

Abstract
Traumatic brain injury (TBI) is a major cause of death and disability worldwide. It produces diffuse axonal injury (DAI), which contributes to cognitive impairment, but effective disease-modifying treatment strategies are missing. We have recently developed a rat model of closed skull TBI that reproduces human TBI consequences, including DAI and clinical sequelae such as memory impairment. Here, we investigated whether sleep modulation after trauma has an impact on DAI and memory outcome. We assessed cognition with the novel object recognition test and stained for amyloid precursor protein, a DAI marker. We found that both sleep induction and restriction acutely after TBI enhanced encephalographic slow-wave activity, markedly reduced diffuse axonal damage in the cortex and hippocampus, and improved memory impairment 2 weeks after trauma. These results suggest that enhancing slow-wave sleep acutely after trauma may have a beneficial disease-modifying effect in subjects with acute TBI.
SIGNIFICANCE STATEMENT Traumatic brain injury (TBI) is a clinically important entity. Cognitive deficits belong to the most prevalent chronic posttraumatic symptoms, most likely due to diffuse axonal injury (DAI). A growing body of evidence suggests a role of sleep in the clearance of waste products in the brain, possibly including amyloid precursor protein (APP), a marker of DAI. In this study, we provide evidence that enhancement of slow-wave oscillatory activity in the delta-frequency range decreases the APP-immunoreactivity and preserves cognitive abilities after trauma, potentially offering novel, noninvasive treatment options for traumatic injury.
Keywords: diffuse axonal damage, memory, sleep, traumatic brain injury
Introduction
Traumatic brain injury (TBI) is a major public health concern affecting ∼12% of the general population worldwide and resulting in high rates of death and disability. The most common causes of TBI include violence, vehicle accidents, and falls (Styrke et al., 2007). TBI is currently interpreted as a disease process (Masel and DeWitt, 2010), presenting not only with acute manifestations, but also with long-lasting symptoms, potentially originated by progressive axonal injury (Bramlett and Dietrich, 2002, Inglese et al., 2005). Studies suggest that diffuse axonal injury (DAI) is an important contributor to posttraumatic cognitive impairment (Li et al., 2006, Kraus et al., 2007, Rostami et al., 2012), which is one of the most prevalent symptoms after TBI (Hall et al., 2005). Immunoreactivity to APP has been established as a gold standard for the detection of DAI. In addition to APP, pathological molecules that are linked to enhanced endoplasmic reticulum (ER) stress and to unfolded protein response, both of which are associated with neuronal death and with neurodegenerative disorders, have also been found to accumulate in the traumatized brain. Among these are activating transcription factor 4 (ATF-4), an ER stress marker, and ubiquitin (UB), a small regulatory protein playing a role in degradation of other proteins (Begum et al., 2014). Potentially noxious molecules are cleared via glymphatic pathways in the brain (Iliff et al., 2014, Plog et al., 2015), particularly during sleep and anesthesia, which are both characterized by predominant slow-wave activity (Xie et al., 2013). Indeed, there is an emerging discussion that sleep might serve as a potential treatment for a number of neurological conditions. Sleep restriction, resulting in increased slow-wave activity during subsequent rebound sleep (Borbély et al., 1984, Franken et al., 1991), has been suggested to be beneficial for TBI (Martinez-Vargas et al., 2012) and stroke subjects (Cam et al., 2013). However, not only physiological sleep, but also pharmacological interventions aimed at increasing slow oscillatory encephalographic activity, were linked to APP clearance in an Alzheimer's disease model (Klein et al., 2015) and similar interventions also improved outcome after stroke in rodents (Gao et al., 2008). Therefore, there is increasing evidence that interventions that enhance slow-wave activity could constitute novel, noninvasive treatment options for acute and chronic neurological conditions.
In this study, we aimed to assess whether sleep modulation procedures that induce increased slow-wave immediately after trauma improve diffuse axonal damage and TBI-induced cognitive deficits in a rat model of TBI.
Materials and Methods
Animals.
We used male Sprague Dawley rats (Charles River) weighing 230–270 g and housed them in groups before interventions; afterward, we single housed them with food and water available ad libitum. The animal room temperature was constantly maintained at 21–22°C and animals were kept on a 12 h light/dark cycle starting at 8:00 or 9:00 A.M. according to season. All experiments were approved by the veterinary office of the Canton Zurich and conducted according to the local guidelines for the care and use of laboratory animals.
Surgeries.
We performed all surgical procedures under deep anesthesia by inhalation of isoflurane (4.5% for induction, 2.5% for maintenance) and subsequent analgesia with buprenorphine (0.05 mg/kg, s.c.). We monitored animals daily over the first week after surgeries and weekly thereafter.
EEG/EMG headset implantation.
Implantation of EEG/EMG electrodes was performed as described previously (Buchele et al., 2015).
TBI.
After 8–10 d of recovery, rats weighed 300–350 g and underwent closed skull TBI in the prefrontocortical area of the brain using the adapted weight-drop model (Buchele et al., 2015). In sham animals, all procedures were identical except for the traumatic hit.
Cardiac perfusion.
Thirty days after TBI or sham procedures, animals underwent cardiac perfusion as described previously (Buchele et al., 2015). We immediately harvested the brains for histological evaluations. After perfusion, brains were cryoprotected by sequential sucrose steps, frozen in dry ice, and finally stored at −80°C until further use.
Behavior: novel object recognition test.
All animals underwent behavioral testing at baseline and 14 d after sham or TBI (Fig. 1A) to assess the effect of sleep modulation on posttraumatic memory impairment. To examine declarative memory performance, we used the novel object recognition test as described previously (Ennaceur and Delacour, 1988, Buchele et al., 2015). We manually scored video-recorded exploratory behavior and calculated the preference ratio, defined as time spent on exploration of a novel object relative to the total exploration time of both objects, for each group and time point tested and compared it against chance performance (0.5) using a one-sample t test. Animals showing signs of anxiety (n = 2) were excluded from analysis.
Figure 1.

Sleep modulation after TBI. A, Time schedule of modulation interventions and behavioral testing. TBI and sham animals were divided into three groups: Ctrl, SI, and SR animals. For 5 d after TBI induction, animals received 2×/d intraperitoneal saline injections (Ctrl), 2× day SO injections (400 mg/kg, i.p.; SI), or 2×/d intraperitoneal saline injections plus 6 h per day gentle handling (SR). B, Exemplary traces representing the four characterized behavioral states: wake, NREM, PS, and SWS-like state. C, Exemplary EEG traces showing the effect of SO administration on EEG and of sleep restriction on sleep rebound EEG.
Sleep modulation interventions.
To evaluate the effect of sleep modulation on histological outcome and memory performance, we divided 25 sham and 25 TBI animals into three groups. Each group received two intraperitoneal injections per day (1 h after lights on and 1 h after lights off). The control (Ctrl) and sleep-restricted (SR) groups were treated with saline and the sleep-induced (SI) group was treated with sodium oxybate (SO; Xyrem-GHB salt, 400 mg/kg; UCB Pharma) (Lettieri and Fung, 1979, Nava et al., 2001). We performed sleep restriction by gentle handling (6 h daily, starting at 2 h after lights on; Tobler and Jaggi, 1987). The sleep modulation procedures started the day after TBI or sham procedures and lasted 5 d (Fig. 1A). Duration of treatment was determined by our previous studies in this model (Büchele et al., 2015), in which we detected a cognitive deficit at 7 d post-TBI, leaving a possible window for interventions of ∼5 d. One week was granted for washout before behavioral testing.
EEG recordings and scoring.
To verify the effect of sleep modulation on EEG spectra, we conducted bilateral EEG/EMG recordings in an additional group of TBI animals (n = 15) and performed visual scoring of acquired EEG as described previously (Büchele et al., 2015). We recorded the animals before TBI induction (baseline) and during the last 24 h of interventions. All scoring was performed manually and the scorer was blinded as to group. EEG and EMG were sampled at 200 Hz and signals were amplified, filtered, and converted into analog-to-digital signals. Hardware EMBLA and Somnologica-3 software (Medcare Flaga) were used. Activity in the 50 Hz band was discarded from the analysis because of power line artifacts. Power spectra of the EEG were obtained by a discrete Fourier transformation (range: 0.75–25 Hz; frequency resolution: 0.25 Hz bins; time resolution: consecutive 4 s epochs; window function: hamming). In accordance with earlier studies involving SO treatment, we identified four behavioral states: wake, paradoxical sleep (PS, REM), non-REM sleep (NREM), and a slow-wave sleep (SWS)-like state induced by SO (also referred to as drug- induced state; Vienne et al., 2010; Fig. 1B). Because the SWS-like state is characterized by predominance of low frequencies on EEG and high-amplitude waves (similar to NREM sleep), we pooled together physiological NREM sleep and SWS-like state for further analysis and refer to both states as SWS. Epochs were assigned to a specific vigilance state when more than half of the epoch fulfilled the criteria for that state. After visual scoring of acquired traces, we exported data indicating duration of vigilance state, bout number, and average bout duration for each behavioral state. This information was used to calculate the fragmentation index, which is expressed as the number of bouts/total number of epochs, with higher index values expressing higher fragmentation of the behavioral state. Exemplary traces of effect of SO on EEG and recovery sleep after SR are presented in Figure 1C.
Spectral analysis.
We previously excluded artifacts by visual review of the raw data. We calculated the relative power spectral density as the summarized power in the delta (0.5–4 Hz) frequency band for the dark phase and light phase separately. Delta power reflects the high-amplitude, low-frequency oscillatory activity (Tobler et al., 1997) and is conceptualized to mirror the depth of sleep. To reduce interindividual variability, the absolute spectral band power (in square microvolts) was normalized to the sum of the corresponding full spectral density in each behavioral state (1–100 Hz). All signal processing and analysis were performed as described using MATLAB (The MathWorks).
Histological evaluation: immunofluorescence.
DAI has been evidenced by APP immunoreactive (APP-IR) axonal swellings (Gentleman et al., 1993). To evaluate the effect of sleep modulation on DAI in our model, we performed immunohistochemical analyses of APP on rat 40 μm coronal brain cryostat sections as described previously (Büchele et al., 2015). Briefly, we performed standard immunofluorescence staining with a primary antibody against APP (mouse anti-APP, MAB348, 1:300; Millipore), followed by secondary antibody incubation (Alexa Fluor 488-coupled anti-mouse secondary antiserum, 1:500; Life Technologies). To evaluate other potential mechanisms behind impairment, additional stainings following the same protocol using UB (1:500, sc-9133; Santa Cruz Biotechnology) and ATF-4 (an ER stress marker; 1:500, sc-200; Santa Cruz Biotechnology) antibodies were conducted. Because these proteins are expressed in the same time window as APP (Begum et al., 2014), it is likely that they also play a role in the chronic functional detriment (e.g., by increasing misfolded protein response, promoting cell death, or decreasing protein degradation). A control experiment using the exact same procedures except for the addition of the primary antibody was performed to assess the degree of unspecific staining. Fluorescent (APP) pictures were taken using a Zeiss Axio Imager M2 microscope.
Histological evaluation: APP and other protein quantifications.
To determine the amount of accumulated APP in cortex and hippocampus, one experimenter blinded to experimental conditions performed APP quantification on previously stained rat brain sections. An APP-immunoreactive cell was defined by the presence of a positive signal both in the soma and in a connected axon, with a length ≥25 μm. We performed a semiquantitative analysis using a Zeiss Axio Imager M2 microscope equipped with a Ludl MAC 6000 stage (Ludl Electronic Products) and MicroBrightField Stereo Investigator 10.5 software (MBF Bioscience).
We measured the areas of interest by tracing the outlines of cortex and hippocampus throughout the anterior–posterior axis between −2.28 to −6.36 mm from bregma using a 2.5× objective. A grid measuring 250 × 250 μm was superimposed and a 20× objective was used to identify APP-IR cells within the brain areas of interest. A square within the grid was considered as positive when at least three positive APP-IR cells were found. We divided the total number of positive squares within the structure studied (cortex or hippocampus) of one section by the area of the structure, yielding a number of positive squares per square millimeter. We calculated the average of all analyzed sections and defined this value as the APP trauma index (total number of positive squares per square millimeter divided by the number of sections analyzed). To aid visualization of the APP trauma index in hippocampus, we multiplied the values by factor of 100,000. We used the same method for the assessment of ATF-4 and UB immunoreactivity.
Results
Vigilance states proportions after sleep modulation
To confirm the effect of both sleep manipulation methods on vigilance states (Fig. 2A), state fragmentation (Fig. 2B), and EEG power spectra (Fig. 2C) in TBI rats, we performed EEG/EMG recordings at baseline and during the last 24 h of sleep interventions. At baseline, there was no difference in the amount of time spent in each of the behavioral states between the groups (Ctrl, SI, and SR) in both light and dark periods (data not shown). During the last day of the intervention, the SR animals showed an increase in time spent awake during the light period (when sleep restriction took place; one-way ANOVA, F(2,12) = 80.4, p < 0.001; Tukey HSD post hoc SR*Ctrl, p < 0.001; SI*Ctrl, p > 0.05). During the dark period, both SR and SI groups spent less time awake compared with control animals (one-way ANOVA, F(2,12) = 7.7, p < 0.01; post hoc SR*Ctrl, p < 0.05; SI*Ctrl, p < 0.01). Over 24 h, only the SI group spent less time awake compared with controls (one-way ANOVA, F(2,12) = 17.1, p < 0.001; post hoc SI*Ctrl, p < 0.001). The sleep-restricted rats showed a reduction in time spent in SWS during the light period (when SR took place) compared with control animals, whereas no difference was found between SI and Ctrl animals (one-way ANOVA, F(2,12) = 23.8, p < 0.001; post hoc Ctrl*SR, p < 0.001; Ctrl*SI, p > 0.05). During the dark period, we observed an increase in time spent in SWS in both SI and SR rats (sleep rebound in the latter animals; one-way ANOVA, F(2,12) = 16.1, p < 0.001; post hoc Ctrl*SR, p < 0.01; Ctrl*SI, p < 0.001). Over 24 h, duration of SWS was similar in SR and controls animals, but increased in the SI group (one-way ANOVA, F(2,12) = 12.3, p < 0.01; post hoc Ctrl*SR, p > 0.05; Ctrl*SI, p < 0.01). Only SR animals showed a difference in time spent in PS, with a decrease during the light period with sleep restriction (one-way ANOVA, F(2,12) = 10.5, p < 0.01; post hoc Ctrl*SR, p < 0.05) and an increase during the dark period with sleep rebound (one-way ANOVA, F(2,12) = 10.5 p < 0.01; post hoc SR*Ctrl, p < 0.01; Fig. 2A).
Figure 2.
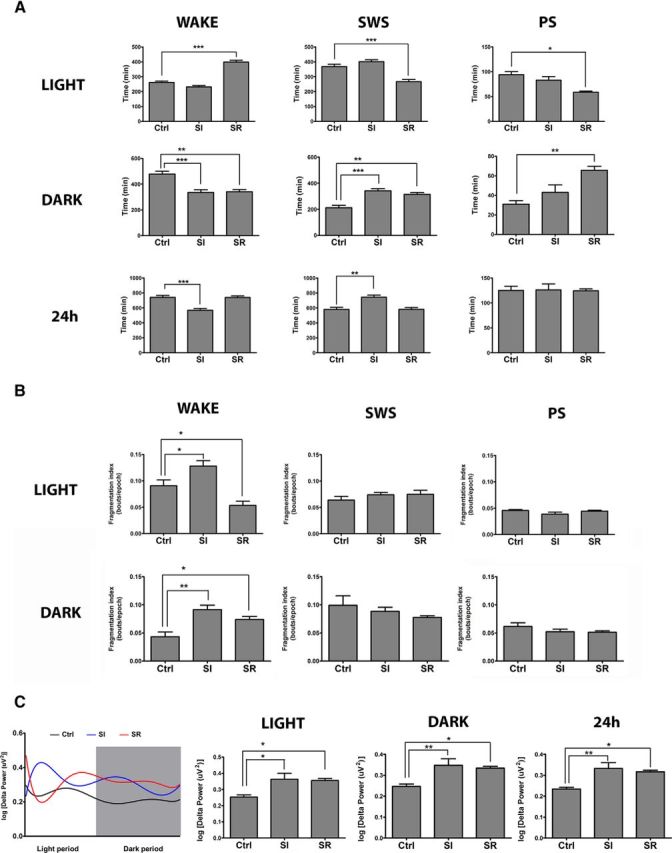
Effect of sleep modulation on vigilance states, fragmentation, and delta power after TBI. A, Proportions of vigilance states during last day of sleep modulation (TBI only). Light period: Only the SR group showed increase time spent awake with subsequent decrease in time spent in SWS and PS compared with Ctrl (wake: p < 0.001, SWS: p < 0.001, PS: p < 0.05). During the dark period, both sleep-modulated groups showed a decrease in time spent in wake compared with control animals (p < 0.05) and an increase in time spent in SWS (p < 0.05); only the SR group showed an increase in the time spent in PS (p < 0.01). Over the 24 h, only the SI group showed increased time spent in SWS (p < 0.01) and decreased time spent awake (p < 0.001). B, Effect of sleep modulation on fragmentation of behavioral states. Differences in fragmentation index (bout/epoch) were only apparent during wake, whereas in the light period, the SR group showed more stable wake (p < 0.05) and the SI group showed more fragmented wake (p < 0.05) compared with controls. During the dark period, both of the groups had more fragmented wake compared with controls (SI: p < 0.01, SR: p < 0.05). C, Effect of sleep modulation on delta power. Both the SI and SR groups had a bigger proportion of delta power during SWS in the light period (p < 0.05), dark period (SI: p < 0.01, SR p < 0.05), and over 24 h (SI: p < 0.01, SR p < 0.05) compared with controls. Data presented in 10 min bins shows dynamic changes in delta power over the 24-h recording period.
Vigilance states fragmentation after sleep modulation
At baseline, fragmentation of vigilance states was similar in all groups (data not shown). During the last day of sleep modulation in the light period, wake fragmentation index was lower in SR animals, in contrast to SI animals, which showed increased wake fragmentation compared with control animals (one-way ANOVA, F(2,12) = 14.6, p < 0.05; post hoc SR*Ctrl, p < 0.05; SI*Ctrl, p < 0.05). During the dark period, wake was more fragmented in both sleep modulation groups compared with control animals (one-way ANOVA, F(2,12) = 10.6, p < 0.01; post hoc SR*Ctrl, p < 0.05; SI*Ctrl, p < 0.01). No difference was observed in fragmentation of SWS or PS in any of the groups (Fig. 2B).
Delta power after sleep modulation
Both modulated groups had an increase in delta power proportion in the light (one-way ANOVA, F(2,11) = 5.6, p < 0.05; post hoc SR*Ctrl, p < 0.05; SI*Ctrl, p < 0.05) and dark period (one-way ANOVA, F(2,11) = 7.7, p < 0.01; post hoc Ctrl*SR, p < 0.05; Ctrl*SI, p < 0.01), as well as over 24 h (one-way ANOVA, F(2,11) = 6.3, p < 0.05; post hoc SR*Ctrl, p < 0.05; SI*Ctrl, p < 0.05). To aid visualization of the effect of sleep manipulation on delta power, we divided the 24 h recording period into 10 min bins (Fig. 2C).
Cognitive performance after acute sleep modulation
To assess the effect of sleep modulation on TBI induced-cognitive impairment, we performed a novel object recognition test (Ennaceur and Delacour, 1988, Büchele et al., 2015). At baseline, before trauma, all animals showed intact memory acquisition/retrieval and performed above the chance level (one-sample t test, all scores compared with chance level of 0.5, p < 0.05). At 14 d after trauma, all sham groups showed preserved cognitive abilities, as evidenced by performances above chance level. In TBI animals, however, only the sleep-induced and sleep-restricted groups performed above chance level, whereas control animals evidenced impaired memory (one-sample t test, all scores compared with chance level of 0.5; sham: Ctrl, SR, SI, p < 0.05; TBI: Ctrl, p > 0.05; SR, SI, p < 0.05; Fig. 3).
Figure 3.

Effect of sleep modulation on cognitive performance in novel object recognition test (NORT) after TBI. All charts show recognition index compared with chance level (0.5). Before TBI induction (baseline, BL), all animals showed intact cognitive abilities (p < 0.05). TBI resulted in a decrease of memory performance at 14 d after trauma, but cognitive abilities were spared in both the SI and SR TBI animals; the sham groups continued performing equally well after the surgical intervention (sham Ctrl: p < 0.05, sham SI: p < 0.05, sham SR: p < 0.05, TBI Ctrl: p > 0.05, TBI SI: p < 0.05, TBI SR: p < 0.05).
APP, UB, and ATF-4 expression after sleep modulation
To assess the effect of sleep modulation after TBI on posttraumatic DAI, we performed APP immunofluorescence on cortical and hippocampal sections obtained from cryosectioned rat brains (Fig. 4A,C). The TBI control group revealed significantly higher APP expression than sham animals in both cortex and hippocampus (factorial ANOVA; F(1,23) = 6.1131, p < 0.05). APP quantification results showed that, compared with TBI control animals, both sleep induction and sleep restriction resulted in markedly lower APP expression after TBI both in cortex and hippocampus (factorial ANOVA; F(2,23) = 11.268, p < 0.001; post hoc Tukey HSD: Ctrl*SI, p < 0.01; Ctrl*SR, p < 0.01; Fig. 4B,D). There was no significant difference between sham control animals and any of the sham-treated groups.
Figure 4.

Effect of sleep modulation on histological markers after TBI. A, C, Representative images from sham and TBI animals (all groups) showing cortical (A) and hippocampal (C) APP staining. Only the non-sleep-modulated control TBI group showed marked axonal staining. B, D, Quantification of APP-immunoreactive cells in cortex (B) and hippocampus (D). All charts show the number of positive grid squares of one section divided by the area of cortex (number of positive squares per square millimeter of cortex). The TBI control group shows the highest APP trauma index (reflecting the highest expression of APP), which is significantly different from the sham group (p < 0.05). Sleep modulation resulted in decrease of APP expression in cortex and hippocampus, as indicated by the decreased APP trauma index in the sleep-induced TBI group (p < 0.01) and the sleep-restricted TBI group (p < 0.01). E, Quantification of ATF-4-immunoreactive cells in cortex. No difference was noted in ATF-4 immunoreactivity between the groups. F, Quantification of UB immunoreactivity in cortex. There was no difference in UB immunoreactivity between the groups.
No differences were detected in immunoreactivity of ATF-4 and UB between the groups at 30 d after trauma (ATF-4: factorial ANOVA, F(2,19)=.94259, p > 0.05; post hoc Tukey HSD: Ctrl*SI, p > 0.05; Ctrl*SR, p > 0.05; UB: factorial ANOVA, F(2,19) = 0.09273, p > 0.05; post hoc Tukey HSD: Ctrl*SI, p > 0.05; Ctrl*SD, p > 0.05; Fig. 4E,F). No signal was detected in the unspecific staining controls (data not shown).
Discussion
The results of our study suggest that posttraumatic sleep modulation leading to increases in delta power markedly reduces posttraumatic APP accumulation in the cortex and hippocampus and prevents TBI-induced cognitive impairment.
Cognitive impairment is one of the most prevalent chronic TBI symptoms, present in up to 79% of patients (Hall et al., 2005). Consistent with the previous reports showing posttraumatic memory impairment in rodents (Lyeth et al., 1990, Smith et al., 1991, Hamm et al., 1996, Hoskison et al., 2009, Yu et al., 2009), we found that the non-sleep-modulated TBI group developed cognitive impairment with no apparent discrimination of a novel object after trauma. The impairment is likely mediated by DAI, as evidenced by several observations of white matter damage in human TBI patients (Inglese et al., 2005, Kraus et al., 2007) and in rats (Li et al., 2006). After TBI, axonal transport is interrupted, which results in appearance of axonal swellings, also referred to as “axonal varicosities” or “axonal bulbs” (Chen et al., 2009). Progressive accumulation of transported material and cytoskeletal reorganization can result in secondary axotomy (Christman et al., 1994) and degeneration (Raghupathi, 2004). Moreover, an imaging study in humans revealed that white matter abnormalities in key pathways that link relevant structures such as the frontal lobe, the hippocampal formation, or the fornices are linked to cognitive function after TBI (Kinnunen et al., 2011), suggesting that DAI might be a primary mechanism underlying TBI-induced cognitive deficit. Indeed, we have observed high levels of APP-immunoreactive cells in hippocampus and cortex of non-sleep-modulated TBI animals, replicating previous findings.
Both sleep induction with SO and sleep restriction by gentle handling in rats within the first days after trauma appear to prevent animals from developing cognitive impairment. Moreover, both interventions led to a lower number of APP-immunoreactive cells; that is, reduced diffuse axonal damage compared with the control TBI group. However, at first glance, it seems counterintuitive that pharmacological sleep induction and sleep restriction by gentle handling would result in similar improvements. Based on these results and previous findings (Vienne et al., 2010, Bellesi et al., 2013, Hodor et al., 2015, Plante et al., 2015), we then hypothesized that both conditions might produce deeper sleep; that is, increased delta power. In fact, we found that SI animals spent more time in SWS compared with the control group. The same was found in SR rats, but only in the dark period, whereas this increase in SWS faded away over 24 h. However, the common EEG characteristic in both SI and SR animals was an increase in delta power over 24 h. These findings are consistent with previous findings because GHB has been shown to promote oscillatory activity in the low-frequency (delta) range (Entholzner et al., 1995, Meerlo et al., 2004), which is distinct from physiological sleep (Vienne et al., 2010). Similarly, the longer somebody is being kept awake, the more frequent and larger are this person's slow waves during subsequent sleep (Bellesi et al., 2013). This phenomenon of strong SWS rebound after partial sleep deprivation was also described in rats (Borbély et al., 1984, Franken et al., 1991).
It appears that slow oscillatory activity in the delta-frequency range could be the key to functional improvement after TBI. The mechanism behind slow-wave-facilitated improvement could be linked with enhanced clearance of proteins and other waste products from interstitial space in brain. In a recent study, SWS and ketamine–xylazine anesthesia were associated with enhanced clearance of potentially neurotoxic waste products in mice (Xie et al., 2013), whereas a correlation between amyloid β accumulation and disruption of NREM sleep was observed in Alzheimer's disease patients (Mander et al., 2015), further supporting the potential beneficial effects of enhanced slow-wave activity.
The main limitation of the present investigation is related to relatively high within-group variability. As with many mechanical injuries, the nature of TBI is very heterogeneous. However, the behavioral variability of our model appears to correspond well with the natural heterogeneity of human TBI despite a far greater reproducibility of symptoms compared with the human condition. Furthermore, our histological analyses apart from APP were limited. Nevertheless, histological analyses of UB and ATF-4 immunoreactivity did not yield any differences, suggesting that the observed impairment is probably not mediated by ER stress or defect in degradation, but rather that other pathophysiological mechanisms including slower accumulation rate cannot be ruled out. Further studies will be needed to assess the cellular effects of posttraumatic delta power enhancement to explore potential mechanisms of the slow-wave-induced preservation of cognitive abilities.
In summary, both sleep induction and sleep restriction produced enhanced delta power and preserved cognition after TBI and prevented DAI. Interpreting our findings in the light of recent studies (Kang et al., 2009, Xie et al., 2013, Mander et al., 2015), we believe that the increase in delta power after both interventions resulted in increased waste clearance, including APP, preventing exaggerated secondary injury. Therefore, sleep modulation may constitute a novel neuroprotective approach for treating TBI sequelae on both the cellular and symptomatic levels.
Footnotes
We thank Marco Penner for help with adjustments to the TBI device and Evangelia-Regkina Symeonidou for help with behavioral testing.
The authors declare no competing financial interests.
References
- Begum G, Yan HQ, Li L, Singh A, Dixon CE, Sun D. Docosahexaenoic acid reduces ER stress and abnormal protein accumulation and improves neuronal function following traumatic brain injury. J Neurosci. 2014;34:3743–3755. doi: 10.1523/JNEUROSCI.2872-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellesi M, Pfister-Genskow M, Maret S, Keles S, Tononi G, Cirelli C. Effects of sleep and wake on oligodendrocytes and their precursors. J Neurosci. 2013;33:14288–14300. doi: 10.1523/JNEUROSCI.5102-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borbély AA, Tobler I, Hanagasioglu M. Effect of sleep deprivation on sleep and EEG power spectra in the rat. Behav Brain Res. 1984;14:171–182. doi: 10.1016/0166-4328(84)90186-4. [DOI] [PubMed] [Google Scholar]
- Bramlett HM, Dietrich WD. Quantitative structural changes in white and gray matter 1 year following traumatic brain injury in rats. Acta Neuropathologica. 2002;103:607–614. doi: 10.1007/s00401-001-0510-8. [DOI] [PubMed] [Google Scholar]
- Büchele F, Morawska MM, Schreglmann SR, Penner M, Muser M, Baumann CR, Noain D. Novel rat model of weight drop-induced closed diffuse traumatic brain injury compatible with electrophysiological recordings of vigilance states. J Neurotrauma. 2015 doi: 10.1089/neu.2015.4001. In press. [DOI] [PubMed] [Google Scholar]
- Cam E, Gao B, Imbach L, Hodor A, Bassetti CL. Sleep deprivation before stroke is neuroprotective: A pre-ischemic conditioning related to sleep rebound. Exp Neurol. 2013;247:673–679. doi: 10.1016/j.expneurol.2013.03.003. [DOI] [PubMed] [Google Scholar]
- Chen XH, Johnson VE, Uryu K, Trojanowski JQ, Smith DH. A lack of amyloid beta plaques despite persistent accumulation of amyloid beta in axons of long-term survivors of traumatic brain injury. Brain Pathol. 2009;19:214–223. doi: 10.1111/j.1750-3639.2008.00176.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christman CW, Grady MS, Walker SA, Holloway KL, Povlishock JT. Ultrastructural studies of diffuse axonal injury in humans. J Neurotrauma. 1994;11:173–186. doi: 10.1089/neu.1994.11.173. [DOI] [PubMed] [Google Scholar]
- Ennaceur A, Delacour J. A new one-trial test for neurobiological studies of memory in rats. 1: Behavioral data. Behav Brain Res. 1988;31:47–59. doi: 10.1016/0166-4328(88)90157-X. [DOI] [PubMed] [Google Scholar]
- Entholzner E, Mielke L, Pichlmeier R, Weber F, Schneck H. [EEG changes during sedation with gamma-hydroxybutyric acid] Anaesthesist. 1995;44:345–350. doi: 10.1007/s001010050163. [DOI] [PubMed] [Google Scholar]
- Franken P, Dijk DJ, Tobler I, Borbély AA. Sleep deprivation in rats: effects on EEG power spectra, vigilance states, and cortical temperature. Am J Physiol. 1991;261:R198–R208. doi: 10.1152/ajpregu.1991.261.1.R198. [DOI] [PubMed] [Google Scholar]
- Gao B, Kilic E, Baumann CR, Hermann DM, Bassetti CL. Gamma-hydroxybutyrate accelerates functional recovery after focal cerebral ischemia. Cerebrovasc Dis. 2008;26:413–419. doi: 10.1159/000151683. [DOI] [PubMed] [Google Scholar]
- Gentleman SM, Nash MJ, Sweeting CJ, Graham DI, Roberts GW. Beta-amyloid precursor protein (beta APP) as a marker for axonal injury after head injury. Neurosci Lett. 1993;160:139–144. doi: 10.1016/0304-3940(93)90398-5. [DOI] [PubMed] [Google Scholar]
- Hall RC, Hall RC, Chapman MJ. Definition, diagnosis, and forensic implications of postconcussional syndrome. Psychosomatics. 2005;46:195–202. doi: 10.1176/appi.psy.46.3.195. [DOI] [PubMed] [Google Scholar]
- Hamm RJ, Temple MD, Pike BR, O'Dell DM, Buck DL, Lyeth BG. Working memory deficits following traumatic brain injury in the rat. J Neurotrauma. 1996;13:317–323. doi: 10.1089/neu.1996.13.317. [DOI] [PubMed] [Google Scholar]
- Hodor A, Palchykova S, Gao B, Bassetti CL. Baclofen and gamma-hydroxybutyrate differentially altered behavior, EEG activity and sleep in rats. Neuroscience. 2015;284:18–28. doi: 10.1016/j.neuroscience.2014.08.061. [DOI] [PubMed] [Google Scholar]
- Hoskison MM, Moore AN, Hu B, Orsi S, Kobori N, Dash PK. Persistent working memory dysfunction following traumatic brain injury: evidence for a time-dependent mechanism. Neuroscience. 2009;159:483–491. doi: 10.1016/j.neuroscience.2008.12.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iliff JJ, Chen MJ, Plog BA, Zeppenfeld DM, Soltero M, Yang L, Singh I, Deane R, Nedergaard M. Impairment of glymphatic pathway function promotes tau pathology after traumatic brain injury. J Neurosci. 2014;34:16180–16193. doi: 10.1523/JNEUROSCI.3020-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inglese M, Makani S, Johnson G, Cohen BA, Silver JA, Gonen O, Grossman RI. Diffuse axonal injury in mild traumatic brain injury: a diffusion tensor imaging study. J Neurosurg. 2005;103:298–303. doi: 10.3171/jns.2005.103.2.0298. [DOI] [PubMed] [Google Scholar]
- Kang JE, Lim MM, Bateman RJ, Lee JJ, Smyth LP, Cirrito JR, Fujiki N, Nishino S, Holtzman DM. Amyloid-beta dynamics are regulated by orexin and the sleep-wake cycle. Science. 2009;326:1005–1007. doi: 10.1126/science.1180962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinnunen KM, Greenwood R, Powell JH, Leech R, Hawkins PC, Bonnelle V, Patel MC, Counsell SJ, Sharp DJ. White matter damage and cognitive impairment after traumatic brain injury. Brain. 2011;134:449–463. doi: 10.1093/brain/awq347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein C, Mathis C, Leva G, Patte-Mensah C, Cassel JC, Maitre M, Mensah-Nyagan AG. gamma-Hydroxybutyrate (Xyrem) ameliorates clinical symptoms and neuropathology in a mouse model of Alzheimer's disease. Neurobiol Aging. 2015;36:832–844. doi: 10.1016/j.neurobiolaging.2014.10.003. [DOI] [PubMed] [Google Scholar]
- Kraus MF, Susmaras T, Caughlin BP, Walker CJ, Sweeney JA, Little DM. White matter integrity and cognition in chronic traumatic brain injury: a diffusion tensor imaging study. Brain. 2007;130:2508–2519. doi: 10.1093/brain/awm216. [DOI] [PubMed] [Google Scholar]
- Lettieri JT, Fung HL. Dose-dependent pharmacokinetics and hypnotic effects of sodium gamma-hydroxybutyrate in the rat. J Pharmacol Exp Ther. 1979;208:7–11. [PubMed] [Google Scholar]
- Li S, Kuroiwa T, Ishibashi S, Sun L, Endo S, Ohno K. Transient cognitive deficits are associated with the reversible accumulation of amyloid precursor protein after mild traumatic brain injury. Neurosci Lett. 2006;409:182–186. doi: 10.1016/j.neulet.2006.09.054. [DOI] [PubMed] [Google Scholar]
- Lyeth BG, Jenkins LW, Hamm RJ, Dixon CE, Phillips LL, Clifton GL, Young HF, Hayes RL. Prolonged memory impairment in the absence of hippocampal cell death following traumatic brain injury in the rat. Brain Res. 1990;526:249–258. doi: 10.1016/0006-8993(90)91229-A. [DOI] [PubMed] [Google Scholar]
- Mander BA, Marks SM, Vogel JW, Rao V, Lu B, Saletin JM, Ancoli-Israel S, Jagust WJ, Walker MP. beta-amyloid disrupts human NREM slow waves and related hippocampus-dependent memory consolidation. Nat Neurosci. 2015;18:1051–1057. doi: 10.1038/nn.4035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Vargas M, Estrada Rojo F, Tabla-Ramon E, Navarro-Arguelles H, Ortiz-Lailzon N, Hernandez-Chavez A, Solis B, Martinez Tapia R, Perez Arredondo A, Morales-Gomez J, Gonzalez-Rivera R, Nava-Talavera K, Navarro L. Sleep deprivation has a neuroprotective role in a traumatic brain injury of the rat. Neurosci Lett. 2012;529:118–122. doi: 10.1016/j.neulet.2012.09.037. [DOI] [PubMed] [Google Scholar]
- Masel BE, DeWitt DS. Traumatic brain injury: a disease process, not an event. J Neurotrauma. 2010;27:1529–1540. doi: 10.1089/neu.2010.1358. [DOI] [PubMed] [Google Scholar]
- Meerlo P, Westerveld P, Turek FW, Koehl M. Effects of gamma-hydroxybutyrate (GHB) on vigilance states and EEG in mice. Sleep. 2004;27:899–904. doi: 10.1093/sleep/27.5.899. [DOI] [PubMed] [Google Scholar]
- Nava F, Carta G, Bortolato M, Gessa GL. gamma-Hydroxybutyric acid and baclofen decrease extracellular acetylcholine levels in the hippocampus via GABA(B) receptors. Eur J Pharmacol. 2001;430:261–263. doi: 10.1016/S0014-2999(01)01163-3. [DOI] [PubMed] [Google Scholar]
- Plante DT, Goldstein MR, Cook JD, Smith R, Riedner BA, Rumble ME, Jelenchick L, Roth A, Tononi G, Benca RM, Peterson MJ. Effects of partial sleep deprivation on slow waves during non-rapid eye movement sleep: a high density EEG investigation. Clin Neurophysiol. 2015 doi: 10.1016/j.clinph.2015.10.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plog BA, Dashnaw ML, Hitomi E, Peng W, Liao Y, Lou N, Deane R, Nedergaard M. Biomarkers of traumatic injury are transported from brain to blood via the glymphatic system. J Neurosci. 2015;35:518–526. doi: 10.1523/JNEUROSCI.3742-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghupathi R. Cell death mechanisms following traumatic brain injury. Brain Pathol. 2004;14:215–222. doi: 10.1111/j.1750-3639.2004.tb00056.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rostami E, Davidsson J, Ng KC, Lu J, Gyorgy A, Walker J, Wingo D, Plantman S, Bellander BM, Agoston DV, Risling M. A model for mild traumatic brain injury that induces limited transient memory impairment and increased levels of axon related serum biomarkers. Front Neurol. 2012;3:115. doi: 10.3389/fneur.2012.00115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith DH, Okiyama K, Thomas MJ, Claussen B, McIntosh TK. Evaluation of memory dysfunction following experimental brain injury using the Morris water maze. J Neurotrauma. 1991;8:259–269. doi: 10.1089/neu.1991.8.259. [DOI] [PubMed] [Google Scholar]
- Styrke J, Stålnacke BM, Sojka P, Björnstig U. Traumatic brain injuries in a well-defined population: epidemiological aspects and severity. J Neurotrauma. 2007;24:1425–1436. doi: 10.1089/neu.2007.0266. [DOI] [PubMed] [Google Scholar]
- Tobler I, Jaggi K. Sleep and EEG spectra in the Syrian hamster (Mesocricetus auratus) under baseline conditions and following sleep deprivation. J Comp Physiol A. 1987;161:449–459. doi: 10.1007/BF00603970. [DOI] [PubMed] [Google Scholar]
- Tobler I, Deboer T, Fischer M. Sleep and sleep regulation in normal and prion protein-deficient mice. J Neurosci. 1997;17:1869–1879. doi: 10.1523/JNEUROSCI.17-05-01869.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vienne J, Bettler B, Franken P, Tafti M. Differential effects of GABAB receptor subtypes, {gamma}-hydroxybutyric acid, and baclofen on EEG activity and sleep regulation. J Neurosci. 2010;30:14194–14204. doi: 10.1523/JNEUROSCI.3145-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie L, Kang H, Xu Q, Chen MJ, Liao Y, Thiyagarajan M, O'Donnell J, Christensen DJ, Nicholson C, Iliff JJ, Takano T, Deane R, Nedergaard M. Sleep drives metabolite clearance from the adult brain. Science. 2013;342:373–377. doi: 10.1126/science.1241224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu S, Kaneko Y, Bae E, Stahl CE, Wang Y, van Loveren H, Sanberg PR, Borlongan CV. Severity of controlled cortical impact traumatic brain injury in rats and mice dictates degree of behavioral deficits. Brain Res. 2009;1287:157–163. doi: 10.1016/j.brainres.2009.06.067. [DOI] [PubMed] [Google Scholar]