

Abstract
Rapid reorganization and stabilization of the actin cytoskeleton in dendritic spines enables cellular processes underlying learning, such as long-term potentiation (LTP). Dendritic spines are enriched in exceptionally short and dynamic actin filaments, but the studies so far have not revealed the molecular mechanisms underlying the high actin dynamics in dendritic spines. Here, we show that actin in dendritic spines is dynamically phosphorylated at tyrosine-53 (Y53) in rat hippocampal and cortical neurons. Our findings show that actin phosphorylation increases the turnover rate of actin filaments and promotes the short-term dynamics of dendritic spines. During neuronal maturation, actin phosphorylation peaks at the first weeks of morphogenesis, when dendritic spines form, and the amount of Y53-phosphorylated actin decreases when spines mature and stabilize. Induction of LTP transiently increases the amount of phosphorylated actin and LTP induction is deficient in neurons expressing mutant actin that mimics phosphorylation. Actin phosphorylation provides a molecular mechanism to maintain the high actin dynamics in dendritic spines during neuronal development and to induce fast reorganization of the actin cytoskeleton in synaptic plasticity. In turn, dephosphorylation of actin is required for the stabilization of actin filaments that is necessary for proper dendritic spine maturation and LTP maintenance.
SIGNIFICANCE STATEMENT Dendritic spines are small protrusions from neuronal dendrites where the postsynaptic components of most excitatory synapses reside. Precise control of dendritic spine morphology and density is critical for normal brain function. Accordingly, aberrant spine morphology is linked to many neurological diseases. The actin cytoskeleton is a structural element underlying the proper morphology of dendritic spines. Therefore, defects in the regulation of the actin cytoskeleton in neurons have been implicated in neurological diseases. Here, we revealed a novel mechanism for regulating neuronal actin cytoskeleton that explains the specific organization and dynamics of actin in spines. The better we understand the regulation of the dendritic spine morphology, the better we understand what goes wrong in neurological diseases.
Keywords: actin cytoskeleton, dendritic spines, LTP, phosphorylation, spinogenesis, synaptic plasticity
Introduction
The majority of excitatory synapses in the CNS exist on dendritic spines. Dendritic spines are found in varying sizes and shapes, but mostly have a spherical head and a thin narrow neck. Synaptic activity is translated into structural changes in dendritic spines (Yuste and Bonhoeffer, 2001), so spines offer a means to regulate synaptic signaling on a synapse-by-synapse basis. Accordingly, aberrant spine morphology is linked to many neurological diseases (Fiala et al., 2002; Penzes et al., 2011). Spines are highly enriched in actin filaments (F-actin) and dynamic modulation of the actin cytoskeleton is crucial for controlling, not only spine formation and elimination, but also the synapse-activation-induced morphological and structural changes in dendritic spines (Okamoto et al., 2004; Honkura et al., 2008; Bosch et al., 2014). The structure and dynamics of the actin cytoskeleton are under tight control of regulatory machineries that operate in a temporally and spatially controlled manner.
Actin filaments in dendritic spines are exceptionally short and dynamic (Star et al., 2002; Honkura et al., 2008; Hotulainen et al., 2009; Frost et al., 2010; Korobova and Svitkina, 2010). Although we and others have studied the functions of numerous actin-binding proteins systematically in dendritic spines (Hotulainen and Hoogenraad, 2010; Bellot et al., 2014), actin-binding proteins seem not to provide a sufficient explanation for the specific organization of actin in spines. Furthermore, cellular processes underlying learning, such as long-term potentiation (LTP), require rapid reorganization and stabilization of the actin cytoskeleton (Honkura et al., 2008; Bosch et al., 2014). However, the molecular mechanisms underlying actin reorganization and stabilization during LTP are still unclear.
Recent results show that the role of posttranslational modifications in actin regulation is more important than previously thought (Terman and Kashina, 2013). Terman's group has elegantly characterized Mical-induced oxidation of actin and the cellular importance of oxidation (Hung et al., 2011). In addition to oxidation and other redox-related modifications, actin is also known to undergo other modifications, such as arginylation, phosphorylation, and SUMOylation. However, the cellular importance of these other modifications is poorly understood (Terman and Kashina, 2013). One posttranslational modification that in particular shows a high potential to serve as a regulator of actin filament turnover is tyrosine-53 (Y53) phosphorylation. Y53 is highly conserved throughout all actin sequences from various species. Y53 phosphorylation stabilizes the D-loop of actin, which leads to a decreased affinity between actin molecules and shortening of the filaments (Liu et al., 2006; Baek et al., 2008). In Dictyostelium (slime mold), Y53 is dynamically phosphorylated when Dictyostelium amoebae are subjected to stress (Schweiger et al., 1992; Howard et al., 1993; Jungbluth et al., 1994; Jungbluth et al., 1995) and during development (Gauthier et al., 1997; Kishi et al., 1998; Liu et al., 2006). Interestingly, we found that actin is phosphorylated in the brains of mice overexpressing tyrosine receptor kinase B (TrkB; Semenov et al., 2006). Considering that TrkB is the main receptor for brain-derived neurotrophic factor (BDNF) in the CNS and that BDNF/TrkB signaling is important for neuronal cell shape changes, we hypothesized that actin phosphorylation could play a similar role in neuronal actin regulation as in Dictyostelium. In addition, actin phosphorylation has been detected in few other mammalian cell types (Baba et al., 2003; Lim et al., 2004), but studies on the role of actin phosphorylation in mammalian actin regulation are currently lacking.
We hypothesized that actin Y53 phosphorylation is the molecular mechanism leading to the exceptionally dynamic actin filaments in dendritic spines in basal conditions. We further hypothesized that actin phosphorylation and dephosphorylation are molecular mechanisms that control the reorganization and stabilization of the actin cytoskeleton in mammalian neurons during neuronal development and LTP expression.
Materials and Methods
Neuronal cultures, fibroblast cultures, and transfections.
For the primary neuronal cultures, hippocampi or cortex was dissected from embryonic day 18 RccHan (outbred from Wistar Han) rat fetuses of either sex and the tissue was dissociated in 0.05% papain solution. Next, the cells were triturated and suspended in a Ca2+/Mg2+-free HBBS medium containing 1 mm sodium pyruvate, 10 mm HEPES, pH 7.2, and 20 U/ml DNase I (Sigma-Aldrich). The cells were plated at a density of 100,000 cells/coverslip (diameter 13 mm), coated with poly-l-lysine (0.1 mg/ml; Sigma-Aldrich), in neurobasal medium (Invitrogen) supplemented with B-27 (Invitrogen), l-glutamine (Invitrogen), and penicillin–streptomycin (Lonza). Transient transfections were performed as described previously (Hotulainen et al., 2009) on d 13 in vitro (DIV13) using Lipofectamine 2000 (Invitrogen). Before all experiments, cells were examined to verify the presence of a dense network of neurons, ensuring the availability of a proper synaptic network. U2 osteosarcoma (U2OS) cells were maintained in DMEM supplemented with 10% fetal bovine serum (Hyclone), 2 mm l-glutamine (Invitrogen), and penicillin–streptomycin (Lonza). Cells were transfected using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. Twenty-four hours later, hippocampal neurons or U2OS cells were either imaged live or fixed in 4% formaldehyde.
Generating specific pY53-actin antibody and immunofluorescence staining.
A customized peptide CGQKDS-pY-VGDEA (corresponding to aa 48–57 in rat/mouse β-actin) was generated, two rabbits were immunized, and the antibody was purified by Biogenes. For immunofluorescence, both primary hippocampal neurons and U2OS cells were permeabilized using 0.1% Triton X-100 in PBS. Immunofluorescence was performed as described previously (Vartiainen et al., 2000) and the primary pY53-actin antibody was used in a 1:50 dilution. Actin (Tyr-53) phospho-specific antibody was purchased from ECM Biosciences and Synapsin I antibody from Covance and used according to the manufacturer's instructions. Filamentous actin was visualized using Alexa Fluor 594-phalloidin (Invitrogen).
Western blotting and the Phos-tag affinity method.
For the Western blots shown in Figure 1, A–D, DIV14 hippocampal or cortical neurons were first treated for 15 min with or without (controls) 1 mm Na3VO4 to inhibit phosphatases. Cells were rinsed 5× with cold PBS and lysed with lysis buffer containing 50 mm HEPES, pH 7.5, 4% SDS, 300 mm NaCl, 1 mm EDTA, 5.5 mm DTT, and proteinase and phosphatase inhibitors (Complete and Phospho-stop, respectively; Roche). For Phos-tag gels, cells were lysed in lysis buffer containing 50 mm HEPES, pH 7.5, 4% SDS, 300 mm NaCl, and proteinase and phosphatase inhibitors (Complete and Phospho-stop, respectively; Roche).
Figure 1.

Amount of actin Y53 phosphorylation changes during neuronal maturation and upon glycine application in cortical neurons. A, Western blot using pY53-actin-specific antibody shows that actin is phosphorylated in hippocampal neurons and the level of phosphorylation increases with Na3VO4 treatment. The total amount of actin was controlled using AC-15 actin antibody. VAN, 15 min 1 mm Na3VO4 treatment. B, Quantification of blots in A shown as the ratio of pY53-actin/total actin normalized to control. Graph represents four independent experiments. The amount of recognized phosphorylated actin was increased 3.0-fold by phosphatase inhibition. *p < 0.05, nonparametric Kruskal–Wallis test. C, Similar experiment as in A, but pY53-actin was detected using commercial pY53-actin antibody (ECM, ECM Biosciences) and cortical neuron cultures were used. D, Quantification of blots in C shown as the ratio of pY53-actin/total actin normalized to control. Graph represents five independent experiments. The amount of recognized phosphorylated actin was increased 2.0-fold by phosphatase inhibition. **p < 0.01, nonparametric Kruskal–Wallis test. E, F, Actin and pY53-actin migrated at similar speeds in normal SDS-PAGE gels (E), but phosphorylated actin migrated more slowly in Phos-tag gel (F). Commercial pY53-actin antibody (ECM Biosciences) detected three different bands in Phos-tag gel but no bands the size of nonphosphorylated actin. Data represent three independent experiments. G, Cortical neurons were cultured for 7, 14, and 21 DIV and lysates were run on SDS-PAGE and blotted against pY53-actin (ECM Biosciences) or total actin (AC-15). H, Quantification of G revealing that Y53 phosphorylation decreases by DIV21 (relative phosphorylation amounts: DIV7 = 1.0, DIV14 = 1.0, DIV21 = 0.2). Graph represents three analyzed blots (two different antibodies) from two independent experiments. *p < 0.05, nonparametric Kruskal–Wallis test. I, Freshly diluted 200 μm glycine in HBS was applied to DIV14 cortical cultures for 3 min after a 10 min incubation with blockers TTX, strychnine, and picrotoxin. Cells were then washed using HBS and lysed after a 10- or 30 min incubation in HBS. Samples were blotted against pAkt, Akt, pY53-actin (ECM), and total actin. J, pAkt levels were increased 1.9-fold 10 and 30 min after glycine application. K, The amount of pY53-actin was increased 2.1-fold 10 min after glycine application, but the increase in phosphorylation diminished after 30 min (1.4-fold increase compared with preinduction). Graph represents four independent experiments. *p < 0.05, nonparametric Kruskal–Wallis test. Data are represented as mean ± SEM.
For the Western blots shown in Figure 1I, freshly diluted 200 μm glycine in HBS was applied to DIV14 cortical cultures for 3 min after a 10 min incubation with the blockers TTX (0.5 μm, Abcam), strychnine (1 μm, Sigma-Aldrich), and picrotoxin (50 μm, Abcam). Cells were then washed in HBS and lysed after a 10 or 30 min incubation in HBS.
For Western blots shown in Figure 7B, CA1 of hippocampal slices were frozen in liquid nitrogen 35 min after LTP induction and lysed as cultured neurons. Samples were run on normal 10% SDS-PAGE gels (Biorad) or on 8% SDS-PAGE gels containing 0.1% (w/v) SDS, 100 μm phos-tag (Wako Chemicals), and 400 μm MnCl2. After the run, Phos-tag gels were soaked in transfer buffer with 5 mm EDTA for 10 min.
Figure 7.

Phosphorylation level of endogenous actin increases during LTP formation in acute hippocampal slices. A, LTP was induced in rat hippocampal slices similar to the experiment presented in Figure 6 (circles). The CA1s of the slices were frozen in liquid nitrogen 35 min after LTP induction. In addition to slices with LTP induction, we collected Western blot samples from control slices recorded without LTP induction (triangles). At 32 min, the fEPSP was potentiated 55 ± 2% [n = 8 slices (8 rats)] in LTP-induced slices and 4 ± 2% [n = 5 (5 rats)] in slices without LTP induction, p = 0.003. **p < 0.01, nonparametric Kruskal–Wallis test. B, The success of LTP induction was further controlled by measuring the known LTP-responsive Akt Ser473 phosphorylation (Racaniello et al., 2010) and pY53-actin levels were detected with pY53-actin antibodies (ECM Biosciences). C, pAkt levels were increased 2.7-fold 35 min after LTP induction. D, Western blotting against pY53-actin revealed that actin phosphorylation was increased 2.3-fold in LTP-induced slices compared with control samples. Graph represents five independent experiments. *p < 0.05, nonparametric Wilcoxon signed-rank test. Data are represented as mean ± SEM.
Gels were blotted on a nitrocellulose or PVDF filter following standard Western blotting protocols. Western blots were blocked using 2% BSA in TBS. The amount of pY53-actin was compared with control lysates by Western blot using rabbit anti-pY53-actin antibodies (homemade and from ECM Biosciences). The total amount of actin was controlled using mouse monoclonal AC-15 actin antibody (Sigma-Aldrich). pAkt was detected by rabbit Phospho-Akt (Ser473; D9E) XP antibody and the total amount of Akt by rabbit anti-Akt antibody, both from Cell Signaling Technology.
Plasmid and virus constructs.
pEGFP-N1 (GFP) and mCherry-C1 (mCherry) plasmids were purchased from Clontech Laboratories. Human GFP-β-actin (Dopie et al., 2012) and mCherry-β-actin plasmids were gifts from Maria Vartiainen (University of Helsinki, Finland) and Martin Bähler (Westfalian Wilhelms-University, Münster, Germany), respectively. SAP97-mCherry was a gift from Lotta von Ossowski and Kari Keinänen (University of Helsinki, Finland; Saarikangas et al., 2015). Actin constructs with mutated tyr-53 residues were cloned using the following oligos: Y53E forward: AAGTGGGCGACGAGGCCCAG, Y53A forward: CCGTGGGCGACGAGGCCCAG, Y53E/Y53A reverse: CGGAGTCCTTCTGACCCATACCCAC and amplified by whole plasmid PCR. The mutations were verified by sequencing. To create virus constructs wt-, Y53E-, and Y53A-GFP-actin were cleaved with AgeI and BamHI from the plasmids and inserted into pLenSyn1Ver2-vector. For virus production, ∼50–75% confluent HEK-293T cells on a 10 cm plate were transfected using 300 μl DMEM, 0.75 μg of the envelope plasmid pMD2.G, 2.25 μg of the packing plasmid pPAX2, 3.0 μg of the plasmid pLEN-SYN1 carrying the actin construct Y53E, Y53A or wt actin, and 18 μg of FUGENE HD (Promega). After a 48 h incubation, the virus was concentrated by centrifugation from the medium.
Confocal imaging: fixed and live.
Imaging was performed on either a Leica TCS SP5 or a Zeiss LSM 710 upright confocal microscope. A 63× 1.3 numerical aperture (NA) objective lens was used to image the fixed samples. For live-cell recordings, the microscopes were equipped with a temperature-controlled chamber and CO2 supply. Live-cell recordings were performed at 37°C and 5% CO2 using a 63× 0.9 NA water-dipping objective. Image files were processed using LAS-AF (Leica Microsystems), ZEN 2012 (Carl Zeiss), ImageJ, Imaris (Bitplane), and Photoshop CS4 (Adobe).
Analysis of PSD density, morphology, and dynamics of dendritic spines.
For analysis of spines, serial image files corresponding to z-stacks of 20–30 optical sections per dendritic segment were directly processed using NeuronStudio, a software package specifically designed for spine detection and analysis (Rodriguez et al., 2008). For all spine analyses, GFP was in preference to mCherry because of its higher signal-to-noise ratio. Detailed analysis was performed as described in Bertling et al. (2012). Classification of spines was done using rules defined by Rodriguez et al. (2008). All spines detected by the software were verified manually. Branched spine density was counted from the same images used in the morphology analysis. Spines with extra protrusions from the spine neck or the spine head were counted and divided by the length of the dendritic segment. The mean value of separate images is shown. To measure the relative localization of mCherry-(wt/Y53E/Y53A)-actin to the spine head, the maximum intensity of actin in the spine head was compared with the intensity of the adjacent dendrite. The result was normalized to the intensity distribution of free GFP along the same line.
To analyze PSD density, SAP97-mCherry was cotransfected with GFP-(wt/Y53E/Y53A)-actin. Clearly separable dots of SAP97 were counted and divided by the length of the dendritic segment. PSD widths were analyzed by measuring the maximum widths of SAP97 dots manually using ImageJ line measurements.
To analyze the density of presynaptic contacts, DIV13 hippocampal neuronal cultures were transfected using GFP-(wt/Y53E/Y53A)-actin, fixed at DIV14, and stained using anti-synapsin antibody. Clear contacts between synapsin-stained presynaptic contacts and dendritic protrusions in GFP-(wt/Y53E/Y53A)-actin expressing neurons were counted from confocal images and divided by the length of the dendritic segment. Presynaptic contact widths were measured manually using ImageJ line measurements.
The motility assay was performed as described in Bertling et al. (2012). Briefly, time-lapse series were taken as 3D stacks at 2 min intervals. Five consecutive images were analyzed using NeuronStudio. Spine head diameter and length were calculated as the average of these five measurements. The values were compared between adjacent time points and the mean percentage changes in the head diameter/length measured for individual spines were designated as motility indices. Only images taken from free GFP were used to diminish errors caused by specific localizations of proteins. With many stubby type spines, the separation of the head fluctuation next to the dendrite was not within the range of the microscope's resolution. In addition, some thin spines appeared or disappeared within the 10 min recording. These were excluded from the analysis. Several experiments were averaged to obtain the final value of each condition.
FRAP.
Fluorescence recovery after photobleaching (FRAP) was performed as described in Koskinen et al. (2012). For the FRAP assays, neurons were transfected using GFP-(wt/Y53E/Y53A)-actin or GFP-wt-actin together with mCherry-(wt/Y53E/Y53A)-actin. Only spines with distinct heads were selected for analysis. The frame was imaged three times before photobleaching. Photobleaching was induced by 5 scans (total bleach time 3.117 s) of a region of interest (a single spine) with ∼2.2 mW of laser power at the sample (488 nm). Imaging of the frame was resumed immediately after the completion of photobleaching and continued every 2–5 s for 100 s. FRAP recordings were analyzed by measuring the fluorescence intensity of the bleached spine and then normalizing this value to a fluorescence intensity value taken from a control area to account for overall bleaching of the whole cell that occurs during imaging. Each intensity value was then divided by the prebleach value recorded. The values from different spine heads of each analyzed cell were averaged. The sizes of stable and dynamic pools were manually measured from the mean curves (Koskinen et al., 2014). In addition, the recovery half-time (t1/2) was determined from these plots by determining the time point at which the dynamic component reached half of the value of full recovery.
Viral transductions and slice preparation for in vivo analysis.
Morphological analysis of cells in the CA1 (cornu ammonis area 1) in rat hippocampus was performed for fixed brains expressing lentivirally transduced actin constructs (wt-, Y53E-, or Y53A-actin). The virus was injected at postnatal day 1 (P1)–P4 at 3 sites in the hippocampus and morphological analysis was performed for brains fixed at age P15–P19. For that, rats were anesthetized and transcardially perfused using cold PBS, followed by cold 4% paraformaldehyde in PBS. Brains were postfixed overnight in 4% formaldehyde in PBS at 4°C, cryoprotected using a 10–20-30% sucrose–PBS gradient over 3 d, frozen in Tissue-Tek OCT (Sakura), and stored at −80°C before cutting. Sections were cut 35 μm thick in the cryostat at −20°C and stored in antifreeze solution (Hoffman et al., 2008). For staining, transfected sections were washed in 3% BSA (Sigma-Aldrich) in PBS (PBS:BSA) and incubated for 30 min in 0.5% Triton X-100 (Sigma-Aldrich) in PBS:BSA for permeabilization. Sections were then incubated with primary anti-GFP antibody (1:500; Roche) overnight at 4°C. After several washes in PBS:BSA, sections were incubated in Alexa Fluor-488-conjugated anti-mouse secondary antibody (1:1000; Invitrogen) or Alexa Fluor-594-conjugated anti-mouse secondary antibody (1:1000; Invitrogen) for 2 h, washed in PBS, and mounted on glass coverslips using Immu-Mount medium (Thermo Scientific). Transduction efficiency was determined with DAPI counterstaining.
Acute slice preparation and electrophysiological recordings.
Acute hippocampal coronal slices were prepared from lentivirally transduced rats at P16–P29 using standard methods (see Clarke et al., 2012; Luchkina et al., 2014). For estimating the efficiency of transduction, slices were examined using a fluorescence microscope before electrophysiological recordings. Only slices with high transduction efficiency (30–35% of cells transduced) were selected for recordings. To prevent recurrent excitatory activity, the CA3 area was removed surgically. For electrophysiological recordings, slices were placed in an interface recording chamber and perfused using artificial CSF (ACSF) containing the following (in mm): 124 NaCl, 3 KCl, 1.25 NaH2PO4, 1 MgSO4, 26 NaHCO3, 15 d-glucose, and 2 CaCl2) at a rate of 1.5–2 ml/min at 30–32°C.
Field EPSPs (fEPSPs) were recorded from the stratum radiatum in area CA1 using ACSF-filled glass electrodes. Synaptic responses were evoked at 0.05 Hz by stimulating a bipolar electrode (nickel-chromium wire) positioned within Schaffer collateral commissural fibers. Evoked responses were collected for offline and online analysis using WinLTP software (Anderson and Collingridge, 2007; www.winltp.com). The slope of the rising phase of fEPSPs (20–70%) was used as a measure of synaptic efficacy. The baseline stimulation intensity was adjusted to yield 40–60% of the maximal intensity of the fEPSP in the appearance of a population spike.
For input/output (I/O) recordings, three stimulation intensities below the baseline value and three above were applied. The amplitude of the presynaptic volley was plotted against the fEPSP slope for different stimulation intensities. To study paired-pulse facilitation (PPF), two pulses with paired-pulse intervals of 20–150 ms were given. The PPF ratio was calculated by dividing the slope of the second fEPSP with the slope of the first. Theta-frequency stimulation (TFS) used for LTP induction consisted of four-pulse bursts at 100 Hz separated by 200 ms and repeated five times. The recording was continued for at least 40 min after the TFS.
Statistical analysis.
All statistical analysis was done in SPSS statistics software versions 20–23 (IBM). One-way ANOVA with Bonferroni's post hoc test was used to compare the groups when the data were normally distributed. Nonparametric Kruskal–Wallis test or Wilcoxon signed-rank tests were used to compare the groups when the data were non-normally distributed. The Dunn–Bonferroni post hoc method was used with the Kruskal–Wallis test when needed. p < 0.05 was considered statistically significant (*p < 0.05; **p < 0.01, ***p < 0.001).
Results
Amount of actin Y53 phosphorylation changes during neuronal maturation and upon glycine application in cortical neurons
To determine whether actin is phosphorylated at Y53 in neurons, we generated a pY53-actin-specific (i.e., actin phosphorylated at Y53) antibody. The antibody was generated against a 12-aa-long peptide containing phosphorylated Y53 and adjacent amino acids from rat β-actin. The antibody recognized a protein with the size of actin (43 kDa) in Western blot analysis of primary hippocampal or cortical neuron lysates (Fig. 1). Phosphatase inhibition (Na3VO4) increased (3.0, p = 0.014) the amount of recognized phosphorylated protein, although the total amount of actin remained the same (Fig. 1A,B). A similar increase (2.0, p = 0.005) in Y53-actin phosphorylation upon phosphatase inhibition was detected with a commercially available pY53-actin antibody (Fig. 1C,D). Next, we used phosphate-affinity gel electrophoresis to separate phosphorylated actin from nonphosphorylated actin (Kinoshita et al., 2008). Our results revealed that, whereas actin and pY53-actin migrated at similar speeds in normal SDS-PAGE gels (Fig. 1E), phosphorylated actin migrated more slowly than nonphosphorylated actin in the Phos-tag gel (Fig. 1F). Using antibodies against total actin, no bands were detected at the level of the slowly migrating bands detected by pY53 antibodies in the Phos-tag gel, indicating that the relative amount of pY53-actin was low (∼<5%). The pY53-actin antibody detected three different bands, indicating that pY53-actin is probably cophosphorylated at other sites too (Fig. 1F). Importantly, the pY53-actin antibody did not detect anything at the same level as the main actin band, showing its specificity for phosphorylated actin (Fig. 1F). From these experiments, we concluded that both pY53-actin antibodies can distinguish pY53-actin from nonphosphorylated actin.
Next, we evaluated whether the level of Y53 phosphorylation changes during neuronal maturation in vitro. We cultured cortical neurons for 7, 14, and 21 DIV and ran lysates on SDS-PAGE gels. This analysis revealed that actin is phosphorylated during the first 2 weeks of neuronal development, but the level of Y53 phosphorylation decreases by DIV21 (relative level of phosphorylation: DIV7 = 1.0, DIV14 = 1.0, DIV21 = 0.2, p < 0.05; Fig. 1G,H).
Protein phosphorylation is proposed to be one of the main signaling systems that trigger synaptic changes underlying LTP (Racaniello et al., 2010). Therefore, we used glycine application previously used to induce LTP chemically (Shahi and Baudry, 1993; Lu et al., 2001), first confirming the previously observed (Racaniello et al., 2010) increase in Akt Ser473 phosphorylation. pAkt levels were increased 1.9-fold 10 and 30 min after adding glycine (Fig. 1I,J). The amount of pY53-actin increased 2.1-fold (p = 0.021) 10 min after glycine application, but the increase in phosphorylation diminished after 30 min (1.4-fold increase compared with preinduction; Fig. 1I,K). The amount of actin Y53 phosphorylation decreases during neuronal maturation and increases transiently upon glycine application.
The subcellular distribution of phosphorylated actin was tested by immunofluorescence analysis. Anti-pY53-actin antibody showed punctate staining in DIV10 hippocampal neurons (Fig. 2A). At DIV14, both pY53-actin antibodies detected the highest pY53-actin levels in local dots in spine heads (Fig. 2B,C). Data obtained with pY53-actin-specific antibodies demonstrated that actin Y53 phosphorylation is strictly controlled both spatially and temporally.
Figure 2.
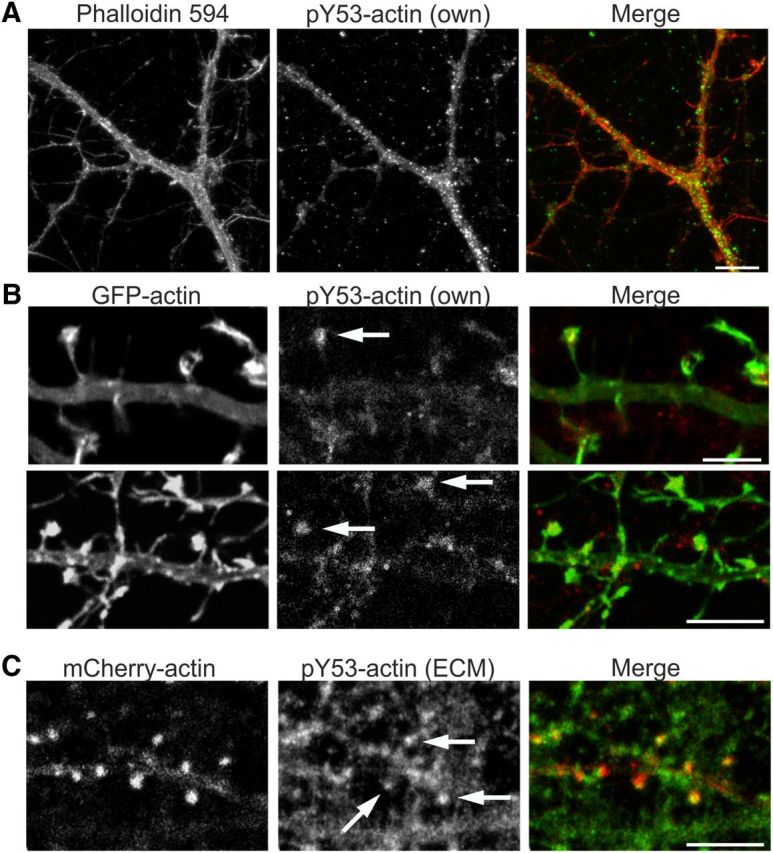
Actin Y53 phosphorylation is a strictly localized phenomenon. A, Immunofluorescence staining of primary DIV10 hippocampal low-density culture using anti-pY53-actin specific antibody. Y53-phosphorylated actin is located throughout dendrites and filopodial protrusions in a punctate manner. Scale bar, 15 μm. B, Immunofluorescence staining of primary DIV14 hippocampal neurons using pY53-actin specific antibody shows the localization of pY53-actin to dendritic spine heads. Cells are transfected with GFP-actin to highlight a single dendrite. Images from one confocal layer (z = 0.450 μm) are shown. Arrows indicate staining in spine heads. Scale bars, 5 μm. C, Immunofluorescence staining of mCherry-actin-transfected primary DIV14 hippocampal neurons using pY53-actin antibody (ECM Biosciences). Images from one confocal layer (z = 0.450 μm) are shown. Antibody staining shows clear localization in spine heads (arrows). Scale bars, 5 μm. Data in A–C represent five independent experiments.
Low levels of Y53E-actin expression enhance the development of lamellipodia and hinder the establishment of stress fibers
Biochemically, actin Y53 phosphorylation stabilizes the D-loop of actin and thus weakens actin–actin affinity (Liu et al., 2006). To study the effects of Y53 phosphorylation on actin and cellular dynamics, we generated an actin construct mimicking actin phosphorylation (Y53E) and a construct that cannot be phosphorylated (Y53A). Although substitution of tyrosine (Y) to glutamic acid (E) does not mimic tyrosine phosphorylation structure in a perfect manner (Fig. 3A), glutamic acid has been used successfully in numerous studies to introduce a negative charge to mimic phosphorylated forms of proteins (Cassens et al., 2010, Ting et al., 2015, Xia et al., 2008). In addition, we substituted Y with alanine (A) to abolish the possibility of phosphorylation at this site (Fig. 3A). Mutated actins were expressed as GFP fusion proteins and GFP-wt-actin was used as a control (Choidas et al., 1998).
Figure 3.
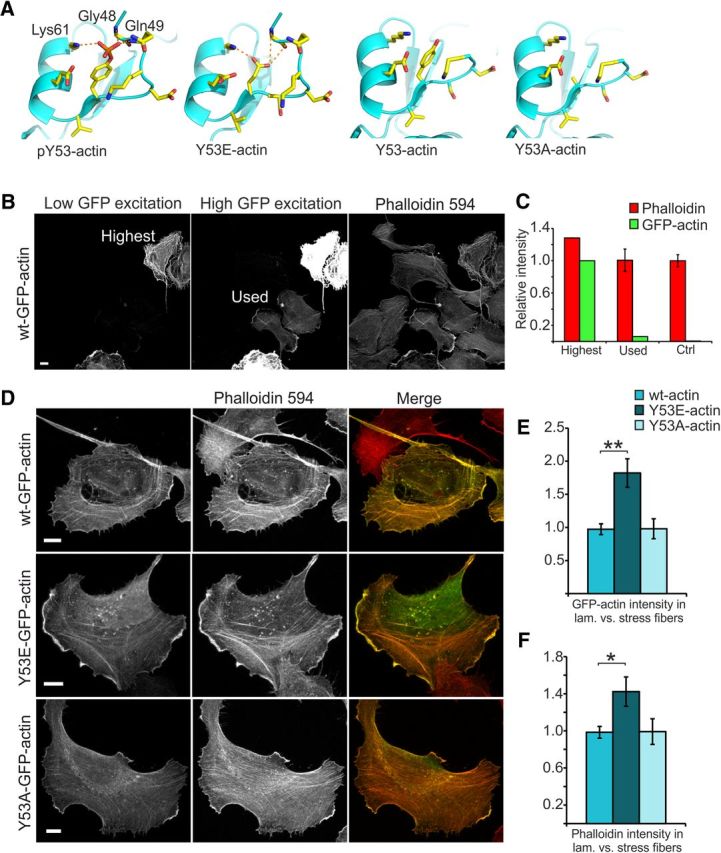
Low levels of Y53E-actin expression enhance the development of lamellipodia and hinder the establishment of stress fibers. A, Actin structures visualizing Y53 and adjacent amino acids. Generated mutations (Y53E and Y53A) visualized by modeled structures. Y53 phosphorylation results in the formation of hydrogen bonds with amino acids Gly48, Gln49, and Lys61, stabilizing the D-loop. Y53E mutation generates a negative charge mimicking the phosphorylated form of actin. Putative hydrogen bonds are indicated in the image. In the nonphosphorylated form and the Y53A mutation, the D-loop is flexible and thus not seen in the structure. Images were generated using the program PyMOL (http://pymol.sourceforge.net/) starting from the structures PDB 3CI5 (pY53-actin) and PDB 3CI5 (nonphosphorylated actin; Baek et al., 2008). B, Images taken of U2OS cells with high and low laser powers reveal different levels in GFP-actin expression and how GFP-actin expression levels affect phalloidin staining intensity. Scale bar, 10 μm. C, When the GFP intensity in the high-expressing cell shown in B is set to 100%, ImageJ intensity analysis reveals that the low-expressing cells used in these experiments express only 6% of this maximum expression level. The intensity of phalloidin staining in these low-expressing cells is the same as in nontransfected controls. D, F-actin staining (phalloidin-594) of U2OS cells expressing GFP-(wt/Y53E/ Y53A)-actin. Scale bars, 10 μm. E, GFP-actin constructs are distributed differently between lamellipodial actin and stress fibers: wt ratio = 1.0 (similar intensity in lamellipodium and in stress fibers); Y53E ratio = 1.8; Y53A ratio = 1.0. Data in E and F represent n(wt) = 17, n(Y53E) = 14, n(Y53A) = 11 cells pooled from two independent experiments. **p < 0.01 one-way ANOVA with Bonferroni's post hoc test. F, Overexpression of GFP-Y53E-actin causes a shift toward lamellipodial actin: wt ratio = 1.0 (similar intensity in lamellipodium and in stress fibers), Y53E ratio = 1.4, Y53A ratio = 1.0. *p < 0.05 one-way ANOVA with Bonferroni's post hoc test. Data are represented as mean ± SEM.
Throughout our experiments, we used low to moderate levels of expression of GFP constructs to avoid possible adverse effects caused by high overexpression (Fig. 3B). We estimated the amount of GFP-actin in these low-expressing cells by comparing their average GFP fluorescence intensity with that of highly expressing cells (Fig. 3C). Low-expressing cells exhibited only 6% of the GFP intensity detected for high-expressing cells. Because it is theoretically possible that all endogenous actin molecules are replaced by GFP-actin, we set the level of replacement for high-expressing cells to 100%. From this, we can estimate that low-expressing cells would have a maximum of 6% of the total cellular actin GFP-tagged. However, it is plausible that high-expressing cells have ∼20% of actin GFP-tagged, which would mean that low-expressing cells only have 1–2% of actin GFP-tagged. This level is similar to GFP-actin levels (1–3% of total actin) measured in transgenic GFP-actin mice (Vaezi et al., 2002; Gurniak and Witke, 2007). In neuron experiments, we used neurons that expressed GFP-actin at similar intensity levels as the low-expressing cells described here.
We then investigated whether mutant actin constructs can be incorporated into native actin structures and the possible effects that they might have on these structures in human U2OS cells. Both mutant constructs colocalized with F-actin (Fig. 3D). Comparison of the maximum intensity of GFP-actin (both wt and mutants) in lamellipodia and stress fibers showed that GFP-Y53E-actin has a preference for lamellipodial localization [wt ratio = 1.0 (similar intensity in lamellipodium and in stress fibers); Y53E ratio = 1.8, p = 0.001; Y53A ratio = 1.0; Fig. 3E]. Correspondingly, GFP-Y53E-actin expression significantly increased the ratio of phalloidin staining intensity in lamellipodia versus stress fibers compared with cells expressing GFP-wt-actin [wt ratio = 1.0 (similar intensity in lamellipodia and stress fibers), Y53E ratio = 1.4, p = 0.024; Y53A ratio = 1.0; Fig. 3F]. GFP- Y53A-actin localization and the lamellipodia/stress fiber ratio of phalloidin staining intensity were similar to wt actin (Fig. 3D–F). These results suggest that mutated actin constructs can be incorporated into the endogenous actin cytoskeleton and that Y53E-actin has a preference for localizing to the dynamic actin meshwork in lamellipodia. Furthermore, low-level Y53E-actin expression enhances the development of lamellipodia and hinders the establishment of stress fibers.
Expression of mutant actin mimicking pY53-actin increases the proportion of mushroom-shaped dendritic spines
We expressed mutated GFP-actin constructs for 24 h in primary cultures of hippocampal neurons and analyzed their localization and associated morphological changes at DIV14 (Fig. 4A). To analyze morphological changes induced by the expression of mutated actin isoforms, we transfected DIV13 hippocampal neurons with mCherry-tagged actin constructs together with free GFP. The following day (DIV14), neurons were fixed and imaged on a confocal microscope. GFP images were used for spine density and morphology analysis using NeuronStudio software (Rodriguez et al., 2008). Neurons expressing Y53E-actin exhibited a higher number of mushroom-shaped spines compared with control neurons expressing wt actin (proportion of mushroom spines: wt = 54%, Y53E = 61%, p = 0.049; Fig. 4B). There were no significant changes in total spine densities (Fig. 4C) or in spine head widths between these three constructs (wt = 0.55 μm, Y53E = 0.54 μm, Y53A = 0.52 μm), but the expression of Y53E-actin reduced the proportion of spines with a head diameter that is <0.2 μm (p = 0.045; Fig. 4D). The clearest defect in Y53A-actin expressing neurons was a 3-fold increase in the density of branched spines (branched spine density: wt = 0.03; Y53A = 0.10 spines/μm, p < 0.001; Fig. 4E).
Figure 4.
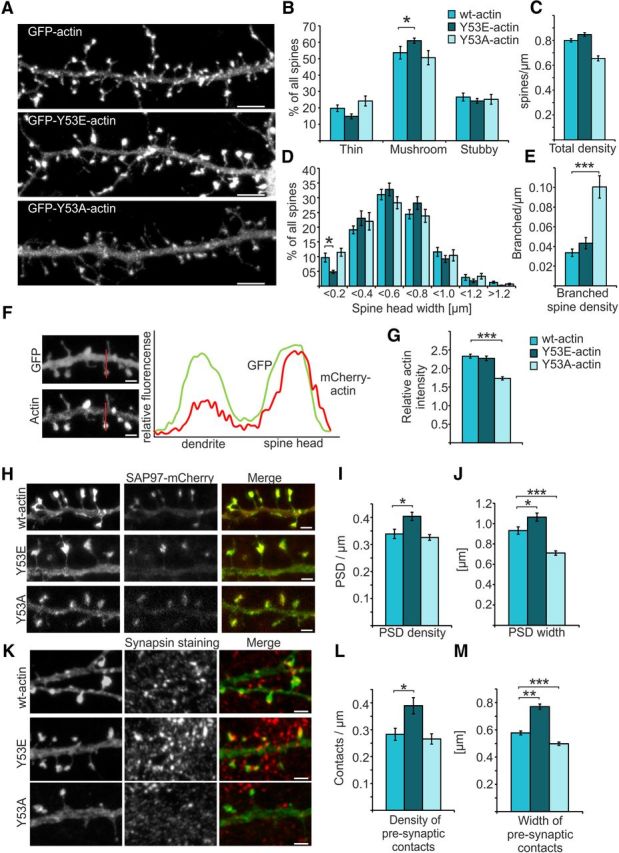
Expression of mutant actin mimicking pY53-actin increases the proportion of mushroom-shaped dendritic spines. A, GFP-actin-, GFP-Y53E-actin-, and GFP-Y53A-actin-expressing primary hippocampal neurons at DIV14. Scale bars, 5 μm. B, C, Quantification of spine morphology from neurons cotransfected with mCherry-actins and free GFP revealed an increased proportion of mushroom spines in mCherry-Y53E-actin expressing neurons. Proportions of thin, mushroom, and stubby spine morphologies: wt: 20% thin, 54% mushroom, 27% stubby, total density = 0.80 spines/μm; Y53E: 15% thin, 61% mushroom, 24% stubby, total density = 0.83 spines/μm; Y53A: 24% thin, 51% mushroom, 25% stubby, total density = 0.66 spines/μm. Data in B–E represent n(wt) = 15 cells, 986 spines, 1342 μm of dendrite; n(Y53E) = 19 cells, 1522 spines, 1998 μm of dendrite; n(Y53A) = 15 cells, 1119 spines, 1587 μm of dendrite pooled from four independent experiments.*p < 0.05 one-way ANOVA with Bonferroni's post hoc test. D, Size distribution of spines analyzed in B and C. Spines are grouped with 0.2 μm intervals. Spines with small heads (<0.2 μm) were significantly reduced in Y53E-actin-expressing neurons. E, GFP-Y53A-actin induces branching of spine heads. Branched spine densities: wt = 0.03, Y53E = 0.04; Y53A = 0.10 spines/μm. ***p < 0.001 one-way ANOVA with Bonferroni's post hoc test. F, Free GFP and mCherry-actin expressed in a primary hippocampal neuron. Expression plot profile was generated from the line shown in the picture. Distribution of actin between spine and dendrite was measured and normalized to GFP signal. Scale bars, 1 μm. G, mCherry-Y53A-actin showed a reduced concentration in spine heads compared with wt- and mCherry-Y53E-actin. Head versus dendrite localizations: wt = 2.3, Y53E = 2.3, Y53A = 1.7. Data represent n = 10 cells, 100 spines for each actin construct pooled from two independent experiments. ***p < 0.001 one-way ANOVA with Bonferroni's post hoc test. H, PSD densities and sizes of PSDs of GFP-(wt/Y53E/Y53A)-actin expressing cells were analyzed based on PSD labeling by SAP97-mCherry. Scale bars, 1 μm. I, Quantification shows that PSD density was significantly increased in cells expressing GFP-Y53E-actin. Density of PSDs: wt = 0.34 PSD/μm, n = 17 cells, 1707 μm of dendrite; Y53E = 0.40 PSD/μm, n = 14 cells, 1394 μm of dendrite; Y53A = 0.33 PSD/μm, n = 14 cells, 1227 μm of dendrite. Data are pooled from two independent experiments. *p < 0.05 one-way ANOVA with Bonferroni's post hoc test. J, Quantification of PSD widths shows that PSD sizes were significantly increased in cells expressing GFP-Y53E-actin together with SAP97-mCherry, whereas, in cells expressing GFP-Y53A-actin, PSD accumulations were significantly smaller. PSD width: wt = 0.9 μm, n = 10 cells, 270 spines with PSD; Y53E = 1.1 μm, n = 10 cells, 235 spines with PSD; Y53A = 0.7 μm, n = 10 cells, 341 spines with PSD. Data are pooled from two independent experiments. *p < 0.05, ***p < 0.001, one-way ANOVA with Bonferroni's post hoc test. K, Presynaptic site densities and widths of synapsin staining next to GFP-(wt/Y53E/Y53A)-actin expressing spines. Scale bars, 1 μm. L, Quantification shows that presynaptic site density was significantly increased in cells expressing GFP-Y53E-actin. Densities: wt = 0.28 presynaptic contacts/μm, n = 14 cells, 1070 μm of dendrite; Y53E = 0.39, n = 12 cells, 835 μm of dendrite; Y53A = 0.27, n = 13 cells, 1031 μm of dendrite. Data are pooled from three independent experiments. *p < 0.05 one-way ANOVA with Bonferroni's post hoc test. M, Quantification of the width of presynaptic contacts shows that widths were significantly increased in cells expressing GFP-Y53E-actin, whereas, in cells expressing GFP-Y53A-actin, synapsin accumulations were significantly reduced. wt = 0.58 μm, n = 6 cells, 241 pre synaptic sites; Y53E = 0.77 μm, n = 4 cells, 269 presynaptic sites; Y53A = 0.50 μm, n = 8 cells, 252 presynaptic sites. Data are pooled from two independent experiments. **p < 0.01, ***p < 0.001, one-way ANOVA with Bonferroni's post hoc test. Data are represented as mean ± SEM.
To quantify the localization of wt and mutant actins in dendritic spines, we measured the fluorescence intensity of mCherry-(wt/Y53E/ Y53A)-actin in spines and in adjacent dendrites and normalized the difference using the intensity distribution of free GFP (Fig. 4F). Wt and mCherry-Y53E-actin localized to spine heads in a similar manner, whereas the localization of mCherry-Y53A-actin was lower [head/dendrite ratio: wt = 2.3; Y53E = 2.3; Y53A = 1.7 (p < 0.001); Fig. 4F,G]. Together, the expression of Y53E-actin increases the amount of mushroom spines and Y53A-actin expression results in the formation of branched spines.
We further tested whether the expression of the mutated actin constructs affects spine maturation and synapse formation. We analyzed the number of spines that displayed postsynaptic density (SAP97-mCherry) as a sign of spine maturation, as we did in earlier studies (Saarikangas et al., 2015). As expected based on the increased mushroom spine density (Fig. 4B), expression of the Y53E-actin increased the density of PSDs [wt = 0.34 PSD/μm; Y53E = 0.40 PSD/μm (p = 0.012); Y53A = 0.33 PSD/μm; Fig. 4H,I]. Moreover, the PSDs were significantly larger in cells expressing GFP-Y53E-actin and smaller in cells with GFP-Y53A-actin than in cells expressing GFP-wt-actin [wt = 0.9 μm; Y53E = 1.1 μm (p = 0.03); Y53A = 0.7 μm (p < 0.001); Fig. 4H,J]. The postsynaptic density must equilibrate with the presynaptic bouton to maintain its structure over longer time periods (Meyer et al., 2014). Therefore, we analyzed the densities and sizes of presynaptic contacts. Staining of presynaptic contacts by synapsin revealed that postsynaptic expression of GFP-Y53E-actin increased the density of presynaptic contacts [wt = 0.28 presynaptic contacts/μm; Y53E = 0.39 (p = 0.001); Y53A = 0.27], as well as the width of presynaptic contacts [wt = 0.58 μm; Y53E = 0.77 μm (p < 0.001); Fig. 4K–M]. Conversely, expression of Y53A-actin significantly decreased the average size of presynaptic contacts compared with wt actin [Y53A = 0.50 μm (p = 0.002); Fig. 4K–M]. Therefore, short 1 d Y53E-actin expression promotes—and Y53A-actin expression hinders—dendritic spine maturation and synapse formation.
Expression of mutant pY53-mimicking actin increases the turnover rate of actin filaments and spine head dynamics
To study the turnover of actin filaments in dendritic spines in cultured neurons, we used the FRAP assay, which we have previously optimized for dendritic spine actin (Koskinen et al., 2012, 2014; Koskinen and Hotulainen, 2014). The FRAP assay measures the rate of addition of new actin monomers to the ends of actin filaments. The relative sizes of the dynamic and stable pools can be calculated from the FRAP recovery curves (Koskinen and Hotulainen, 2014). The dynamic pool has a turnover time of ∼40 s, whereas the stable pool turnover time is much longer, at ∼17 min (Honkura et al., 2008). We first recorded FRAP in DIV14 cultured hippocampal neurons expressing GFP-actin, GFP-Y53E-actin, or GFP-Y53A-actin (Fig. 5A,B). Cells expressing low to moderate amounts of GFP-actin and displaying spines with comparable size and morphology were selected for analyses. The relative size of the stable F-actin pool was significantly increased and decreased in cells expressing GFP-Y53A-actin and GFP-Y53E-actin, respectively, compared with wt GFP-actin expressing cells [stable pool fraction size of wt = 11%, Y53E = 6% (p = 0.023), Y53A = 20% (p < 0.001); Fig. 5C]. Analysis of the recovery curves demonstrated that actin filaments in the dynamic pool containing GFP-Y53E-actin recovered faster than those containing GFP-actin or GFP-Y53A-actin [recovery half-time of dynamic pool, wt = 8.8 s, Y53E = 5.5 s (p < 0.001), Y53A = 7.7 s; Fig. 5D]. To determine whether the effects of these mutants on actin dynamics are dominant over wt actin in spines, we performed the same FRAP assay on cells expressing GFP-wt-actin together with mCherry-tagged wt-, Y53E-, and Y53A-actin. Expression of mutant actins changed the sizes of the slow F-actin pools measured by following GFP-wt-actin, similarly to what was seen in experiments in which the recovery of the mutant protein was followed directly [stable pool size: wt = 13%; Y53E = 7% (p = 0.001); Y53A = 20% (p = 0.002; Fig. 5E,F]. However, the turnover time of the GFP-wt-actin dynamic pool was not significantly changed (Fig. 5G). Therefore, these results suggest that Y53E-actin decreases the size of the stable F-actin pool, whereas Y53A-actin stabilizes actin filaments in dendritic spines.
Figure 5.
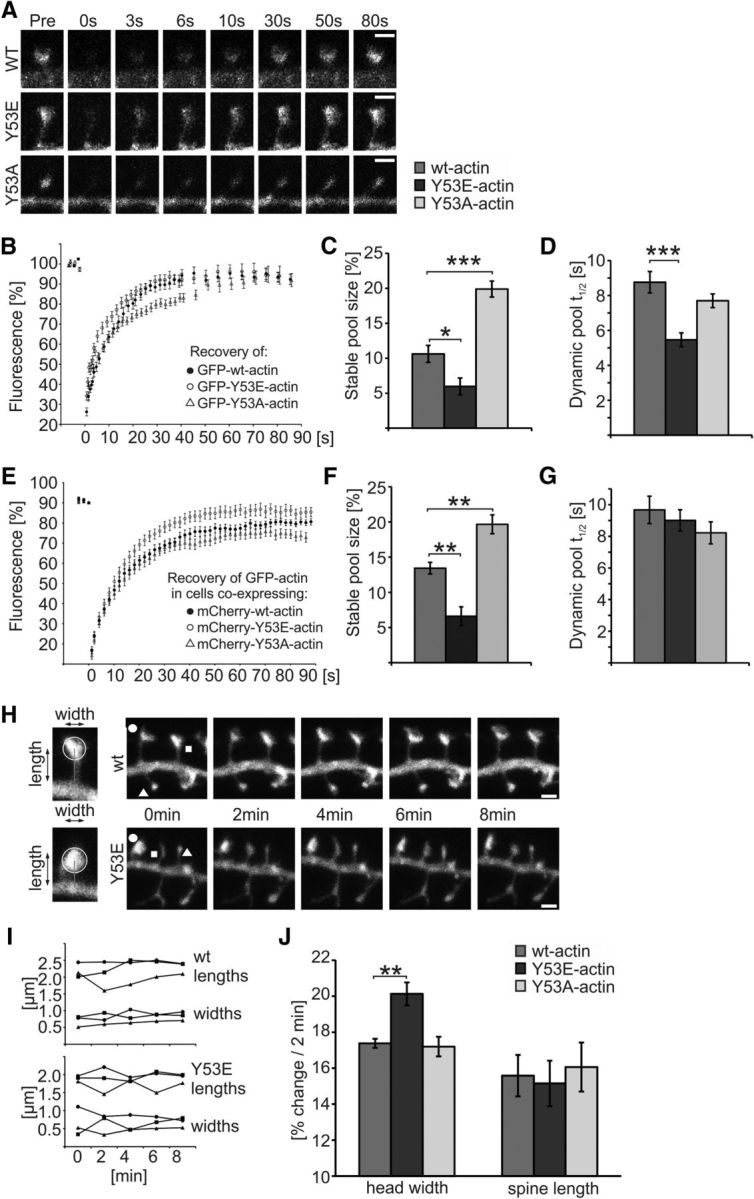
Expression of mutant actin mimicking pY53-actin increases the turnover rate of actin filaments and spine head dynamics. A, Example images presenting fluorescence recovery of GFP-(wt/Y53E/Y53A)-actin in dendritic spines of primary DIV14 hippocampal neurons. B, Analyses of FRAP assays show the mean GFP-actin fluorescence recovery curves of GFP-(wt/Y53E/Y53A)-actin. C, Stable pool sizes measured from the mean FRAP curves of individual cells show that GFP-Y53E-actin has a smaller, whereas GFP-Y53A-actin has a larger, stable-F-actin pool fraction. Stable pool size: wt = 11%, Y53E = 6%, Y53A = 20%. *p < 0.05, ***p < 0.001, one-way ANOVA with Bonferroni's post hoc test. D, GFP-Y53E-actin recovery half-time is significantly shorter, whereas GFP-Y53A-actin shows no significant change compared with GFP-wt-actin. Recovery half-time of dynamic pool: wt: 8.8 s; Y53E: 5.5 s; Y53A: 7.7 s. Data in C and D represent n(wt) = 14 cells, 51 spines, n(Y53E) = 14 cells, 50 spines, n(Y53A) = 14 cells, 55 spines pooled from three independent experiments. ***p < 0.001, one-way ANOVA with Bonferroni's post hoc test. E, Analyses of FRAP assays show the mean GFP-actin fluorescence recovery curves in dendritic spines of primary DIV14 hippocampal neurons coexpressing mCherry-tagged wt-, Y53E-, or Y53A-actin. F, Stable pool sizes measured from the mean FRAP curves of individual cells show that GFP-actin in spines coexpressing mCherry-Y53E-actin have a smaller stable-F-actin pool fraction, whereas spines coexpressing mCherry-Y53A-actin have a larger stable-F-actin pool fraction. Stable pool size: wt = 13%, Y53E = 7%, Y53A = 20%. **p < 0.01 one-way ANOVA with Bonferroni's post hoc test. G, GFP-actin recovery half-time of dynamic F-actin pool is not significantly different in cells expressing mCherry-(wt/Y53E/Y53A)-actin together with GFP-actin. Recovery half-time of dynamic pool: wt: 9.7 s; Y53E: 9.0 s; Y53A: 8.2 s. Data in F and G represent n(wt) = 15 cells, 58 spines, n(Y53E) = 14 cells, 56 spines, n(Y53A) = 15 cells, 59 spines pooled from four independent experiments. H, Representative images used in spine length and width analyses. Images of GFP fluorescence taken at 2 min intervals. Scale bars, 1 μm. I, Analysis of spine motility by measuring the mean head width and spine length fluctuation at 2 min intervals. Symbols indicating the time points refer to the spines marked with corresponding symbols in H. J, Fluctuation percentages were averaged to give a motility index for cells expressing mCherry-(wt/Y53E/Y53A)-actin together with GFP. The magnitude of spine head movement was larger in cells expressing Y53E-actin compared with wt-actin (wt: 17%, n = 6 cells, 340 spines; Y53E: 20%, n = 5 cells, 375 spines; Y53A: 17%, n = 6 cells, 420 spines). No significant change in spine length fluctuation was observed (wt: 16%, Y53E: 15%, Y53A: 16%). Data are pooled from three independent experiments. **p < 0.01 one-way ANOVA with Bonferroni's post hoc test. Data are represented as mean ± SEM.
To measure spine dynamics, we used a motility analysis that we developed recently (Bertling et al., 2012; Koskinen et al., 2014). For these analyses, 5 3D confocal images were collected at 2 min intervals (Fig. 5H). The spine head diameter and length of each spine at each time point were analyzed using NeuronStudio (Fig. 5I). This analysis revealed that the change in the width of a spine head within 2 min was significantly larger in mCherry-Y53E-actin expressing neurons compared with wt neurons (wt = 17%, Y53E = 20%, p = 0.009; Fig. 5J). The expression of mCherry-Y53A-actin resulted in similar spine dynamics as the expression of mCherry-wt-actin (Fig. 5J). No significant changes in spine length fluctuations were observed. These results indicate that the expression of Y53E-actin increases the short-term dynamics of dendritic spine heads.
Expression of mutant pY53-mimicking actin in the rat hippocampus decreases dendritic spine density and impairs LTP in CA1 pyramidal neurons
To elucidate the importance of actin phosphorylation in vivo, we expressed wt actin and actin mutants in the rat hippocampus using a lentiviral vector and analyzed dendritic spine density and morphology, as well as synaptic function in transduced CA1 pyramidal neurons. Actin constructs were transduced into the hippocampus on P1–P4 (Fig. 6A, left). For imaging experiments, we used slices with sparse labeling of neurons to image individual dendrites (Fig. 6A, center). For fEPSP recordings, only slices with a high transduction efficiency (30–35%) were used (Fig. 6A, right). A transfection efficiency of that magnitude was shown to be sufficient to detect changes in plasticity in our earlier studies (Luchkina et al., 2014).
Figure 6.
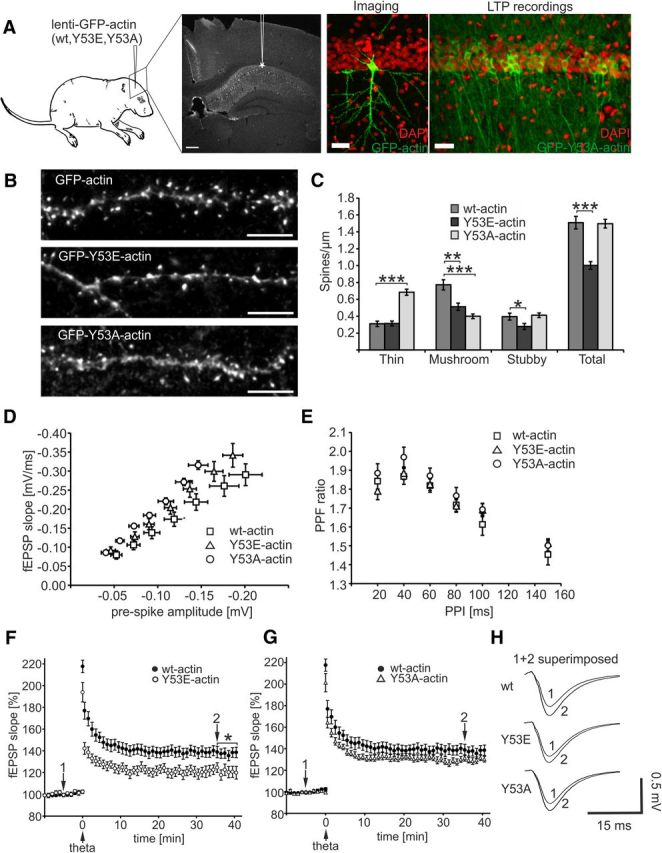
Expression of Y53E-actin in rat hippocampus decreases dendritic spine density and attenuates LTP formation. A, Lentiviral transduction was performed at P1–P4 in rat hippocampi. Higher-magnification confocal images of the CA1 area show transduction efficiency used for imaging and for LTP recordings. DAPI nuclear staining is shown in red, GFP-actin fluorescence in green. Transduction efficiency in slices used for LTP recording is ∼35%. Scale bars, 10 μm. B, Dendrites of CA1 pyramidal hippocampal neurons at P15–P19 expressing GFP-tagged wt-, Y53E-, or Y53A-actin. Similar to wt, GFP-Y53E-actin localizes to spines, whereas GFP-Y53A-actin shows shaft localization. Scale bars, 5 μm. C, Quantification of spine morphology reveals that the total spine number is diminished in neurons expressing GFP-Y53E-actin. Expression of GFP-Y53A-actin shifts spine type distribution to thin spines without altering the total spine density: wt: thin = 0.31, mushroom = 0.77, stubby = 0.40, total = 1.51 spines/μm; n = 11 cells, 1235 spines, 821 μm dendrite, pooled from 3 rats; Y53E: thin = 0.31, mushroom = 0.51, stubby = 0.28, total = 1.00 spines/μm; n = 10 cells, 795 spines, 746 μm dendrite, pooled from 3 rats; Y53A: thin = 0.68, mushroom = 0.40, stubby = 0.41, total = 1.50 spines/μm; n = 10 cells, 964 spines, 637 μm dendrite, pooled from 3 rats. **p < 0.01, ***p < 0.001, one-way ANOVA with Bonferroni's post hoc test. D, I/O ratios did not differ between slices expressing different actin constructs. The number of recordings from slices expressing wt-, Y53E-, and Y53A-actin constructs were 13 (13 rats), 13 (10 rats), and 17 (12 rats), respectively. E, No significant differences in the PPF were found between the wt-, Y53E-, and Y53A-actin construct groups [n(wt) = 12 (12 rats), n(Y53E) = 14 (13 rats), and n(Y53A) = 14 (12 rats)]. F, G, Pooled data from recordings of fEPSPs in the CA1 of hippocampal slices expressing wt-actin (F, G), Y53E-actin (F),and Y53A-GFP-actin (G). A stimulation interval of 20 s was used and every third response is shown in the graph. TFS was applied at 0 min. At 35–40 min, the fEPSP was potentiated 37 ± 4% [n = 18 slices (13 rats)] in wt-actin-expressing slices, 22 ± 4% [n = 11 (8 rats)] in Y53E-actin-expressing slices, and 30 ± 2% [n = 21 (12 rats)] in Y53A-actin-expressing slices. *p < 0.05, Kruskal–Wallis test with Dunn–Bonferroni post hoc method. H, Superimposed average fEPSP responses at −5 min (1) and 35 min (2) for all groups. Data are represented as mean ± SEM.
Spine morphology and density were analyzed at P15–P19, so the expression time was considerably longer compared with the 1 d expression performed in cultured neurons. GFP-Y53E-actin strongly localized to dendritic spine heads in vivo and spine heads were round and well formed (Fig. 6B). The analysis revealed that the expression of the GFP-Y53E-actin significantly decreased overall spine density, especially reducing the amount of mushroom and stubby spines [Y53E: thin = 0.31, mushroom = 0.51 (p = 0.001), stubby = 0.28 (p = 0.029), total = 1.00 spines/μm (p < 0.001)], compared with cells expressing GFP-wt-actin (wt: thin = 0.31, mushroom = 0.77, stubby = 0.40, total = 1.50 spines/μm; Fig. 6C). The spine density in cells expressing GFP-Y53A-actin was unchanged, but the spine type distribution was shifted toward smaller spine heads, resulting in an increase in the number of thin spines (spine head width <0.35 μm) [Y53A: thin = 0.68 (p < 0.001), mushroom = 0.40 (p < 0.001), stubby = 0.41, total = 1.50 spines/μm; Fig. 6C]. In summary, the long-term expression of GFP-Y53E-actin in the rat hippocampus decreased overall spine density.
To determine whether actin mutants affect synaptic function, we performed fEPSP recordings in the CA1 stratum radiatum for virally transduced rat hippocampi at P16–P29. The I/O ratios did not differ between slices expressing the different actin constructs, implicating that the basic synaptic properties were similar in the transduced slices regardless of the actin construct expressed (Fig. 6D). Next, we studied PPF, a form of short-term plasticity reflecting neurotransmitter release probability. PPF is generally used as a measure of presynaptic function (Zucker, 1989; Lauri et al., 2007). No significant differences in the PPF were found between the GFP-(wt/Y53E/ Y53A)-actin construct groups (Fig. 6E). This result corroborates the idea that basal synaptic transmission was unaltered in wt-, Y53E-, and Y53A-actin expressing rats. We then examined LTP, the principal model for studying cellular mechanisms of long-term synaptic strengthening (Bliss and Collingridge, 1993). Synaptic responses were evoked at 0.05 Hz by stimulating a bipolar electrode positioned within Schaffer collateral commissural fibers. fEPSPs were recorded in the stratum radiatum in area CA1 and LTP was induced by TFS stimulation. We found a significantly smaller potentiation of the fEPSP slope after LTP induction in the slices expressing Y53E-actin (time point 35–40 min: 22 ± 4%, p = 0.013) compared with slices expressing wt or Y53A-actin (37 ± 4% and 30 ± 2%, respectively; Fig. 6F–H). Therefore, actin phosphorylation is associated with an impaired capacity of CA1 synapses to express LTP.
Next, we investigated whether the phosphorylation level of endogenous actin changes during LTP expression in acute slices. The experimental protocol used in the experiments shown in Figure 6, F and G, was used (Fig. 7A). The CA1 area from both the control slices receiving only low-frequency stimulation and the slices undergoing theta-frequency LTP induction were frozen in liquid nitrogen 35 min after the LTP induction, a time point when we detected a significant difference in the level of LTP in the slices expressing Y53E-actin (Fig. 6). In addition to slices with LTP induction, we collected samples from control slices recorded without LTP induction (baseline). Moreover, we measured the known LTP-responsive Akt Ser473 phosphorylation (pAkt) levels (Racaniello et al., 2010). pAkt levels were increased 2.7-fold 35 min after LTP induction (Fig. 7B,C). Western blotting against pY53-actin revealed that actin phosphorylation was increased 2.3-fold (p = 0.046) in LTP-induced slices compared with control slices (Fig. 7B,D). These results show that the level of actin phosphorylation increases during LTP expression.
Discussion
The current paradigm is that actin-binding proteins dictate what structures are built from actin molecules. However, recent studies have shown that actin posttranslational modifications are powerful mechanisms that change the properties of actin in a temporally and spatially controlled manner (Terman and Kashina, 2013). Despite the identification of 35 different phosphorylation sites, the role of actin phosphorylation in higher eukaryotes is poorly understood (Terman and Kashina, 2013). Actin Y53 phosphorylation has been best characterized in the slime molds Dictyostelium and Physarum, which are proposed to have their own machinery for phosphorylation-dependent actin regulation. Interestingly, Anaplasma phagocytophilum, the agent of human anaplasmosis, uses actin phosphorylation to modify the host cell's actin cytoskeleton (Sultana et al., 2010). Because actin, especially Y53, is highly conserved, it would be surprising if mammalian cells did not use the regulatory possibilities facilitated by actin phosphorylation.
Here, we show that actin is Y53-phosphorylated in mammalian neurons (Figs. 1, 2). Based on its subcellular localization and rapid increase upon phosphatase inhibition (Figs. 1, 2), actin phosphorylation seems to be strictly controlled both spatially and temporally. To understand the cellular importance of actin phosphorylation, we used actin mutants that either mimic phosphorylation (Y53E) or cannot be phosphorylated (Y53A). These constructs have also been used in earlier studies in Dictyostelium (Liu et al., 2006; Liu et al., 2010; Shu et al., 2010). The expression of the construct mimicking Y53-actin phosphorylation increased actin filament turnover rate and cell dynamics in neurons (Fig. 5). These results fit well with the properties reported for phosphorylated actin (Baek et al., 2008) indicating that the glutamic acid substitution indeed structurally mimics the properties of phosphorylated tyrosine. We propose that Y53E-actin creates points with weaker actin–actin contacts along the filament. When filaments break at these points, individual filaments become shorter and the increase in the numbers of both plus and minus ends enhances the filament turnover rate (Figure 5; Fig. 8A). Conversely, expression of the phosphorylation-deficient mutant reduced actin dynamics (Figure 5). In previous studies, Y53A-actin mutation has been reported to produce biochemically similar effects as Y53E-actin (Liu et al., 2010). However, these effects were seen in experiments with purified proteins in which all actin monomers were mutated. It is plausible that low expression levels of Y53A-actin result in more stable actin filaments. Indeed, Shu et al. (2010) reported that the effect of the Y53A mutation was mostly reversed by copolymerization with wt-actin.
Figure 8.
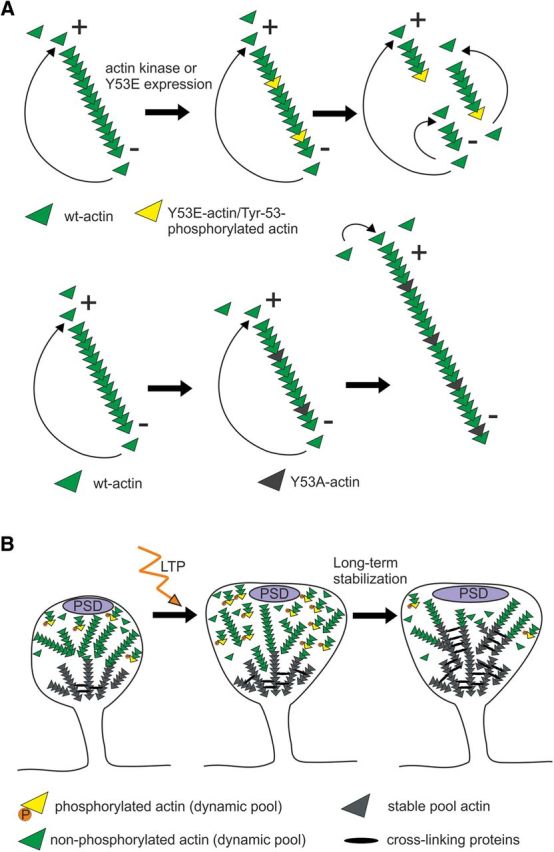
Model figure illustrating the function of mutant actin constructs and the role of Y53 phosphorylation during dendritic spine maturation and LTP formation. A, Working model for the effects of the Y53E- and Y53A-actin expression on actin filament turnover. In a cellular context, there is a net addition of monomers at the plus end and net dissociation at the pointed end, leading to filament elongation occurring mainly at the plus end. The constant incorporation and dissociation of monomers at filament ends results in a “treadmilling” effect. Y53E-actin creates points with weaker actin–actin binding along the filament. When filaments break at these points, the filaments become shorter and the increase in numbers of both plus and minus ends enhances the turnover rate. Y53A-actin creates higher affinity binding with adjacent actin monomers, thus slowing down the depolymerization rate at minus ends. This “pointed end capping” increases filament length. B, During spine morphogenesis, actin filaments are maintained highly dynamic by Y53 phosphorylation (spine on left). High-frequency synapse activation induces reorganization, polymerization, and stabilization of actin filaments. We propose that actin phosphorylation serves as a mechanism to break actin filaments into shorter filaments to facilitate the reorganization of the actin cytoskeleton (spine in the middle). In turn, dephosphorylation is required for the stabilization of actin filaments during LTP maintenance or during dendritic spine maturation (spine on right).
In Y53A-actin expressing neurons, the proportion of branched spines was increased (Fig. 4). A similar phenotype was observed when the actin-depolymerizing agent cofilin-1 was knocked down by siRNA (Hotulainen et al., 2009) or when actin dynamics were decreased through the inhibition of myosin II contractility (Ryu et al., 2006; Koskinen et al., 2014). It is probable that, in all of these cases, the actin filaments are too long. It is important to note that, with transient Y53A-actin expression, the expected proportion of exogenous actin mutants is ∼2% of total actin (Fig. 3B; Vaezi et al., 2002; Gurniak and Witke, 2007), so 98% of all actin molecules are still available for phosphorylation. Because the low expression of Y53E-actin was sufficient to induce significant changes in the actin cytoskeleton and neuronal function, the Y53A-expression-induced cellular effects cannot be fully explained solely by the inhibition of phosphorylation. We propose that Y53A-actin increases the affinity between adjacent actin monomers, making it more difficult for terminal monomers to depolymerize at the minus ends compared with wt-actin. This “capping” slows down the depolymerization rate and increases filament length (Fig. 8A). Accordingly, these studies suggest that, if, during development, actin filaments are too long, too stable, or both, dendritic spine morphogenesis is abnormal and spines exhibit protrusions from the spine heads. Therefore, actin phosphorylation supports spine head growth and dynamic intrinsic fluctuations.
The expression of Y53E-actin induced faster spine dynamics, a faster turnover rate of the dynamic actin filament pool, and a smaller stable F-actin pool (Fig. 5). In contrast, Y53A-actin expression increased the stable F-actin pool size, but did not affect the turnover rate of the F-actin dynamic pool or spine dynamics (Fig. 5). These data led us to conclude that the dynamic pool, and not the stable pool, mediates spine dynamics, at least in the short term. We proposed earlier that during neuronal maturation fast spine dynamics provide the optimal method for searching the proper morphology (Koskinen et al., 2014). When the mature morphology is obtained, the dynamics slow down to provide the optimal conditions for maintaining the given spine morphology (Koskinen et al., 2014; Hlushchenko et al., 2016). Consistent with these ideas, our results suggest that short, 1 d Y53E-actin expression increased the number of spines with more mature mushroom morphology, indicating that Y53E-actin expression helps to find the proper morphology of the spines (Figs. 4A,C, 8B). However, when Y53E-actin was expressed over 2 weeks, dendritic spine density was decreased (Fig. 6B,C), indicating that Y53E-actin expression induced deficiency in the long-term dendritic spine stabilization (Fig. 8B). Evidently, the 24 h expression of the Y53E-actin was too short to induce a loss of dendritic spines. Moreover, our Western blot data from neuronal cultures of different ages showed a significant decrease in actin phosphorylation as neurons matured and spines stabilized (Fig. 1G,H). Therefore, although short-term expression of Y53E-actin promoted dendritic spine morphogenesis, dephosphorylation and stabilization of the dendritic spine actin cytoskeleton are required for dendritic spine maintenance over longer time periods (Fig. 8B). These results also show that cells cannot rescue the spine loss through increasing the rate of formation of new spines, but existing spines need to be properly stabilized.
Activation of synapses causes highly regulated and remarkably rapid changes in spine head size and longevity (Matsuzaki et al., 2004; Okamoto et al., 2004). LTP is proposed to be the structural counterpart of memory formation (Nabavi et al., 2014). The current leading view is that, during LTP induction and expression, actin is first rapidly reorganized through actin severing and new actin filaments are polymerized, leading to the growth of spine heads (Okamoto et al., 2004; Honkura et al., 2008; Bosch et al., 2014; Hlushchenko et al., 2016). Chen et al. (2015) further emphasized the importance of actin filament severing. Using two-photon correlation spectroscopy, they showed that, in chemical LTP, actin filaments of the dynamic F-actin pool are cut into shorter filaments to quickly reorganize and expand the actin cytoskeleton within the spine head (Chen et al., 2015). In chemical LTP, the increase in short filaments was visible after 30 min of LTP induction (Chen et al., 2015). Many studies suggest that, after reorganization, actin filaments are gradually restabilized between 7 and 120 min after LTP induction (Honkura et al., 2008; Bosch et al., 2014; Hlushchenko et al., 2016). Our results suggest that actin phosphorylation could provide a mechanism to induce the breaking of actin filaments into shorter filaments during LTP induction. In turn, dephosphorylation is required to restabilize filaments later in LTP. Our glycine application experiments suggested that actin phosphorylation is transiently increased after chemical LTP induction and this high phosphorylation level is then decreased during LTP stabilization (Fig. 1I–K). The LTP evoked by theta stimulation in acute hippocampal slices resulted in a 2.3-fold increase in actin phosphorylation 35 min after LTP induction (Fig. 7). We only measured one time point after theta stimulation, but we hypothesize that, in acute slices, similarly to cultured neurons, actin phosphorylation is highest soon after LTP induction. After the reorganization stage, actin is slowly dephosphorylated and actin filaments are subsequently stabilized (Fig. 8B). This idea is supported by the fact that actin phosphorylation significantly decreased during neuronal maturation when actin filaments are stabilized, similar to LTP-induced stabilization (Honkura et al., 2008; Bosch et al., 2014; Koskinen et al., 2014; Fig. 1I–K). Impaired LTP in Y53E-actin-expressing CA1 pyramidal neurons (Fig. 6F–H) might mean that the actin filaments cannot be cut shorter and respond to LTP induction because most of the filaments are already short. Therefore, less change in actin structures can be obtained and LTP is impaired. Consistent with this evidence, we also observed a significant increase in the number and size of postsynaptic and presynaptic sites in neurons expressing Y53E-actin in cultures (Fig. 4H–M). These results indicate that synapses are already stronger in these neurons under basal conditions compared with wt neurons. Because small spines are preferential sites for LTP induction (Matsuzaki et al., 2004), it is logical that Y53E-actin-expressing neurons respond inefficiently to LTP induction. In addition, when actin cannot be dephosphorylated, actin filaments cannot be stabilized, causing the deficiency in LTP maintenance.
In conclusion, we discovered a novel mechanism of actin regulation that underlies the exceptionally dynamic actin cytoskeleton in dendritic spines. Actin phosphorylation can serve as a molecular switch to regulate fast changes in actin filament turnover rate in spine morphogenesis and synaptic plasticity. In the future, it will be important to identify and characterize the kinases and phosphatases regulating actin phosphorylation and dephosphorylation during these processes. In addition, it would be interesting to test whether actin phosphorylation plays a role in learning processes or in neurological disorders associated with cognitive dysfunction. If so, actin phosphorylation could provide a novel treatment target for neurological diseases.
Footnotes
This work was supported by the Academy of Finland (Grant SA 252302 to E.B., and Grant SA 125867 and Grant 266351 to P.H. and Grant SA 257486 to E.C.), the University of Helsinki (a 3-year grant to P.H.), ERC Grant No 322742 –iPLASTICITY to E.C. and the Sigrid Juselius Foundation (E.C.). We thank Ville Paavilainen for creating structural images for Figure 3; Ville Paavilainen, Maria Vartiainen, Olaya Llano, and Claudio Rivera for valuable comments; Amr Abou Elezz for English proofreading; Maria Vartiainen, Martin Bähler, Lotta von Ossowski, and Kari Keinänen for kindly providing GFP-actin, mCherry-actin, and SAP97-mCherry plasmid constructs; and Seija Lågas and Outi Nikkilä for hippocampal neuron dissociation.
The authors declare no competing financial interests.
References
- Anderson WW, Collingridge GL. Capabilities of the WinLTP data acquisition program extending beyond basic LTP experimental functions. J Neurosci Methods. 2007;162:346–356. doi: 10.1016/j.jneumeth.2006.12.018. [DOI] [PubMed] [Google Scholar]
- Baba T, Fusaki N, Shinya N, Iwamatsu A, Hozumi N. Actin tyrosine dephosphorylation by the Src homology 1-containing protein tyrosine phosphatase is essential for actin depolymerization after membrane IgM cross-linking. J Immunol. 2003;170:3762–3768. doi: 10.4049/jimmunol.170.7.3762. [DOI] [PubMed] [Google Scholar]
- Baek K, Liu X, Ferron F, Shu S, Korn ED, Dominguez R. Modulation of actin structure and function by phosphorylation of Tyr-53 and profilin binding. Proc Natl Acad Sci U S A. 2008;105:11748–11753. doi: 10.1073/pnas.0805852105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellot A, Guivernau B, Tajes M, Bosch-Morató M, Valls-Comamala V, Muñoz FJ. The structure and function of actin cytoskeleton in mature glutamatergic dendritic spines. Brain Res. 2014;1573:1–16. doi: 10.1016/j.brainres.2014.05.024. [DOI] [PubMed] [Google Scholar]
- Bertling E, Ludwig A, Koskinen M, Hotulainen P. Methods for three-dimensional analysis of dendritic spine dynamics. Methods Enzymol. 2012;506:391–406. doi: 10.1016/B978-0-12-391856-7.00043-3. [DOI] [PubMed] [Google Scholar]
- Bliss TV, Collingridge GL. A synaptic model of memory: Long-term potentiation in the hippocampus. Nature. 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- Bosch M, Castro J, Saneyoshi T, Matsuno H, Sur M, Hayashi Y. Structural and molecular remodeling of dendritic spine substructures during long-term potentiation. Neuron. 2014;82:444–459. doi: 10.1016/j.neuron.2014.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassens C, Kleene R, Xiao MF, Friedrich C, Dityateva G, Schafer-Nielsen C, Schachner M. Binding of the receptor tyrosine kinase TrkB to the neural cell adhesion molecule (NCAM) regulates phosphorylation of NCAM and NCAM-dependent neurite outgrowth. J Biol Chem. 2010;285:28959–28967. doi: 10.1074/jbc.M110.114835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JH, Kellner Y, Zagrebelsky M, Grunwald M, Korte M, Walla PJ. Two-photon correlation spectroscopy in single dendritic spines reveals fast actin filament reorganization during activity-dependent growth. PLoS One. 2015;10:e0128241. doi: 10.1371/journal.pone.0128241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choidas A, Jungbluth A, Sechi A, Murphy J, Ullrich A, Marriott G. The suitability and application of a GFP-actin fusion protein for long-term imaging of the organization and dynamics of the cytoskeleton in mammalian cells. Eur J Cell Biol. 1998;77:81–90. doi: 10.1016/S0171-9335(98)80075-7. [DOI] [PubMed] [Google Scholar]
- Clarke VR, Collingridge GL, Lauri SE, Taira T. Synaptic kainate receptors in CA1 interneurons gate the threshold of theta-frequency-induced long-term potentiation. J Neurosci. 2012;32:18215–18226. doi: 10.1523/JNEUROSCI.2327-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dopie J, Skarp KP, Rajakylä EK, Tanhuanpää K, Vartiainen MK. Active maintenance of nuclear actin by importin 9 supports transcription. Proc Natl Acad Sci U S A. 2012;109:E544–E552. doi: 10.1073/pnas.1118880109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiala JC, Spacek J, Harris KM. Dendritic spine pathology: cause or consequence of neurological disorders? Brain Res Rev. 2002;39:29–54. doi: 10.1016/S0165-0173(02)00158-3. [DOI] [PubMed] [Google Scholar]
- Frost NA, Shroff H, Kong H, Betzig E, Blanpied TA. Single-molecule discrimination of discrete perisynaptic and distributed sites of actin filament assembly within dendritic spines. Neuron. 2010;67:86–99. doi: 10.1016/j.neuron.2010.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gauthier ML, Lydan MA, O'Day D, Cotter AD. Endogenous autoinhibitors regulate changes in actin tyrosine phosphorylation during Dictyostelium spore germination. Cell Signal. 1997;9:79–83. doi: 10.1016/S0898-6568(96)00113-1. [DOI] [PubMed] [Google Scholar]
- Gurniak CB, Witke W. HuGE, a novel GFP-actin-expressing mouse line for studying cytoskeletal dynamics. Eur J Cell Biol. 2007;86:3–12. doi: 10.1016/j.ejcb.2006.08.007. [DOI] [PubMed] [Google Scholar]
- Hlushchenko I, Koskinen M, Hotulainen P. Dendritic spine actin dynamics in neuronal maturation and synaptic plasticity. Cytoskeleton. 2016 doi: 10.1002/cm.21280. In press. [DOI] [PubMed] [Google Scholar]
- Hoffman GE, Le WW, Sita LV. The importance of titrating antibodies for immunocytochemical methods. Curr Protoc Neurosci. 2008 doi: 10.1002/0471142301.ns0212s45. Chapter 2: Unit 2.12. [DOI] [PubMed] [Google Scholar]
- Honkura N, Matsuzaki M, Noguchi J, Ellis-Davies GC, Kasai H. The subspine organization of actin fibers regulates the structure and plasticity of dendritic spines. Neuron. 2008;57:719–729. doi: 10.1016/j.neuron.2008.01.013. [DOI] [PubMed] [Google Scholar]
- Hotulainen P, Hoogenraad CC. Actin in dendritic spines: connecting dynamics to function. J Cell Biol. 2010;189:619–629. doi: 10.1083/jcb.201003008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotulainen P, Llano O, Smirnov S, Tanhuanpää K, Faix J, Rivera C, Lappalainen P. Defining mechanisms of actin polymerization and depolymerization during dendritic spine morphogenesis. J Cell Biol. 2009;185:323–339. doi: 10.1083/jcb.200809046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard PK, Sefton BM, Firtel RA. Tyrosine phosphorylation of actin in Dictyostelium associated with cell-shape changes. Science. 1993;259:241–244. doi: 10.1126/science.7678470. [DOI] [PubMed] [Google Scholar]
- Hung RJ, Pak CW, Terman JR. Direct redox regulation of F-actin assembly and disassembly by Mical. Science. 2011;334:1710–1713. doi: 10.1126/science.1211956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jungbluth A, von Arnim V, Biegelmann E, Humbel B, Schweiger A, Gerisch G. Strong increase in the tyrosine phosphorylation of actin upon inhibition of oxidative phosphorylation: correlation with reversible rearrangements in the actin skeleton of Dictyostelium cells. J Cell Sci. 1994;107:117–125. doi: 10.1242/jcs.107.1.117. [DOI] [PubMed] [Google Scholar]
- Jungbluth A, Eckerskorn C, Gerisch G, Lottspeich F, Stocker S, Schweiger A. Stress-induced tyrosine phosphorylation of actin in Dictyostelium cells and localization of the phosphorylation site to tyrosine-53 adjacent to the DNase I binding loop. FEBS Lett. 1995;375:87–90. doi: 10.1016/0014-5793(95)01165-B. [DOI] [PubMed] [Google Scholar]
- Kinoshita E, Kinoshita-Kikuta E, Matsubara M, Yamada S, Nakamura H, Shiro Y, Aoki Y, Okita K, Koike T. Separation of phosphoprotein isotypes having the same number of phosphate groups using phosphate-affinity SDS-PAGE. Proteomics. 2008;8:2994–3003. doi: 10.1002/pmic.200800243. [DOI] [PubMed] [Google Scholar]
- Kishi Y, Clements C, Mahadeo DC, Cotter DA, Sameshima M. High levels of actin tyrosine phosphorylation: correlation with the dormant state of Dictyostelium spores. J Cell Sci. 1998;111:2923–2932. doi: 10.1242/jcs.111.19.2923. [DOI] [PubMed] [Google Scholar]
- Korobova F, Svitkina T. Molecular architecture of synaptic actin cytoskeleton in hippocampal neurons reveals a mechanism of dendritic spine morphogenesis. Mol Biol Cell. 2010;21:165–176. doi: 10.1091/mbc.E09-07-0596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koskinen M, Hotulainen P. Measuring F-actin properties in dendritic spines. Front Neuroanat. 2014;8:74. doi: 10.3389/fnana.2014.00074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koskinen M, Bertling E, Hotulainen P. Methods to measure actin treadmilling rate in dendritic spines. Methods Enzymol. 2012;505:47–58. doi: 10.1016/B978-0-12-388448-0.00011-5. [DOI] [PubMed] [Google Scholar]
- Koskinen M, Bertling E, Hotulainen R, Tanhuanpää K, Hotulainen P. Myosin IIb controls actin dynamics underlying the dendritic spine maturation. Mol Cell Neurosci. 2014;61:56–64. doi: 10.1016/j.mcn.2014.05.008. [DOI] [PubMed] [Google Scholar]
- Lauri SE, Palmer M, Vesikansa A, Segerstrale M, Taira T, Collingridge GL. Presynaptic mechanisms of expression of LTP at CA1 synapses in the hippocampus. Neuropharmacology. 2007;52:1–11. doi: 10.1016/j.neuropharm.2006.06.017. [DOI] [PubMed] [Google Scholar]
- Lim YP, Wong CY, Ooi LL, Druker BJ, Epstein RJ. Selective tyrosine hyperphosphorylation of cytoskeletal and stress proteins in primary human breast cancers: implications for adjuvant use of kinase-inhibitory drugs. Clin Cancer Res. 2004;10:3980–3987. doi: 10.1158/1078-0432.CCR-03-0663. [DOI] [PubMed] [Google Scholar]
- Liu X, Shu S, Hong MS, Levine RL, Korn ED. Phosphorylation of actin Tyr-53 inhibits filament nucleation and elongation and destabilizes filaments. Proc Natl Acad Sci U S A. 2006;103:13694–13699. doi: 10.1073/pnas.0606321103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Shu S, Hong MS, Yu B, Korn ED. Mutation of actin Tyr-53 alters the conformations of the DNase I-binding loop and the nucleotide-binding cleft. J Biol Chem. 2010;285:9729–9739. doi: 10.1074/jbc.M109.073452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu W, Man H, Ju W, Trimble WS, MacDonald JF, Wang YT. Activation of synaptic NMDA receptors induces membrane insertion of new AMPA receptors and LTP in cultured hippocampal neurons. Neuron. 2001;29:243–254. doi: 10.1016/S0896-6273(01)00194-5. [DOI] [PubMed] [Google Scholar]
- Luchkina NV, Huupponen J, Clarke VR, Coleman SK, Keinänen K, Taira T, Lauri SE. Developmental switch in the kinase dependency of long-term potentiation depends on expression of GluA4 subunit-containing AMPA receptors. Proc Natl Acad Sci U S A. 2014;111:4321–4326. doi: 10.1073/pnas.1315769111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuzaki M, Honkura N, Ellis-Davies GC, Kasai H. Structural basis of long-term potentiation in single dendritic spines. Nature. 2004;429:761–766. doi: 10.1038/nature02617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer D, Bonhoeffer T, Scheuss V. Balance and stability of synaptic structures during synaptic plasticity. Neuron. 2014;82:430–443. doi: 10.1016/j.neuron.2014.02.031. [DOI] [PubMed] [Google Scholar]
- Nabavi S, Fox R, Proulx CD, Lin JY, Tsien RY, Malinow R. Engineering a memory with LTD and LTP. Nature. 2014;511:348–352. doi: 10.1038/nature13294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto K, Nagai T, Miyawaki A, Hayashi Y. Rapid and persistent modulation of actin dynamics regulates postsynaptic reorganization underlying bidirectional plasticity. Nat Neurosci. 2004;7:1104–1112. doi: 10.1038/nn1311. [DOI] [PubMed] [Google Scholar]
- Penzes P, Cahill ME, Jones KA, VanLeeuwen JE, Woolfrey KM. Dendritic spine pathology in neuropsychiatric disorders. Nat Neurosci. 2011;14:285–293. doi: 10.1038/nn.2741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Racaniello M, Cardinale A, Mollinari C, D'Antuono M, De Chiara G, Tancredi V, Merlo D. Phosphorylation changes of CaMKII, ERK1/2, PKB/Akt kinases and CREB activation during early long-term potentiation at Schaffer collateral-CA1 mouse hippocampal synapses. Neurochem Res. 2010;35:239–346. doi: 10.1007/s11064-009-0047-0. [DOI] [PubMed] [Google Scholar]
- Rodriguez A, Ehlenberger DB, Dickstein DL, Hof PR, Wearne SL. Automated three-dimensional detection and shape classification of dendritic spines from fluorescence microscopy images. PLoS One. 2008;3:e1997. doi: 10.1371/journal.pone.0001997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu J, Liu L, Wong TP, Wu DC, Burette A, Weinberg R, Wang YT, Sheng M. A critical role for myosin IIb in dendritic spine morphology and synaptic function. Neuron. 2006;49:175–182. doi: 10.1016/j.neuron.2005.12.017. [DOI] [PubMed] [Google Scholar]
- Saarikangas J, Kourdougli N, Senju Y, Chazal G, Segerstråle M, Minkeviciene R, Kuurne J, Mattila PK, Garrett L, Hölter SM, Becker L, Racz I, Hans W, Klopstock T, Wurst W, Zimmer A, Fuchs H, Gailus-Durner V, Hrabě de Angelis M, von Ossowski L, Taira T, Lappalainen P, Rivera C, Hotulainen P. MIM-induced membrane bending promotes dendritic spine initiation. Dev Cell. 2015;33:644–659. doi: 10.1016/j.devcel.2015.04.014. [DOI] [PubMed] [Google Scholar]
- Schweiger A, Mihalache O, Ecke M, Gerisch G. Stage-specific tyrosine phosphorylation of actin in Dictyostelium discoideum cells. J Cell Sci. 1992;102:601–609. doi: 10.1242/jcs.102.3.601. [DOI] [PubMed] [Google Scholar]
- Semenov A, Goldsteins G, Castrén E. Phosphoproteomic analysis of neurotrophin receptor TrkB signaling pathways in mouse brain. Cell Mol Neurobiol. 2006;26:163–175. doi: 10.1007/s10571-006-9023-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahi K, Baudry M. Glycine-induced changes in synaptic efficacy in hippocampal slices involve changes in AMPA receptors. Brain Res. 1993;627:261–266. doi: 10.1016/0006-8993(93)90329-L. [DOI] [PubMed] [Google Scholar]
- Shu S, Liu X, Kriebel PW, Hong MS, Daniels MP, Parent CA, Korn ED. Expression of Y53A-actin in Dictyostelium disrupts the cytoskeleton and inhibits intracellular and intercellular chemotactic signaling. J Biol Chem. 2010;285:27713–27725. doi: 10.1074/jbc.M110.116277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Star EN, Kwiatkowski DJ, Murthy VN. Rapid turnover of actin in dendritic spines and its regulation by activity. Nat Neurosci. 2002;5:239–246. doi: 10.1038/nn811. [DOI] [PubMed] [Google Scholar]
- Sultana H, Neelakanta G, Kantor FS, Malawista SE, Fish D, Montgomery RR, Fikrig E. Anaplasma phagocytophilum induces actin phosphorylation to selectively regulate gene transcription in Ixodes scapularis ticks. J Exp Med. 2010;207:1727–1743. doi: 10.1084/jem.20100276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terman JR, Kashina A. Post-translational modification and regulation of actin. Curr Opin Cell Biol. 2013;25:30–38. doi: 10.1016/j.ceb.2012.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ting PY, Johnson CW, Fang C, Cao X, Graeber TG, Mattos C, Colicelli J. Tyrosine phosphorylation of RAS by ABL allosterically enhances effector binding. FASEB J. 2015;29:3750–3761. doi: 10.1096/fj.15-271510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaezi A, Bauer C, Vasioukhin V, Fuchs E. Actin cable dynamics and Rho/Rock orchestrate a polarized cytoskeletal architecture in the early steps of assembling a stratified epithelium. Dev Cell. 2002;3:367–381. doi: 10.1016/S1534-5807(02)00259-9. [DOI] [PubMed] [Google Scholar]
- Vartiainen M, Ojala PJ, Auvinen P, Peränen J, Lappalainen P. Mouse A6/twinfilin is an actin monomer-binding protein that localizes to the regions of rapid actin dynamics. Mol Cell Biol. 2000;20:1772–1783. doi: 10.1128/MCB.20.5.1772-1783.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia F, Li J, Hickey GW, Tsurumi A, Larson K, Guo D, Yan SJ, Silver-Morse L, Li WX. Raf activation is regulated by tyrosine 510 phosphorylation in Drosophila. PLoS Biol. 2008;6:e128. doi: 10.1371/journal.pbio.0060128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuste R, Bonhoeffer T. Morphological changes in dendritic spines associated with long-term synaptic plasticity. Annu Rev Neurosci. 2001;24:1071–1089. doi: 10.1146/annurev.neuro.24.1.1071. [DOI] [PubMed] [Google Scholar]
- Zucker RS. Short-term synaptic plasticity. Annu Rev Neurosci. 1989;12:13–31. doi: 10.1146/annurev.ne.12.030189.000305. [DOI] [PubMed] [Google Scholar]