

Abstract
Lipocalin-2 (LCN2) is a member of the highly heterogeneous secretory protein family of lipocalins and increases in its levels can contribute to neurodegeneration in the adult brain. However, there are no reports on the role of LCN2 in Parkinson's disease (PD). Here, we report for the first time that LCN2 expression is increased in the substantia nigra (SN) of patients with PD. In mouse brains, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) treatment for a neurotoxin model of PD significantly upregulated LCN2 expression, mainly in reactive astrocytes in both the SN and striatum. The increased LCN2 levels contributed to neurotoxicity and neuroinflammation, resulting in disruption of the nigrostriatal dopaminergic (DA) projection and abnormal locomotor behaviors, which were ameliorated in LCN2-deficient mice. Similar to the effects of MPTP treatment, LCN2-induced neurotoxicity was also observed in the 6-hydroxydopamine (6-OHDA)-treated animal model of PD. Moreover, treatment with the iron donor ferric citrate (FC) and the iron chelator deferoxamine mesylate (DFO) increased and decreased, respectively, the LCN2-induced neurotoxicity in vivo. In addition to the in vivo results, 1-methyl-4-phenylpyridinium (MPP+)-induced neurotoxicity in cocultures of mesencephalic neurons and astrocytes was reduced by LCN2 gene deficiency in the astrocytes and conditioned media derived from MPP+-treated SH-SY5Y neuronal enhanced glial expression of LCN2 in vitro. Therefore, our results demonstrate that astrocytic LCN2 upregulation in the lesioned DA system may play a role as a potential pathogenic factor in PD and suggest that inhibition of LCN2 expression or activity may be useful in protecting the nigrostriatal DA system in the adult brain.
SIGNIFICANCE STATEMENT Lipocalin-2 (LCN2), a member of the highly heterogeneous secretory protein family of lipocalins, may contribute to neuroinflammation and neurotoxicity in the brain. However, LCN2 expression and its role in Parkinson's disease (PD) are largely unknown. Here, we report that LCN2 is upregulated in the substantia nigra of patients with PD and neurotoxin-treated animal models of PD. Our results suggest that LCN2 upregulation might be a potential pathogenic mechanism of PD, which would result in disruption of the nigrostriatal dopaminergic system through neurotoxic iron accumulation and neuroinflammation. Therefore, inhibition of LCN2 expression or activity may be useful in protecting the nigrostriatal dopaminergic projection in PD.
Keywords: astrocyte, lipocalin-2, neurodegeneration, neuroinflammation, Parkinson's disease
Introduction
Parkinson's disease (PD) is a common neurodegenerative disorder characterized by progressive degeneration of the dopaminergic (DA) projection between the substantia nigra (SN) and the striatum and by a decrease in striatal dopamine, which is associated with clinical movement disorders such as resting tremor, limb rigidity, bradykinesia, and postural instability (Braak et al., 2003; Kim et al., 2011; Kim et al., 2015). These motor manifestations can be treated successfully for a limited period either with drugs that restore DA function or, more recently, with deep-brain stimulation. However, there is no treatment that forestalls the deterioration attributable to progressive neurodegeneration, suggesting that novel strategies to inhibit neurodegeneration should also be considered as potential therapeutics for PD.
Although the etiology of PD is not fully understood, accumulating evidence suggests that neuroinflammatory processes, which are mainly mediated by activated microglia and astrocytes, are crucial for the initiation and progression of PD (McGeer et al., 2005; Hirsch and Hunot, 2009; Menza et al., 2010; Niranjan, 2014). Therefore, the control of glial activation may be useful in prevention and treatment of the degeneration of the nigrostriatal DA projection in the adult brain. Characterization of the action mechanisms of the endogenous biomolecules involved in neuroinflammation may be necessary to find a novel therapeutic strategy for PD based on such control.
Lipocalin-2 (LCN2), also known as 24p3 and neutrophil gelatinase-associated lipocalin (Borregaard and Cowland, 2006), is a member of the highly heterogeneous secretory protein family of lipocalins and plays a variety of physiological roles such as regulation of cell differentiation (Miharada et al., 2008; Shashidharamurthy et al., 2013; Ferreira et al., 2015), apoptotic cell death (Chien et al., 2012; Lin et al., 2012), and cellular uptake of iron (Devireddy et al., 2005; Lee et al., 2012; Jha et al., 2015). LCN2 has been implicated in glial activation in the brain (Lee et al., 2007; Lee et al., 2012; Zamanian et al., 2012) and its expression can be increased in reactive astrocytes and microglia (Ferreira et al., 2015; Jha et al., 2015). LCN2 gene knock-out (KO) exerted neuroprotective effects in animal models of ischemic stroke (Jin et al., 2014) and experimental autoimmune encephalomyelitis (EAE) (Nam et al., 2014). However, Berard et al. (2012) reported a detrimental effect of LCN2 gene deficiency in the EAE model (Berard et al., 2012). In the two previous studies, different amounts of myelin oligodendrocyte glycoprotein were used to induce EAE and animals in different disease conditions were compared. Nevertheless, the role of LCN2 in EAE remains controversial. Moreover, our group and others reported that reactive astrocytes could secrete LCN2, resulting in neurotoxic effects in vitro (Lee et al., 2012; Bi et al., 2013). Choy et al. (2015) showed that LCN2 expression was increased in the SN after medial forebrain bundle (MFB) transection in vivo, suggesting that an increase in LCN2 levels might be related to PD pathogenesis (Choy et al., 2015). However, there are no studies of LCN2 expression in the brains of PD patients or its role in animal models of PD.
In the present study, we investigated the following: (1) whether LCN2 expression is altered in the SN of patients with PD and in the nigrostriatal DA system of the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-treated animal model of PD, (2) whether the alteration of LCN2 expression contributes to neurodegeneration, and (3) how LCN2 is involved in the pathophysiological mechanisms of PD. Similar to MPTP-treated mice, we found LCN2 expression to be upregulated and associated with neurotoxicity in the 6-hydroxydopamine (6-OHDA)-treated animal model of PD. Our results suggest that elevated LCN2 expression may be a universal mechanism of neurodegeneration in the nigrostriatal DA system involved in PD.
Materials and Methods
Institutional review of human protocols
Human tissue experiments were approved by the Bioethics Committee, Institutional Review Board of Kyungpook National University Industry Foundation (IRB no. 2013-0016).
Human brain tissue
Frozen brain tissues were obtained from the Victorian Brain Bank Network, supported by The Florey Institute of Neuroscience and Mental Health and The Alfred and the Victorian Forensic Institute of Medicine, and funded by Australia's National Health and Medical Research Council and Parkinson's Victoria. The levels of tyrosine hydroxylase (TH) and LCN2 expression in the human SN tissues (Table 1) were analyzed by Western blotting.
Table 1.
Human postmortem tissues used for Western blot analysis in Figure 1
| Case no. | Diagnosis | PMI | Age | Tissue |
|---|---|---|---|---|
| V12/019 | Control | 25 | 81.2 | SN |
| 03/833 | 57 | 79.3 | ||
| 04/250 | 31.5 | 79.6 | ||
| 07/239 | 19 | 78.8 | ||
| 04/034 | 26.5 | 73.7 | ||
| 04/112 | 22 | 73.5 | ||
| 06/144 | 24 | 69.4 | ||
| V12/008 | PD | 30 | 81.1 | SN |
| 09/292 | 59 | 79.9 | ||
| 10/002 | 39 | 79.5 | ||
| V12/012 | 20 | 79.5 | ||
| V11/073 | 65 | 80.1 | ||
| 05/413 | 45 | 72.7 | ||
| 08/319 | 32.5 | 70 |
PMI, Postmortem interval (h).
Mice and animal care procedures
Male C57BL/6 mice (8–12 weeks old) were obtained from Samtaco and LCN2 KO mice in a C57BL/6 background were kindly provided by Drs. Kiyoshi Mori (Kyoto University, Japan) and Shizuo Akira (Osaka University, Japan). All animal experiments were performed in accordance with the approved animal protocols and guidelines established by the Animal Care Committee at Kyungpook National University (no. KNU 2015-0010).
LCN2 KO mice
LCN2 KO mice were back-crossed for 8–10 generations into a C57BL/6 background to generate homozygous and heterozygous animals, as described previously (Flo et al., 2004). The deletion of the LCN2 gene was confirmed by PCR analysis of genomic DNA (Nairz et al., 2009). Animals used in the present study were acquired and cared for in accordance with the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health.
MPTP injection
For MPTP treatment, mice received four intraperitoneal injections of MPTP (20 mg/kg free base; Sigma-Aldrich) dissolved in saline at 2 h intervals and the brain tissues were analyzed at the indicated time points for each experiment.
Traditional reverse transcription PCR (RT-PCR) and quantitative real time-PCR (qRT-PCR)
Total RNA from the ventral midbrain and striatum of saline or MPTP-injected mice was isolated with QIAzol (QIAGEN). First-strand cDNA was synthesized from total RNA using a first-strand cDNA synthesis kit (MBI Fermentas) according to the manufacturer's instructions. The primers used in RT-PCR analyses of mouse LCN2 and GAPDH were as follows: LCN2, 5′-ATGTCACCTCCATCCTGGTC-3′ (forward) and 5′-CACACTCACCACCCATTCAG-3′ (reverse); GAPDH, 5′-ACCACAGTCCATGCCATCAC-3′ (forward), 5′-TCCACCACCCTGTTGCTGTA-3′ (reverse). The primers used in qRT-PCR analyses of mouse LCN2 and GAPDH were as follows: LCN2, 5′-CCCCATCTCTGCTCACTGTC-3′ (forward) and 5′-TTTTTCTGGACCGCATTG-3′ (reverse); GAPDH, 5′-TGGGCTACACTGAGCACCAG-3′ (forward), 5′-GGGTGTCG-CTGTTGAAGTCA-3′ (reverse). Real-time RT-PCR was performed using the One-step SYBR PrimeScript RT-PCR kit (Perfect Real Time; Takara Bio) and the ABI Prism 7000 sequence detection system (Applied Biosystems) according to the manufacturer's instructions.
Western blotting analysis
Animal or human tissue samples were homogenized in a protein lysis buffer (20 mm HEPES, pH 7.0, 20 mm NaCl, 10% glycerol, 0.5% Triton X-100) containing protease and phosphatase inhibitor mixture (Roche) and centrifuged at 4°C for 20 min at 14,000 × g. The supernatant was transferred to a fresh tube and the concentration was determined using the BCA kit (Thermo Scientific). An equal amount of protein for each sample was loaded onto an SDS-PAGE gel (10–12%). Proteins separated by gel electrophoresis were transferred onto polyvinylidene difluoride membranes (Millipore) using an electrophoretic transfer system (Bio-Rad Laboratories) and the membranes were incubated overnight at 4°C with the following primary antibodies: goat anti-LCN 2 (1:500; R&D Systems), rabbit anti-TH (1:1000; Millipore), mouse anti-TNF-α, rabbit anti-IL-1β (1:1000; Santa Cruz Biotechnology), mouse anti-α-tubulin (1:2000; Sigma-Aldrich), and mouse anti-β-actin (1:2000; Thermo Scientific). After washing, the membranes were incubated with horseradish peroxidase-conjugated specific secondary antibodies (1:2000; Sigma-Aldrich) for 1 h. The blots were developed with ECL Western-blotting detection reagents (Thermo Scientific), and analyzed on the MicroChemi system (DNR Bio-imaging Systems).
Intranigral injection of materials
After anesthetization with ketamine mixture, mice were placed in a stereotaxic apparatus (Kopf Instruments). For the immunohistochemical analysis of the SN shown in Figure 4, each mouse received an unilateral injection of saline as a control, ferric citrate (FC, 2.5 mm), or deferoxamine mesylate (DFO, 50 mm) into the left SN [anteroposterior (AP), −2.9 mm; mediolateral (ML), 1.3 mm; dorsoventral (DV), −4.2 mm relative to the bregma, according to the atlas of Paxinos and Watson] using a 26-gauge Hamilton syringe attached to an automated microinjector and then received an unilateral injection of recombinant LCN2 (1 μg), a mixture of recombinant LCN2 and FC, or a mixture of recombinant LCN2 and DFO into the right SN (AP, −2.9 mm; ML, −1.3 mm; DV, −4.2 mm). For the immunohistochemical analyses of the SN and behavioral test shown in Figure 3, each mouse received a bilateral injection of recombinant LCN2 protein or denatured LCN2 protein (1 μg) in the right and left SN. Infusions were made at a rate of 0.2 μl/min for 10 min. After each injection, the needle was left in place for an additional 5 min before being retracted slowly. Animals were killed and analyzed at the indicated time points for each experiment.
Figure 4.

Iron uptake upregulation enhances LCN2-mediated neurotoxicity in the SN. To clarify whether the function of LCN2 as an iron transporter contributes to neurotoxicity in the SN, we injected LCN2, FC, DFO, a mixture of LCN2 and FC, or a mixture of LCN2 and DFO into the SN and measured neurotoxicity by TH immunostaining 6 d after each treatment. Scale bar, 500 μm. Similar to the neurotoxicity in the SN shown in Figure 3, LCN2 treatment induced a significant decrease in the number of TH-positive neurons. In addition, FC alone also induced a loss of DA neurons in the SN and its neurotoxicity was enhanced significantly by cotreatment with FC and LCN2. However, LCN2-induced neurotoxicity was attenuated significantly by cotreatment with LCN2 and DFO, which showed no neurotoxicity on their own. *p < 0.01 and **p < 0.001 versus saline-treated mice; #p < 0.05 between the indicated group (one-way ANOVA; n = 5 for each group).
Figure 3.

LCN2 upregulation induces disruption of the nigrostriatal DA projection in vivo. A, Double immunofluorescence staining for LCN2 receptor (LCN2R; green) and TH (red) in the SN of a mouse brain reveals that the DA neurons express LCN2R. Scale bar, 50 μm. Arrows indicate colocalization of LCN2R and TH-positive neurons. B, Schematic diagram of the experimental design to determine whether LCN2 upregulation by treatment with recombinant LCN2 induces disruption of the nigrostriatal DA projection in vivo. C, D, Disruption of the nigrostriatal DA projection (SN, C; striatum, D) examined by TH staining is apparent at 6 d after LCN2 injection into the SN of the mouse brain. Denatured LCN2 (Denat. LCN2) has no neurotoxic effect. Scale bar, 500 μm. C, Histogram results showing the number of TH-positive neurons counted using a stereological technique in the entire SN. **p < 0.01 and ##p < 0.01 versus saline-treated mice and LCN2-treated mice, respectively (one-way ANOVA; n = 4 for untreated mice, n = 5 for other groups). D, Histogram showing the optical density of TH-positive fibers after immunoperoxidase staining in the striatum. **p < 0.01 and ##p < 0.01 versus saline-treated mice and LCN2-treated mice, respectively (one-way ANOVA; n = 4 for untreated mice, n = 5 for other groups). Consistent with the results obtained by immunoperoxidase staining, immunofluorescence staining for TH showed that LCN2 treatment induced a significant loss of TH-positive fibers in the striatum compared with controls. *p < 0.05 and #p < 0.05 versus saline-treated mice and LCN2-treated mice, respectively (one-way ANOVA; n = 4 for untreated mice, n = 5 for other groups). E, Locomotor deficits were assessed by pole test performance. *p < 0.05 and #p < 0.05 versus saline-treated mice and LCN2-treated mice, respectively (one-way ANOVA; n = 5 for untreated mice, n = 8 for other groups). F, Motor deficits were estimated by the latency to fall in the rotarod test. **p < 0.01 and ##p < 0.01 versus saline-treated mice and LCN2-treated mice, respectively (one-way ANOVA; n = 3 for untreated mice, n = 4 for other groups). G, Total distance traveled in 5 min and velocity were measured in the open-field test. #p < 0.05 versus LCN2-treated mice (t test; n = 3 for untreated mice, n = 4 for other groups).
Intrastriatal injection of 6-OHDA
For the 6-OHDA-treated mouse model of PD, mice were anesthetized using a ketamine mixture while immobilized in a stereotaxic frame (Kopf Instruments). Each mouse was pretreated with an intraperitoneal injection of 20 mg/kg desipramine 30 min before 6-OHDA administration, followed by a unilateral injection of 6-OHDA (Sigma-Aldrich; 5 μg/μl in 0.9% NaCl/0.02% ascorbate) into the right striatum (AP, +0.9 mm; ML, −2.2 mm; DV, −2.5 mm) at a rate of 0.5 μl/min for a total dose of 15 μg as described previously (Kim et al., 2012; Nam et al., 2015a; Kim et al., 2016). After each injection, the needle was left in place for 5 min before being retracted slowly.
Immunohistochemical staining
As described previously with some modifications (Kim et al., 2011; Kim et al., 2015), animals were transcardially perfused and fixed and the brain sections (30 μm thick) were rinsed in PBS and then incubated for 48 h with the following primary antibodies: rabbit anti-TH (1:1000; Millipore), goat anti-LCN2 (1:400; R&D Systems), rabbit anti-ionized calcium-binding adapter molecule 1 (Iba-1, 1:400; Wako), rabbit anti-glial fibrillary acidic protein (GFAP, 1:1000; Dako), rabbit anti-LCN2R (1:200; Sigma-Aldrich), and rabbit anti-NeuN (Novus Biologicals). For immunohistochemical staining, the sections were incubated with biotin-conjugated secondary antibodies for 1 h at room temperature, followed by an avidin-biotin complex kit (Vector Laboratories) for 1 h, and then the signal was detected by a DAB kit (Vector Laboratories). For immunofluorescence staining, the sections incubated with primary antibodies were rinsed and incubated with FITC and Cy3-conjugated secondary antibodies (1:200; Jackson Laboratories). The stained tissues were analyzed by confocal and bright-field microscopy (Axio Imager; Carl Zeiss).
Stereological estimation of TH-positive neurons and Iba-1- and GFAP-positive cells
Stereological cell counting was performed as described previously (Kim et al., 2011; Kim et al., 2015; Nam et al., 2015b; Shin et al., 2015). The total numbers of TH-positive neurons in the various animal groups were determined using the optical fractionator method under a bright-field microscope (Olympus, BX51) and Stereo Investigator software (MBF Bioscience). This unbiased stereological method of cell counting is not affected by either the reference volume or the size of the counted elements (neurons). For the numbers of Iba-1- and GFAP-positive cells, sections stained with Iba-1 or GFAP antibodies were analyzed stereologically using the optical fractionator method as described above with some modifications for the total area of the striatum in the mouse brain (Kim et al., 2012). Microglia and astrocytes were expressed quantitatively as the percentage of the number of Iba-1- and GFAP-positive cells, respectively, relative to the control.
Quantitative determination of striatal TH-positive fibers
Densitometric analyses of the mouse striatum were performed as described previously with some modifications (Kim et al., 2011; Nam et al., 2015a). Briefly, an average of eight coronal sections of the striatum were captured at 1.25× magnification and the fiber density was measured using the Science Lab 2001 Image Gauge (Fujifilm). To clarify the results of the measurement of striatal optical density after immunoperoxidase staining, the quantitative data obtained by immunofluorescence staining were added as supporting results. TH-positive fiber innervation in the striatum was expressed quantitatively as the percentage of the optical density and fluorescence intensity relative to the control.
Behavioral testing
Pole test.
The pole test for bradykinesia was performed as described previously (Kim et al., 2014; Kim et al., 2015). Briefly, mice were placed head-upward at the top of a rough-surfaced pole (8 mm in diameter and 55 cm in height). Performance was measured as the total time it took each mouse to arrive at the floor from the top. Before the actual test, mice were trained in 5 trials/d for 3 d. The locomotor activity of each mouse was evaluated as the average of 5 trials performed at 6 d after MPTP and LCN2 administration.
Rotarod test.
The rotarod test was performed as described previously with slight modifications (Rozas et al., 1998). Mice were trained on the rotarod apparatus (3 cm rod diameter; Ugo Basile) at a fixed speed of 10 rpm for 600 s once daily for 3 consecutive days. Six days after the injection, performance on the rod was evaluated at a constant acceleration rate of 4–40 rpm in 300 s. Two consecutive trials were performed at 60 min intervals.
Open-field test.
The open-field test was performed as described previously (Jang et al., 2013). In brief, 6 d after the injection, mice were placed individually in the corner of an arena (40 × 40 × 40 cm) enclosed in white acrylic walls. After a 1 min adaptation period, animal behaviors such as the total distance traveled (in centimeters) and velocity (in centimeters per second) were recorded for 5 min using a video camera connected to a PC. Then, the alteration of locomotor activity was analyzed offline by a video-tracking software (ABCLab). The arena was wiped between trials with 70% ethyl alcohol. To minimize stress levels, tests were performed under a low-illumination red light.
Cell culture
Primary mesencephalic DA neurons.
Mesencephalic DA neurons were isolated from the ventral midbrains of embryonic day 16.5 (E16.5) mice as described previously (Choi et al., 2013). Briefly, tissues were homogenized in Hank's balanced salt solution (HBSS; Invitrogen) containing collagenase/DNase (Sigma-Aldrich). Resulting homogenates were passed through a nylon cell strainer and centrifuged at 500 × g for 10 min. The pellets were dissociated mechanically by gentle pipetting and the dissociated cells were seeded onto plates coated with poly-d-lysine (Sigma-Aldrich) in Neurobasal medium containing 2 mm l-glutamine (Sigma-Aldrich), penicillin–streptomycin, nerve growth factor (Invitrogen), N2 supplement (Invitrogen), and B27 supplement (Invitrogen). Cells were used for experiments after being cultured for 7 d. Cells were plated in 24-well plates (2 × 105 cells/well) or 6-well plates (5 × 105 cells/well) and washed 1 h later to remove the unattached cells. The purity of the mesencephalic neuron cultures was assessed using the TH antibody, with >90% of cells staining positive.
Mixed glial cultures.
Mixed glial cultures were prepared as described previously with minor modifications (Lee et al., 2009). In brief, the whole brains of 2- or 3-d-old mice were chopped and disrupted mechanically using a nylon mesh. The cells obtained were seeded in culture flasks and grown at 37°C in a 5% CO2 atmosphere in DMEM supplemented with 10% FBS and 100 U/ml penicillin–streptomycin (Invitrogen). Culture media were changed initially after 5 d and then every 3 d. The cells were used after being cultured for 14–21 d.
Primary astrocyte cultures.
Primary astrocytes were obtained by shaking mixed glial cultures at 200 rpm overnight. The culture media were discarded and astrocytes were dissociated using trypsin-EDTA (Invitrogen) and collected by centrifugation at 1200 rpm for 10 min. Primary astrocyte cultures were grown and maintained in DMEM supplemented with 10% FBS and penicillin–streptomycin.
SH-SY5Y human neuroblastoma.
SH-SY5Y cells were obtained from the American Type Culture Collection and cultured in DMEM supplemented with 10% FBS and 100 U/ml penicillin/streptomycin at 37°C in a humidified atmosphere of 5% CO2. Cell differentiation was induced by the addition of 10 μm all-trans retinoic acid (RA; Sigma-Aldrich) dissolved in ethanol as a 10 mm stock solution. The cells were fed fresh medium containing RA every other day. On day 5 of differentiation, the cells were washed 3 times with DMEM and incubated with MPP+ (1 μm) at the indicated time points.
Astrocyte–neuron cocultures.
Mesencephalic neurons were plated at a density of 2 × 105 cells per well in 24-well companion plates and allowed to settle at 37°C in a 5% CO2 atmosphere for 7 d. Primary astrocytes were separately plated at a density of 2 × 105 cells per well in cell culture inserts (0.4 μm pore size; BD Biosciences) and allowed to settle at 37°C in a 5% CO2 atmosphere overnight. The mesencephalic neurons were pretreated with MPP+ (1 μm), and cell culture inserts containing astrocytes were transferred into wells containing the mesencephalic neurons. After 24 h of incubation, the cell culture inserts were removed and the viability of neurons was measured.
Assessment of neuronal viability
Cocultures of mesencephalic neurons and astrocytes, which were produced by adding culture inserts containing astrocytes into wells of neuronal cultures, were treated with MPP+ (1 μm) for 24 h and the culture inserts containing the astrocytes were removed 24 h after the MPP+ treatment. The neuronal viability was measured using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay as described previously (Lee et al., 2009). In brief, MTT (0.5 mg/ml; Sigma-Aldrich) was added after removing the culture media and the neurons were incubated at 37°C in a 5% CO2 incubator. Insoluble formazan crystals induced by the MTT treatment were dissolved completely in DMSO and the absorbance at 570 nm was measured using a microplate reader (Molecular Devices). To clarify the results of the MTT assays, a lactate dehydrogenase (LDH) release assay was also performed as described previously (Hwang et al., 2010). For total LDH determination, cells were incubated in 96-well plates with the indicated concentrations of recombinant LCN2 (1, 2, or 4 μg/ml) or 100 μm MPP+ for 4 d. The cell suspension was centrifuged at 4000 × g for 5 min at 4°C and the supernatant was collected. The LDH contents of supernatant aliquots were determined using a cytotoxicity assay kit according to the manufacturer's instructions (Takara). Absorbance was read at 440 nm and LDH-expressed cytotoxicity (%) was calculated using the following formula: (supernatant value − blank value)/[(supernatant value − blank value) + (upper control value − blank value)] × 100%.
Measurement of LCN2 protein levels
The levels of LCN2 protein in culture media were determined using the mouse Lipocalin-2/NGAL DuoSet ELISA kit (R&D Systems) according to the manufacturer's instructions. The optical densities of the standards and samples were measured by subtracting the readings at 540 nm from the readings at 450 nm using Softmax software (Molecular Devices).
Gelatin zymography
Conditioned media were collected from differentiated SH-SY5Y cells treated with MPP+ (100 μm) for 24 h and gelatin zymography was performed to measure the activities of matrix metalloproteinase (MMP)-2 and MMP-9. Twenty microliters of culture medium was loaded onto an 8% SDS-PAGE gel containing 1 mg/ml gelatin under nonreducing conditions. After electrophoresis, the gel was washed 2 times in 2.5% Triton X-100 for 20 min at 22–24°C to remove the SDS. The gel was washed another 2 times in deionized water at 22–24°C and incubated in reaction buffer (50 mm Tris, pH 7.5, 10 mm CaCl2, 150 mm NaCl, and 2 μm ZnCl2) for 24 h at 37°C. After the reaction buffer incubation, the gel was stained with 1% Coomassie Brilliant Blue R-250 for 1 h at 22–24°C, followed by incubation in destaining buffer (10% MeOH, 5% AcOH). An MMP-9 standard was used at a concentration of 100 ng/ml (R&D Systems). MMP-2 and MMP-9 activities were visualized as clear bands in the blue background of stained undigested gelatin.
Statistical analysis
All data were analyzed using GraphPad Prism version 5.01 software. All data are expressed as mean ± SEM. The statistical analysis was performed by comparing the means of different groups using the unpaired two-tailed Student's t test, one-way ANOVA with Tukey's multiple-comparisons test, and two-way ANOVA with the post hoc Bonferroni test. P < 0.05 was considered significant.
Results
Upregulation of LCN2 expression in the SN of patients with PD
To examine the alteration of TH and LCN2 expression in the SN of patients with PD compared with that in age-matched controls, we measured the TH and LCN2 protein levels by Western blotting of SN samples obtained from postmortem PD and age-matched control brains (Table 1). Our results showed that TH expression was decreased significantly in the SN of patients with PD compared with age-matched controls (Fig. 1A; p = 0.024), whereas LCN2 expression was increased significantly in the SN of patients with PD compared with age-matched controls (Fig. 1A; p = 0.039). These observations suggest that the loss of DA neurons might be associated with an increase in LCN2 expression in the adult brain. However, further comparative analyses using larger numbers of PD patients and age-matched controls will be necessary to find any statistically significant correlations between the level of LCN2 expression and the clinical history of each patient (e.g., disease duration, cause of death, dementia score, and family history). Such information on the limited number of samples used in this study was incomplete (data not shown).
Figure 1.

LCN2 upregulation in the brains of patients with PD and MPTP-treated mice. A, Western blot analyses of TH and LCN2 in SN tissues from patients with PD and age-matched controls. TH and LCN2 expression is significantly decreased and increased, respectively, in the SN of patients with PD compared with age-matched controls (p = 0.024 for TH; p = 0.039 for LCN2; t test; n = 7 for each group). B, LCN2 mRNA levels in the striatum and SN of mouse brains after MPTP treatment were determined by qRT-PCR (top) and traditional RT-PCR (bottom). LCN2 expression is increased significantly 24 h after MPTP administration compared with saline-treated controls. *p < 0.05 and **p < 0.01 (one-way ANOVA; n = 3 for each group). C, Western blot analysis of LCN2 protein expression in the striatum (left) and SN (right) of mouse brains. Similar to the mRNA levels, the expression of LCN2 protein is significantly upregulated at 24 h compared with saline-treated controls. *p < 0.05 and **p < 0.01 (one-way ANOVA; n = 3 for each group). D, Comparison of LCN2 mRNA levels in the SN, striatum (STR), hippocampus (HIP), and cerebral cortex (CC) after MPTP administration in mice. The upregulation of LCN2 mRNA is more apparent in the nigrostriatal DA system than the HIP and CC. *p < 0.05 and **p < 0.01 versus saline-treated controls (one-way ANOVA; n = 3 for each group; ns, not significant).
LCN2 upregulation in the MPTP-treated animal model of PD
The rodent model of PD using the neurotoxic substance MPTP, which targets the nigrostriatal DA system specifically, has been useful in establishing the possible cellular and molecular underpinnings of this neurodegenerative condition (Moretto and Colosio, 2013). To clarify whether the MPTP-treated animal model of PD can be used to determine the role of LCN2 upregulation in neurodegeneration, we first examined the alteration of LCN2 expression in the nigrostriatal DA system after intraperitoneal injections of MPTP and saline as a control. Similar to the patient data, there were significant increases in the LCN2 mRNA (Fig. 1B; *p < 0.05 and **p < 0.01 vs saline controls) and protein (Fig. 1C; *p < 0.05 and **p < 0.01 vs saline controls) levels, which were determined by PCR and Western blotting, respectively, in both the SN and striatum at 24 h after the first MPTP treatment. Moreover, the LCN2 upregulation induced by MPTP administration was more apparent in the nigrostriatal DA system, including the SN and striatum, than in other brain areas such as the hippocampus and cerebral cortex (Fig. 1D), suggesting that the animal model of PD induced by MPTP treatment can be used to study the role of LCN2 in PD pathogenesis.
Localization of LCN2 protein in reactive astrocytes
To determine the cellular localization of LCN2 protein in the striatum and SN of MPTP-treated mice, double immunofluorescence staining for LCN2 and GFAP or Iba-1 in the brain was performed 24 h after the MPTP administration. Consistent with the results of Western blotting, LCN2 upregulation was observed in both the striatum and SN of the MPTP-treated mice (Fig. 2A). The LCN2 staining was localized mainly within reactive astrocytes, even though some activated microglia also showed LCN2 overexpression (Fig. 2B,C). Consistent with the results in Figure 2A, saline treatment did not cause an increase in LCN2 expression or glial activation (Fig. 2B,C). Moreover, there was no LCN2 upregulation in neurons in the striatum and SN after MPTP treatment (data not shown). These results suggest that LCN2 upregulation in the brains of MPTP-treated mice is associated with glial activation and reactive astrocytes are the major cell type expressing LCN2.
Figure 2.

LCN2 upregulation in the glia after MPTP administration. A, Immunofluorescence staining of LCN2 in the striatum and SN of mouse brains at 24 h after MPTP treatment. Note that LCN2 expression is upregulated in both the striatum and SN of the MPTP-treated mouse. Scale bar, 50 μm. B, C, Colocalization of LCN2 expression with glial cells in the striatum (B) and SN (C) by double immunofluorescence staining. LCN2 (red) upregulation after MPTP administration is mainly localized within GFAP-positive astrocytes (green) in both the striatum and SN and partially in the Iba-1-positive microglia (green) at 24 h after injection. Scale bar, 50 μm. Arrows indicate colocalization of LCN2 and astrocytes/microglia.
LCN2 neurotoxicity in the nigrostriatal DA system
As described in a previous study, LCN2 binds to a cell surface receptor (LCN2R, also known as 24p3R) to regulate cell death and survival (Devireddy et al., 2005; Lee et al., 2012; Jha et al., 2015). As shown in Figure 3A, we confirmed that nigral DA neurons expressed LCN2R, suggesting that glial LCN2 upregulation might contribute to the degeneration of DA neurons. Therefore, we evaluated the neurotoxic effects of recombinant LCN2 protein in the nigrostriatal DA system of the mouse brain (Fig. 3B). A significant loss of the nigrostriatal DA projection was observed 6 d after an intranigral injection of LCN2 protein, but not in the untreated or saline-treated controls (Fig. 3C,D). The administration of 1 μg of LCN2 protein was found to significantly reduce the number of DA neurons and the optical density/fluorescence intensity of DA fibers. These were expressed as the number of TH-positive neurons and the percentage of the optical density and fluorescence intensity relative to the control in the SN and striatum, respectively (*p < 0.05 and **p < 0.01 vs saline controls). There was no neurotoxicity caused by treatment with denatured LCN2 protein (Fig. 3C,D; #p < 0.05 and ##p < 0.01 vs LCN2-treated mice, but no significance vs controls). Consistent with the disruption of the DA projection, LCN2-injected mice showed significant behavioral impairments as demonstrated by the pole test (Fig. 3E; *p < 0.05 vs saline-injected controls) and rotarod test (Fig. 3F; **p < 0.01 vs saline-injected controls), but not in denatured LCN2-treated mice (Fig. 3E,F; #p < 0.05 and ##p < 0.01 vs LCN2-treated mice, but no significance vs controls). Moreover, the open-field test showed that there was a significant difference as demonstrated by Student's t test between LCN2- and denatured LCN2-treated mice (Fig. 3G; #p < 0.05 vs LCN2-treated mice). These data suggest that LCN2-induced neurotoxicity could lead to abnormal locomotor activities associated with the lesioned DA system.
LCN2 plays a role in transferring iron into cells (Devireddy et al., 2005). Moreover, intracellular iron accumulation is involved directly in PD pathogenesis (Zecca et al., 2004; Hare et al., 2013; Ward et al., 2014; Ayton et al., 2015; Hare et al., 2015). Therefore, to investigate whether LCN2 upregulation contributes to neurodegeneration through intracellular iron accumulation in DA neurons, we evaluated the neurotoxic effects of recombinant LCN2 protein in the presence of an iron supplement or an iron chelator in the nigrostriatal DA system of mouse brains. Intranigral cotreatment with the iron donor FC and LCN2 enhanced the LCN2-induced loss of DA neurons in the SN significantly (#p < 0.05 vs LCN2 alone), whereas LCN2-induced neurotoxicity was attenuated by cotreatment with the iron chelator DFO (Fig. 4; #p < 0.05 vs LCN2 alone). Together, these results suggest that LCN2 upregulation contributes to neurodegeneration in the nigrostriatal DA system and intracellular iron accumulation may be an important mechanism underlying LCN2 neurotoxicity in the adult brain.
LCN2 deficiency protects the nigrostriatal DA projection against MPTP neurotoxicity in vivo
To determine whether the control of LCN2 expression might be useful in protecting the nigrostriatal DA system in the adult brain, we evaluated MPTP-induced neurotoxicity in LCN2-deficient mice compared with normal mice. At 7 d after treatment with saline or MPTP, MPTP-injected WT mice showed an apparent loss of the nigrostriatal DA projection compared with saline-treated controls, which was attenuated significantly in LCN2-deficient (KO) mice (Fig. 5B,C). MPTP treatment reduced the number of DA neurons (**p < 0.001) and the optical density/fluorescence intensity of the DA fibers (**p < 0.001) significantly when the number of TH-positive neurons and the density/intensity of the TH-positive fibers were quantified. These were expressed as the percentage of the optical density and fluorescence intensity relative to the control in the SN and striatum, respectively. However, LCN2-deficient mice were protected significantly against the MPTP-induced TH-neuronal loss and terminal loss in the SN and striatum, respectively (#p < 0.05 and ##p < 0.01, respectively, vs MPTP-treated WT mice). Consistent with the results of immunohistochemical staining, the abnormal locomotor behaviors induced by MPTP neurotoxicity were ameliorated significantly in LCN2-deficient mice as demonstrated by the pole test (Fig. 5D; #p < 0.05 vs MPTP-treated WT mice), rotarod test (Fig. 5E; #p < 0.05 vs MPTP-treated WT mice), and open-field test (Figure 5F; ##p < 0.01 vs MPTP-treated WT mice, t test). There was no significant difference in the baseline performance between WT mice and LCN2-deficient mice (data not shown). Therefore, these results suggest that the control of LCN2 upregulation in pathophysiological conditions may be used to protect the nigrostriatal DA system in the adult brain. The results also support an important role of LCN2 in PD pathogenesis.
Figure 5.

LCN2 deficiency protects the nigrostriatal DA projection in the MPTP-treated model of PD. A, Diagram of the experimental design to determine whether LCN2 gene ablation affects MPTP neurotoxicity. B, Representative images of TH immunoreactivity in SN sections and the number of TH-positive neurons. Scale bar, 500 μm. **p < 0.001 and ##p < 0.01 versus saline-treated WT mice and MPTP-treated WT mice, respectively (two-way ANOVA; n = 8 for each group). C, Representative images of TH-positive fibers in striatum sections, their optical density, and fluorescence intensity. Scale bar, 500 μm. **p < 0.001 and ##p < 0.01 in immunoperoxidase staining versus saline-treated controls and MPTP-treated WT mice, respectively (two-way ANOVA; n = 5 for each group). **p < 0.001 and #p < 0.05 in immunofluorescence staining versus saline-treated controls and MPTP-treated WT mice, respectively (two-way ANOVA; n = 5 for each group). D, Ablation of the LCN2 gene alleviates the MPTP-induced behavioral deficit, which was measured by pole test performance. *p < 0.01 and #p < 0.05 versus saline-treated WT mice and MPTP-treated WT mice, respectively (two-way ANOVA; n = 9 for each group). E, Motor deficits were measured using the rotarod test. *p < 0.01 and #p < 0.05 versus saline-treated WT mice and MPTP-treated WT mice, respectively (two-way ANOVA; n = 4 for saline-treated WT mice, n = 5 for other groups). F, Total distance traveled in 5 min and velocity were measured using the open-field test. ##p < 0.01 versus MPTP-treated WT mice (t test; n = 3 for saline-treated WT mice, n = 4 for other groups).
LCN2 deficiency inhibits glial activation in the MPTP-treated model of PD
Glial activation is a well known pathological feature of the brains of PD patients and animal models of PD (Teismann et al., 2003; Hirsch and Hunot, 2009; Rappold and Tieu, 2010; Cabezas et al., 2014). To investigate whether LCN2 deficiency in the MPTP-treated model of PD inhibits glial activation and subsequent production of neurotoxic proinflammatory cytokines, we evaluated the total number of microglia and astrocytes in both the striatum and SN after MPTP administration. We also examined the changes in interleukin-1-β (IL-1β) and tumor necrosis factor-α (TNF-α) expression in the brains of LCN2-deficient mice compared with WT mice. At 3 d after the MPTP injection, brain sections were immunostained with anti-Iba-1 and anti-GFAP antibodies to label the microglia and astrocytes, respectively. As shown in the high-magnification images of Figure 6, A and B, MPTP-induced neurotoxicity was accompanied by morphological changes in microglia and astrocytes. In addition, we observed that the numbers of microglia and astrocytes were increased significantly in both the striatum and SN of MPTP-treated WT mice (Fig. 6A,B; **p < 0.001 vs saline-treated WT mice). However, consistent with the mild morphological changes in the glia, the increases in the numbers of microglia and astrocytes were attenuated significantly in the MPTP-treated LCN2-deficient mice compared with the MPTP-treated WT mice (Fig. 6A,B; #p < 0.05, ##p < 0.01, and ###p < 0.001 vs MPTP-treated WT mice). Moreover, Western blot analyses of TNF-α and IL-1β expression showed that the significant increases in the levels of both neurotoxic cytokines in the SN of WT mice after MPTP treatment (Fig. 6C; *p < 0.01 vs saline-treated WT mice) were absent in the LCN2-deficient mice (Fig. 6C; #p < 0.05 vs MPTP-treated WT mice). These data suggest that LCN2 might be an important trigger of glial activation and neuroinflammation in the MPTP-treated model of PD.
Figure 6.

MPTP-induced glial activation is attenuated in LCN2-deficient mice. A, Images of striatal immunohistochemical staining with anti-Iba-1 and anti-GFAP antibodies for microglia and astrocytes, respectively, after saline or MPTP treatment. Scale bars, 100 and 25 μm (higher-magnification images). The histograms indicate the number of microglia (top) and astrocytes (bottom) in the striatum. **p < 0.001 versus saline-treated WT mice; ###p < 0.001 and #p < 0.05 versus MPTP-treated WT mice (two-way ANOVA; n = 4 for saline groups, n = 5 for MPTP groups). B, Nigral immunohistochemical images of staining with anti-Iba-1 and anti-GFAP antibodies after saline or MPTP treatment. Scale bars, 100 and 25 μm (higher-magnification images). Similar to the results in the striatum, increases in the numbers of microglia and astrocytes are observed in the SN of the WT mouse brains after MPTP treatment compared with the saline-treated controls. However, those increases are reduced significantly in LCN2-deficient mice. **p < 0.001 versus saline-treated WT mice; #p < 0.05 and ##p < 0.01 versus MPTP-treated WT mice (two-way ANOVA; n = 4 for saline groups, n = 5 for MPTP groups). C, Western blot analysis of TNF-α and IL-1β expression in the SN of mouse brains 3 d after MPTP treatment. Representative gel images show that MPTP administration induces significant increases in TNF-α and IL-1β expression in the SN of WT mice (p < 0.01 vs saline-treated WT mice). However, the upregulation of the cytokine levels in the SN of WT mice is attenuated in the SN of LCN2-deficient mice. *p < 0.01 versus saline-treated WT mice; #p < 0.05 versus MPTP-treated WT mice (two-way ANOVA; n = 3 for each group).
LCN2 deficiency protects against 6-OHDA neurotoxicity in vivo
To confirm the upregulation of LCN2 in the lesioned DA system and its contribution to neurodegeneration in the nigrostriatal DA system of the adult brain, we examined the neuroprotective effects of LCN2 deficiency against 6-OHDA-induced neurotoxicity in the nigrostriatal DA projections of LCN2-null mice. Similar to the upregulation of LCN2 expression after MPTP treatment, Western blot analyses of WT brains showed that an intrastriatal injection of 6-OHDA induced a significant increase in LCN2 expression in both the striatum and SN compared with intact and vehicle-injected controls (Fig. 7B; **p < 0.01 and ***p < 0.001 vs vehicle controls) and double immunofluorescence staining for LCN2 and GFAP or Iba-1 showed that the increases in LCN2 expression after 6-OHDA treatment were localized within astrocytes and microglia (Fig. 7C), suggesting that LCN2 upregulation in the brain might be associated with glial activation. The neurotoxicity and movement impairments after 6-OHDA treatment were attenuated significantly in LCN2-deficient mice (Fig. 7D–G). These results suggest that the neurotoxicity in the nigrostriatal DA projection induced by elevated LCN2 expression might be a common feature of animal models of PD and the increased LCN2 levels in the lesioned DA system might be a potential pathogenic factor in PD.
Figure 7.
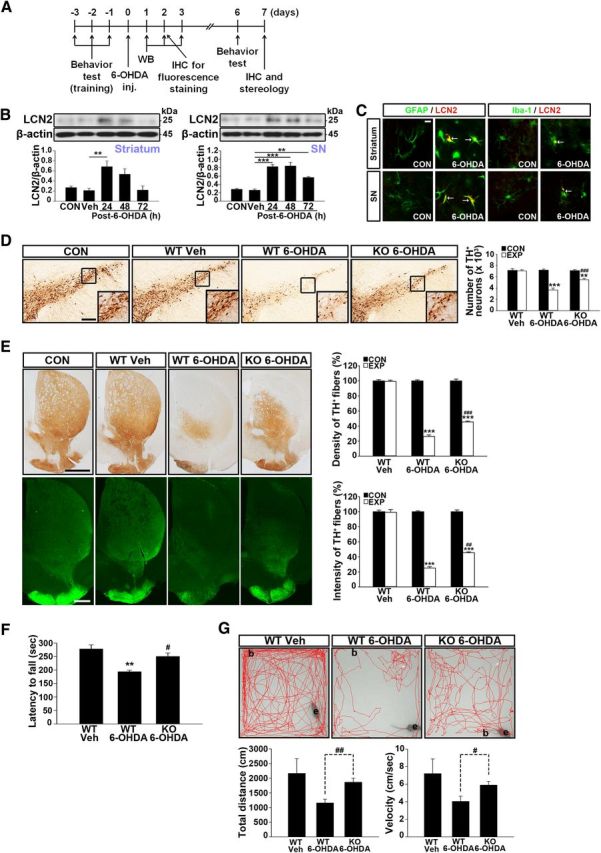
LCN2 deficiency protects the nigrostriatal DA projection in the 6-OHDA-treated animal model of PD. A, Diagram of the experimental design to determine whether intrastriatal injection of 6-OHDA induces LCN2 upregulation in the nigrostriatal DA system in the adult brain. B, Western blot analyses of the levels of LCN2 expression in the striatum and SN of mouse brains. ***p < 0.001 and **p < 0.01 in the striatum and SN, respectively, versus vehicle controls (one-way ANOVA; n = 4 for each group). C, Colocalization of LCN2 expression with glial cells in the striatum and SN by double immunofluorescence staining 48 h after 6-OHDA treatment. Scale bar, 10 μm. Arrows indicate colocalization of LCN2 and astrocytes/microglia. D, Number of TH-positive neurons in the SN. Scale bar, 200 μm. ***p < 0.001 and **p < 0.01 versus vehicle controls; ###p < 0.001 versus 6-OHDA-treated WT mice (one-way ANOVA; n = 4 for each group). CON, Contralateral control side; EXP, ipsilateral injected side. E, Optical densities (ODs) of TH-positive fibers in the striatum. Scale bars, 1000 μm (top) and 500 μm (bottom). ***p < 0.001 in immunoperoxidase staining versus vehicle controls; ###p < 0.001 in immunoperoxidase staining versus 6-OHDA-treated WT mice; ***p < 0.001 in immunofluorescence staining versus vehicle controls; ##p < 0.01 in immunofluorescence staining versus 6-OHDA-treated WT mice (one-way ANOVA; n = 4 for each group). F, G, Movement impairment analyses by the rotarod and open-field tests. **p < 0.01 and #p < 0.05 in the results of the rotarod test (F) versus vehicle-treated mice and 6-OHDA-treated WT mice, respectively (one-way ANOVA; n = 4 for each group). #p < 0.05 and ##p < 0.01 in the results of the open-field test (G) versus 6-OHDA-treated WT mice (t test; n = 4 for each group).
Astrocytic LCN2 contributes to MPP+-induced neurotoxicity in vitro
To clarify further whether astrocytic LCN2 contributes to neurotoxicity in DA neurons, we compared MPP+-induced neurotoxicity in cocultures of mesencephalic neurons and normal astrocytes with toxicity in cocultures of mesencephalic neurons and astrocytes obtained from LCN2-deficient mice. MPP+ at a concentration of 1 μm induced significant neuronal death in mesencephalic-neuron-enriched cultures at 48 h after the treatment compared with untreated controls (Fig. 8A; *p < 0.01). We cocultured neurons with astrocytes by transferring culture inserts containing astrocytes into wells with neuron-enriched cultures at 24 h after the MPP+ treatment. We measured neuronal viability using the MTT assay after coculturing the cells for another 24 h. Our results showed that the reduction in neuronal viability was greater in the cocultures of mesencephalic neurons and normal astrocytes than in the pure neuron-enriched cultures (Fig. 8A; ##p < 0.01). However, the decrease in neuronal viability was attenuated significantly in the cocultures of mesencephalic neurons and LCN2-deficient astrocytes (Fig. 8A; #p < 0.05 vs cocultures of mesencephalic neurons and normal astrocytes). Therefore, these results suggest that astrocyte-derived LCN2 contributes to the death of DA neurons in vitro. In addition, we confirmed the direct neurotoxicity of LCN2 by treatment of SH-SY5Y neuronal cultures differentiated by 10 μm RA and expressing DA markers with recombinant LCN2 protein (1, 2, or 4 μg/ml) or MPP+ (100 μm) as a positive control for neurotoxicity. Cell viability was evaluated by the MTT and LDH assays at 4 d after treatment with LCN2 or MPP+ (Fig. 8B; *p < 0.05 and **p < 0.01 vs untreated group). Moreover, we observed that treatment with conditioned medium (CM) derived from RA-differentiated SH-SY5Y neuronal cultures pretreated with MPP+ induced a significant increase in LCN2 release from mixed glial cultures compared with CM derived from untreated SH-SY5Y cultures (Fig. 9A,C). Furthermore, we observed that LCN2 expression, at both the mRNA (Fig. 9D) and protein (Fig. 9E) levels, was significantly upregulated in the glial cultures treated with CM derived from SH-SY5Y neuronal cultures after MPP+ treatment compared with CM derived from untreated SH-SY5Y cultures. MPP+ treatment alone had no effect on LCN2 release in SH-SY5Y neuronal cultures (Fig. 9B). These results indicate that the secretory factors released from MPP+-damaged DA neurons may induce glial LCN2 expression and secretion.
Figure 8.

LCN2 contributes to neurotoxicity in the MPP+-treated model of PD in vitro. A, Astrocytes isolated from WT or LCN2-deficient mice were cocultured with mesencephalic neurons treated with MPP+ for 24 h. Neuronal viability was measured using the MTT assay after 24 h of coculture. MPP+ induced significant neurotoxicity at 48 h after the treatment in mesencephalic-neuron-enriched cultures and the neurotoxicity was increased significantly in the cocultures with WT astrocytes. However, the addition of LCN2-deficient astrocytes induced lower neurotoxicity compared with cocultures of mesencephalic neurons and WT astrocytes. *p < 0.01 and **p < 0.001 versus untreated mesencephalic DA neurons; #p < 0.05 and ##p < 0.01 between the indicated groups. B, LCN2 treatment induces direct neurotoxicity in SH-SY5Y cultures. SH-SY5Y cells were differentiated by treatment with 10 μm RA for 5 d and then the cells were treated with LCN2 (1, 2, or 4 μg/ml) or MPP+ (100 μm) as a positive control for neurotoxicity. Cell viability was evaluated by MTT and LDH assays 4 d after the LCN2 or MPP+ treatment. *p < 0.05 and **p < 0.01, significantly different from untreated control (one-way ANOVA; n = 3).
Figure 9.

Conditioned media derived from MPP+-treated neuronal cultures induce an increase in LCN2 expression in glial cultures. A, Diagram of the experimental design. B, SH-SY5Y neurons differentiated by treatment with 10 μm RA for 5 d were exposed to 100 μm MPP+ and the levels of LCN2 expression in the culture medium were measured by ELISA at 24 h after the MPP+ treatment (t test; n = 3). C, Mixed glial cultures including astrocytes and microglia were treated with CM derived from untreated or MPP+-treated neuronal cultures and the levels of LCN2 protein in the culture medium were measured by ELISA at 24 h after the CM treatment. **p < 0.01, significantly different from the untreated CM control (t test; n = 3). D, E, Increased levels of LCN2 mRNA and protein in the mixed glia were confirmed by traditional RT-PCR (D) and Western blotting (E), respectively, 24 h after the CM treatment.
It has been reported previously that MMP-9 is released from damaged DA neurons (Lorenzl et al., 2004) and induces glial activation (Annese et al., 2015). Similar to these reports, we observed that MPP+ treatment induced an increase in MMP-9 activity, as measured by gelatin zymography, in SH-SY5Y neuronal cultures (Fig. 10A). Moreover, consistent with the increase in LCN2 release in glial cultures after treatment with CM derived from SH-SY5Y neuronal cultures after MPP+ treatment (Fig. 9C), MMP-9 treatment induced an increase in LCN2 release in mixed glial cultures (Fig. 10B) and astrocyte-enriched cultures (Fig. 10C). These results suggest that MMP-9 derived from the damaged neurons may stimulate the production of glial LCN2 further to amplify neurotoxicity (Fig. 11).
Figure 10.

Increases in LCN2 expression in mixed glial cultures and astrocyte-enriched cultures after treatment with MMP-9. A, SH-SY5Y neurons differentiated by treatment with 10 μm RA for 5 d were exposed to 100 μm MPP+ and the levels of MMP-9 zymogen in the culture medium were measured by gelatin zymography 24 h after the MPP+ treatment. The levels of MMP-9 were increased significantly in the SH-SY5Y neuronal cultures treated with MPP+ compared with normal controls. *p < 0.05, significantly different from untreated controls (t test; n = 4). B, C, Mixed glia and astrocyte-enriched cultures were treated with MMP-9 (100 and 500 ng/ml) and the levels of LCN2 protein in the culture medium were measured by ELISA 24 h after the MMP-9 treatment. The levels of LCN2 were increased significantly in both glial cultures (B) and astrocyte-enriched cultures (C) treated with MMP-9 compared with normal controls. *p < 0.05, significantly different from untreated controls (one-way ANOVA; n = 3). Lipopolysaccharide (LPS; 100 ng/ml) and interferon gamma (IFN-γ; 50 units/ml) were used as positive controls for LCN2 induction. **p < 0.01 and *p < 0.05, significantly different from the untreated control (one-way ANOVA; n = 3).
Figure 11.

Schematic representation of LCN2-mediated neurotoxicity in PD. Damaged DA neurons may release biomolecules such as MMP-9, which contributes to glial activation, resulting in LCN2 upregulation in reactive astrocytes and activated microglia. In addition to glial activation, which can produce neurotoxic cytokines such as TNF-α and IL-1β, the upregulation of LCN2 may induce direct neurotoxicity via excessive iron delivery into DA neurons, resulting in the disruption of the nigrostriatal DA system in the adult brain.
Discussion
Neurodegenerative movement disorders such as PD are the result of a progressive loss of neurons and axons in the CNS, leading to motor dysfunction (Kim et al., 2011; Kim et al., 2012). Although the main cause of PD remains elusive, it is well known that CNS inflammation and immune activation play major roles in the pathophysiology of PD. Neuroinflammatory activities resulting in neurodegeneration are mostly mediated by resident innate immune cells such as activated microglia and reactive astrocytes, which are able to produce reactive oxygen intermediates, nitric oxide, and inflammatory cytokines (McGeer et al., 2005; Hirsch and Hunot, 2009; Menza et al., 2010; Niranjan, 2014). Therefore, an understanding of the neuroinflammatory mechanisms and key biomolecules that control glial activation is indispensable for developing a novel therapeutic strategy for the protection of the nigrostriatal DA projection in the PD brain.
Astrocytes are the most abundant glial cells in the mammalian brain (Chung et al., 2010) and play both beneficial and detrimental roles in PD (Teismann et al., 2003; McGeer and McGeer, 2008; Rappold and Tieu, 2010; Cabezas et al., 2014). Astrocytes play important roles in multiple brain processes such as the promotion of neurovascular coupling, glutamate uptake and release, pH control, dopamine metabolism, and production of glutathione and superoxide dismutases with antioxidant effects (Danbolt, 2001; Teismann et al., 2003; Cabezas et al., 2014). In addition, astrocytes provide the optimal microenvironment for neuronal function by exerting active control of the cerebral blood flow (Mulligan and MacVicar, 2004; Gordon et al., 2008) and the extracellular concentrations of neurotransmitters (Danbolt, 2001). Moreover, astrocytes promote the survival and maintenance of DA neurons through secretion of various neurotrophic factors in the SN (Teismann et al., 2003; McGeer and McGeer, 2008). However, the presence of reactive astrocytes in the brains of patients with PD is one of the key features of the disease (Teismann et al., 2003; Rappold and Tieu, 2010; Cabezas et al., 2014). In experimental models of PD using neurotoxic substances such as 6-OHDA and MPTP, reactive astrocytes have been implicated in causing oxidative stress and releasing cytokines that trigger degeneration of DA neurons (Liberatore et al., 1999; Rappold and Tieu, 2010; Lazzarini et al., 2013). At the same time, reactive astrocytes also display beneficial characteristics in these models of PD (Voutilainen et al., 2009; Rappold and Tieu, 2010; Yokoyama et al., 2011). In addition, LCN2 produced by activated astrocytes in response to neurodegeneration contributes to neurotoxicity in vitro, suggesting that reactive astrocytes might have neurotoxic properties such as secretion of neurotoxic molecules (Lee et al., 2012; Bi et al., 2013). Reactive astrocytes also contribute to neuronal impairment through induction of neurotoxic factors in β-amyloid-treated astrocytic/neuronal cocultures (Malchiodi-Albedi et al., 2001). Together, these results indicate that, even though the crosstalk between damaged neurons and reactive astrocytes remains poorly understood, reactive astrocytes may play detrimental roles in PD under specific pathological conditions. Such conditions include gliosis induced by neuronal injury or toxic insults (Rappold and Tieu, 2010) and the simultaneous loss of neuroprotective functions and gain of neurotoxic properties (Bi et al., 2013). Moreover, reactive astrocytes can produce soluble factors that stimulate microglial activation, resulting in neurodegeneration (Rohl and Sievers, 2005; Ovanesov et al., 2008). Therefore, the characterization of the action mechanisms of reactive-astrocyte-derived molecules involved in neurotoxicity and neuroinflammation may be important for finding a novel therapeutic strategy for PD.
Here, we report that LCN2 upregulation is observed in the SN of patients with PD (Fig. 1A). Moreover, we demonstrate that an increase in astrocytic LCN2 in the nigrostriatal DA system may be a potential pathogenic mechanism in PD. MPTP administration significantly upregulated LCN2 expression, mainly in the activated astrocytes, in the SN and striatum of the mouse brain (Fig. 2) and its upregulation was more apparent in the nigrostriatal DA system than in any other brain areas, such as the hippocampus and cerebral cortex (Fig. 1D). LCN2 upregulation was also observed in the 6-OHDA-treated animal model of PD (Fig. 7), suggesting that its upregulation might be a universal feature of PD models. In addition to the observations of LCN2 upregulation, LCN2 neurotoxicity was confirmed by the intranigral injection of recombinant LCN2 protein, resulting in the disruption of the nigrostriatal DA projection in vivo (Fig. 3), and by reductions in neurotoxicity and movement impairments induced by MPTP (Fig. 5) and 6-OHDA (Fig. 7) in LCN2-deficient mice. These results suggest that LCN2 may be an important pathogenic biomolecule involved in PD. The results are consistent with a recent report showing an increase in LCN2 expression in the SN after MFB transection in vivo (Choy et al., 2015). However, the precise role of LCN2 in PD is still largely unknown.
LCN2 can play a role as an iron transport protein, regulating intracellular iron accumulation by binding to its receptor, LCN2R (Devireddy et al., 2005; Lee et al., 2012; Jha et al., 2015). We show that DA neurons express LCN2R (Fig. 3A). Moreover, intracellular iron accumulation is a well characterized feature of the SN of patients with PD (Zecca et al., 2004; Ward et al., 2014; Ayton et al., 2015) and animal models of PD (Hare et al., 2013). To evaluate a possible link between LCN2-induced neurotoxicity and intracellular iron accumulation, we investigated whether treatment with an iron donor or an iron chelator could alter the neurotoxicity induced by LCN2 upregulation. As described in Figure 4, the LCN2-induced neurotoxicity increased in the presence of the iron donor, FC, but decreased in the presence of the iron chelator, DFO, in the mouse brain (Fig. 4). These results suggest that an increase in the labile iron pool, at least in part, might be associated with the LCN2-induced neurotoxicity in vivo.
In addition to demonstrating LCN2 upregulation in the MPTP-treated model of PD and its neurotoxicity in the nigrostriatal DA projection in vivo, we investigated whether LCN2 deficiency in the MPTP-treated model of PD prevented glial activation and the ensuing production of neurotoxic cytokines such as IL-1β and TNF-α. LCN2 upregulation has been shown to mediate glial activation, resulting in neurodegeneration (Lee et al., 2007; Ferreira et al., 2015; Jha et al., 2015). Previous immunohistochemical studies have shown that numerous activated microglia, characterized by enlarged cell bodies with short processes, are present in the neurotoxin-treated SN of various animal models of PD (Choi et al., 2003; Whitton, 2007; Kim et al., 2011) and in the SN of patients with PD (Hirsch et al., 1998). Furthermore, under neuropathological conditions, activated microglia produce a spectrum of potentially neurotoxic molecules, including reactive oxygen species, which contribute to the oxidative stress and death of DA neurons (Choi et al., 2003; Block and Hong, 2005; Kim et al., 2010). Similar to microglial activation, reactive astrocytes could produce neurotoxic cytokines such as IL-1β and TNF-α in animal models of PD (Hunot et al., 1999; Yokoyama et al., 2011). Therefore, the characterization of endogenous biomolecules associated with glial activation, resulting in neurodegeneration through oxidative stress and proinflammatory mechanisms, is important for the development of PD therapies. Our experiments showed that glial activation in the MPTP-treated model of PD was ameliorated in LCN2-deficient mice (Fig. 6A,B). Consistent with the morphological activation of glia, Western blot analyses showed that the significant increases in the levels of neurotoxic inflammatory cytokines such as TNF-α and IL-1β were inhibited in LCN2-deficient mice (Fig. 6C). These findings show that an increase in LCN2 levels may be involved in the glial activation and production of neurotoxic inflammatory cytokines, suggesting that LCN2 upregulation after neurotoxicity in the brain may be a key trigger of neuroinflammation contributing to further severe disruption of the nigrostriatal DA system.
To investigate further whether lesioned neurons affect LCN2 expression in glia, we measured the LCN2 protein levels in the culture media of mixed glial cultures after treatment with CM derived from SH-SY5Y neuronal cultures. Our results showed that treatment with CM from MPP+-treated SH-SY5Y cultures induced glial expression and secretion of LCN2 (Fig. 9). Moreover, MPP+-induced neurotoxicity was significantly greater in cocultures of mesencephalic neurons and WT astrocytes than in cocultures of mesencephalic neurons and LCN2-deficient astrocytes (Fig. 8A). Therefore, these results suggest that the damaged neurons stimulate the production of glial LCN2; however, LCN2-inducing factors derived from the damaged neurons remain to be determined in vivo. It has been reported previously that MMP-9 can be released from damaged DA neurons (Lorenzl et al., 2004) and induce glial activation involved in PD (Annese et al., 2015). Moreover, LCN2 protein is known to interact directly with MMP-9, resulting in an LCN2/MMP-9 protein complex (Yan et al., 2001). We therefore investigated whether glial LCN2 expression is altered by treatment with MMP-9 in vitro. Our data suggest that MMP-9 is a strong candidate for the activator of glial LCN2 expression in the adult brain because MMP-9 treatment induced an increase in the LCN2 protein levels in glial cultures (Fig. 10B,C). Therefore, our results suggest for the first time that endogenous factors such as MMP-9 released from damaged DA neurons may induce astrocytic LCN2 expression and secretion in the PD brain, which may have a direct neurotoxicity with the induction of neurotoxic inflammation, thereby playing a central role in PD pathogenesis (Fig. 11).
In conclusion, LCN2 upregulation in the SN of patients with PD might be a potential pathogenic mechanism leading to further disruption of the nigrostriatal DA system through neurotoxic iron accumulation and neuroinflammation in PD. Astrocytes, possibly stimulated by MMP-9 released from damaged DA neurons, are the major source of LCN2 in the nigrostriatal DA system of the adult brain. Therefore, the control of LCN2 expression or its activity in the adult brain may be important for the protection of the nigrostriatal DA projection in PD and the development of inhibitors of LCN2-induced neurotoxicity and neuroinflammation will be useful for the treatment of PD.
Footnotes
This work was supported by the Korea Healthcare Technology R&D Project, Ministry of Health and Welfare, Republic of Korea (Grants HI14C3331, and HI15C1928), the Basic Science Research Program through the National Research Foundation (NRF) funded by the Korean Ministry of Education, Science, and Technology (MEST Grants 2008-0062282 and 2015R1A2A1A10051958), and by the NRF (Grant 2014R1A1A2056508 funded by the Korean Ministry of Education).
The authors declare no competing financial interests.
References
- Annese V, Herrero MT, Di Pentima M, Gomez A, Lombardi L, Ros CM, De Pablos V, Fernandez-Villalba E, De Stefano ME. Metalloproteinase-9 contributes to inflammatory glia activation and nigro-striatal pathway degeneration in both mouse and monkey models of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced Parkinsonism. Brain Struct Funct. 2015;220:703–727. doi: 10.1007/s00429-014-0718-8. [DOI] [PubMed] [Google Scholar]
- Ayton S, Lei P, Hare DJ, Duce JA, George JL, Adlard PA, McLean C, Rogers JT, Cherny RA, Finkelstein DI, Bush AI. Parkinson's disease iron deposition caused by nitric oxide-induced loss of beta-amyloid precursor protein. J Neurosci. 2015;35:3591–3597. doi: 10.1523/JNEUROSCI.3439-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berard JL, Zarruk JG, Arbour N, Prat A, Yong VW, Jacques FH, Akira S, David S. Lipocalin 2 is a novel immune mediator of experimental autoimmune encephalomyelitis pathogenesis and is modulated in multiple sclerosis. Glia. 2012;60:1145–1159. doi: 10.1002/glia.22342. [DOI] [PubMed] [Google Scholar]
- Bi F, Huang C, Tong J, Qiu G, Huang B, Wu Q, Li F, Xu Z, Bowser R, Xia XG, Zhou H. Reactive astrocytes secrete lcn2 to promote neuron death. Proc Natl Acad Sci U S A. 2013;110:4069–4074. doi: 10.1073/pnas.1218497110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Block ML, Hong JS. Microglia and inflammation-mediated neurodegeneration: multiple triggers with a common mechanism. Prog Neurobiol. 2005;76:77–98. doi: 10.1016/j.pneurobio.2005.06.004. [DOI] [PubMed] [Google Scholar]
- Borregaard N, Cowland JB. Neutrophil gelatinase-associated lipocalin, a siderophore-binding eukaryotic protein. Biometals. 2006;19:211–215. doi: 10.1007/s10534-005-3251-7. [DOI] [PubMed] [Google Scholar]
- Braak H, Del Tredici K, Rub U, de Vos RA, Jansen Steur EN, Braak E. Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol Aging. 2003;24:197–211. doi: 10.1016/S0197-4580(02)00065-9. [DOI] [PubMed] [Google Scholar]
- Cabezas R, Avila M, Gonzalez J, El-Bachá RS, Báez E, García-Segura LM, Jurado Coronel JC, Capani F, Cardona-Gomez GP, Barreto GE. Astrocytic modulation of blood brain barrier: perspectives on Parkinson's disease. Front Cell Neurosci. 2014;8:211. doi: 10.3389/fncel.2014.00211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chien MH, Ying TH, Yang SF, Yu JK, Hsu CW, Hsieh SC, Hsieh YH. Lipocalin-2 induces apoptosis in human hepatocellular carcinoma cells through activation of mitochondria pathways. Cell Biochem Biophys. 2012;64:177–186. doi: 10.1007/s12013-012-9370-1. [DOI] [PubMed] [Google Scholar]
- Choi SH, Joe EH, Kim SU, Jin BK. Thrombin-induced microglial activation produces degeneration of nigral dopaminergic neurons in vivo. J Neurosci. 2003;23:5877–5886. doi: 10.1523/JNEUROSCI.23-13-05877.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi WS, Kim HW, Xia Z. Preparation of primary cultured dopaminergic neurons from mouse brain. Methods Mol Biol. 2013;1018:61–69. doi: 10.1007/978-1-62703-444-9_6. [DOI] [PubMed] [Google Scholar]
- Choy YJ, Hong SY, Pack SJ, Woo RS, Baik TK, Song DY. Changes of gene expression of Gal3, Hsp27, Lcn2, and Timp1 in rat substantia nigra following medial forebrain bundle transection using a candidate gene microarray. J Chem Neuroanat. 2015;66–67:10–18. doi: 10.1016/j.jchemneu.2015.03.003. [DOI] [PubMed] [Google Scholar]
- Chung YC, Ko HW, Bok E, Park ES, Huh SH, Nam JH, Jin BK. The role of neuroinflammation on the pathogenesis of Parkinson's disease. BMB Rep. 2010;43:225–232. doi: 10.5483/BMBRep.2010.43.4.225. [DOI] [PubMed] [Google Scholar]
- Danbolt NC. Glutamate uptake. Prog Neurobiol. 2001;65:1–105. doi: 10.1016/S0301-0082(00)00067-8. [DOI] [PubMed] [Google Scholar]
- Devireddy LR, Gazin C, Zhu X, Green MR. A cell-surface receptor for lipocalin 24p3 selectively mediates apoptosis and iron uptake. Cell. 2005;123:1293–1305. doi: 10.1016/j.cell.2005.10.027. [DOI] [PubMed] [Google Scholar]
- Ferreira AC, Da Mesquita S, Sousa JC, Correia-Neves M, Sousa N, Palha JA, Marques F. From the periphery to the brain: Lipocalin-2, a friend or foe? Prog Neurobiol. 2015;131:120–136. doi: 10.1016/j.pneurobio.2015.06.005. [DOI] [PubMed] [Google Scholar]
- Flo TH, Smith KD, Sato S, Rodriguez DJ, Holmes MA, Strong RK, Akira S, Aderem A. Lipocalin 2 mediates an innate immune response to bacterial infection by sequestrating iron. Nature. 2004;432:917–921. doi: 10.1038/nature03104. [DOI] [PubMed] [Google Scholar]
- Gordon GR, Choi HB, Rungta RL, Ellis-Davies GC, MacVicar BA. Brain metabolism dictates the polarity of astrocyte control over arterioles. Nature. 2008;456:745–749. doi: 10.1038/nature07525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hare DJ, Adlard PA, Doble PA, Finkelstein DI. Metallobiology of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine neurotoxicity. Metallomics. 2013;5:91–109. doi: 10.1039/c2mt20164j. [DOI] [PubMed] [Google Scholar]
- Hare DJ, Arora M, Jenkins NL, Finkelstein DI, Doble PA, Bush AI. Is early-life iron exposure critical in neurodegeneration? Nat Rev Neurol. 2015;11:536–544. doi: 10.1038/nrneurol.2015.100. [DOI] [PubMed] [Google Scholar]
- Hirsch EC, Hunot S. Neuroinflammation in Parkinson's disease: a target for neuroprotection? Lancet Neurol. 2009;8:382–397. doi: 10.1016/S1474-4422(09)70062-6. [DOI] [PubMed] [Google Scholar]
- Hirsch EC, Hunot S, Damier P, Faucheux B. Glial cells and inflammation in Parkinson's disease: a role in neurodegeneration? Ann Neurol. 1998;44:S115–120. doi: 10.1002/ana.410440717. [DOI] [PubMed] [Google Scholar]
- Hunot S, Dugas N, Faucheux B, Hartmann A, Tardieu M, Debre P, Agid Y, Dugas B, Hirsch EC. FcepsilonRII/CD23 is expressed in Parkinson's disease and induces, in vitro, production of nitric oxide and tumor necrosis factor-alpha in glial cells. J Neurosci. 1999;19:3440–3447. doi: 10.1523/JNEUROSCI.19-09-03440.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang J, Lee S, Lee JT, Kwon TK, Kim DR, Kim H, Park HC, Suk K. Gangliosides induce autophagic cell death in astrocytes. Br J Pharmacol. 2010;159:586–603. doi: 10.1111/j.1476-5381.2009.00563.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang E, Kim JH, Lee S, Kim JH, Seo JW, Jin M, Lee MG, Jang IS, Lee WH, Suk K. Phenotypic polarization of activated astrocytes: the critical role of lipocalin-2 in the classical inflammatory activation of astrocytes. J Immunol. 2013;191:5204–5219. doi: 10.4049/jimmunol.1301637. [DOI] [PubMed] [Google Scholar]
- Jha MK, Lee S, Park DH, Kook H, Park KG, Lee IK, Suk K. Diverse functional roles of lipocalin-2 in the central nervous system. Neurosci Biobehav Rev. 2015;49:135–156. doi: 10.1016/j.neubiorev.2014.12.006. [DOI] [PubMed] [Google Scholar]
- Jin M, Kim JH, Jang E, Lee YM, Soo Han H, Woo DK, Park DH, Kook H, Suk K. Lipocalin-2 deficiency attenuates neuroinflammation and brain injury after transient middle cerebral artery occlusion in mice. J Cereb Blood Flow Metab. 2014;34:1306–1314. doi: 10.1038/jcbfm.2014.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim BW, Koppula S, Park SY, Kim YS, Park PJ, Lim JH, Su Kim I, Choi DK. Attenuation of neuroinflammatory responses and behavioral deficits by Ligusticum officinale (Makino) Kitag in stimulated microglia and MPTP-induced mouse model of Parkinsons disease. J Ethnopharmacol. 2014;164:388–397. doi: 10.1016/j.jep.2014.11.004. [DOI] [PubMed] [Google Scholar]
- Kim BW, Koppula S, Kumar H, Park JY, Kim IW, More SV, Kim IS, Han SD, Kim SK, Yoon SH, Choi DK. alpha-Asarone attenuates microglia-mediated neuroinflammation by inhibiting NF kappa B activation and mitigates MPTP-induced behavioral deficits in a mouse model of Parkinson's disease. Neuropharmacology. 2015;97:46–57. doi: 10.1016/j.neuropharm.2015.04.037. [DOI] [PubMed] [Google Scholar]
- Kim HD, Jeong KH, Jung UJ, Kim SR. Naringin treatment induces neuroprotective effects in a mouse model of Parkinson's disease in vivo, but not enough to restore the lesioned dopaminergic system. J Nutr Biochem. 2016;28:140–146. doi: 10.1016/j.jnutbio.2015.10.013. [DOI] [PubMed] [Google Scholar]
- Kim SR, Chung ES, Bok E, Baik HH, Chung YC, Won SY, Joe E, Kim TH, Kim SS, Jin MY, Choi SH, Jin BK. Prothrombin kringle-2 induces death of mesencephalic dopaminergic neurons in vivo and in vitro via microglial activation. J Neurosci Res. 2010;88:1537–1548. doi: 10.1002/jnr.22318. [DOI] [PubMed] [Google Scholar]
- Kim SR, Chen X, Oo TF, Kareva T, Yarygina O, Wang C, During M, Kholodilov N, Burke RE. Dopaminergic pathway reconstruction by Akt/Rheb-induced axon regeneration. Ann Neurol. 2011;70:110–120. doi: 10.1002/ana.22383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SR, Kareva T, Yarygina O, Kholodilov N, Burke RE. AAV transduction of dopamine neurons with constitutively active Rheb protects from neurodegeneration and mediates axon regrowth. Mol Ther. 2012;20:275–286. doi: 10.1038/mt.2011.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazzarini M, Martin S, Mitkovski M, Vozari RR, Stühmer W, Bel ED. Doxycycline restrains glia and confers neuroprotection in a 6-OHDA Parkinson model. Glia. 2013;61:1084–1100. doi: 10.1002/glia.22496. [DOI] [PubMed] [Google Scholar]
- Lee S, Lee J, Kim S, Park JY, Lee WH, Mori K, Kim SH, Kim IK, Suk K. A dual role of lipocalin 2 in the apoptosis and deramification of activated microglia. J Immunol. 2007;179:3231–3241. doi: 10.4049/jimmunol.179.5.3231. [DOI] [PubMed] [Google Scholar]
- Lee S, Park JY, Lee WH, Kim H, Park HC, Mori K, Suk K. Lipocalin-2 is an autocrine mediator of reactive astrocytosis. J Neurosci. 2009;29:234–249. doi: 10.1523/JNEUROSCI.5273-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Lee WH, Lee MS, Mori K, Suk K. Regulation by lipocalin-2 of neuronal cell death, migration, and morphology. J Neurosci Res. 2012;90:540–550. doi: 10.1002/jnr.22779. [DOI] [PubMed] [Google Scholar]
- Liberatore GT, Jackson-Lewis V, Vukosavic S, Mandir AS, Vila M, McAuliffe WG, Dawson VL, Dawson TM, Przedborski S. Inducible nitric oxide synthase stimulates dopaminergic neurodegeneration in the MPTP model of Parkinson disease. Nat Med. 1999;5:1403–1409. doi: 10.1038/70978. [DOI] [PubMed] [Google Scholar]
- Lin CW, Tseng SW, Yang SF, Ko CP, Lin CH, Wei LH, Chien MH, Hsieh YS. Role of lipocalin 2 and its complex with matrix metalloproteinase-9 in oral cancer. Oral Dis. 2012;18:734–740. doi: 10.1111/j.1601-0825.2012.01938.x. [DOI] [PubMed] [Google Scholar]
- Lorenzl S, Calingasan N, Yang L, Albers DS, Shugama S, Gregorio J, Krell HW, Chirichigno J, Joh T, Beal MF. Matrix metalloproteinase-9 is elevated in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced parkinsonism in mice. Neuromolecular Med. 2004;5:119–132. doi: 10.1385/NMM:5:2:119. [DOI] [PubMed] [Google Scholar]
- Malchiodi-Albedi F, Domenici MR, Paradisi S, Bernardo A, Ajmone-Cat MA, Minghetti L. Astrocytes contribute to neuronal impairment in beta A toxicity increasing apoptosis in rat hippocampal neurons. Glia. 2001;34:68–72. doi: 10.1002/glia.1041. [DOI] [PubMed] [Google Scholar]
- McGeer EG, Klegeris A, McGeer PL. Inflammation, the complement system and the diseases of aging. Neurobiol Aging. 2005;26:94–97. doi: 10.1016/j.neurobiolaging.2005.08.008. [DOI] [PubMed] [Google Scholar]
- McGeer PL, McGeer EG. Glial reactions in Parkinson's disease. Mov Disord. 2008;23:474–483. doi: 10.1002/mds.21751. [DOI] [PubMed] [Google Scholar]
- Menza M, Dobkin RD, Marin H, Mark MH, Gara M, Bienfait K, Dicke A, Kusnekov A. The role of inflammatory cytokines in cognition and other non-motor symptoms of Parkinson's disease. Psychosomatics. 2010;51:474–479. doi: 10.1176/appi.psy.51.6.474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miharada K, Hiroyama T, Sudo K, Danjo I, Nagasawa T, Nakamura Y. Lipocalin 2-mediated growth suppression is evident in human erythroid and monocyte/macrophage lineage cells. J Cell Physiol. 2008;215:526–537. doi: 10.1002/jcp.21334. [DOI] [PubMed] [Google Scholar]
- Moretto A, Colosio C. The role of pesticide exposure in the genesis of Parkinson's disease: epidemiological studies and experimental data. Toxicology. 2013;307:24–34. doi: 10.1016/j.tox.2012.11.021. [DOI] [PubMed] [Google Scholar]
- Mulligan SJ, MacVicar BA. Calcium transients in astrocyte endfeet cause cerebrovascular constrictions. Nature. 2004;431:195–199. doi: 10.1038/nature02827. [DOI] [PubMed] [Google Scholar]
- Nairz M, Theurl I, Schroll A, Theurl M, Fritsche G, Lindner E, Seifert M, Crouch ML, Hantke K, Akira S, Fang FC, Weiss G. Absence of functional Hfe protects mice from invasive Salmonella enterica serovar Typhimurium infection via induction of lipocalin-2. Blood. 2009;114:3642–3651. doi: 10.1182/blood-2009-05-223354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nam JH, Leem E, Jeon MT, Jeong KH, Park JW, Jung UJ, Kholodilov N, Burke RE, Jin BK, Kim SR. Induction of GDNF and BDNF by hRheb(S16H) transduction of SNpc neurons: neuroprotective mechanisms of hRheb(S16H) in a model of Parkinson's disease. Mol Neurobiol. 2015a;51:487–499. doi: 10.1007/s12035-014-8729-2. [DOI] [PubMed] [Google Scholar]
- Nam JH, Park ES, Won SY, Lee YA, Kim KI, Jeong JY, Baek JY, Cho EJ, Jin M, Chung YC, Lee BD, Kim SH, Kim EG, Byun K, Lee B, Woo DH, Lee CJ, Kim SR, Bok E, Kim YS, Ahn TB, Ko HW, Brahmachari S, Pletinkova O, Troconso JC, Dawson VL, Dawson TM, Jin BK. TRPV1 on astrocytes rescues nigral dopamine neurons in Parkinson's disease via CNTF. Brain. 2015b;138:3610–3622. doi: 10.1093/brain/awv297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nam Y, Kim JH, Seo M, Jin M, Jeon S, Seo JW, Lee WH, Bing SJ, Jee Y, Lee WK, Park DH, Kook H, Suk K. Lipocalin-2 protein deficiency ameliorates experimental autoimmune encephalomyelitis: the pathogenic role of lipocalin-2 in the central nervous system and peripheral lymphoid tissues. J Biol Chem. 2014;289:16773–16789. doi: 10.1074/jbc.M113.542282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niranjan R. The role of inflammatory and oxidative stress mechanisms in the pathogenesis of Parkinson's disease: focus on astrocytes. Mol Neurobiol. 2014;49:28–38. doi: 10.1007/s12035-013-8483-x. [DOI] [PubMed] [Google Scholar]
- Ovanesov MV, Ayhan Y, Wolbert C, Moldovan K, Sauder C, Pletnikov MV. Astrocytes play a key role in activation of microglia by persistent Borna disease virus infection. J Neuroinflammation. 2008;5:50. doi: 10.1186/1742-2094-5-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rappold PM, Tieu K. Astrocytes and therapeutics for Parkinson's disease. Neurotherapeutics. 2010;7:413–423. doi: 10.1016/j.nurt.2010.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohl C, Sievers J. Microglia is activated by astrocytes in trimethyltin intoxication. Toxicol Appl Pharmacol. 2005;204:36–45. doi: 10.1016/j.taap.2004.08.007. [DOI] [PubMed] [Google Scholar]
- Rozas G, López-Martin E, Guerra MJ, Labandeira-García JL. The overall rod performance test in the MPTP-treated-mouse model of Parkinsonism. J Neurosci Methods. 1998;83:165–175. doi: 10.1016/S0165-0270(98)00078-8. [DOI] [PubMed] [Google Scholar]
- Shashidharamurthy R, Machiah D, Aitken JD, Putty K, Srinivasan G, Chassaing B, Parkos CA, Selvaraj P, Vijay-Kumar M. Differential role of lipocalin 2 during immune complex-mediated acute and chronic inflammation in mice. Arthritis Rheum. 2013;65:1064–1073. doi: 10.1002/art.37840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin WH, Jeon MT, Leem E, Won SY, Jeong KH, Park SJ, McLean C, Lee SJ, Jin BK, Jung UJ, Kim SR. Induction of microglial toll-like receptor 4 by prothrombin kringle-2: a potential pathogenic mechanism in Parkinson's disease. Sci Rep. 2015;5:14764. doi: 10.1038/srep14764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teismann P, Tieu K, Cohen O, Choi DK, Wu DC, Marks D, Vila M, Jackson-Lewis V, Przedborski S. Pathogenic role of glial cells in Parkinson's disease. Mov Disord. 2003;18:121–129. doi: 10.1002/mds.10332. [DOI] [PubMed] [Google Scholar]
- Voutilainen MH, Back S, Porsti E, Toppinen L, Lindgren L, Lindholm P, Peranen J, Saarma M, Tuominen RK. Mesencephalic astrocyte-derived neurotrophic factor is neurorestorative in rat model of Parkinson's disease. J Neurosci. 2009;29:9651–9659. doi: 10.1523/JNEUROSCI.0833-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward RJ, Zucca FA, Duyn JH, Crichton RR, Zecca L. The role of iron in brain ageing and neurodegenerative disorders. Lancet Neurol. 2014;13:1045–1060. doi: 10.1016/S1474-4422(14)70117-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitton PS. Inflammation as a causative factor in the aetiology of Parkinson's disease. Br J Pharmacol. 2007;150:963–976. doi: 10.1038/sj.bjp.0707167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan L, Borregaard N, Kjeldsen L, Moses MA. The high molecular weight urinary matrix metalloproteinase (MMP) activity is a complex of gelatinase B/MMP-9 and neutrophil gelatinase-associated lipocalin (NGAL). Modulation of MMP-9 activity by NGAL. J Biol Chem. 2001;276:37258–37265. doi: 10.1074/jbc.M106089200. [DOI] [PubMed] [Google Scholar]
- Yokoyama H, Uchida H, Kuroiwa H, Kasahara J, Araki T. Role of glial cells in neurotoxin-induced animal models of Parkinson's disease. Neurol Sci. 2011;32:1–7. doi: 10.1007/s10072-010-0424-0. [DOI] [PubMed] [Google Scholar]
- Zamanian JL, Xu L, Foo LC, Nouri N, Zhou L, Giffard RG, Barres BA. Genomic analysis of reactive astrogliosis. J Neurosci. 2012;32:6391–6410. doi: 10.1523/JNEUROSCI.6221-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zecca L, Stroppolo A, Gatti A, Tampellini D, Toscani M, Gallorini M, Giaveri G, Arosio P, Santambrogio P, Fariello RG, Karatekin E, Kleinman MH, Turro N, Hornykiewicz O, Zucca FA. The role of iron and copper molecules in the neuronal vulnerability of locus coeruleus and substantia nigra during aging. Proc Natl Acad Sci U S A. 2004;101:9843–9848. doi: 10.1073/pnas.0403495101. [DOI] [PMC free article] [PubMed] [Google Scholar]