

Abstract
Although injured axons usually do not regenerate in the adult CNS, parallel fibers (PFs) regenerate synaptic connections onto cerebellar Purkinje cells (PCs). In this study, we investigated the role of GluD2 in this regenerative process after PF transection using GluD2-knock-out (KO) mice. All dendritic spines on distal dendrites were innervated by PFs in sham-operated wild-type controls, whereas one-third were devoid of innervation in GluD2-KO mice. In both genotypes, a steep drop in the number of PF synapses occurred with a reciprocal surge in the number of free spines on postlesion day 1, when the PF territory aberrantly expanded toward the proximal dendrites. In wild-type mice, the territory and number of PF synapses were nearly fully restored to normal on postlesion day 7, although PF density remained low. Moreover, presynaptic and postsynaptic elements were markedly enlarged, and the PF terminal-to-PC spine contact ratio increased from 1:1 to 1:2 at most synapses. On postlesion day 30, the size and contact ratio of PF synapses returned to sham-operated control values and PF density recovered through the sprouting and elongation of PF collaterals. However, GluD2-KO mice showed neither a hypertrophic response nor territorial restoration 7 d postlesion, nor the recovery of PF axons or synapses on postlesion day 30. This suggests that PF wiring regenerates initially by inducing hypertrophic responses in surviving synaptic elements (hypertrophic phase), followed by collateral formation by PF axons and retraction of PF synapses (remodeling phase). Without GluD2, no transition to these regenerative phases occurs.
SIGNIFICANCE STATEMENT The glutamate receptor GluD2 expressed at parallel fiber (PF)–Purkinje cell (PC) synapses regulates the formation and maintenance of the synapses. To investigate the role of GluD2 in their extraordinarily high regenerative capacity, the process after surgical transection of PFs was compared between wild-type and GluD2-knock-out mice. We discovered that, in wild-type mice, PF synapses regenerate initially by inducing hypertrophic responses in surviving synaptic elements, and then by sprouting and elongation of PF collaterals. Subsequently, hypertrophied PF synapses remodel into compact synapses. In GluD2-knock-out mice, PF wiring remains in the degenerative phase, showing neither a hypertrophic response nor recovery of PF axons or synapses. Our finding thus highlights that synaptic connection in the adult brain can regenerate with aid of GluD2.
Keywords: cerebellum, climbing fiber, GluD2, parallel fiber, Purkinje cell, synapse regeneration
Introduction
Dendrites of Purkinje cells (PCs) are arborized in the parasagittal plane of the cerebellum, and consist of two compartments, distal and proximal, that differ in the origin of their inputs as well as in the mode of innervation. Spiny branchlets in the distal compartment are innervated by 105–106 parallel fibers (PFs), T-shaped granule cell axons running in the transverse plane, but each PF usually forms only one synapse onto a given PC (Palay and Chan-Palay, 1974; Napper and Harvey, 1988; Kurihara et al., 1997). In comparison, a single climbing fiber (CF) originating from the inferior olivary nucleus forms hundreds of synapses onto a single PC by twisting around the dendritic shafts in the proximal compartment (Crepel et al., 1981; Rossi et al., 1993; Ichikawa et al., 2002). The segregated innervation territories of the two orthogonal afferents and the mono-innervation by single CFs are the two distinguishing features of excitatory synaptic wiring on PCs, and are established in the first 3–4 postnatal weeks in rodents (Altman, 1972; Watanabe and Kano, 2011). These salient features are thought to be the neural basis for information processing and integration underlying cerebellar motor function and learning (Ito, 2012).
Various molecular mechanisms regulate the formation and maturation of PC circuits (Yuzaki, 2009; Watanabe and Kano, 2011; Hashimoto and Kano, 2013). Of these, the glutamate receptor GluD2 (GluRδ2), which is exclusively concentrated on the postsynaptic membrane at PF–PC synapses (Takayama et al., 1995; Landsend et al., 1997), plays a key role in synapse formation. Ablation of the Grid2 gene (which encodes GluD2) causes a severe reduction of PF–PC synapses and results in numerous spines lacking innervation, ie, free spines (Guastavino et al., 1990; Kashiwabuchi et al., 1995; Kurihara et al., 1997; Lalouette et al., 2001; Takeuchi et al., 2005). This input-selective synaptic strengthening is mediated by a tripartite molecular complex comprised of GluD2, Cbln1, and neurexin, in which Cbln1 released from PFs interacts with GluD2 on PC spines and neurexin on PF terminals (Matsuda et al., 2010; Uemura et al., 2010). This molecular interaction is also important for suppressing aberrant extension of the CF territory; without it, CFs invade distal dendrites to take over free spines on innervating and neighboring PCs (Ichikawa et al., 2002; Hirai et al., 2005). Furthermore, this GluD2–Cbln1–neurexin system is essential for maintaining the PC circuitry in adulthood (Takeuchi et al., 2005; Ito-Ishida et al., 2008; Torashima et al., 2009; Miyazaki et al., 2010).
Axons injured in the adult mammalian CNS of higher species do not usually regenerate (Dusart et al., 2005; Maier and Schwab, 2006). But intriguingly, PF synapses on PCs rapidly regenerate after surgical transection of PFs. Chen and Hillman (1982) were the first to show that, in the adult rat cerebellum, degenerating PF terminals and free spines almost completely disappear within 5 d after surgical transection, and that PF synapses are restored by takeover of preexisting PC spines and new synapse formation. However, the molecular mechanisms underlying this high regenerative ability remain unknown. In the present study, we investigated whether GluD2 plays a role in the regeneration of PF synapses using electron microscopic reconstruction of dendritic innervation and of the territories of PF-transected PCs. We show in wild-type mice that the regeneration of PF–PC synapses proceeds in three distinct phases: the degenerative, hypertrophic, and remodeling phases. In GluD2-knock-out (KO) mice, lesioned PF wiring remains in the degenerative phase, without shifting to the hypertrophic and remodeling phases of regeneration.
Materials and Methods
Animals.
Wild-type (C57BL/6N) and grid2 (GluD2)-KO mice of either sex at 2 months of age were used in the present study. GluD2-KO mice were obtained by mating heterozygous pairs, which had been produced and maintained on a pure C57BL/6N genetic background (Yamasaki et al., 2011). The genotype was determined by PCR, as reported previously (Takeuchi et al., 2005). All experiments were performed according to guidelines laid down and approved by the animal care and welfare committees of Sapporo Medical University and Hokkaido University. Three wild-type and three GluD2-KO mice were analyzed for each qualitative and quantitative analysis at each time point.
PF transection.
Under anesthesia with chloral hydrate (350 mg/kg body weight, i.p.), a transfolial incision was made in the cerebellar vermis (lobules 4–7) to produce PF-transected “cerebellar islands.” A pair of incisions, with a 2.0 mm intervening space, were made by inserting a microknife to a depth of 0.6 mm from the pial surface, 30° to the parasagittal plane (Fig. 1A). Then, 1, 7, and 30 d later, mice under chloral hydrate anesthesia were perfused transcardially with 4% paraformaldehyde in 0.1 m phosphate buffer (PB), pH 7.4, for light microscopic immunohistochemical analyses, or with 4% paraformaldehyde and 0.1% glutaraldehyde in 0.1 m PB for electron microscopic reconstruction. After excision from the skull, brains were immersed overnight in the same fixative. All analyses were done for cerebellar islands on lobule 6 in PF-transected mice or the corresponding region in sham-operated mice.
Figure 1.

Surgical transection of PFs. A, Illustration of the dorsal cerebellar view and slanted double incisions. B, C, Immunofluorescence for calbindin (green) and VGluT1 (red) in a coronal cerebellar section 7 d after PF transection. Both fluorescent signals are disrupted at the double incisions (arrows). Cerebellar islands between double incisions were used for analysis. C, An enlarged image of the boxed region in B. D, E, Injection of BDA into cerebellar islands or the corresponding cerebellar portions (yellow dots) in sham-operated control (D) and PF-transected (E) mice. Note that tracts of BDA-labeled PFs do not extend beyond the incision in PF-transected mice (E). Scale bars: B, D, E, 500 μm; C, 100 μm.
Tracer labeling of PFs.
To follow the regeneration of PF synapses, biotinylated dextran amine (BDA; molecular weight, 10,000; Invitrogen) was injected into the vermis (lobule 6) of PF-transected mice 2 d before transcardial perfusion. Briefly, under chloral hydrate anesthesia, a 1-MΩ-resistance glass pipette, filled with 0.5 μl of 10% BDA in PBS, pH 7.4, was inserted into the left side of the cortical islands, and BDA was iontophoretically applied with a 7 μA positive current for 20 min (current parameters: 700 ms on and 1300 ms off; Ichikawa et al., 2011). To visualize BDA-labeled PFs, coronal microslicer sections were immersed overnight in streptavidin–peroxidase conjugate (Nichirei) diluted with PB containing 0.5% Triton X-100 for light microscopy and visualized with 3,3′-diaminobenzidine (DAB). Using a light microscope (AX-80, Olympus), the density of PF varicosities was measured from >20 labeled PFs from each mouse and averaged. The p value was calculated using ANOVA with Dunnett's test (see Fig. 3G).
Figure 3.

Sprouting and elongation of PC collaterals. A–F, Phase-contrast light microscopic images of BDA-labeled PF trajectory in sham-operated control (A, D) and PF-transected mice on postlesion days 7 (B, E) and 30 (C, F) in wild-type (A–C) and GluD2-KO (D–F) genotypes. Arrowheads indicate varicosities or boutons along BDA-labeled PFs, while arrows indicate the branching point of PF collaterals. C, Inset, A different focal plane of the boxed region in the center, showing numerous varicosities along a long collateral of a BDA-labeled PF. G, Bar graphs showing the density of PF varicosities per mm of BDA-labeled PF (mean ± SD, n = 3 mice for each). No significant difference in density after PF lesioning was found in either wild-type (left) or GluD2-KO (right) mice. The density of PF varicosities, at ∼160/mm, is consistent with a previous study in the cat (Brand et al., 1976). Scale bars, 10 μm.
Immunohistochemistry.
For immunofluorescence, sections (50 μm thick) were made on a microslicer (MAN52, Campden) and were prepared in the parasagittal and coronal planes. Sections were incubated overnight at room temperature in a mixture of rabbit calbindin antiserum (1:10,000; Nakagawa et al., 1998) and guinea pig vesicular glutamate transporter type 2 (VGluT2) antibody (1.0 μg/ml; Miyazaki et al., 2003) for parasagittal sections (see Fig. 4A–H) or rabbit calbindin antiserum and guinea pig vesicular glutamate transporter type 1 (VGluT1) antibody (1.0 μg/ml; Miyazaki et al., 2003) for coronal sections (Fig. 1B,C). Subsequently, sections were incubated in a mixture of AlexaFluor 594-labeled anti-guinea pig IgG and AlexaFluor 488-labeled anti-rabbit IgG for 2 h (1:200; Invitrogen). Coronal images were obtained on a fluorescence microscope (BZ-9000, Keyence), and parasagittal images were captured using a confocal scanning laser microscope (Radiance 2100, Zeiss) equipped with a Ar/Kr laser and a Plan-Apochromat (40×/1.0 or 60×/1.4, oil-immersion) objective lens (Zeiss). To avoid cross talk between fluorophores, fluorescent signals were acquired sequentially using the 488 and 568 nm excitation laser lines. Five images (512 × 512 pixels; pinhole size, 1.6 mm) at different levels along the z-axis were compiled into single images. The heights of CFs and the molecular layer were measured using 10 images from each mouse, and averaged. Statistical differences between sham-operated control and PF-transected mice were assessed using ANOVA with Dunnett's test, whereas those between wild-type and GluD2-KO mice were assessed using Student's t test (see Fig. 4I–K).
Figure 4.
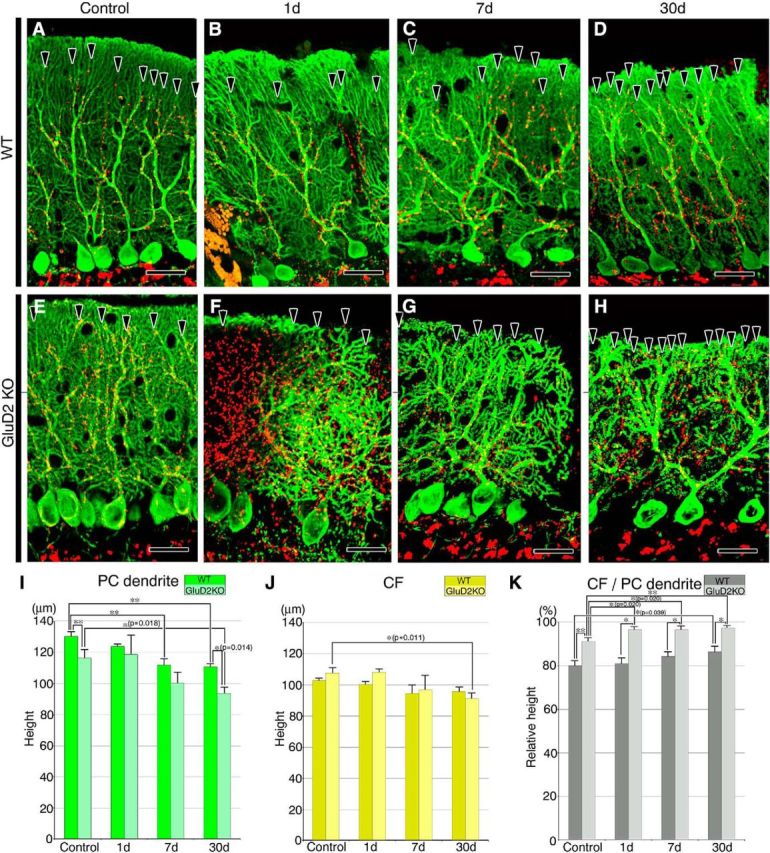
Changes in molecular layer thickness and CF reach after PF transection. A–H, Double-immunofluorescence for calbindin (green) and VGluT2 (red) in sham-operated control (A, E), and PF-transected mice on postlesion days 1 (B, F), 7 (C, G), and 30 (D, H) in wild-type (A–D) and GluD2-KO (E–H) genotypes. Arrowheads indicate the distal tips of VGluT2-labeled CF terminals. I–K, Bar graphs showing the vertical height to the tips of calbindin-positive PC dendrites (ie, molecular layer thickness; I) or to the tips of VGluT2-labeled CF terminals (ie, CF reach; J), and the CF reach relative to molecular layer thickness (K; mean ± SD, n = 3 mice for each). Differences between sham-operated control and PF-transected mice in each genotype were assessed using ANOVA with Dunnett's test, while those between wild-type and GluD2-KO mice at each time point were assessed using Student's t test. **p < 0.01, *p < 0.05 (actual p values are indicated in the parenthesis). Scale bars, 25 μm.
For immunoelectron microscopic labeling of CFs, parasagittal microslicer sections were incubated overnight with guinea pig VGluT2 antibody (1.0 μg/ml) and peroxidase-labeled guinea pig IgG antibody (1:200, Jackson Immunoresearch), and visualized with DAB. To label GABAergic terminals, sections were incubated overnight with rabbit VIAAT antibody (1.0 μm/ml), and subjected to silver-enhanced pre-embedding immunogold microscopy using 1.4 nm gold particle-conjugated rabbit IgG antibody (1:100; Nanoprobes) and the HQ silver kit (Nanoprobes).
Serial electron microscopy and 3D reconstruction.
To measure the density, size, and contact ratio of PFs, 10 sets of five serial electron microscopic images were taken from superficial portions of the molecular layer (0∼40 μm depth from the pia mater) for each of three PF-transected or sham-operated mice. We measured the short diameter of PF terminals and the density of PF axons. The p value was calculated using ANOVA with Dunnett's test (Fig. 2I–L).
Figure 2.

Changes in PFs and PF synapses after surgical transection. A–H, Electron micrographs of the superficial molecular layer in sham-operated control (A, E) and PF-transected mice on postlesion days 1 (B, F), 7 (C, G), and 30 (D, H) in wild-type (A–D) and GluD2-KO (E–H) genotypes. PF axons and terminals are pseudocolored in red and orange, respectively, whereas PC spines in contact with PF terminals or lacking synaptic contact (ie, free spines) are in blue or green, respectively. Pairs of arrowheads indicate the PSD. I–L, Bar graphs showing the density of PF axons per square micrometer of the molecular layer (I), the percentage of PC spines in contact with PF terminals (J), the size (short diameter) of PF terminals (K), and the contact ratio of PC spines per PF terminal (L; mean ± SD, n = 3 mice for each). Left and right bars represent scores in wild-type and GluD2-KO mice, respectively. Differences between sham-operated control and PF-transected mice in each genotype were assessed using ANOVA with Dunnett's test. **p < 0.01, *p < 0.05 (actual p value is indicated in the parenthesis). Scale bars, 500 nm.
To reconstruct 3D images of PC dendrites, we prepared sets of 1000–1400 ultrathin sections (100 nm thick) in the horizontal plane. A ribbon of serial sections, each consisting of at least 20–30 sections, was mounted on a single-slot copper grid (1 × 2 mm) supported with a formvar membrane. Electron micrographs were taken from every second section at an original magnification of 15,000×. Using electron microscopic images, the path length (L) of curved or leaning dendrites was calculated using the long (DL) and short (DS) diameters of dendritic profiles and using the thickness (t) and number (n) of sections: L = DL/DS × t × n. From the reconstructed images, we obtained the mean number and density of synapses and free spines, as well as the composition of three dendritic segments innervated by CFs only, CFs plus PFs, and PFs only. The p value was calculated using ANOVA with Dunnett's test (see Fig. 8).
Figure 8.

Changes after PF transection in the path length of innervation territories and in the number of PF synapses, CF synapses, free spines, and spine-type GABAergic synapses. A, The path length (μm) of whole single dendritic tracts and of PCD-I (blue), PCD-II (cyan), and PCD-III (green). B, The path length of the PF territory (ie, PCD-II + PCD-III). C, PF synapse number in the PF territory. D, The path length of the CF territory (ie, PCD-I + PCD-II). E, CF synapse number in the CF territory. F, The path length of dendrites studded with free spines. G, Free spine number. H, Spine-type GABAergic synapse number. Left and right bars represent scores in wild-type and GluD2-KO mice, respectively. All scores were measured from three mice at each time point for each analysis, and expressed as the mean ± SD. Differences between sham-operated control and PF-transected mice in each genotype were assessed using ANOVA with Dunnett's test. **p < 0.01, *p < 0.05 (actual p values are indicated in the parenthesis).
Results
Surgical transection of PFs
A pair of transfolial incisions were made in the cerebellar vermis of adult wild-type (C57BL/6N) and GluD2-KO mice (Fig. 1A) to investigate the role of the receptor in the degeneration and regeneration of PF–PC synapses in PF-deafferented cerebellar islands. The corresponding cerebellar regions in sham-operated mice were used as experimental controls for each genotype. Transection of PFs was ascertained by the discontinuity at incision sites by immunofluorescence labeling for VGluT1, a marker of PF terminals (Fig. 1B,C), and by anterograde tracer labeling of BDA injected into the cerebellar islands (Fig. 1D,E). Long transverse beams of BDA-labeled PFs extended bilaterally from the injection sites in sham-operated controls (Fig. 1D), whereas in transected mice, the PF beams did not extend beyond the incision sites, even by postlesion day 30 (Fig. 1E). These findings demonstrate the successful transection of PFs and the lack of regeneration beyond the lesion site, at least until postlesion day 30.
Degeneration and regeneration in cerebellar islands
Changes in PF axons, terminals, and synapses were examined in cerebellar islands on postlesion days 1, 7, and 30 by electron microscopy (Fig. 2). In this analysis, we prepared 10 sets of parasagittal serial sections from each mouse, and photos were taken from superficial portions of the molecular layer. Using serial images, we identified PF axons (red) and terminals (orange) running orthogonal to the section plane, and distinguished PC spines forming PF synapses (blue) from those lacking synaptic contact, ie, free spines (green).
In sham-operated controls, most PF terminals in wild-type mice formed synaptic contacts with single PC spines (ie, 1:1 contact), and the postsynaptic density (PSD) was large and well thickened beneath the postsynaptic membrane (Fig. 2A, arrowheads). In contrast, approximately one-third of PC spines were free spines in sham-operated GluD2-KO mouse controls (Fig. 2E,J), a hallmark of synaptic defects in this mutant, and were associated with rudimentary PSD-like condensation, as reported previously (Guastavino et al., 1990; Kurihara et al., 1997; Ichikawa et al., 2002; Takeuchi et al., 2005). On postlesion day 1, transfolial transection caused a marked reduction of PF axons and terminals, with a concomitant emergence or increase of free spines in wild-type and GluD2-KO mice, respectively (Fig. 2B,F). This was reflected in a significant drop in the profile density of PF axons (per μm2 of area) in the molecular layer (Fig. 2I) and in the percentage of PC spines having synaptic contact with PF terminals (Fig. 2J), compared with sham-operated controls (n = 3; p < 0.05, ANOVA with Dunnett's test). Thereafter, genotypic differences became more apparent. In wild-type mice, free spines virtually disappeared (Fig. 2C,J), and PF terminals and PC spines were markedly enlarged on postlesion day 7 (Fig. 2C,K). Furthermore, the PF terminal-to-PC spine contact ratio was increased to 1:2 at most PF–PC synapses (Fig. 2C,L). On postlesion day 30, the profile density of PF axons was increased to 75.2 ± 13.0% of the control level (Fig. 2I), and the size of PF terminals and the PF terminal-to-PC spine contact ratio returned to control levels (Fig. 2D,K,L). In contrast, in GluD2-KO mice, no such hypertrophic response was observed on postlesion day 7 (Fig. 2G,K,L). In addition, >90% of PC spines were free spines (Fig. 2F–H,J), and the profile density of PF axons remained low, at 55.0 ± 3.2% of control level, on postlesion day 30 (Fig. 2I). Thus, regeneration of PF axons and synapses was severely impaired in GluD2-KO mice compared with wild-type animals.
Sprouting, elongation, and varicosity formation in PF collaterals
Light microscopic examination of BDA-labeled PFs on postlesion day 7 confirmed transient hypertrophic responses, ie, thickening of PF axons and enlargement of PF terminals, in wild-type mice (Fig. 3A–C), but not in GluD2-KO mice (Fig. 3D–F). We also found on postlesion day 30 that PFs in wild-type mice often projected long collateral branches bearing a number of varicosities (Fig. 3C), whereas those in GluD2-KO mice gave off a few short branches bearing only one or a few varicosities (Fig. 3F). No significant differences were noted in the density of PF varicosities (per millimeter of BDA-labeled PFs) between sham-operated controls and PF-transected mice on postlesion day 7 or 30 in any genotype (Fig. 3G). Given the lack of PF regeneration beyond the transection site, these findings suggest that the increase in PF density on postlesion day 30 in wild-type mice is achieved mainly by sprouting and elongation of PF collaterals within the cerebellar islands.
Changes in PC dendrites and CF reach
Next, we examined changes in molecular layer thickness and CF reach after PF transection by double-immunofluorescence labeling for calbindin (green) and VGluT2 (red), markers for PCs and CF terminals, respectively. Confocal microscopic images were taken from parasagittal sections prepared from cerebellar islands and the corresponding regions of sham-operated controls (Fig. 4A–H). In sham-operated controls, the thickness of the molecular layer, ie, the mean vertical height to the tips of calbindin-positive PC dendrites, was significantly lower in GluD2-KO mice compared with wild-type mice (Fig. 4I; n = 3; p < 0.01, Student's t test), consistent with our previous report (Kurihara et al., 1997). Nevertheless, the CF reach, ie, the mean vertical height to the tips of VGluT2-labeled CF terminals, was similar in GluD2-KO and wild-type mice (Fig. 4J), suggesting invasion of the CFs to the distal dendritic compartment in GluD2-KO mice (Fig. 4E). As a result, the CF reach relative to molecular layer thickness was significantly elevated in sham-operated GluD2-KO controls compared with wild-type mice (Fig. 4K; p < 0.01, Student's t test).
After PF transection, the molecular layer thickness was reduced significantly and progressively in both genotypes (Fig. 4I), and the reduction in CF reach was mild in both genotypes (Fig. 4J). Accordingly, the CF reach relative to the molecular layer thickness remained significantly high in GluD2-KO mice (92–97%) compared with wild-type mice (80–86%) on each postlesion day examined (Fig. 4K; n = 3; p < 0.05 for each day, Student's t test). Of note, the distal dendrites were severely atrophied in GluD2-KO mice on postlesion days 7 and 30, as reflected by an increase in calbindin-negative space between calbindin-positive dendritic shafts, and atrophied dendrites were associated with more VGluT2-labeled CF terminals (Fig. 4G,H).
Reconstruction of dendritic innervation
To clarify changes in synaptic composition and location, we reconstructed PC dendrites by serial electron microscopy using horizontal sections cut parallel to the pial surface. From each genotype at each time point, we selected three PCs that followed a single dendritic tract from the apical pole of PC somata up to the dendritic tips ascending to the pia mater. We identified CF synapses by the presence of dark immunoperoxidase precipitates for VGluT2 (Fig. 5A) and PF synapses by innervation by VGluT2-negative transverse axons (Fig. 5B). All of the identified CF and PF terminals formed asymmetrical synapses on dendritic spines (Fig. 5A,B). After PF transection, unusual synaptic elements were frequently encountered, including degenerating PF synapses with high electron density (Fig. 5C, right), symmetrical synapses on dendritic spines, and free spines lacking presynaptic contact (Fig. 5C, left and middle). Symmetrical synapses on PC spines were atypical GABAergic synapses by molecular layer interneurons, as their presynaptic terminals were labeled for VIAAT (Fig. 5D).
Figure 5.
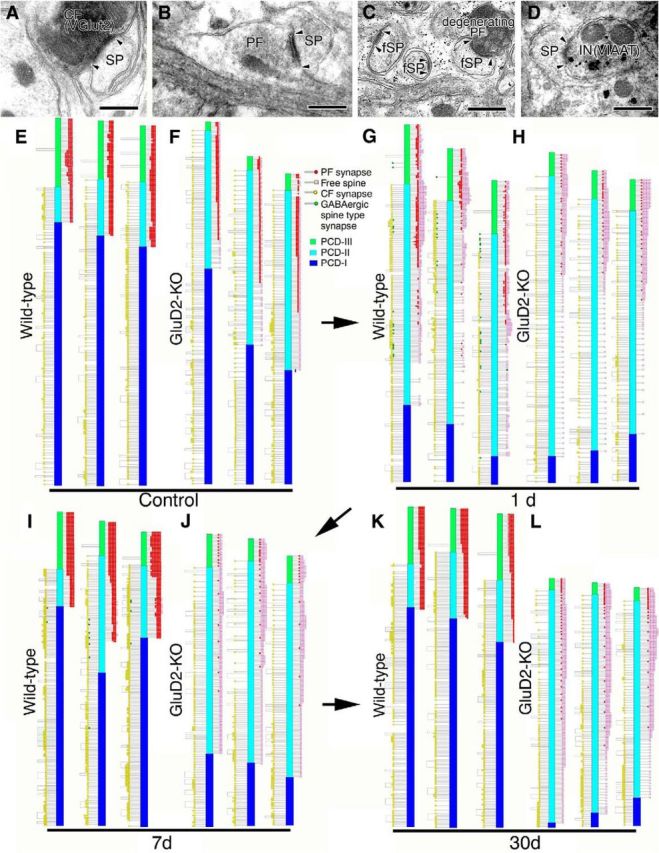
Ultrastructural reconstruction of dendritic innervation. A–D, Electron micrographs showing CF synapse labeled for VGluT2 by immunoperoxidase (A), PF synapse innervated by transverse axons on the transverse plane (B), free spines (fSP) and degenerating PF synapse 1 d after PF transection (C), and inhibitory synapse on PC spine labeled for VIAAT by silver-enhanced immunogold 1 d after PF transection (D). Compared with the well developed PSD in CF and PF synapses (pairs of arrowheads in A, B), the PSD in free spines is small and thin (pairs of arrowheads in C). E–L, Schematic illustrations showing the location of three kinds of spine-type synapses (PF synapse, red; CF synapse, yellow; inhibitory synapse, green) and free spines (pink) along reconstructed single dendritic tracts in sham-operated control (E, F) and PF-transected mice on postlesion days 1 (G, H), 7 (I, J), and 30 (K, L) in wild-type (E, G, I, K) and GluD2-KO (F, H, J, L) genotypes. We also illustrate the three distinct dendritic domains in different colors: PCD-I, blue; PCD-II, cyan; PCD-III, green. Scale bars, 500 nm.
Three kinds of spine-type synapses and free spines were reconstructed along dendritic tracts in wild-type and GluD2-KO PCs (Fig. 5E–L). PF synapses (red dots) and free spines (pink squares) are plotted to the right of each dendrite, whereas CF synapses (yellow circles) and spine-type GABAergic synapses (green) are to the left. We also categorized PC dendrites into three domains according to the location of PF and CF synapses (Fig. 5E–L): PC dendrite-I (PCD-I), innervated by CFs alone (blue columns); PCD-II, innervated by both CFs and PFs (cyan); and PCD-III, innervated by PFs alone (green). Because free spines result from failed synaptic contact with PFs in GluD2-KO mice (Ichikawa et al., 2002; Takeuchi et al., 2005) and also emerge after surgical transection of PFs (Chen and Hillman, 1982; the present study), we assumed that free spines represented PF synapses in categorization of dendritic domains. Short segments from the distal and proximal dendrites were 3D-reconstructed and are presented in Figures 6 and 7, respectively.
Figure 6.

Electron micrographs and 3D reconstructed images of PF synapses on distal dendrites in sham-operated control (A, B, I, J) and PF-transected mice on postlesion days 1 (C, D, K, L), 7 (E, F, M, N) and 30 (G, H, O, P) in wild-type (A–H) and GluD2-KO (I–P) genotypes. In electron micrographs, PC dendrites/spines and PF axons/terminals are pseudocolored in green and orange, respectively. Insets, Enlarged and nonpseudocolored images of PF–PC synapses in boxed regions of pseudocolored images. Pairs of arrowheads and asterisks indicate the PSD and free spines, respectively. Note the emergence of free spines at distal dendrites on postlesion day 1 in wild-type mice (C, D) and their very frequent occurrence on all postlesion days in GluD2-KO mice (K–P). Also, note the hypertrophic changes in PF–PC synapses on postlesion day 7 in wild-type mice (E, F). Scale bars, 500 nm.
Figure 7.

Electron micrographs and 3D reconstructed images of CF synapses on proximal dendrites in sham-operated control (A, B, I, J) and PF-transected mice on postlesion days 1 (C, D, K, L), 7 (E, F, M, N), and 30 (G, H, O, P) in wild-type (A–H) and GluD2-KO (I–P) genotypes. In electron micrographs, PC dendrites/spines are pseudocolored in green, whereas CFs are labeled by dark precipitates for VGluT2. Insets, Enlarged images of PF–PC synapses in boxed regions. Pairs of arrowheads and asterisks indicate the PSD and free spines, respectively. Note the emergence of free spines at proximal dendrites on postlesion day 1 in wild-type mice (C, D) and their frequent occurrence on all postlesion days in GluD2-KO mice (K–P). Scale bars, 500 nm.
Changes in PF and CF innervation onto PCs
Using the reconstructed data, we analyzed differences in PC innervation, including differences in territories, and measured the path length of each dendritic domain in GluD2-KO and wild-type mice. Consistent with our previous study (Ichikawa et al., 2002), the PCD-II domain was abnormally elongated with reciprocal shortening of the PCD-I and PCD-III domains in sham-operated GluD2-KO control mice compared with wild-type mice (Fig. 8A). Moreover, reflecting progressive thinning of the molecular layer (Fig. 4I), the total path length of traced PC dendrites was significantly shorter on postlesion days 7 and 30 compared with sham-operated GluD2-KO and wild-type controls (Fig. 8A; n = 3; p < 0.05 for each day, ANOVA with Dunnett's test).
In wild-type mice, the mean path length of the PF territory, ie, the summed length of the PCD-II and PCD-III domains, was significantly longer on postlesion day 1 (Fig. 8B; n = 3; p < 0.01, ANOVA with Dunnett's test), when the mean number of PF synapses in the PF territory showed significant reductions (Fig. 8C; n = 3; p < 0.01, ANOVA with Dunnett's test). These changes were transient and returned to control levels on postlesion days 7 and 30 (Fig. 8B,C). GluD2-KO mice showed similar changes on postlesion day 1, ie, elongation of the PF territory and a reduction in the number of PF synapses (Fig. 8B,C). However, the number of PF synapses showed no recovery on postlesion day 7 or 30 (Fig. 8C; p < 0.01 at each time point, ANOVA with Dunnett's test). Thus, the PF territory expands through the massive degeneration of PF synapses in both strains of mice, but whereas these changes are transient in wild-type mice, they persist in the GluD2-KO.
The mean path length of the CF territory, ie, the summed length of the PCD-I and PCD-II domains, showed a mild reduction after PF transection in both genotypes (Fig. 8D). The shortening of the CF territory showed a tendency to correlate with the total path length of PC dendrites (Fig. 8A) and molecular layer thickness (Fig. 4I). After PF transection, no significant changes were found in the number of CF synapses in the CF territory in either genotype, although the values were consistently higher in wild-type mice than in GluD2-KO mice on each postlesion day (Fig. 8E). Thus, the reorganization and restoration of PF innervation of PCs proceeds swiftly and dynamically in a GluD2-dependent manner, whereas changes in CF innervation and territory are subtle, if any, in either strain of mice.
Free spines and atypical GABAergic synapses
Reflecting the steep increase in the number of free spines per dendritic tract (Fig. 8G), the mean path length of dendrites studded with free spines increased strikingly on postlesion day 1 in both strains of mice (Fig. 8F; n = 3; p < 0.01 or 0.05 on postlesion day 1 in wild-type and GluD2-KO mice, respectively, ANOVA with Dunnett's test). Most free spines had disappeared by postlesion day 7 and were completely gone by postlesion day 30 in wild-type mice (Fig. 8G). In comparison, in GluD2-KO mice, free spines were numerous along substantial lengths of PC dendrites on postlesion days 7 and 30 (Fig. 8F,G; n = 3; p < 0.01 on each postlesion day, ANOVA with Dunnett's test).
Atypical GABAergic synapses on PC spines were not found in sham-operated wild-type or GluD2-KO control mice (Fig. 5E,F). On postlesion days 1 and 7, they appeared in wild-type mice but not in GluD2-KO mice (Fig. 8H). These findings indicate that the rapid disappearance of free spines and the transient appearance of atypical GABAergic synapses are GluD2-dependent processes.
Discussion
Animal models of perturbed cerebellar circuitry have provided important insight into the mechanisms underlying the developmental regulation and plasticity of synaptic wiring (Altman and Anderson, 1972; Strata and Rossi, 1998; Sotelo, 2004; Yuzaki, 2009; Watanabe and Kano, 2011). In the present study, we examined the regeneration of PF wiring by reconstructing dendritic innervation onto PCs after surgical transection of PFs. The findings revealed striking differences between wild-type and GluD2-KO mice following PF transection, highlighting the pivotal role of GluD2 in PF regenerative rewiring.
Comparison with previous studies
Chen and Hillman (1982) documented the rapid regeneration of PF–PC synapses. Collapsed PF ghosts, phagosomes in Bergmann glia and free spines emerge and peak within a day after PF transection. Dual innervation of single spines by degenerating and normal PFs or by two normal PFs also appear in transected cerebellar islands. Because 1:1 contact of presynaptic and postsynaptic structures is typical of normal PF–PC synapses (∼90% of synapses; Palay and Chan-Palay, 1974; Kurihara et al., 1997), the emergence of dual innervation is considered to represent rapid regeneration of the synapse. Thereafter, PC spines enlarge, elongate, and branch approximately postlesion day 4, and giant spines in contact with enlarged PF terminals prevail on postlesion days 5–30. Enlarged PF terminals often contact multiple spines. Our present observations in mice are consistent with Chen and Hillman's (1982) study in rats, except that we did not observe innervation of single spines by dual PF terminals. In the current study, we also quantitatively evaluated changes in innervation along dendritic trees.
The three phases of PF regenerative wiring
Here, we propose three distinct phases following PF lesioning in wild-type mice: the (1) degenerative, (2) hypertrophic, and (3) remodeling phases (Fig. 9).
Figure 9.

Summary schematic illustrating the three distinct phases of regenerative PF wiring in wild-type mice (top) and the arrested regeneration in GluD2-KO mice (bottom). Three dendritic domains are colored in blue for PCD-I, cyan for PCD-II, and green for PCD-III. In wild-type mice, PF axons and synapses massively degenerate after PF transection. In this degenerative phase, numerous free spines emerge and the PF territory expands aberrantly. Then, PF synapses enlarge, and there is an increase in the contact ratio with PC spines (to 1:2) in the hypertrophic phase. In this phase, the number and density of PF synapses nearly recover to normal levels, and free spines nearly completely disappear, but PF density remains low. In the remodeling phase, PF density increases, at least partly through sprouting and elongation of PF collaterals, and PF synapses reacquire a compact structure and a 1:1 contact ratio with PC spines. This suggests that competitive synaptic wiring by PFs and CFs onto PCs is regained in wild-type mice. In contrast, in the GluD2-KO mice, the degenerative phase persists, without a shift to the hypertrophic or remodeling phases of PF–PC regenerative rewiring.
Reflecting massive degeneration, the number of PF–PC synapses dropped steeply, whereas there was a reciprocal increase in the number of free spines on postlesion day 1. At this time point, the PF territory, which is confined to the distal one-third of dendritic trees in sham-operated adult wild-type controls (Ichikawa et al., 2002, 2016; Fig. 5E), markedly elongated to encompass the proximal dendritic portions. This also occurred, to an even greater extent, for free spines. These observations suggest that free spines are produced not only by detachment of degenerating PF terminals, but also by de novo protrusion from proximal dendrites, and that some of these free spines on proximal dendrites are innervated by PFs. Aberrant interneuron innervation of PC spines in the expanded PF territory also suggests that regenerative responses start as immediately as degeneration does. Nevertheless, the reduction of PF synapse number to less than half that in sham-operated controls indicates that degeneration exceeds regeneration on postlesion day 1. Importantly, all events in this degenerative phase, including aberrant expansion of the PF territory, elongation of the PCD-II domain, massive emergence of free spines, and aberrant spine innervation by interneurons, recapitulate the developmental events in the early postnatal cerebellum (Kurihara et al., 1997; Ichikawa et al., 2016). Therefore, disorganized synaptic circuitry in the degenerative phase mirrors the immature synaptic circuitry during development.
In wild-type mice on postlesion days 7 and 30, most measured parameters of PF wiring returned to control levels, with retraction of the PF territory, restoration of PF synapse number, and disappearance of free spines and spine-type interneuron synapses. Nevertheless, there were significant differences between the two postlesion days. For example, although the size of PF terminals and PC spines and the PF terminal-to-PC spine contact ratio were increased on postlesion day 7, they returned to control levels by postlesion day 30. In addition, the profile density of PF axons remained low on postlesion day 7, but substantially recovered by postlesion day 30, when long PF collaterals with numerous varicosities were observed. These findings suggest that PF synapses regenerate first via hypertrophic responses in surviving PFs (the hypertrophic phase), and second, through an increase in PF density by collateral sprouting and elongation (the remodeling phase). In the latter phase, the shape, size, and connectivity of PF synapses are also restored to a relatively normal compact architecture.
Role of GluD2 in PF synapse regeneration
The most notable difference in GluD2-KO mice was the lack of any sign of PF synapse regeneration. On postlesion day 7, GluD2-KO mice showed no hypertrophic response. Moreover, the PF territory remained elongated, and free spines were further increased in number. This continued until postlesion day 30, when the density of PF axon profiles remained low, suggesting that GluD2 is essential for the regeneration of PF innervation and territory after lesioning.
The connectivity of PF synapses is strengthened and maintained by synaptic adhesion systems comprised of GluD2, Cbln1 and neurexin (Guastavino et al., 1990; Kashiwabuchi et al., 1995; Kurihara et al., 1997; Hirai et al., 2005; Takeuchi et al., 2005; Matsuda et al., 2010; Uemura et al., 2010). GluD2-KO and Cbln1-KO mice share many common phenotypes, including the severe loss of PF–PC synapses, mismatched alignment of presynaptic and postsynaptic structures at PF–PC synapses, emergence of free spines, distal invasion of the CF territory, persistent multiple CF innervation of PCs, impaired long-term depression at PF–PC synapses, and ataxia (Kashiwabuchi et al., 1995; Ichikawa et al., 2002; Hirai et al., 2005; Miyazaki et al., 2010). Importantly, cerebellar administration of recombinant Cbln1 alleviated these phenotypic defects in Cbln1-KO mice (Ito-Ishida et al., 2008). Furthermore, administration of recombinant Cbln1 to cultured Cbln1-KO cerebellar neurons induced the formation of PF protrusions, which often form enlarged circular structures encapsulating PC spines, facilitated the accumulation of synaptic vesicles and GluD2, and promoted the maturation of PF–PC synapses (Ito-Ishida et al., 2012). Therefore, rapid regeneration of PF wiring is likely mediated by the interaction of GluD2 with Cbln1 released from surviving and regenerating PFs, as in cerebellar development (Yuzaki, 2009; Watanabe and Kano, 2011). Without GluD2, this interaction is disrupted and PF wiring remains in the degenerative phase.
Expansion of the PF territory in the degenerative phase
Robust expansion of the PF territory occurred in the degenerative phase in both strains of mice, whereas the subsequent territorial retraction occurred only in wild-type mice. Recent studies provide insight into how these territorial changes might be produced. In normal development, PF and CF territories on PC dendrites are highly overlapping until postnatal day 15 in mice, and then segregated through the elimination of PF synapses from proximal dendrites (Ichikawa et al., 2016). This territorial segregation is arrested in mutant mice defective in the type-1 metabotropic glutamate receptor (mGluR1) or the P/Q-type voltage-dependent Ca2+ channel CaV2.1 (Miyazaki et al., 2004, 2012; Ichikawa et al., 2016). In PCs, mGluR1 is readily activated by PF stimulation and induces inositol 1,4,5-trisphosphate-mediated Ca2+ release in local dendrites (Finch and Augustine, 1998; Takechi et al., 1998), whereas CaV2.1 is activated upon strong depolarization by CF firing and mediates global Ca2+ influx in the whole dendritic tree (Kano et al., 1992; Konnerth et al., 1992; Regehr and Mintz, 1994). Ectopic PF innervation of proximal dendrites also occurs following deprivation of CF input to PCs (Desclin and Escubi, 1974; Sotelo et al., 1975; Rossi et al., 1991; Cesa et al., 2003, 2005) or blockade of electrical activity in the cerebellar cortex (Bravin et al., 1999; Morando et al., 2001; Cesa et al., 2003, 2007). These findings suggest that PF and CF activities inducing local and global Ca2+ dynamics in PC dendrites work in concert to promote PF synapse elimination from proximal dendrites. Therefore, expansion of the PF territory in the degenerative phase might be caused by a lack of mGluR1 activation resulting from the massive loss of PF synapses in both strains of mice. The subsequent retraction of the PF territory in wild-type mice may be triggered by the restoration of mGluR1 activation following PF synapse regeneration.
High regenerative capacity of PF wiring
Although axotomy of adult central neurons induces sprouting from proximal axon stumps and upregulation of growth-associated genes, these initial responses are transitory and are rapidly followed by growth arrest and the formation of retraction bulbs (Maier and Schwab, 2006). The major obstacle to regeneration is the presence of neurite growth inhibitory factors and molecules in the environment in the adult CNS, several of which are upregulated after injury, and associated with myelin and glial scars (Fawcett et al., 2012). In comparison, PFs are unmyelinated axons, and oligodendrocytes and astrocytes that form myelin and glial scars are lacking in the molecular layer of the cerebellum (Palay and Chan-Palay, 1974). Instead, the Bergmann glia, specialized astrocytes in the molecular layer, thoroughly enwrap the somatodendritic elements of PCs and support their metabolism and function (Yamada and Watanabe, 2002). These unique environments in the cerebellar molecular layer may also help GluD2 work for rapid and effective regeneration of PF–PC wiring. Therefore, our findings shed light on the role of synaptic adhesion systems in the regeneration of synaptic connection in the adult brain.
Footnotes
This work was supported by Grants-in-Aid for Scientific Research (24220007 to M.W.; 17500233 and 24500413 to R.I.) and also by the Brain Mapping by Integrated Neurotechnologies for Disease Studies (Brain/MINDS) from AMED. We thank H. Okushima and E. Suzuki, Laboratory of Electron Microscopy, School of Medicine, Sapporo Medical University, for technical assistance.
The authors declare no competing financial interests.
References
- Altman J. Postnatal development of the cerebellar cortex in the rat: II. Phases in the maturation of Purkinje cells and of the molecular layer. J Comp Neurol. 1972;145:399–463. doi: 10.1002/cne.901450402. [DOI] [PubMed] [Google Scholar]
- Altman J, Anderson WJ. Experimental reorganization of the cerebellar cortex: I. Morphological effects of elimination of all microneurons with prolonged x-irradiation started at birth. J Comp Neurol. 1972;146:355–406. doi: 10.1002/cne.901460305. [DOI] [PubMed] [Google Scholar]
- Brand S, Dahl AL, Mugnaini E. The length of parallel fibers in the cat cerebellar cortex: an experimental light and electron microscopic study. Exp Brain Res. 1976;26:39–58. doi: 10.1007/BF00235248. [DOI] [PubMed] [Google Scholar]
- Bravin M, Morando L, Vercelli A, Rossi F, Strata P. Control of spine formation by electrical activity in the adult rat cerebellum. Proc Natl Acad Sci U S A. 1999;96:1704–1709. doi: 10.1073/pnas.96.4.1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cesa R, Morando L, Strata P. Glutamate receptor δ2 subunit in activity-dependent heterologous synaptic competition. J Neurosci. 2003;23:2363–2370. doi: 10.1523/JNEUROSCI.23-06-02363.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cesa R, Morando L, Strata P. Purkinje cell spinogenesis during architectural rewiring in the mature cerebellum. Eur J Neurosci. 2005;22:579–586. doi: 10.1111/j.1460-9568.2005.04244.x. [DOI] [PubMed] [Google Scholar]
- Cesa R, Scelfo B, Strata P. Activity-dependent presynaptic and postsynaptic structural plasticity in the mature cerebellum. J Neurosci. 2007;27:4603–4611. doi: 10.1523/JNEUROSCI.5617-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Hillman DE. Plasticity of the parallel fiber-Purkinje cell synapse by spine takeover and new synapse formation in the adult rat. Brain Res. 1982;240:205–220. doi: 10.1016/0006-8993(82)90217-7. [DOI] [PubMed] [Google Scholar]
- Crepel F, Delhaye-Bouchaud N, Dupont JL. Fate of the multiple innervation of cerebellar Purkinje cells by climbing fibers in immature control, x-irradiated and hypothyroid rats. Brain Res. 1981;227:59–71. doi: 10.1016/0165-3806(81)90094-8. [DOI] [PubMed] [Google Scholar]
- Desclin JC, Escubi J. Effects of 3-acetylpyridine on the central nervous system of the rat, as demonstrated by silver methods. Brain Res. 1974;77:349–364. doi: 10.1016/0006-8993(74)90627-1. [DOI] [PubMed] [Google Scholar]
- Dusart I, Ghoumari A, Wehrle R, Morel MP, Bouslama-Oueghlani L, Camand E, Sotelo C. Cell death and axon regeneration of Purkinje cells after axotomy: challenges of classical hypotheses of axon regeneration. Brain Res Rev. 2005;49:300–316. doi: 10.1016/j.brainresrev.2004.11.007. [DOI] [PubMed] [Google Scholar]
- Fawcett JW, Schwab ME, Montani L, Brazda N, Müller HW. Defeating inhibition of regeneration by scar and myelin components. Handb Clin Neurol. 2012;109:503–522. doi: 10.1016/B978-0-444-52137-8.00031-0. [DOI] [PubMed] [Google Scholar]
- Finch EA, Augustine GJ. Local calcium signalling by inositol-1,4,5-trisphosphate in Purkinje cell dendrites. Nature. 1998;396:753–756. doi: 10.1038/25541. [DOI] [PubMed] [Google Scholar]
- Guastavino JM, Sotelo C, Damez-Kinselle I. Hot-foot murine mutation: behavioral effects and neuroanatomical alterations. Brain Res. 1990;523:199–210. doi: 10.1016/0006-8993(90)91488-3. [DOI] [PubMed] [Google Scholar]
- Hashimoto K, Kano M. Synapse elimination in the developing cerebellum. Cell Mol Life Sci. 2013;70:4667–4680. doi: 10.1007/s00018-013-1405-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirai H, Pang Z, Bao D, Miyazaki T, Li L, Miura E, Parris J, Rong Y, Watanabe M, Yuzaki M, Morgan JI. Cbln1 is essential for synaptic integrity and plasticity in the cerebellum. Nat Neurosci. 2005;8:1534–1541. doi: 10.1038/nn1576. [DOI] [PubMed] [Google Scholar]
- Ichikawa R, Miyazaki T, Kano M, Hashikawa T, Tatsumi H, Sakimura K, Mishina M, Inoue Y, Watanabe M. Distal extension of climbing fiber territory and multiple innervation caused by aberrant wiring to adjacent spiny branchlets in cerebellar Purkinje cells lacking glutamate receptor δ2. J Neurosci. 2002;22:8487–8503. doi: 10.1523/JNEUROSCI.22-19-08487.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichikawa R, Yamasaki M, Miyazaki T, Konno K, Hashimoto K, Tatsumi H, Inoue Y, Kano M, Watanabe M. Developmental switching of perisomatic innervation from climbing fibers to basket cell fibers in cerebellar Purkinje cells. J Neurosci. 2011;31:16916–16927. doi: 10.1523/JNEUROSCI.2396-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichikawa R, Hashimoto K, Miyazaki T, Uchigashima M, Yamasaki M, Aiba A, Kano M, Watanabe M. Territories of heterologous inputs onto Purkinje cell dendrites are segregated by mGluR1-dependent parallel fiber synapse elimination. Proc Natl Acad Sci U S A. 2016;113:2282–2287. doi: 10.1073/pnas.1511513113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito M. Cerebellum: the brain for an implicit self. Upper Saddle River, NJ: Pearson Education; 2012. [Google Scholar]
- Ito-Ishida A, Miura E, Emi K, Matsuda K, Iijima T, Kondo T, Kohda K, Watanabe M, Yuzaki M. Cbln1 regulates rapid formation and maintenance of excitatory synapses in mature cerebellar Purkinje cells in vitro and in vivo. J Neurosci. 2008;28:5920–5930. doi: 10.1523/JNEUROSCI.1030-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito-Ishida A, Miyazaki T, Miura E, Matsuda K, Watanabe M, Yuzaki M, Okabe S. Presynaptically released Cbln1 induces dynamic axonal structural changes by interacting with GluD2 during cerebellar synapse formation. Neuron. 2012;76:549–564. doi: 10.1016/j.neuron.2012.07.027. [DOI] [PubMed] [Google Scholar]
- Kano M, Rexhausen U, Dreessen J, Konnerth A. Synaptic excitation produces a long-lasting rebound potentiation of inhibitory synaptic signals in cerebellar Purkinje cells. Nature. 1992;356:601–604. doi: 10.1038/356601a0. [DOI] [PubMed] [Google Scholar]
- Kashiwabuchi N, Ikeda K, Araki K, Hirano T, Shibuki K, Takayama C, Inoue Y, Kutsuwada T, Yagi T, Kang Y, Aizawa S, Mishina M. Impairment of motor coordination, Purkinje cell synapse formation, and cerebellar long-term depression in GluRδ2 mutant mice. Cell. 1995;81:245–252. doi: 10.1016/0092-8674(95)90334-8. [DOI] [PubMed] [Google Scholar]
- Konnerth A, Dreessen J, Augustine GJ. Brief dendritic calcium signals initiate long-lasting synaptic depression in cerebellar Purkinje cells. Proc Natl Acad Sci U S A. 1992;89:7051–7055. doi: 10.1073/pnas.89.15.7051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurihara H, Hashimoto K, Kano M, Takayama C, Sakimura K, Mishina M, Inoue Y, Watanabe M. Impaired parallel fiber→Purkinje cell synapse stabilization during cerebellar development of mutant mice lacking the glutamate receptor δ2 subunit. J Neurosci. 1997;17:9613–9623. doi: 10.1523/JNEUROSCI.17-24-09613.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalouette A, Lohof A, Sotelo C, Guénet J, Mariani J. Neurobiological effects of a null mutation depend on genetic context: comparison between two hotfoot alleles of the delta-2 ionotropic glutamate receptor. Neuroscience. 2001;105:443–455. doi: 10.1016/S0306-4522(01)00193-2. [DOI] [PubMed] [Google Scholar]
- Landsend AS, Amiry-Moghaddam M, Matsubara A, Bergersen L, Usami S, Wenthold RJ, Ottersen OP. Differential localization of δ glutamate receptors in the rat cerebellum: coexpression with AMPA receptors in parallel fiber-spine synapses and absence from climbing fiber-spine synapses. J Neurosci. 1997;17:834–842. doi: 10.1523/JNEUROSCI.17-02-00834.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier IC, Schwab ME. Sprouting, regeneration and circuit formation in the injured spinal cord: factors and activity. Philos Trans R Soc Lond B Biol Sci. 2006;361:1611–1634. doi: 10.1098/rstb.2006.1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda K, Miura E, Miyazaki T, Kakegawa W, Emi K, Narumi S, Fukazawa Y, Ito-Ishida A, Kondo T, Shigemoto R, Watanabe M, Yuzaki M. Cbln1 is a ligand for an orphan glutamate receptor δ2, a bidirectional synapse organizer. Science. 2010;328:363–368. doi: 10.1126/science.1185152. [DOI] [PubMed] [Google Scholar]
- Miyazaki T, Fukaya M, Shimizu H, Watanabe M. Subtype switching of vesicular glutamate transporters at parallel fibre-Purkinje cell synapses in developing mouse cerebellum. Eur J Neurosci. 2003;17:2563–2572. doi: 10.1046/j.1460-9568.2003.02698.x. [DOI] [PubMed] [Google Scholar]
- Miyazaki T, Hashimoto K, Shin HS, Kano M, Watanabe M. P/Q-type Ca2+ channel α1A regulates synaptic competition on developing cerebellar Purkinje cells. J Neurosci. 2004;24:1734–1743. doi: 10.1523/JNEUROSCI.4208-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazaki T, Yamasaki M, Takeuchi T, Sakimura K, Mishina M, Watanabe M. Ablation of glutamate receptor GluRδ2 in adult Purkinje cells causes multiple innervation of climbing fibers by inducing aberrant invasion to parallel fiber innervation territory. J Neurosci. 2010;30:15196–15209. doi: 10.1523/JNEUROSCI.0934-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazaki T, Yamasaki M, Hashimoto K, Yamazaki M, Abe M, Usui H, Kano M, Sakimura K, Watanabe M. Cav2.1 in cerebellar Purkinje cells regulates competitive excitatory synaptic wiring, cell survival, and cerebellar biochemical compartmentalization. J Neurosci. 2012;32:1311–1328. doi: 10.1523/JNEUROSCI.2755-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morando L, Cesa R, Rasetti R, Harvey R, Strata P. Role of glutamate δ-2 receptors in activity-dependent competition between heterologous afferent fibers. Proc Natl Acad Sci U S A. 2001;98:9954–9959. doi: 10.1073/pnas.171098398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa S, Watanabe M, Isobe T, Kondo H, Inoue Y. Cytological compartmentalization in the staggerer cerebellum, as revealed by calbindin immunohistochemistry for Purkinje cells. J Comp Neurol. 1998;395:112–120. doi: 10.1002/(SICI)1096-9861(19980525)395:1<112::AID-CNE8>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- Napper RM, Harvey RJ. Number of parallel fiber synapses on an individual Purkinje cell in the cerebellum of the rat. J Comp Neurol. 1988;274:168–177. doi: 10.1002/cne.902740204. [DOI] [PubMed] [Google Scholar]
- Palay SL, Chan-Palay V. Cerebellar cortex, cytology and organization. Berlin: Springer; 1974. [Google Scholar]
- Regehr WG, Mintz IM. Participation of multiple calcium channel types in transmission at single climbing fiber to Purkinje cell synapses. Neuron. 1994;12:605–613. doi: 10.1016/0896-6273(94)90216-X. [DOI] [PubMed] [Google Scholar]
- Rossi F, van der Want JJ, Wiklund L, Strata P. Reinnervation of cerebellar Purkinje cells by climbing fibres surviving a subtotal lesion of the inferior olive in the adult rat: II. Synaptic organization on reinnervated Purkinje cells. J Comp Neurol. 1991;308:536–554. doi: 10.1002/cne.903080404. [DOI] [PubMed] [Google Scholar]
- Rossi F, Borsello T, Vaudano E, Strata P. Regressive modifications of climbing fibres following Purkinje cell degeneration in the cerebellar cortex of the adult rat. Neuroscience. 1993;53:759–778. doi: 10.1016/0306-4522(93)90622-M. [DOI] [PubMed] [Google Scholar]
- Sotelo C. Cellular and genetic regulation of the development of the cerebellar system. Prog Neurobiol. 2004;72:295–339. doi: 10.1016/j.pneurobio.2004.03.004. [DOI] [PubMed] [Google Scholar]
- Sotelo C, Hillman DE, Zamora AJ, Llinás R. Climbing fiber deafferentation: its action on Purkinje cell dendritic spines. Brain Res. 1975;98:574–581. doi: 10.1016/0006-8993(75)90374-1. [DOI] [PubMed] [Google Scholar]
- Strata P, Rossi F. Plasticity of the olivocerebellar pathway. Trends Neurosci. 1998;21:407–413. doi: 10.1016/S0166-2236(98)01305-8. [DOI] [PubMed] [Google Scholar]
- Takayama C, Nakagawa S, Watanabe M, Mishina M, Inoue Y. Light- and electron-microscopic localization of the glutamate receptor channel δ2 subunit in the mouse Purkinje cell. Neurosci Lett. 1995;188:89–92. doi: 10.1016/0304-3940(95)11403-J. [DOI] [PubMed] [Google Scholar]
- Takechi H, Eilers J, Konnerth A. A new class of synaptic response involving calcium release in dendritic spines. Nature. 1998;396:757–760. doi: 10.1038/25547. [DOI] [PubMed] [Google Scholar]
- Takeuchi T, Miyazaki T, Watanabe M, Mori H, Sakimura K, Mishina M. Control of synaptic connection by glutamate receptor δ2 in the adult cerebellum. J Neurosci. 2005;25:2146–2156. doi: 10.1523/JNEUROSCI.4740-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torashima T, Iizuka A, Horiuchi H, Mitsumura K, Yamasaki M, Koyama C, Takayama K, Iino M, Watanabe M, Hirai H. Rescue of abnormal phenotypes in δ2 glutamate receptor-deficient mice by the extracellular N-terminal and intracellular C-terminal domains of the δ2 glutamate receptor. Eur J Neurosci. 2009;30:355–365. doi: 10.1111/j.1460-9568.2009.06841.x. [DOI] [PubMed] [Google Scholar]
- Uemura T, Lee SJ, Yasumura M, Takeuchi T, Yoshida T, Ra M, Taguchi R, Sakimura K, Mishina M. Trans-synaptic interaction of GluRδ2 and Neurexin through Cbln1 mediates synapse formation in the cerebellum. Cell. 2010;141:1068–1079. doi: 10.1016/j.cell.2010.04.035. [DOI] [PubMed] [Google Scholar]
- Watanabe M, Kano M. Climbing fiber synapse elimination in cerebellar Purkinje cells. Eur J Neurosci. 2011;34:1697–1710. doi: 10.1111/j.1460-9568.2011.07894.x. [DOI] [PubMed] [Google Scholar]
- Yamada K, Watanabe M. Cytodifferentiation of Bergmann glia and its relationship with Purkinje cells. Anat Sci Int. 2002;77:94–108. doi: 10.1046/j.0022-7722.2002.00021.x. [DOI] [PubMed] [Google Scholar]
- Yamasaki M, Miyazaki T, Azechi H, Abe M, Natsume R, Hagiwara T, Aiba A, Mishina M, Sakimura K, Watanabe M. Glutamate receptor δ2 is essential for input pathway-dependent regulation of synaptic AMPAR contents in cerebellar Purkinje cells. J Neurosci. 2011;31:3362–3374. doi: 10.1523/JNEUROSCI.5601-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuzaki M. New (but old) molecules regulating synapse integrity and plasticity: Cbln1 and the δ2 glutamate receptor. Neuroscience. 2009;162:633–643. doi: 10.1016/j.neuroscience.2008.12.002. [DOI] [PubMed] [Google Scholar]