristoylation signal peptide (for anchoring to the plasma membrane), the cytoplasmic region of TrkC (aa 454–825), light-inducible interacting domain PHR from cryptochrome (CRY2PHR), P2A peptide sequence, and sequence of the near-infrared fluorescent protein iRFP682. B, Optimization of light intensity to establish appropriate TrkC activation conditions using the Opto-TrkC system. Representative images of PLC-γ1 activation detected with the intensiometric intracellular calcium indicator R-GECO1 in Opto-TrkC-expressing neurons before (gray bar) and after (blue bar) light illumination at DIV8. Opto-TrkC was activated by 488 nm light at a power of 6.5 or 25 μW. Scale bar, 25 μm (applies to all images). C, Validation of light stimulation conditions for mimicking the effects of concentration-dependent NT-3 treatment in DIV8 hippocampal cultured neurons expressing TrkC-mCitrine and pCMV-R-GECO1 without (gray bar) or with (green bar) NT-3 treatment at the indicated concentration (25 or 100 ng/ml). Treatment with 100 ng/ml NT-3, but not 25 ng/ml, significantly increased the normalized R-GECO1 intensity. Scale bar, 20 μm (applies to all images). D, E, Summary graphs showing minimum and maximum values of normalized R-GECO1 intensity after light stimulation (D; quantification of results in B) and the normalized R-GECO1 intensity after treatment with the indicated NT-3 concentration (E; quantification of results in C). All data are shown as means ± SEM (n = 3–5 independent experiments). F, Left, Fluorescence images of DIV8–10 neurons infected with lentiviruses expressing sh-Control (green) or sh-PTPσ (green) at DIV3 and cotransfected with ERK-FusionRed (pseudocolored) and Opto-TrkC. ERK-FusionRed was imaged before and after repetitive stimulation with 488 nm light (five 350-ms pulses at a power of 6.5 μW [low] or 25 μW [high]). Right, Quantification of normalized ERK-FusionRed fluorescence (ERKnuc/ERKcyt) intensities expressed as a percentage before (gray) and after (blue) light illumination. Scale bar, 25 μm (applies to all images). G, Same as F, except that DIV8–10 neurons were cotransfected with mCherry-Akt (PH) before (gray) and after (blue) light illumination. Adjacent bar graphs show the normalized cytosolic intensity of mCherry-Akt (PH). All data are shown as means ± SEM. H, I, Representative images (H) and summary graphs (I) of intracellular calcium levels monitored using the genetically encoded calcium indicator R-GECO1. DIV8–10 neurons were infected with lentiviruses expressing sh-Control (green) or sh-PTPσ (green) at DIV3 and cotransfected with R-GECO1 (pseudocolored) and Opto-TrkC, after which Opto-TrkC was activated at different powers of light. All data are shown as means ± SEM. Scale bar, 25 μm (applies to all images). J, K, Representative images (J) and summary bar graphs (K) demonstrating that light stimulation itself does not alter the basal intensity of ERK-fusionRed, mCherry-Akt(PH) or R-GECO1 in cultured hippocampal neurons at DIV8. Scale bar, 20 μm (applies to all images). L, M, Representative immunoblot images (L) and summary graphs (M) quantifying basal levels of pPLC-γ1, PLC-γ1, pAkt, Akt, pERK1/2, and ERK1/2 in HEK293T cells transfected with EGFP, pCMV-R-GECO1, mCherry-Akt(PH), or ERK-fusionRed. All data are shown as means ± SEM (n = 3 independent experiments).
The shRNA lentiviral vector against NT-3 was generated using oligonucleotides targeting the rat NT-3 sequence, 5′-GCA AAC CTA CGT CCG AGC ACT-3′ (Yang et al., 2012b). pVL1393-hNT-3, encoding full-length (aa 1-257) human NT-3 (GenBank No. NM_001102654), was subcloned into BamHI and XbaI sites in the pVL1393 (BD Biosciences) vector containing a thrombin cleavage site (LVPRGS). The following constructs were described previously: IgC-PTPσ (Ko et al., 2015b), l-309 sh-PTPσ (Yim et al., 2013), pGW1-LRRTM2-mVenus (Ko et al., 2009a), mCherry-Akt (PH; Yang et al., 2012a), ERK-mFusionRed (Chang et al., 2014), and pCMV-R-GECO1 (purchased from Addgene; Zhao et al., 2011).
Antibodies.
Synapsin-2 fusion proteins (JK014; pGEX-KG Synapsin-2a; a gift from Dr. Thomas Südhof, Stanford University) were produced from BL21 Escherichia coli, purified, and used to immunize rabbits. The following commercially available antibodies were purchased: mouse monoclonal anti-HA (clone HA-7; Covance), mouse monoclonal anti-β-actin (clone AC-74; Sigma-Aldrich), goat polyclonal anti-EGFP (Rockland), rabbit monoclonal anti-TrkC (clone C44H5; Cell Signaling Technology), rabbit polyclonal anti-pTrkC (Tyr820; Novus Biologicals), rabbit polyclonal anti-pERK1/2 (Cell Signaling Technology), rabbit polyclonal anti-ERK1/2 (Cell Signaling Technology), rabbit polyclonal anti-pPLC-γ1 (Tyr783; Cell Signaling Technology), mouse monoclonal anti-PLC-γ1 (clone 610027; BD Biosciences), rabbit polyclonal anti-Akt (Cell Signaling Technology), rabbit polyclonal anti-pAkt (Ser473; Cell Signaling Technology), mouse monoclonal anti-pCREB (clone 1B6; Cell Signaling Technology), rabbit monoclonal anti-CREB (Cell Signaling Technology), mouse monoclonal anti-neuroligin-2 (clone 5E6; Synaptic Systems), and mouse monoclonal anti-MAP2 (clone AP-20; Sigma-Aldrich). Rabbit polyclonal anti-Shank1 (#1172) was a gift from Dr. Eunjoon Kim (KAIST, Korea).
Saturation ligand-binding assays.
Transfected HEK293T cells, growing on 24-well plates in DMEM containing 50 mm HEPES-NaOH, pH 7.4, 2 mm CaCl2, 2 mm MgCl2, and 0.1% bovine serum albumen (BSA), were incubated with the indicated concentrations of recombinant NT-3 protein (added as serial dilutions) for 16 h at 4°C with gentle agitation. Excess recombinant protein was removed by washing cells 3 times with cold DMEM, after which cells were fixed with 4% paraformaldehyde for 10 min on ice. The cells were washed again with cold PBS and then blocked by incubating with PBS containing 3% BSA at room temperature for 1 h. Thereafter, cells were incubated for 1 h at room temperature with horseradish peroxidase (HRP)-conjugated rabbit anti-human IgG antibody (Sigma-Aldrich), diluted 1:40,000 in blocking solution. The cells were washed three times with blocking solution and once with PBS, and then a colorimetric 3,3′,5,5′-tetramethylbenzidine peroxidase enzyme immunoassay (Bio-Rad) was performed according to the manufacturer's instructions. Briefly, the provided HRP substrate solution was added to each well and samples were incubated at room temperature for 15 min with vigorous agitation until a blue color developed. Each reaction was stopped by adding an equal volume of 0.5 m sulfuric acid, which produced a yellow color. Absorbance at 450 nm was measured using a Benchmark Plus microplate spectrophotometer (Bio-Rad) and ligand concentration was plotted against the difference in absorbance between plasmid and mock-transfected cells. A dissociation constant (Kd) value was calculated from the data obtained from three independent experiments using a Scatchard analysis.
Cell-surface binding assays.
IgC-fusion proteins of PTPσ were produced from HEK293T cells as described previously (Boucard et al., 2012; Ko et al., 2015b). Soluble IgC protein was purified using protein A-Sepharose beads (GE Healthcare), eluted, and immediately neutralized with 1 m Tris-HCl, pH 8.0. Transfected HEK293T cells expressing HA-tagged TrkC or the indicated TrkC point mutants were incubated with 0.2 μm IgC or IgC-PTPσ. Images were acquired using a confocal microscope (LSM700; Zeiss).
Surface biotinylation assays in rat neurons.
Cultured cortical rat neurons treated with NT-3 for 2 h at the indicated concentrations were washed twice with ice-cold PBS, incubated with 1 mg/ml Sulfo-NHS-LC-biotin (Pierce) in ice-cold PBS for 30 min on ice, rinsed briefly once with 0.1 m glycine in PBS, and incubated with 0.1 m glycine/PBS for 15 min at room temperature to complete quenching of biotin reactions. Cells were then scraped with 1 ml of PBS, resuspended in lysis buffer (1% Triton X-100, 0.2% SDS, 5 mm EDTA, 2 mm DTT, and protease inhibitors), and incubated for 30 min at 4°C. After clearing by centrifugation, the extracts were incubated with streptavidin agarose (Pierce) for 4 h at 4°C. Beads were washed five times with lysis buffer and surface proteins were eluted with the sample buffer and analyzed by immunoblotting with anti-TrkC (1 μg/ml), anti-NL-2 (1:1000), or anti-actin (1:2000) antibodies, followed by the appropriate secondary antibodies.
Recombinant human NT-3 expression and purification.
Human NT-3 protein was expressed in High Five insect cells (Invitrogen) using the pVL1393-hNT-3 vector, as described by the manufacturer. Briefly, High Five cells transfected with the human NT-3 P4 baculovirus were incubated for 3 d and then pelleted by centrifugation. The supernatants, containing processed NT-3 and a C-terminal protein A tag, were pooled and affinity purified using IgG-Sepharose column chromatography (GE Healthcare). NT-3-bound resin was washed with buffer consisting of 20 mm Tris-HCl, pH 8.0, and 200 mm NaCl and then treated with thrombin (0.5% v/v) in washing buffer at 4°C overnight to remove the C-terminal protein A tag. Processed NT-3 protein was then eluted, concentrated to 1.5 mg/ml, and exchanged into Dulbecco's PBS (Welgene) by passing through illustra Nap-5 columns (GE Healthcare).
Production of recombinant lentiviruses and quantitative RT-PCR analyses.
Recombinant lentiviruses were produced by triply transfecting HEK293T cells with three plasmids, l-309 vectors (L-309 sh-PTPσ, l-309 sh-NT-3, or l-309 sh-TrkC), psPAX, and pMD2G, using FuGene-6 (Roche) as described previously (Um et al., 2014). Lentiviruses were harvested 72 h after transfection by collecting the media from transfected HEK293T cells and briefly centrifuging at 1000 × g to remove cellular debris. High-density cultured hippocampal neurons at 4 d in vitro (DIV4) or DIV5 in 24-well tissue culture plates were infected with 300 μl of conditioned medium per well and harvested at DIV12 or DIV13 for quantitative RT-PCR analyses, as described previously (Ko et al., 2011). The following primers were used for quantitative RT-PCR analyses: NT-3, 5′-GCA ACA GAC ACA GAA CTA-3′; TrkC, 5′-GCC ACA AGC CAG ACA CAT A-3′; and PTPσ, 5′-GCC ACA CAC CTT CTA TAA T-3′ (Yim et al., 2013).
Heterologous synapse-formation assays.
HEK293T cells were transfected with pcDNA3.1-rTrkC, pGW1-LRRTM2-mVenus (LRRTM2), or EGFP alone (control) using FuGene (Roche). After 48 h, transfected HEK293T cells were trypsinized, seeded onto cultured hippocampal neurons at DIV9, and incubated with 25 or 100 ng/ml NT-3 or BDNF (R&D Systems), as indicated, for 48 h. Cells were then double immunostained with antibodies against EGFP and synapsin at DIV11, as described previously (Ko et al., 2009b). Images were acquired by confocal microscopy (LSM700; Zeiss). For quantification, the contours of transfected HEK293T cells were chosen as the region of interest. The fluorescence intensities of synapsin puncta, normalized with respect to the area of each HEK293T cell, were quantified for both red and green channels using MetaMorph Software.
Primary neuronal culture, transfections, infections, immunocytochemistry, and image acquisition and analyses.
Hippocampal or cortical rat cultures were prepared from embryonic day 18 rat embryos, as described previously (Ko et al., 2006). For immunocytochemistry, cultured rat neurons were fixed with 4% paraformaldehyde/4% sucrose for 10 min at room temperature, permeabilized with 0.2% Triton X-100 in PBS for 5 min at 4°C, blocked with 3% horse serum/0.1% crystalline grade BSA in PBS for 30 min at room temperature, and incubated with the indicated primary and secondary antibodies in blocking solution for 1 h at room temperature. For overexpression of TrkC, hippocampal neurons at DIV10 were transfected with HA-tagged, full-length TrkC wild-type (WT) or point mutants, together with pEGFP-N1, and immunostained at DIV14. For knock-down (KD) of TrkC, hippocampal neurons were cotransfected with l-309 sh-TrkC at DIV8 and immunostained at DIV14. For KD of TrkC, NT-3 or PTPσ, hippocampal rat neurons were infected with the corresponding shRNA lentiviruses at DIV3-4 and immunostained with antibodies against synapsin and microtubule-associated protein (MAP2) at DIV12-13. NT-3 at the indicated concentration was added daily to transfected or infected cultured rat neurons at DIV8 or DIV10, as indicated. Transfected or infected neurons were chosen randomly and images were acquired using a confocal microscope (LSM700; Carl Zeiss) with a 63× objective lens; all image settings were kept constant. Z-stack images obtained through confocal microscopy were converted to maximal projection and analyzed to obtain the size and density of presynaptic terminals using MetaMorph software (Molecular Devices). Dendritic branching was assessed using the Sholl Analysis plugin for ImageJ downloaded from the Anirvan Ghosh Laboratory at the University California, San Diego. All analyses were performed in a blinded manner.
Semiquantitative immunoblotting in cultured rat neurons.
Cultured hippocampal rat neurons were prepared and 25 or 100 ng/ml NT-3 was added at DIV14. At the indicated times after NT-3 addition, neuron lysates were prepared and analyzed by immunoblotting with the indicated antibodies. Western blot signals were detected using an enhanced chemiluminescence system (GE Healthcare) and quantified using ImageJ software. Ratios of phosphorylated protein to total protein were normalized to 1.0. Values from control and experimental groups at various time points were compared using one-way ANOVA followed by post hoc Tukey's test. A p-value <0.05 was taken as significant.
OptoTrk imaging, light stimulation, and data analysis.
Cultured hippocampal rat neurons were transfected with Lipofectamine LTX and Plus reagents (Invitrogen) according to the manufacturer's instructions. Briefly, 500 ng of DNA was mixed with 0.5 μl of LTX and Plus reagents in 100 μl of Opti-MEM (Invitrogen) in 2 wells of a 24-well plate. After 24 h, transfected neurons were preincubated with extracellular buffer solution containing the following (in mm): (25 HEPES, 119 NaCl, 2.5 KCl, 2 CaCl2, 2 MgCl2, and 30 glucose for 30 min. Live-cell imaging was performed using a Nikon A1R confocal microscope mounted on a Nikon Eclipse Ti body equipped with CFI Plan Apochromat VC objectives (60/1.4 numerical aperture oil-immersion lenses) and a Chamlide TC system and maintained at 37°C and 5% CO2 (Live Cell Instrument). Green (488 nm) and red (561 nm) lasers were used for acquisition of images (512 × 512 pixels, 0.41 μm/pixel). Photoactivation was performed with a 488 nm laser through a Galvano scanner incorporated in a hybrid confocal scan head containing a high-speed hyperselector (Nikon). The laser power for light stimulation, measured with an optical power meter (ADCMT), was 5–25 μW, corresponding to 6.5–32.47 mW/cm2. Image data were analyzed using Nikon imaging software (NIS-element AR 64-bit version 4.10; Laboratory Imaging).
Statistics.
All data are expressed as means ± SEM. All experiments were repeated using at least three independent cultures and data were evaluated statistically using one-way ANOVA followed by post hoc Tukey's test or Student's t test, as indicated.
Results
NT-3 increases TrkC binding affinity for PTPσ and enhances TrkC-mediated presynaptic differentiation
To determine whether NT-3-induced TrkC dimerization affects TrkC binding affinity for PTPσ, we first produced highly purified NT-3 recombinant protein from Sf9 insect cells (data not shown) and performed a quantitative cell-surface-binding assay that allowed calculation of approximate binding affinities between TrkC and PTPσ. We incubated HEK293T cells transfected with full-length TrkC with recombinant PTPσ at concentrations ranging from 6.25 to 200 nm without or with NT-3 (25 or 100 ng/ml; Fig. 1A). Assuming a single, independent TrkC-binding site for PTPσ, we computed the net binding of the PTPσ recombinant protein at a given concentration and analyzed binding using the Michaelis–Menten equation. This analysis yielded a Kd of TrkC for PTPσ of 78.13 ± 15.15 nm without NT-3 treatment, 39.06 ± 14.26 nm with 25 ng/ml NT-3, and 36.36 ± 8.09 nm with 100 ng/ml NT-3 (Fig. 1A). These data indicate that NT-3 increases the binding affinity of TrkC for PTPσ, possibly by inducing TrkC dimerization. We next performed heterologous synapse-formation assays to determine whether the increased binding of TrkC to PTPσ induced by NT-3 manifests enhanced presynaptic differentiation activity (Fig. 1B–E). To this end, HEK293T cells expressing HA-tagged TrkC were cocultured with hippocampal neurons from DIV10 to DIV12 without or with NT-3 (Fig. 1B,D). Consistent with its ability to increase the binding affinity of TrkC for PTPσ (Fig. 1A), NT-3 significantly enhanced clustering of synapsin puncta in axons of cocultured hippocampal neurons regardless of its concentration (Fig. 1B–E). In contrast, BDNF treatment did not facilitate the synaptogenic activity of TrkC, suggesting that the NT-3 effect is specific (Fig. 1D,E). In addition, NT-3 induced no alteration of dendritic complexity in neurons (Fig. 1F,G). Together, these results suggest that NT-3-induced dimerization of TrkC positively regulates presynaptic differentiation organized by the TrkC–PTPσ synaptic adhesion complex. Recently, Ammendrup-Johnsen et al. (2015) reported similar observations indicating that NT-3 promotes PTPσ-mediated presynaptic differentiation.
Figure 1.
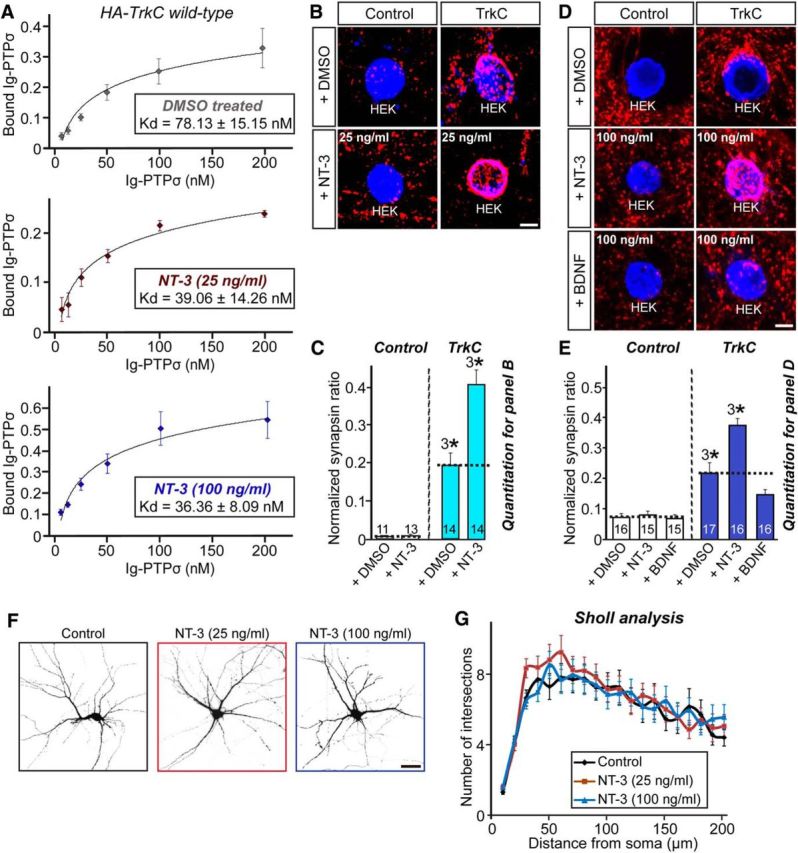
NT-3 promotes TrkC binding to PTPσ to trigger TrkC-mediated presynaptic differentiation. A, Saturation binding of Ig-PTPσ to HA-TrkC expressed in HEK293T cells in the absence or presence of NT-3 (25 or 100 ng/ml). Kds were calculated from three independent experiments. All data are shown as means ± SEM. B, Representative images of the heterologous synapse-formation activities of TrkC. Neurons were cocultured for 2 d with HEK293T cells expressing EGFP alone (Control) or HA-TrkC (TrkC) in the absence (+DMSO) or presence of 25 ng/ml NT-3, and then stained with antibodies against EGFP or HA (blue) and synapsin (red). Scale bar, 10 μm (applies to all images). C, Synapse-forming activity in B was quantified by measuring the ratio of synapsin staining intensity (red) to HA immunoreactivity intensity (blue). All data are shown as means ± SEM (3*p < 0.001; ANOVA with post hoc Tukey's test). D, E, Same as B and C except that the NT-3 or BDNF concentration was 100 ng/ml (3*p < 0.001; ANOVA with post hoc Tukey's test). Scale bar, 10 μm (applies to all images). F, G, Representative images (F) and quantification results (G) demonstrating no alteration in dendritic branch number in NT-3-treated cultured neurons. Hippocampal neurons at DIV7 were treated at the indicated concentration of NT-3 and analyzed by Sholl analysis at DIV10. Scale bar, 60 μm (applies to all images). All data are shown as means ± SEM. n denotes the number of neurons as follows: Control, n = 20; NT-3 (25 ng/ml), n = 21; and NT-3 (100 ng/ml), n = 21.
NT-3 is required for TrkC-mediated presynaptic differentiation
To address whether NT-3 is required for the synaptogenic activity of TrkC, we first developed shRNA lentiviruses that specifically knocked down NT-3 and then infected cultured neurons with lentiviruses expressing these NT-3-targeting shRNAs (sh-NT-3) or an empty shRNA vector (sh-Control) and performed heterologous synapse-formation assays using HEK293T cells expressing HA-TrkC, HA-LRRTM2 (leucine-rich repeat transmembrane protein 2), or EGFP alone (control; Fig. 2A,B). TrkC strongly recruited synapsin to the vicinity of HEK293T cells in control neurons, but did not do so in NT-3-deficient neurons (Fig. 2A,B). In contrast, LRRTM2 strongly induced synapsin clustering in both control and NT-3-deficient neurons (Fig. 2A,B). Together, these data suggest that NT-3 is specifically required for the synaptogenic activity of TrkC responsible for induction of presynaptic differentiation.
Figure 2.

NT-3 is required for TrkC-mediated presynaptic differentiation. A, Representative images of the heterologous synapse-formation activities of TrkC in NT-3-deficient neurons. Neurons were infected with lentiviruses expressing sh-Control or sh-NT-3 at DIV3 and then cocultured from DIV10 to DIV12 with HEK293T cells expressing EGFP alone (Control), HA-TrkC (TrkC), or HA-LRRTM2 (LRRTM2). Neurons were stained with antibodies against EGFP or HA (blue) and synapsin (red). Scale bar, 10 μm (applies to all images). B, Synapse-formation activity in A was quantified by measuring the ratio of synapsin staining intensity (red) to HA/EGFP intensity (blue). Note that the normalized synapsin ratio values presented in Figures 1 and 2 cannot be compared because they were obtained from different sets of experiments. All data are shown as means ± SEM (3*p < 0.001; ANOVA with post hoc Tukey's test).
NT-3 modulates TrkC-mediated synapse number in a concentration-dependent manner
The observation that NT-3 augments TrkC-mediated presynaptic differentiation in heterologous synapse-formation assays through increased binding to PTPσ prompted us to next investigate whether NT-3 also regulates postsynaptic development mediated by TrkC (Fig. 3). Cultured hippocampal neurons were transfected with EGFP alone (control) or together with TrkC WT at DIV10, incubated with or without NT-3 at the indicated concentration, and then immunostained with anti-synapsin and anti-EGFP antibodies at DIV14 (Fig. 3A,B). Chronic treatment with 25 ng/ml NT-3 for 3 d significantly increased the synapse density of control hippocampal neurons (Fig. 3A,B). Overexpression of TrkC WT in cultured neurons strongly increased the synapse density in a control condition (i.e., without additional NT-3 treatment; Fig. 3A,B). Strikingly, the synaptogenic effects of NT-3 observed at 10 and 25 ng/ml were completely abrogated in both control and TrkC-expressing neurons by treatment with 100 ng/ml NT-3 (Fig. 3A,B). An examination of the postsynaptic excitatory marker protein Shank1 yielded similar results (Fig. 3C,D). To address how treatment with NT-3 at a concentration of 100 ng/ml abolished TrkC synaptogenic activity, we performed surface biotinylation experiments in cultured hippocampal neurons (Fig. 4A,B). We found that treatment with 100 ng/ml NT-3 markedly decreased the surface level of TrkC in neurons, indicating that this treatment may cause TrkC endocytosis, followed by degradation. Strikingly, treatment with 25 ng/ml NT-3 also significantly decreased the level of surface TrkC, albeit to a lesser extent than treatment with 100 ng/ml NT-3, suggesting that NT-3 treatment induces endocytosis of TrkC, but TrkC endocytosis and TrkC synaptogenic activity are not strictly correlated (Fig. 4A,B). Together, these results suggest that NT-3 exerts its enhancing effect on synapse number in a concentration-dependent manner. Although complete mechanistic details remain to be defined, it is likely that treatment with NT-3 at 100 ng/ml (for 4 h) induces TrkC degradation, leading to disruption of TrkC-mediated synapse-promoting activity (see below).
Figure 3.
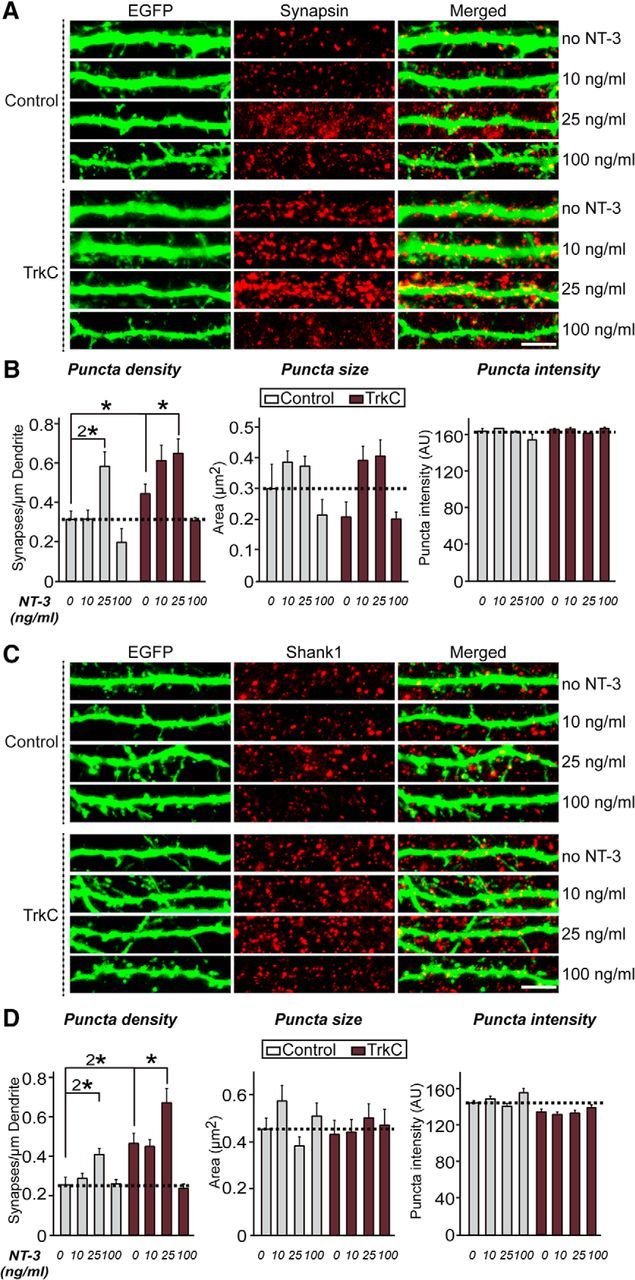
NT-3 affects TrkC-mediated synapse density differentially and in a concentration-dependent manner. A, Cultured hippocampal neurons were transfected with an EGFP (Control) or TrkC WT expression vector at DIV10 and then treated daily with the indicated concentration of NT-3 for 3 d. The transfected neurons were analyzed at DIV14 by double immunofluorescence using antibodies to EGFP (green) and synapsin (a presynaptic marker; red). Scale bar, 10 μm (applies to all images). B, Summary graphs of the effects of TrkC overexpression in neurons on synaptic marker density (left), synaptic marker size (middle), and synaptic marker intensity (right) measured using synapsin as a synaptic marker. Two or three dendrites per transfected neurons were analyzed and group averaged. All data are shown as means ± SEM (*p < 0.05, 2*p < 0.01; ANOVA with post hoc Tukey's test). “n” denotes the number of neurons as follows: Control/0 ng/ml NT-3, n = 25; Control/10 ng/ml NT-3, n = 19; Control/25 ng/ml NT-3, n = 14; Control/100 ng/ml, n = 19; TrkC/0 ng/ml NT-3, n = 24; TrkC/10 ng/ml NT-3, n = 15; TrkC/25 ng/ml NT-3, n = 20; and TrkC/100 ng/ml NT-3, n = 26. C, D, Representative images (C) and summary bar graphs (D) of the effects of TrkC overexpression in neurons on Shank1 (a postsynaptic marker; red) puncta. Scale bar, 10 μm (applies to all images). All data are shown as means ± SEM (*p < 0.05, 2*p < 0.01; ANOVA with post hoc Tukey's test). “n” denotes the number of neurons as follows: Control, 0 ng/ml NT-3, n = 23; Control, 10 ng/ml NT-3, n = 23; Control, 25 ng/ml NT-3, n = 20; Control, 100 ng/ml, n = 21; TrkC, 0 ng/ml NT-3, n = 20; TrkC, 10 ng/ml NT-3, n = 20; TrkC, 25 ng/ml NT-3, n = 19; and TrkC, 100 ng/ml NT-3, n = 20.
Figure 4.
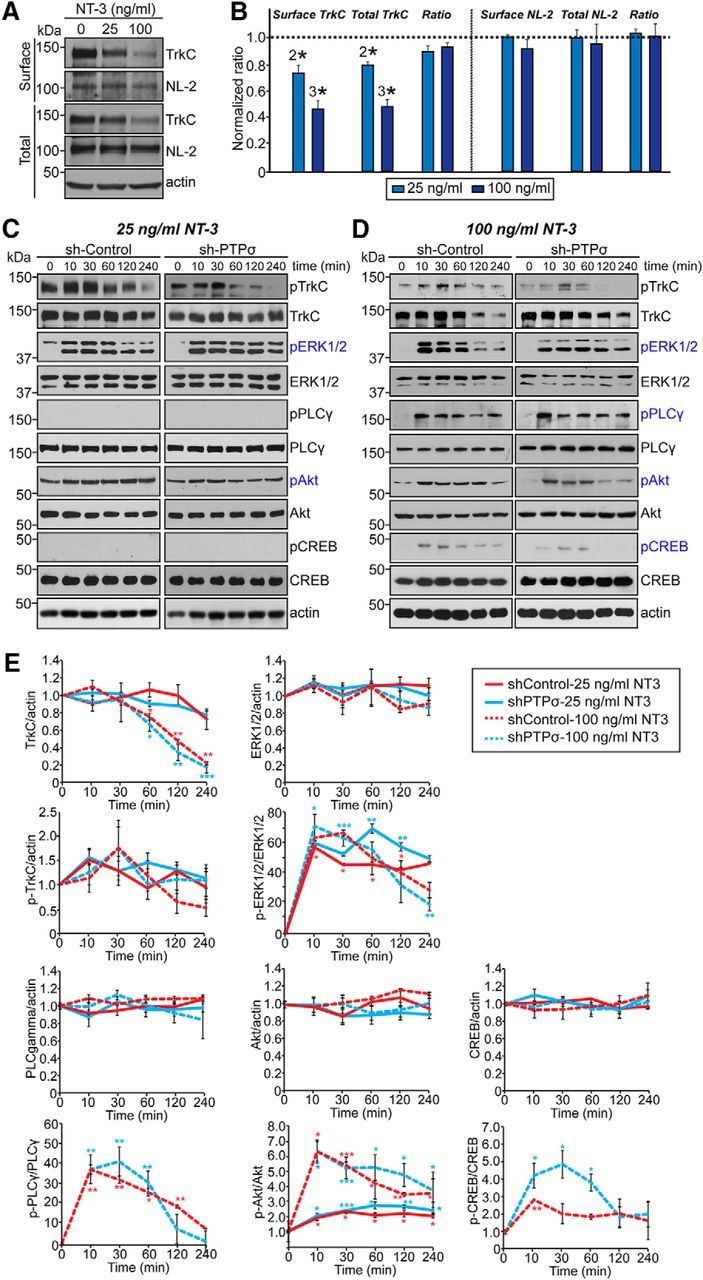
NT-3 activates distinct signaling components downstream of TrkC in a time- and concentration-dependent, but PTPσ-independent, manner. A, B, Representative immunoblot images (A) and summary bar graphs (B) quantifying the surface expression levels of endogenous TrkC and neuroligin-2 (NL-2) in cultured cortical neurons 2 h after NT-3 treatment. Individual band intensities were normalized to that for the untreated condition for each experimental group. All data are shown as means ± SEM (2*p < 0.01; 3*p < 0.001; ANOVA with post hoc Tukey's test; n = 3 independent experiments). C, D, Cultured cortical neurons were infected with lentiviruses expressing sh-Control or sh-PTPσ at DIV3 and then treated at DIV8 with 25 ng/ml (A) or 100 ng/ml (B) of NT-3 for the indicated times. Levels of TrkC downstream signaling proteins were examined by semiquantitative immunoblotting 10, 30, 60, 120, and 240 min after NT-3 treatment using the indicated antibodies. E, Quantification of protein levels from C and D. Summary graphs were plotted from three to five independent experiments. All data are shown as means ± SEM (*p < 0.05, **p < 0.01, ***p < 0.001; ANOVA with post hoc Tukey's test). Statistical significance was determined based on the differences between the zero time point and different individual time points.
NT-3 differentially activates TrkC intracellular signaling pathways in an NT-3-concentration-dependent, but PTPσ-independent, manner
To determine how different concentrations of NT-3 trigger either synaptogenic or synaptotoxic effects on synapse density, we first investigated changes in intracellular signaling pathways induced by NT-3 treatment using semiquantitative immunoblotting of cultured cortical neurons (Fig. 4C–E). To this end, we treated cultured DIV8 cortical neuron with NT-3 and collected neuron lysates at the indicated time points and immunoblotted with antibodies targeting various intracellular kinases known to be downstream targets of Trk family receptor tyrosine kinases (Huang and Reichardt, 2003; Fig. 4C–E). Notably, NT-3 at a concentration of 100 ng/ml caused an abrupt decrease in the total amount of TrkC protein between 120 and 240 min after treatment (Fig. 4C–E). These results are consistent with our finding that treatment with 100 ng/ml NT-3 compromised TrkC-promoted synapse density (Fig. 3). Moreover, the proportion of Tyr820-phosphorylated TrkC remained relatively constant at this same NT-3 concentration, suggesting that the remaining TrkC protein is still responsive to NT-3 (Fig. 4C–E). In contrast, treatment with 25 ng/ml NT-3 did not diminish total TrkC protein levels, again consistent with results of our transfection experiments (Figs. 4C–E, Fig. 3). In addition, we found that ERK1/2 was robustly and similarly activated by treatment with both 25 and 100 ng/ml NT-3, whereas Akt was activated to a greater extent by 100 ng/ml NT-3 and PLC-γ1 and CREB kinases were activated only at an NT-3 concentration of 100 ng/ml (Fig. 4C–E). It was shown previously that LAR is required for activation of BDNF-induced TrkB and its downstream targets in cultured neurons (Yang et al., 2006). Therefore, we also investigated whether NT-3-induced activation of TrkC and its intracellular targets similarly requires PTPσ. We found no quantitative differences in the total expression or activation profiles of TrkC or its downstream targets between PTPσ-deficient neurons (expressing sh-PTPσ) and control neurons (expressing sh-Control; Fig. 4C–E). To confirm that PTPσ is not involved in NT-3-induced TrkC activation per se, we next used the recently developed Opto-Trk, an optogenetic approach based on Arabidopsis thaliana cryptochrome 2 in which light stimulation is used to activate neurotrophin signaling (Chang et al., 2014). Cultured hippocampal neurons were infected with lentiviruses expressing either sh-Control or sh-PTP at DIV3, transfected with Opto-TrkC (Fig. 5A) and genetically encoded signaling sensors at DIV7–9, and imaged at DIV8–10 (Fig. 5). We used an mCherry-tagged pleckstrin homology (PH) domain of Akt [mCherry-Akt(PH)] to monitor activation of the phosphoinositide 3-kinase (PI3K)-Akt signaling pathway, fusionRed-tagged ERK [ERK-fusionRed] to monitor activation of the ERK1/2 signaling pathway, and R-GECO1 to monitor the activation of PLC-γ1 and intracellular calcium levels (Fig. 5B–I). To mirror the concentration-dependent effects of NT-3 observed in biochemical and cellular experiments using the Opto-Trk approach (Figs. 3, 4), we optimized the experimental conditions by first stimulating transfected neurons expressing Opto-TrkC and R-GECO1 with different powers of light (Fig. 5B,D). We found that light stimulation at a power of 6 μW corresponded to an NT-3 concentration of 25 ng/ml, causing a robust increase in ERK1/2 activity with a minimal increase in intracellular calcium level, whereas stimulation at a power of 25 μW induced robust calcium increases, mimicking the effects of 100 ng/ml NT-3 (Fig. 5B–E; data not shown). Consistent with this, treatment with 100 ng/ml NT-3, but not 25 ng/ml, led to a significant increase in intracellular calcium level, monitored by R-GECO1 intensity (Fig. 5C,E). Next, stimulation at either light intensity led to translocation of ERK-fusionRed into the nucleus and mCherry-Akt(PH) into the plasma membrane from the cytosol and increased the intensity of R-GECO1 signals in transfected neurons (Fig. 5F–I). However, none of these sensors increased baseline levels of Akt, ERK, or PLC-γ1 in cultured neurons when singly expressed with or without light stimulation (Fig. 5J,K) or in HEK293T cells when expressed alone (Fig. 5L,M). Again, PTPσ was not required for any of the signaling pathways examined. Together, the results of these two independent experimental approaches consistently indicate that TrkC activates distinct signaling pathways that are dependent on NT-3 concentration, but independent of PTPσ-binding activity.
TrkC promotes synaptic development through extracellular binding and intracellular signaling cascades
To confirm that activation of intracellular signaling pathways is required for TrkC-mediated synaptic development, we next investigated whether point mutations in TrkC alter TrkC-induced increases in synapse number (Figs. 6, 7). We first generated a variety of TrkC point mutants in which binding to the extracellular ligand PTPσ (D240A/D242A) or NT-3 (N366A/T369A) is specifically disrupted (Takahashi et al., 2011; Coles et al., 2014) or that are unable to activate intracellular signaling pathways because of elimination of the NPQY516 adaptor-binding site (Y516F), inhibition of kinase activity (D679N), inactivation of PLC-γ1-mediated protein kinase C activation (Y820F; Tauszig-Delamasure et al., 2007), or the presence of a mutation found in sporadic cancer patients (E543D; Genevois et al., 2013; Fig. 6A). We then tested whether these TrkC mutants exhibited comparable surface expression and PTPσ binding (Fig. 6B,C). All TrkC mutants were targeted well to the cell surface when expressed in HEK293T cells (Fig. 6B). In addition, recombinant Ig-PTPσ robustly bound to HEK293T cells expressing various TrkC mutants, except TrkC D240A/D242A, results that are consistent with those reported in a recent structural study (Coles et al., 2014; Fig. 6C).
Figure 6.
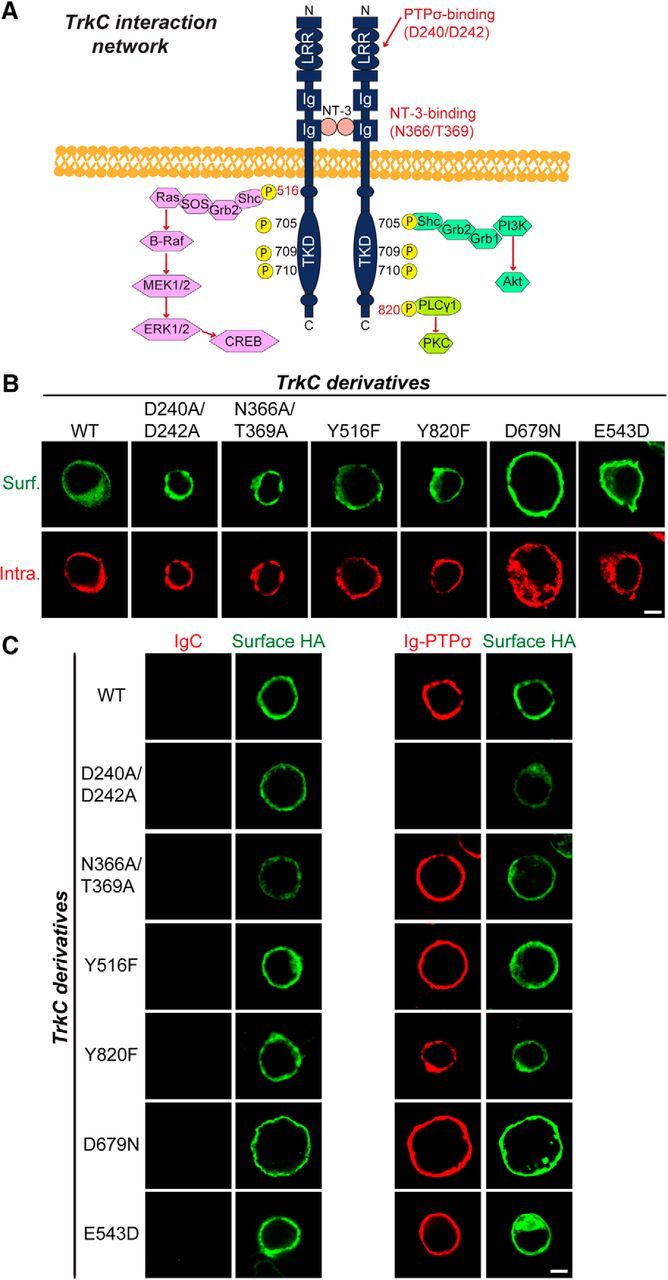
Characterization of surface expression levels and PTPσ-binding properties of TrkC point mutants used in the current study. A, Schematic depiction of the major intracellular signaling pathways downstream of TrkC. Tyrosine 516 (Y516) is located in the activation loop of the TrkC tyrosine kinase domain, which forms a PTB-binding motif (NPXpY) known to bind to Shc and FRS2 adaptor proteins. Tyrosine 820 (Y820) binds to PLC-γ1. Mutation of aspartate (D) to asparagine (N) at residue 679 abolishes the kinase activity of TrkC. Mutation of D240 and D242 to alanines eliminates binding to PTPσ, whereas mutation of N366 and T369 to alanines abolishes binding to NT-3. B, HEK293T cells expressing HA-tagged TrkC WT or its various point mutants were immunostained with mouse anti-HA antibodies (green) and detected with FITC-conjugated anti-mouse secondary antibodies under nonpermeabilized conditions, followed by permeabilization of cells. Cells were stained with rabbit anti-HA antibodies (red), followed by Cy3-conjugated anti-rabbit secondary antibodies. Scale bar, 10 μm (applies to all images). C, Representative images of cell-surface-binding assays. HEK293T cells expressing HA-tagged TrkC WT or its point mutants were incubated with 10 μg/ml control IgC or Ig-PTPσ and then analyzed by immunofluorescence imaging of Ig-fusion proteins (red) and HA antibodies (green). Scale bar, 10 μm (applies to all images).
Figure 7.
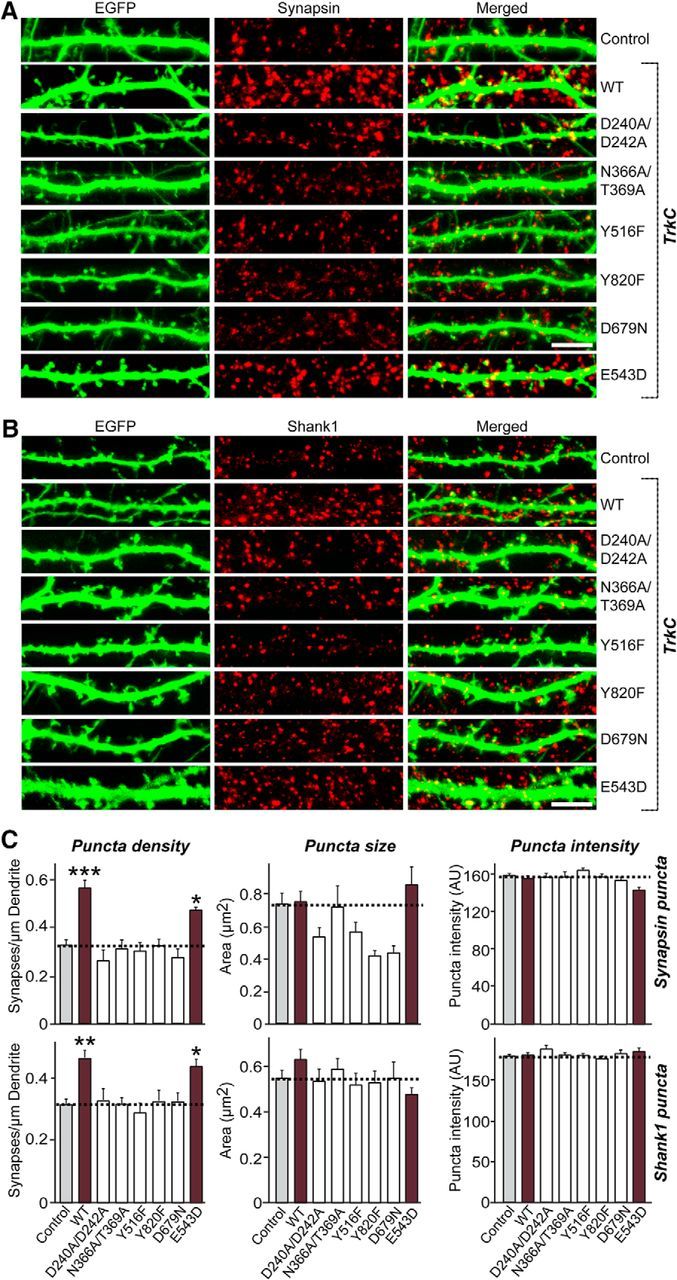
TrkC extracellular binding, intracellular signaling, and kinase activity is required for TrkC-mediated regulation of synapse density. A, B, Cultured hippocampal neurons were transfected with EGFP (Control), TrkC WT, or the indicated TrkC point mutants at DIV10 and analyzed at DIV14 by double immunofluorescence with antibodies to EGFP (green) and synapsin (A; a presynaptic marker; red) or Shank1 (B; a postsynaptic marker; red). Scale bar, 10 μm (applies to all images). C, Summary graphs of the effects of TrkC overexpression in neurons on puncta density (left), puncta size (middle), or puncta intensity (right) as measured using synapsin (A) or Shank1 (B) as synaptic markers. Two or three dendrites per transfected neuron were analyzed and group averaged. All data are shown as means ± SEM (*p < 0.05, **p < 0.01, ***p < 0.001; ANOVA with post hoc Tukey's test). “n” denotes the number of neurons as follows: Control/synapsin, n = 39; TrkC WT/synapsin, n = 37; TrkC D240A/D242A/synapsin, n = 17; TrkC N366A/T369A/synapsin, n = 16; TrkC Y516F/synapsin, n = 24; TrkC Y820F/synapsin, n = 25; TrkC D679N/synapsin, n = 17; TrkC E543D/synapsin, n = 16; Control/Shank1, n = 36; TrkC WT/ Shank1, n = 32; TrkC D240A/D242A/Shank1, n = 28; TrkC N366A/T369A/Shank1, n = 30; TrkC Y516F/Shank1, n = 30; TrkC Y820F/Shank1, n = 23; TrkC D679N/Shank1, n = 26; and TrkC E543D/Shank1, n = 25.
Next, we transfected DIV10 hippocampal cultured neurons with TrkC WT or various point mutants and stained them for the presynaptic and postsynaptic markers synapsin and Shank1, respectively, and EGFP (Fig. 7). Overexpression of TrkC WT significantly increased the density of synapses labeled with anti-synapsin and anti-Shank1 antibodies compared with control neurons expressing EGFP alone (control; Fig. 7A–C). Strikingly, overexpression of TrkC D240A/D242A or N366A/T369A markedly compromised the synapse-boosting effects of TrkC WT overexpression (Fig. 7A–C). In addition, overexpression of TrkC Y516F, TrkC Y820F, or TrkC D679N, but not TrkC E543D, also significantly compromised TrkC WT effects (Fig. 7A–C). Collectively, these data suggest that TrkC extracellular interactions with both NT-3 and PTPσ, major intracellular signaling pathways, and kinase activity contribute to the regulation of TrkC-mediated synapse density.
TrkC-dependent competition regulates synapse number
NT-3 was recently reported to be involved in TrkC-dependent, competitive dendritic morphogenesis in cerebellar Purkinje neurons (Joo et al., 2014). To explore this possibility, we first screened various TrkC nucleotide sequences for effective KD sequences, together with a previously validated sequence (Takahashi et al., 2011; Fig. 8A,B). We found that two newly screened sequences, designated T4-CDS and T7-UTR, and the previously validated sequence, designated T9-CDS (identical to the sh-TrkC#2 construct used in Takahashi et al., 2011) were quite effective in reducing TrkC mRNA levels, as measured by quantitative RT-PCR (Fig. 8B). We also validated the KD synapse phenotype, showing that TrkC-KD leads to a significant decrease in synapse number, but not dendritic complexity, in cultured hippocampal neurons (Fig. 8C–F). The molecular replacement approach, which allows simultaneous RNA interference-mediated KD of endogenous TrkC and expression of various mutant forms of recombinant TrkC in a single neuron, confirmed the interpretation that TrkC requires both extracellular ligand-binding activity and activation of intracellular signaling (Fig. 9). To determine whether a similar NT-3/TrkC-dependent competition mechanism operates in hippocampal neurons, we transfected these cells with TrkC-KD vector at DIV8 or infected them with TrkC-KD lentiviruses at DIV3-4 and immunostained transfected or infected neurons with antibodies against synapsin and EGFP (for transfected neurons) or synapsin and MAP2 (for infected neurons) at DIV14 (Fig. 10A,B). Surprisingly, lentiviral infection-mediated global TrkC-KD did not decrease synapse density; in contrast, the sparse TrkC-KD achieved by transfection robustly reduced synapse density, which was completely rescued by reexpression of shRNA-resistant TrkC WT (Fig. 10A–D). These results are reminiscent of the normal Purkinje cell morphogenesis observed in global TrkC-KO mice (Joo et al., 2014). However, unlike reexpression of shRNA-resistant TrkC WT, global NT-3 KD did not restore the deficit in synapse density observed in sparse TrkC-KD (Fig. 10C,D), suggesting that the NT-3/TrkC-dependent synapse competition mechanism does not operate in hippocampal neurons. Strikingly, global NT-3 KD profoundly reduced synapse density in MAP2-positive dendrites compared with control neurons (Fig. 10A,B). These results are consistent with finding that NT-3 is required for TrkC-mediated increases in synapse number and suggest that the absence of NT-3 does not nonspecifically deplete presynaptic vesicle pools (Fig. 2).
Figure 8.
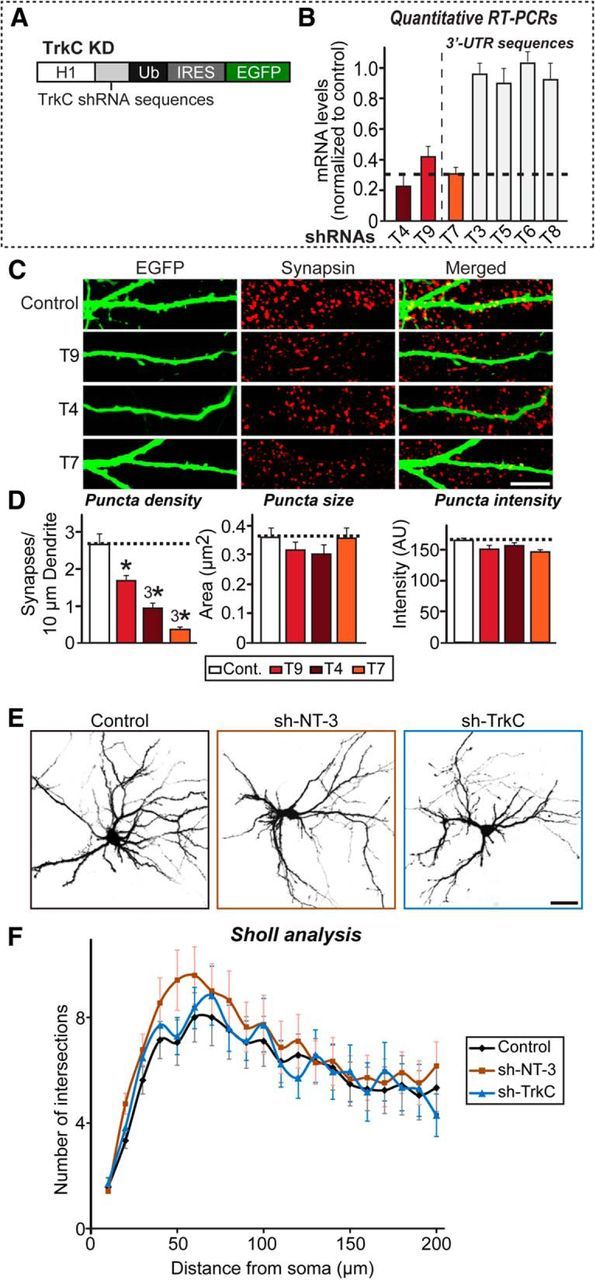
Characterization of TrkC KD used in the current study. A, Design of lentiviral shRNA vectors for KD of TrkC. H1, Human H1 promoter; IRES, internal ribosome entry sequence; Ub, ubiquitin promoter. B, Levels of TrkC mRNA were measured at DIV12–13 by quantitative RT-PCR in cultured cortical neurons infected at DIV3 with lentiviruses expressing the indicated candidate TrkC shRNAs (T3–T9). Dashed line indicates 70% KD cutoff level for tests of biological effects. C, Rat hippocampal neurons were transfected with lentiviral vectors expressing shRNAs targeting TrkC (T4, T7, or T9) or control vectors lacking shRNA expression (sh-Control) at DIV8 and analyzed at DIV14 by double immunofluorescence with antibodies to EGFP (green) and synapsin (red). Scale bar, 10 μm (applies to all images). D, Summary graphs of the effects of lentiviral vectors expressing shRNAs targeting TrkC (T4, T7, or T9) in neurons on puncta density (left), puncta size (middle), and puncta intensity (right) measured using synapsin as a synaptic marker. All data are shown as means ± SEM (*p < 0.05, 3*p < 0.001; ANOVA with post hoc Tukey's test). E, F, Representative images (E) and quantification results (F) demonstrating no alteration in dendritic branch number in NT-3- or TrkC-deficient cultured neurons. Hippocampal neurons were infected with lentiviruses expressing sh-Control (Control), sh-NT-3, or sh-TrkC at DIV3 and analyzed at DIV11 using Sholl analysis. All data are shown as means ± SEM. “n” denotes the number of neurons as follows: sh-Control, n = 21; sh-NT-3, n = 22; and sh-TrkC, n = 17. Scale bar, 60 μm (applies to all images).
Figure 9.
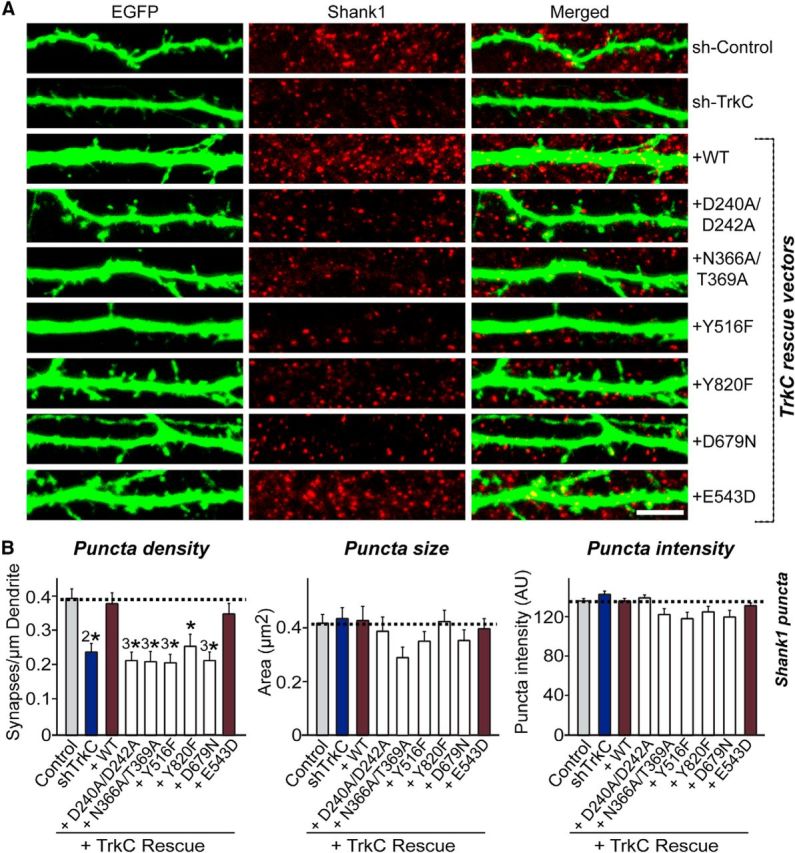
Replacement of endogenous TrkC with exogenous TrkC WT, but not with TrkC mutants, rescues TrkC-mediated regulation of postsynapse density. A, Cultured hippocampal neurons were transfected with a lentiviral vector expressing sh-Control, sh-TrkC (TrkC KD) or coexpressing sh-TrkC and various TrkC constructs (WT and point mutants presented in Figs. 6 and 7) at DIV8 and analyzed at DIV14 by double immunofluorescence with antibodies to EGFP (green) and Shank1 (a postsynaptic marker; red). Scale bar, 10 μm (applies to all images). Note that the TrkC shRNA sequence used here targets 3′-UTRs of rat TrkC (see Fig. 8B). B, Summary graphs of the effects of TrkC molecular replacement in neurons on puncta density (left), puncta size (middle), and puncta intensity (right) measured using Shank1 as a synaptic marker. Two or three dendrites per transfected neuron were analyzed and group averaged. All data are shown as means ± SEM (*p < 0.05, 2*p < 0.01, 3*p < 0.001; ANOVA with post hoc Tukey's test). “n” denotes the number of neurons as follows: sh-Control/Shank1, n = 28; sh-TrkC/Shank1, n = 20; TrkC WT rescue/Shank1, n = 28; TrkC D240A/D242A rescue/Shank1, n = 23; TrkC N366A/T369A rescue/Shank1, n = 21; TrkC Y516F rescue/Shank1, n = 22; TrkC Y820F rescue/Shank1, n = 21; TrkC D679N rescue/Shank1, n = 21; and TrkC E543D rescue/Shank1, n = 25.
Figure 10.
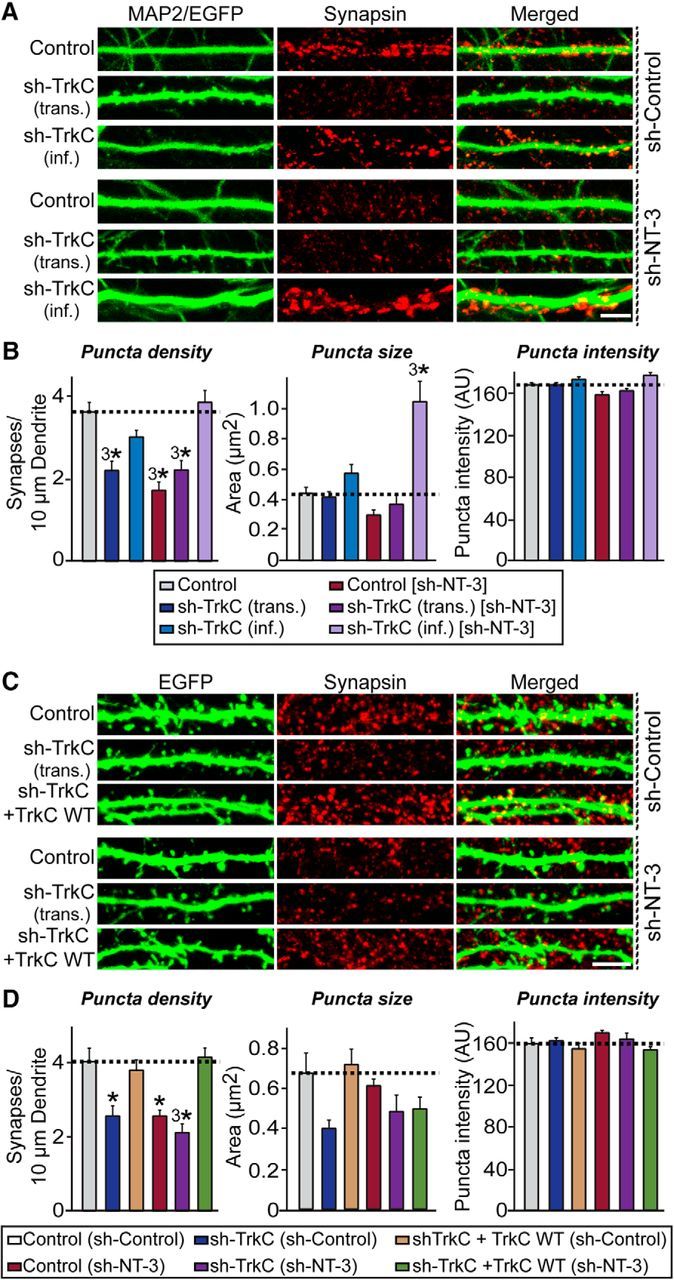
The relative amount of TrkC dictates synapse number through an interneuronal competition mechanism. A, Cultured hippocampal neurons were infected with lentiviruses expressing sh-TrkC [sh-TrkC (inf.)] or control vectors lacking shRNA expression (Control) at DIV3. Control neurons were further transfected with TrkC KD vector (T7) at DIV8 [sh-TrkC (trans.)]. Neurons expressing sh-NT-3 [Control/sh-NT-3, sh-TrkC (trans.)/sh-NT-3, and sh-TrkC (inf.)/sh-NT-3] were tested under identical conditions. Transfected/infected neurons were analyzed at DIV14 by double immunofluorescence with antibodies to EGFP (green) or MAP2 (green) and synapsin (red). Scale bar, 10 μm (applies to all images). Note that synapsin puncta size is dramatically increased in neurons expressing sh-NT-3 and sh-TrkC. B, Quantification of synapsin puncta density (left), synapsin puncta size (middle), and synapsin puncta intensity (right) for images shown in A. All data are shown as means ± SEM (3*p < 0.001; ANOVA with post hoc Tukey's test). “n” denotes the number of neurons as follows: Control/sh-Control, n = 61; sh-TrkC (inf.)/sh-Control, n = 35; sh-TrkC (trans.)/sh-Control, n = 39; Control/sh-NT-3, n = 30; sh-TrkC (inf.)/sh-NT-3, n = 29; and sh-TrkC (trans.)/sh-NT-3, n = 32. C, Rescue experiments demonstrate that the TrkC KD phenotypes presented in A and B are specific. Cultured hippocampal neurons were infected with lentiviruses expressing sh-NT-3 or control vectors lacking shRNA expression (Control) at DIV3 and then further transfected with TrkC KD vector [T7; sh-TrkC (trans.)], Control vector, or cotransfected with sh-TrkC and shRNA-resistant TrkC WT vector (sh-TrkC + TrkC WT) at DIV8. Transfected/infected neurons were then analyzed at DIV14 by double immunofluorescence with antibodies to EGFP (green) and synapsin (red). Scale bar, 10 μm (applies to all images). D, Quantification of synapsin puncta density (left), synapsin puncta size (middle), and synapsin puncta intensity (right) for images shown in C. Note that the puncta size of sh-TrkC neurons is not different from that of control neurons. All data are shown as means ± SEM (*p < 0.05; 3*p < 0.001; ANOVA with post hoc Tukey's test). “n” denotes the number of neurons as follows: Control/sh-Control, n = 15; sh-TrkC/sh-Control, n = 14; sh-TrkC+TrkC WT/sh-Control, n = 14; Control/sh-NT-3, n = 14; sh-TrkC/sh-NT-3, n = 14; and sh-TrkC+TrkC WT/sh-NT-3, n = 15.
Discussion
In the present study, we sought to address the role of NT-3 in regulating synapse development mediated by TrkC receptor tyrosine kinase and its various signaling pathways. To this end, we investigated whether extracellular adhesion events and intracellular signaling pathways of TrkC are responsible for synapse development. We made five key observations.
First, we discovered that NT-3 promotes presynaptic differentiation during heterologous synapse formation by enhancing the binding strength of TrkC with its presynaptic receptor PTPσ (Fig. 1). Interestingly, the positive effect of NT-3 on TrkC-mediated presynaptic differentiation was independent of the concentration of NT-3 (but see below for our interpretation of the results; Fig. 1). These results indicate that dimerization of NT-3 per se is sufficient to facilitate TrkC-mediated presynaptic differentiation. Identical results were also recently reported, although the investigators in that study used only a single concentration (100 ng/ml) of NT-3 (Ammendrup-Johnsen et al., 2015).
Second, NT-3 is necessary for TrkC-mediated presynaptic differentiation, suggesting that NT-3-induced dimerization of TrkC, in addition to direct interaction with PTPσ, is necessary for recruitment of the presynaptic machinery (Fig. 2). This observation is particularly intriguing because simple ligand–receptor binding is sufficient to induce the formation of artificial synapses in the case of most reported synaptogenic adhesion molecules, except Slitrk1 (Ko et al., 2009b; Gokce and Südhof, 2013; Um et al., 2014). For example, neuroligin-1 mutations that disrupt neurexin binding, but not those that disrupt dimerization, abolish presynapse-inducing activity in heterologous synapse-formation assays (Ko et al., 2009b). Conversely, a Slitrk1 mutant that still retains PTPσ binding but is incapable of inducing lateral clustering does not induce presynaptic differentiation (Um et al., 2014). Although Slitrk1 is not reported to multimerize and PTPσ triggers local clustering of Slitrk1 in cellular membranes, it remains to be determined whether growth factors such as BDNF and/or NT-3 are capable of inducing multimerization of Slitrk1 (Um et al., 2014). In the presence of BDNF, dimerized TrkB preferentially binds to Slitrk5, displacing its trans-PTPσ-binding mode with a cis-Slitrk5-binding mode (Song et al., 2015).
Third, the effect of NT-3 on TrkC-mediated synapse density depends on its concentration (Fig. 3). The concentration dependence of NT-3 effects is notable because NT-3 secretion is known to be regulated by synaptic activity (Arvanian et al., 2003; Kolarow et al., 2007), which stimulates a distinct signaling pathway and leads to a specific synaptic, and possibly circuit, output. It is also remarkable that postsynaptic neurons adjust the surface level of TrkC according to the local concentration of NT-3 to exert either synaptotrophic or synaptotoxic effects (Figs. 3, 4). A recent report showed that the potentiating effect of NT-3 on recycling presynaptic vesicles in axonal boutons occurs exclusively through postsynaptic TrkC in a tyrosine-kinase-independent manner (Ammendrup-Johnsen et al., 2015). Intriguingly, this latter study used an NT-3 concentration of 100 ng/ml, which we found to augment the ability of TrkC to induce presynaptic differentiation (Fig. 1) but also to cause the degradation of endogenous TrkC, decreasing synapse number in control neurons and abrogating synapse-promoting effects in TrkC-overexpressing neurons, probably by decreasing surface and total TrkC levels (Figs. 3, 4). In contrast, a lower concentration of NT-3 (e.g., 25 ng/ml) increased both presynaptic differentiation and synapse number (Figs. 1, 3), suggesting that the synapse-promoting action of NT-3 requires other mechanisms in addition to its TrkC-dimerization-inducing activity. Notably, an NT-3 concentration of 25 ng/ml also significantly decreased surface TrkC levels, implying that the synaptogenic activity of TrkC may not be entirely controlled by TrkC turnover at synaptic membranes.
Fourth, TrkC-induced synaptic development requires molecular interactions with extracellular ligands, activation of all primary intracellular cascades, and TrkC kinase activity (Figs. 4, 5, 6, 7, 8). Because TrkC alters synapse number over an NT-3 concentration range of 10–25 ng/ml (Fig. 3), we initially suspected that distinct sets of intracellular signaling cascades uniquely activated at different NT-3 concentrations might determine whether NT-3 acts as a synaptogenic or synaptotoxic factor. Indeed, at a concentration of 100 ng/ml NT-3 or under strong light-intensity stimulation conditions, quantitative biochemical experiments and optical imaging analyses using Opto-TrkC showed that the levels of phosphorylated PLC-γ1 or PLC-γ1-mediated calcium, respectively, were elevated, effects that were not observed at 25 ng/ml NT-3 or weak light-intensity stimulation conditions (Figs. 4, 5). These results, however, should not be simply interpreted to mean that an increase in intracellular calcium level is solely responsible for the synaptotoxic effects of high concentrations of NT-3, because blocking phosphorylation at Tyr820 by a point mutation completely abolished the increase in synapse number induced by TrkC WT (Figs. 7, 8). Instead, it is reasonable to infer that transcriptional activation of a unique set of effector genes downstream of intracellular calcium elevation underlies the synaptotoxic effects of NT-3. In support of this idea, only 100 ng/ml NT-3 resulted in CREB kinase activation, which could be induced by PLC-γ1-mediated calcium transients (Fig. 4). A full understanding of the signaling pathways underlying NT-3-mediated effects on synapse density will require unraveling the transcriptional mechanisms that are specifically activated by higher concentrations of NT-3. Notably, activation of the three signaling cascades investigated in the current study did not require the presence of PTPσ, suggesting that NT-3-induced TrkC dimerization is sufficient to elicit the intracellular signaling outcomes necessary for regulating synapse density; this outcome is distinct from presynaptic differentiation, which is exclusively dependent on the presence of PTPσ (Figs. 4, 5). However, in addition to NT-3-binding activity and activation of intracellular signaling pathways, TrkC regulation of synapse density also requires binding to PTPσ (Figs. 7, 8). Moreover, TrkC kinase activity is also required for the observed effects of TrkC on synapse density, although the present study did not reveal the identity of substrates involved. This is in contrast to other roles of TrkC in the recycling of presynaptic vesicles and presynaptic differentiation, where there is no such requirement for kinase activity (Takahashi et al., 2011; Ammendrup-Johnsen et al., 2015). Collectively, our findings indicate that a combination of multiple, and seemingly confounding, molecular mechanisms including presynaptic differentiation, synaptic adhesion, synapse maturation and plasticity, and activation of synaptic signaling pathways are involved in TrkC-mediated synapse development.
Fifth, we found that TrkC controls synapse density in cultured hippocampal neurons through an intercellular competitive mechanism (Fig. 10). The intercellular competitive action of TrkC was recently suggested to regulate dendrite morphogenesis in cerebellar Purkinje neurons (Joo et al., 2014). However, our results point to slightly different actions of TrkC between hippocampus and cerebellum (Fig. 10; Joo et al., 2014). In both hippocampus and cerebellum, NT-3 from presynaptic neurons is required for TrkC-dependent presynaptic differentiation, synapse density, and dendrite morphogenesis, suggesting that the relative level of NT-3 is a limiting factor in the control of synapse development. However, our results suggest that the level of NT-3 secreted by presynaptic neurons and TrkC levels in postsynaptic neurons are both critical for NT-3/TrkC-signaling-dependent synapse development (Fig. 10), although more studies are required to clarify this issue. Similar intercellular competitive mechanisms have been reported for other synaptic cell adhesion molecules, such as ephrin-B3, neuroligin-1, and TrkB (Marshak et al., 2007; McClelland et al., 2010; Kwon et al., 2012). In the case of ephrin-B3 and neuroligin-1, binding to presynaptic EphB2 and neurexins, respectively, is required for the intercellular competition (McClelland et al., 2010; Kwon et al., 2012). Therefore, whether presynaptic PTPσ is also required for the TrkC-dependent competitive mechanism regulating synapse density warrants further investigation. Overall, our results generally support a competition-based synaptogenesis model mediated by synaptic adhesion molecules.
In sum, our study demonstrated that NT-3 exerts its modulatory effects on various aspects of synapse development through its canonical receptor TrkC, together with the TrkC-binding protein PTPσ and TrkC intracellular signaling pathways. Our study also raises a number of questions that need to be addressed. How do postsynaptic neurons express different amounts of TrkC? How do relative levels of TrkC in neighboring neurons regulate synapse density? Does NT-3 function either cell autonomously or non-cell autonomously to regulate synapse development in other brain regions? Does NT-3/TrkC signaling cooperate with other synaptic signaling pathways to regulate synapse development? Addressing these questions will be crucial for expanding our current understanding of how secreted factors participate in modulating synaptic adhesion pathways that are considered to be primarily regulated by classical transmembrane proteins.
Footnotes
This work was supported by the National Research Foundation of Korea (NRF) funded by the Ministry of Science and Future Planning (Grant 2014051826 to J.K.), the Yonsei University Future-Leading Research Initiative of 2014 (J.K.), the Yonsei University Future-Leading Research Initiative of 2015 (J.W.U.), NRF funded by the Ministry of Education, Science and Technology (Grant NRF-2013R1A6A3A04061338 to J.W.U.), Yonsei University College of Medicine (Faculty Research Grant 6-2015-0085 to J.W.U.). S.K., G.C., and S.J. are fellowship awardees of the BK21 PLUS program.
The authors declare no competing financial interests.
References
- Ammendrup-Johnsen I, Naito Y, Craig AM, Takahashi H. Neurotrophin-3 enhances the synaptic organizing function of TrkC-protein tyrosine phosphatase sigma in rat hippocampal neurons. J Neurosci. 2015;35:12425–12431. doi: 10.1523/JNEUROSCI.1330-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arvanian VL, Horner PJ, Gage FH, Mendell LM. Chronic neurotrophin-3 strengthens synaptic connections to motoneurons in the neonatal rat. J Neurosci. 2003;23:8706–8712. doi: 10.1523/JNEUROSCI.23-25-08706.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biederer T, Scheiffele P. Mixed-culture assays for analyzing neuronal synapse formation. Nat Protoc. 2007;2:670–676. doi: 10.1038/nprot.2007.92. [DOI] [PubMed] [Google Scholar]
- Boucard AA, Ko J, Südhof TC. High affinity neurexin binding to cell adhesion G-protein-coupled receptor CIRL1/latrophilin-1 produces an intercellular adhesion complex. J Biol Chem. 2012;287:9399–9413. doi: 10.1074/jbc.M111.318659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang KY, Woo D, Jung H, Lee S, Kim S, Won J, Kyung T, Park H, Kim N, Yang HW, Park JY, Hwang EM, Kim D, Heo WD. Light-inducible receptor tyrosine kinases that regulate neurotrophin signalling. Nat Commun. 2014;5:4057. doi: 10.1038/ncomms5057. [DOI] [PubMed] [Google Scholar]
- Coles CH, Mitakidis N, Zhang P, Elegheert J, Lu W, Stoker AW, Nakagawa T, Craig AM, Jones EY, Aricescu AR. Structural basis for extracellular cis and trans RPTPsigma signal competition in synaptogenesis. Nat Commun. 2014;5:5209. doi: 10.1038/ncomms6209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genevois AL, Ichim G, Coissieux MM, Lambert MP, Lavial F, Goldschneider D, Jarrosson-Wuilleme L, Lepinasse F, Gouysse G, Herceg Z, Scoazec JY, Tauszig-Delamasure S, Mehlen P. Dependence receptor TrkC is a putative colon cancer tumor suppressor. Proc Natl Acad Sci U S A. 2013;110:3017–3022. doi: 10.1073/pnas.1212333110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gokce O, Südhof TC. Membrane-tethered monomeric neurexin LNS-domain triggers synapse formation. J Neurosci. 2013;33:14617–14628. doi: 10.1523/JNEUROSCI.1232-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang EJ, Reichardt LF. Trk receptors: roles in neuronal signal transduction. Annu Rev Biochem. 2003;72:609–642. doi: 10.1146/annurev.biochem.72.121801.161629. [DOI] [PubMed] [Google Scholar]
- Johnson-Venkatesh EM, Umemori H. Secreted factors as synaptic organizers. Eur J Neurosci. 2010;32:181–190. doi: 10.1111/j.1460-9568.2010.07338.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joo W, Hippenmeyer S, Luo L. Neurodevelopment. Dendrite morphogenesis depends on relative levels of NT-3/TrkC signaling. Science. 2014;346:626–629. doi: 10.1126/science.1258996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko J. The leucine-rich repeat superfamily of synaptic adhesion molecules: LRRTMs and Slitrks. Mol Cells. 2012;34:335–340. doi: 10.1007/s10059-012-0113-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko JS, Pramanik G, Um JW, Shim JS, Lee D, Kim KH, Chung GY, Condomitti G, Kim HM, Kim H, de Wit J, Park KS, Tabuchi K, Ko J. PTPsigma functions as a presynaptic receptor for the glypican-4/LRRTM4 complex and is essential for excitatory synaptic transmission. Proc Natl Acad Sci U S A. 2015b;112:1874–1879. doi: 10.1073/pnas.1410138112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko J, Kim S, Chung HS, Kim K, Han K, Kim H, Jun H, Kaang BK, Kim E. SALM synaptic cell adhesion-like molecules regulate the differentiation of excitatory synapses. Neuron. 2006;50:233–245. doi: 10.1016/j.neuron.2006.04.005. [DOI] [PubMed] [Google Scholar]
- Ko J, Fuccillo MV, Malenka RC, Südhof TC. LRRTM2 functions as a neurexin ligand in promoting excitatory synapse formation. Neuron. 2009a;64:791–798. doi: 10.1016/j.neuron.2009.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko J, Zhang C, Arac D, Boucard AA, Brunger AT, Südhof TC. Neuroligin-1 performs neurexin-dependent and neurexin-independent functions in synapse validation. EMBO J. 2009b;28:3244–3255. doi: 10.1038/emboj.2009.249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko J, Soler-Llavina GJ, Fuccillo MV, Malenka RC, Südhof TC. Neuroligins/LRRTMs prevent activity- and Ca2+/calmodulin-dependent synapse elimination in cultured neurons. J Cell Biol. 2011;194:323–334. doi: 10.1083/jcb.201101072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko J, Choii G, Um JW. The balancing act of GABAergic synapse organizers. Trends Mol Med. 2015a;21:256–268. doi: 10.1016/j.molmed.2015.01.004. [DOI] [PubMed] [Google Scholar]
- Kolarow R, Brigadski T, Lessmann V. Postsynaptic secretion of BDNF and NT-3 from hippocampal neurons depends on calcium calmodulin kinase II signaling and proceeds via delayed fusion pore opening. J Neurosci. 2007;27:10350–10364. doi: 10.1523/JNEUROSCI.0692-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon HB, Kozorovitskiy Y, Oh WJ, Peixoto RT, Akhtar N, Saulnier JL, Gu C, Sabatini BL. Neuroligin-1-dependent competition regulates cortical synaptogenesis and synapse number. Nat Neurosci. 2012;15:1667–1674. doi: 10.1038/nn.3256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshak S, Nikolakopoulou AM, Dirks R, Martens GJ, Cohen-Cory S. Cell-autonomous TrkB signaling in presynaptic retinal ganglion cells mediates axon arbor growth and synapse maturation during the establishment of retinotectal synaptic connectivity. J Neurosci. 2007;27:2444–2456. doi: 10.1523/JNEUROSCI.4434-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClelland AC, Hruska M, Coenen AJ, Henkemeyer M, Dalva MB. Trans-synaptic EphB2-ephrin-B3 interaction regulates excitatory synapse density by inhibition of postsynaptic MAPK signaling. Proc Natl Acad Sci U S A. 2010;107:8830–8835. doi: 10.1073/pnas.0910644107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park H, Poo MM. Neurotrophin regulation of neural circuit development and function. Nat Rev Neurosci. 2013;14:7–23. doi: 10.1038/nrc3653. [DOI] [PubMed] [Google Scholar]
- Song M, Giza J, Proenca CC, Jing D, Elliott M, Dincheva I, Shmelkov SV, Kim J, Schreiner R, Huang SH, Castrén E, Prekeris R, Hempstead BL, Chao MV, Dictenberg JB, Rafii S, Chen ZY, Rodriguez-Boulan E, Lee FS. Slitrk5 Mediates BDNF-Dependent TrkB Receptor Trafficking and Signaling. Dev Cell. 2015;33:690–702. doi: 10.1016/j.devcel.2015.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Südhof TC. Neuroligins and neurexins link synaptic function to cognitive disease. Nature. 2008;455:903–911. doi: 10.1038/nature07456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi H, Arstikaitis P, Prasad T, Bartlett TE, Wang YT, Murphy TH, Craig AM. Postsynaptic TrkC and presynaptic PTPsigma function as a bidirectional excitatory synaptic organizing complex. Neuron. 2011;69:287–303. doi: 10.1016/j.neuron.2010.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi H, Katayama K, Sohya K, Miyamoto H, Prasad T, Matsumoto Y, Ota M, Yasuda H, Tsumoto T, Aruga J, Craig AM. Selective control of inhibitory synapse development by Slitrk3-PTPdelta trans-synaptic interaction. Nat Neurosci. 2012;15:389–398. S1–S2. doi: 10.1038/nn.3040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tauszig-Delamasure S, Yu LY, Cabrera JR, Bouzas-Rodriguez J, Mermet-Bouvier C, Guix C, Bordeaux MC, Arumäe U, Mehlen P. The TrkC receptor induces apoptosis when the dependence receptor notion meets the neurotrophin paradigm. Proc Natl Acad Sci U S A. 2007;104:13361–13366. doi: 10.1073/pnas.0701243104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Um JW, Ko J. LAR-RPTPs: synaptic adhesion molecules that shape synapse development. Trends Cell Biol. 2013;23:465–475. doi: 10.1016/j.tcb.2013.07.004. [DOI] [PubMed] [Google Scholar]
- Um JW, Kim KH, Park BS, Choi Y, Kim D, Kim CY, Kim SJ, Kim M, Ko JS, Lee SG, Choii G, Nam J, Heo WD, Kim E, Lee JO, Ko J, Kim HM. Structural basis for LAR-RPTP/Slitrk complex-mediated synaptic adhesion. Nat Commun. 2014;5:5423. doi: 10.1038/ncomms6423. [DOI] [PubMed] [Google Scholar]
- Yang HW, Shin MG, Lee S, Kim JR, Park WS, Cho KH, Meyer T, Heo WD. Cooperative activation of PI3K by Ras and Rho family small GTPases. Mol Cell. 2012a;47:281–290. doi: 10.1016/j.molcel.2012.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Q, Feng B, Zhang K, Guo YY, Liu SB, Wu YM, Li XQ, Zhao MG. Excessive astrocyte-derived neurotrophin-3 contributes to the abnormal neuronal dendritic development in a mouse model of fragile X syndrome. PLoS Genet. 2012b;8:e1003172. doi: 10.1371/journal.pgen.1003172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang T, Massa SM, Longo FM. LAR protein tyrosine phosphatase receptor associates with TrkB and modulates neurotrophic signaling pathways. J Neurobiol. 2006;66:1420–1436. doi: 10.1002/neu.20291. [DOI] [PubMed] [Google Scholar]
- Yim YS, Kwon Y, Nam J, Yoon HI, Lee K, Kim DG, Kim E, Kim CH, Ko J. Slitrks control excitatory and inhibitory synapse formation with LAR receptor protein tyrosine phosphatases. Proc Natl Acad Sci U S A. 2013;110:4057–4062. doi: 10.1073/pnas.1209881110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Araki S, Wu J, Teramoto T, Chang YF, Nakano M, Abdelfattah AS, Fujiwara M, Ishihara T, Nagai T, Campbell RE. An expanded palette of genetically encoded Ca(2; +) indicators. Science. 2011;333:1888–1891. doi: 10.1126/science.1208592. [DOI] [PMC free article] [PubMed] [Google Scholar]