

Abstract
Development of the hippocampal dentate gyrus (DG) in the mammalian brain is achieved through multiple processes during late embryonic and postnatal stages, with each developmental step being strictly governed by extracellular cues and intracellular mechanisms. Here, we show that the maintenance DNA methyltransferase 1 (Dnmt1) is critical for development of the DG in the mouse. Deletion of Dnmt1 in neural stem cells (NSCs) at the beginning of DG development led to a smaller size of the granule cell layer in the DG. NSCs lacking Dnmt1 failed to establish proper radial processes or to migrate into the subgranular zone, resulting in aberrant neuronal production in the molecular layer of the DG and a reduction of integrated neurons in the granule cell layer. Interestingly, prenatal deletion of Dnmt1 in NSCs affected not only the developmental progression of the DG but also the properties of NSCs maintained into adulthood: Dnmt1-deficient NSCs displayed impaired neurogenic ability and proliferation. We also found that Dnmt1 deficiency in NSCs decreased the expression of Reelin signaling components in the developing DG and increased that of the cell cycle inhibitors p21 and p57 in the adult DG. Together, these findings led us to propose that Dnmt1 functions as a key regulator to ensure the proper development of the DG, as well as the proper status of NSCs maintained into adulthood, by modulating extracellular signaling and intracellular mechanisms.
SIGNIFICANCE STATEMENT Here, we provide evidence that Dnmt1 is required for the proper development of the hippocampal dentate gyrus (DG). Deletion of Dnmt1 in neural stem cells (NSCs) at an early stage of DG development impaired the ability of NSCs to establish secondary radial glial scaffolds and to migrate into the subgranular zone of the DG, leading to aberrant neuronal production in the molecular layer, increased cell death, and decreased granule neuron production. Prenatal deletion of Dnmt1 in NSCs also induced defects in the proliferation and neurogenic ability of adult NSCs. Furthermore, we found that Dnmt1 regulates the expression of key extracellular signaling components during developmental stages while modulating intracellular mechanisms for proliferation and neuronal production of NSCs in the adult.
Keywords: dentate gyrus, Dnmt1, epigenetics
Introduction
The mammalian forebrain has two restricted regions where neurogenesis persists into adulthood: the subventricular zone (SVZ) of the lateral ventricles and the subgranular zone (SGZ) of the dentate gyrus (DG) in the hippocampus (Altman and Das, 1965; Lois and Alvarez-Buylla, 1993; Kuhn et al., 1996; Eriksson et al., 1998). Almost all other brain regions cease neuronal production around birth, suggesting that these two regions have distinct developmental trajectories that allow them to maintain their neural stem cell (NSC) pool and neurogenic potential into adulthood from embryonic stages (Kriegstein and Alvarez-Buylla, 2009). In the adult DG, new neurons generated from NSCs residing in the SGZ have been implicated in hippocampus-dependent learning and memory (Imayoshi et al., 2008; Deng et al., 2010). Although our understanding of adult neurogenesis has progressed in the last decade, little is still known about the mechanisms controlling development of the DG that enable neurogenesis to occur even in the adult DG.
DG development begins from the late embryonic stage (around embryonic day (E)15) and lasts until the postnatal stage (postnatal day (P)14-P21). This is achieved through multiple spatiotemporally regulated developmental processes involving NSC migration, differentiation, and morphological change (Altman and Bayer, 1990a, b; Li and Pleasure, 2005). Accumulating studies indicate that cooperation between extracellular cues and intrinsic transcription factors is necessary for each step in DG development (Li and Pleasure, 2005; Shen et al., 2006; Hodge et al., 2012). For example, Wnt and bone morphogenetic protein signaling induce expression of the homeodomain transcription factor Emx2, whose deletion impairs the production and migration of neuronal progenitors (NPs) during embryonic DG development (Theil et al., 2002; Oldekamp et al., 2004). Furthermore, Reelin signaling participates in the migration of granule neurons to their final destination during postnatal DG development (Brunne et al., 2013).
Epigenetic modifications, such as DNA methylation and histone modification, regulate mammalian development, including that in the CNS (Meehan, 2003; Hirabayashi and Gotoh, 2010). During cortical development, epigenetic modifications play a pivotal role in regulating the competence of NSCs for neuronal and glial differentiation by coordinating specific signaling pathways and transcription factors (Hirabayashi and Gotoh, 2010; Murao et al., 2016). For instance, at the onset of the gliogenic phase of NSC differentiation, astrocyte-specific gene promoters undergo DNA demethylation, enabling NSCs to react to astrogliogenic cues (Takizawa et al., 2001; Namihira et al., 2009), whereas neurogenic gene promoters obtain repressive histone modifications, making them unresponsive to Wnt signaling, which induces neuronal differentiation (Hirabayashi et al., 2009). In contrast to the accumulating evidence regarding cortical development, it remains unclear whether and how epigenetic regulation contributes to DG development. DNA methyltransferase 1 (Dnmt1), which is responsible for maintaining DNA methylation during DNA replication, plays essential role in cortical development by preventing precocious astrocyte differentiation in the neurogenic phase (Fan et al., 2001, 2005; Hutnick et al., 2009; Namihira et al., 2009). Recently, we found that DNMT1 is highly expressed in proliferating NSCs in the adult DG, ensuring the survival of newly generated neurons (Noguchi et al., 2015). These findings imply that Dnmt1 also has particular functions in DG development.
We reveal here that Dnmt1 is indispensable for DG development. Deletion of Dnmt1 in NSCs at the beginning of DG development impaired multiple developmental steps, resulting in a smaller granule cell layer (GCL) in adult DGs. NSCs lacking Dnmt1 are mispositioned and failed to establish radial processes. Furthermore, Dnmt1 ablation leads to aberrant neuronal production and increased cell death, ultimately resulting in fewer granule neurons in the GCL. Although Dnmt1-deleted NSCs are maintained into adulthood, they display impaired neurogenic ability and an increased quiescent population. Deletion of Dnmt1 also disrupted the expression of Reelin signaling components and the cell cycle inhibitors p21 and p57, which affect migration and proliferation of NSCs, respectively (Kippin et al., 2005; Brunne et al., 2013; Furutachi et al., 2015).
Materials and Methods
Animals: generation of Nestin-CreERT2; Dnmt1 conditional mutant mice.
For tamoxifen (TAM)-inducible Cre-mediated Dnmt1 deletion in NSCs, Dnmt1flox/flox mice (Fan et al., 2001) were crossed with Nestin-CreERT2 transgenic mice in which CreERT2 is expressed under the Nestin promoter and enhancer (Imayoshi et al., 2006). TAM administration in Nestin-CreERT2; Dnmt1 conditional mutant mice (cKO) inactivates Dnmt1 through deletion of exons 4 and 5 of Dnmt1 in Nestin-expressing NSCs. Either Nestin-CreERT2; Dnmt1flox/+ or CreERT2-negative mice were used as controls. No differences between these mouse genotypes were observed with respect to the brain shape, morphology of DG, or volume of GCL; accordingly, these mice were analyzed together as controls. For in vitro assay of NSCs, ICR background mice were used. All pregnant mice (ICR background) were obtained from SLC. For timed mating, the day of vaginal plug appearance was considered as embryonic day (E) 0.5, and the day of birth was defined as postnatal day (P) 0. Eight- to ten-week-old animals were used as adult mice; both male and female mice were analyzed, with no distinction. All mice used in this study were maintained on a 12 h light/dark cycle with free access to food and water. All animal procedures were in accordance with the animal experimentation guidelines of Nara Institute of Science and Technology, which follow the National Institutes of Health Guide for the care and use of laboratory animals. All efforts were made to minimize animal suffering and to reduce the number of animals used.
TAM and BrdU administration.
TAM (Sigma) was dissolved in sesame oil at 10 mg/ml. Pregnant mice were intraperitoneally administered 2 mg of TAM with 27-gauge needles. For BrdU labeling, neonatal pups were intraperitoneally injected with BrdU (Sigma) dissolved in saline (0.9% NaCl) at a dose of 100 mg/kg.
Tissue preparation.
To prepare embryonic brain, pregnant mice were killed by cervical dislocation on the indicated developmental day, and embryos were perfused successively PBS and ice-cold 4% PFA in PBS, pH 7.2. For preparation of postnatal and adult brains, pups and adult mice were deeply anesthetized with sodium pentobarbital (50 mg/kg i.p.) before perfusion with 4% PFA in PBS. Brains were dissected and postfixed with 4% PFA in PBS overnight at 4°C. For cryoprotection, fixed brains were stored in 15% sucrose in PBS overnight at 4°C and then transferred into 30% sucrose in PBS overnight at 4°C. One side of the brain was embedded in optimal cutting temperature compound (Tissue Tek, Sakura Finetek, 25608–930) and frozen at −80°C for cryosectioning. Frozen brains were serially sectioned with Leica CM 1900 (Leica Microsystems) in the coronal plane at 20 μm (for embryonic brains) or 40 μm (for postnatal and adult brains) thickness. The embryonic brain sections were mounted on Matsunami adhesive slide-coated glass slides (Matsunami Glass, S9441) and stored at −20°C until use. In the preparation of postnatal and adult brain sections, every sixth section was serially collected in individual wells of a 6-well plate containing sterilized PBS, in order from anterior to posterior, and preserved at 4°C.
Cell culture.
We obtained primary postnatal NSCs from mouse at P1. DGs of ICR P1 mice were carefully dissected after stripping of meninges. The tissue was digested with papain (Sigma) at 37°C for 20 min. After centrifugation (200 × g, 5 min), the cell pellet was resuspended in calcium- and magnesium-free HBSS (Sigma), and the suspension was passed through a 40 mm Cell Strainer (BD Falcon). After centrifugation (200 × g, 5 min), the cell pellet was resuspended in N2-supplemented DMEM/F-12 (Invitrogen, 11320-033), containing 10 ng/ml bFGF (PeproTech) and 10 ng/ml EGF (PeproTech). Adult NSCs were collected from the DGs of 8- to 10-week-old mice, as previously described (Noguchi et al., 2015). Both postnatal and adult NSCs were cultured on a poly-L-ornithine/fibronectin-coated dish in N2-supplemented DMEM/F-12, containing 10 ng/ml each of bFGF and EGF, <5% CO2 at 37°C. HEK293T cells were maintained in DMEM with 10% FBS (heat inactivated, Biowest) and gentamicin sulfate solution (100 mg/ml, Nacalai Tesque). To inhibit DNMT activity, the cells were treated with 50 or 200 nm of the DNMT inhibitor RG108 (Wako). To label with 5-ethynyl uridine (EdU), the cells were cultured with 10 μm EdU (Click-iT EdU Imaging Kit, Invitrogen) for 30 min, and stained according to the manufacturer's instructions.
Lentiviral constructs, preparation of lentivirus, and viral infection.
The lentivirus vector (pLLX) used to express short hairpin RNA was generously provided by Drs. Z. Zhou and M. E. Greenberg. pLLX is a dual-promoter lentivirus vector constructed by inserting the U6 promoter-driven shRNA cassette 5′ to the ubiquitin-C promoter in the FUIGW plasmid (Lois et al., 2002; Zhou et al., 2006). pLLX was modified to express GFP together with a puromycin resistance gene under the ubiquitin C promoter. shRNAs for Dnmt1, p21, and p57 lentivirus constructs were generated by inserting oligonucleotides into the HpaI and XhoI sites of pLLX. The following oligonucleotides were used for targeting Dnmt1, p21, and p57 mRNA as previously reported: Dnmt1, ACCAAGCTGTGTAGTACTT (targeting the 3′UTR of Dnmt1 mRNA) (Noguchi et al., 2015); p21, TTAGGACTCAACCGTAATA (targeting the 3′UTR of p21 mRNA) (Fasano et al., 2007); and p57, CGACTTCTTCGCCAAGCGC (targeting the coding region of p57 mRNA) (Zou et al., 2011). The control sequence was GCTTCAATTCGCGCACCTA, which does not exist in either mouse genomic DNA or mRNA. To prepare lentivirus, HEK293T cells were cotransfected with these constructs and lentiviral packaging vectors (pCAG-HIVgp and pCMV-VSV-G-RSV-Rev). The culture supernatants were collected 48 h after transfection, and virus was introduced into NSCs by adding the supernatants to the culture medium. NSCs were infected with lentivirus and treated with puromycin (0.2 μg/ml; Sigma, P8833) 4 d after infection for 3 d. For RNA collection and proliferation analysis, infected NSCs were cultured for 1 week in N2 medium with bFGF and EGF.
Immunocytochemistry.
Cryosections were washed with PBS and blocked for 1 h at room temperature with blocking solution (3% FBS and 0.1% Triton X-100), and incubated overnight at 4°C with primary antibodies diluted in blocking solution. The following primary antibodies were used in this study: rabbit anti-DNMT1 (1:500; Cosmo Bio, BAM-70-203-EX); mouse anti-Ki67 (1:500; BD Biosciences, 550609); goat anti-Sox2 (1:100; Santa Cruz Biotechnology, sc-17320); rabbit anti-Tbr2 (1:500; Abcam, ab23345); mouse anti-Nestin (1:500; Millipore, MAB353); goat anti-DCX (1:100; Santa Cruz Biotechnology, sc-8066); mouse anti-NeuN (1:500; Millipore, MAB377); chick anti-GFAP (1:500; Millipore, AB5541); goat anti-NeuroD (1:100; Santa Cruz Biotechnology, sc-1084); rabbit anti-S100β (1:500; Abcam, ab41548); mouse anti-S100β (1:500; Sigma, S2532); rabbit anti-Active caspase 3 (1:500; R&D Systems, AF835); mouse anti-phospho-histone H3 (Ser10) (pHH3) (1:500; Cell Signaling Technology, 9706); rabbit anti-GFAP (1:500; Sigma, G9269); chick anti-GFP (1:500; Aves Laboratories, GFP-1020); and rat anti-BrdU (1:500; AbD Serotec, OBT0030). For staining of Ki67, Tbr2, pHH3, and DNMT1, antigen retrieval was performed by heating sections in target retrieval solution (DAKO) at 105°C for 15 min before blocking. For detection of BrdU-labeled cells, free-floating sections were treated with 2 m HCl at 37°C for 5 min and washed with PBS before blocking and incubation with primary antibodies. After three washes in PBS, sections were incubated for 2 h with corresponding secondary antibodies: CF488 donkey anti-mouse IgG (H+L), highly cross-adsorbed (1:500; Biotium, 20014); CF543 donkey anti-rabbit IgG (H+L), highly cross-adsorbed (1:500; Biotium, 20038); CF647 donkey anti-goat IgG (H+L), highly cross-adsorbed (1:500; Biotium, 20048); CF647 donkey anti-rabbit IgG (H+L), highly cross-adsorbed (1:500; Biotium, 20047); and CF568 donkey anti-rat IgG (H+L), highly cross-adsorbed (1:500; Biotium, 20092). Hoechst 33258 (1:500; Nacalai Tesque) was used for nuclear staining. After a final rinse with PBS, sections were mounted on glass slides with Immu-Mount (Thermo Scientific, 9990412), and images were taken using a Zeiss LSM 780 confocal microscope. For immunostaining of cells, the cells were fixed with 4% PFA in PBS for 20 min and incubated for 3 h at 4°C with primary antibodies diluted in blocking solution, followed by incubation with the corresponding secondary antibodies. After a final rinse with PBS, samples were mounted, and images were taken using a Leica AF600 fluorescence microscope.
Volumetric analyses and cell counting.
Measurements for volumetric analyses were taken in every sixth 40 μm coronal section stained with Hoechst, as previously described (Ansorg et al., 2012). Briefly, the areas of the dentate GCL were measured using ImageJ software (National Institutes of Health). Volumes (V) were calculated as V = ΣA × i × d, according to Cavalieri's principle, where A = the sum of GCL areas in each section, i = the interval between the sections, and d = the section thickness. Following previous reports (Kronenberg et al., 2003; Steiner et al., 2006), cell counting was performed on every sixth (for counting Type 1 cells), 12th (for counting Ki67+ cells; pHH3+ cells; Active caspase 3+ cells; and DCX+ cells) or 24th (for counting BrdU+ cells; BrdU-labeled marker-positive cells; Sox2+ cells; Tbr2+ cells; and S100β+ cells) coronal section containing DG at the same anatomical level, and marker-positive cells were counted in the series of collected sections throughout the GCL, molecular layer (ML), and hilus of the entire DG. The total number of marker-positive cells in the DG was obtained by multiplying the resultant counts by 6, 12, or 24 (according to the interval between sections). The total number of marker-positive cells in the complete GCL, ML, and hilus of the DG were estimated in a similar way. To assess proliferation of NSCs in vitro, at least 300 GFP-positive cells from one sample (n = 3 per group) were counted.
Real-time PCR.
Total RNA was isolated using Sepasol-RNA I Super G (Nacalai Tesque), according to the manufacturer's instructions, and each sample was reverse-transcribed using a SuperScript VILO cDNA Synthesis Kit (Invitrogen). Quantitative PCRs were performed using a KAPA SYBR Fast qPCR Kit (KAPA Biosystems) with ROX as reference dye. Expression levels of each gene were normalized to GAPDH and calculated relative to the control. Primer sequences for PCRs are available upon request.
Chromatin immunoprecipitation assay.
Chromatin immunoprecipitation (ChIP) was performed as previously described (Asano et al., 2009). Cells were cross-linked in medium containing 1% formaldehyde for 10 min at room temperature, neutralized with 1.5 m glycine for 3 min, and then washed three times with cold PBS. The cells were collected in PBS and pelleted by centrifugation for 20 min at 4°C at 3000 g. The pellet was resuspended in 200 μl of SDS-lysis buffer (1% SDS, 10 mm EDTA, 50 mm Tris-HCl, pH 8.0), and then incubated for 10 min on ice. The suspension was sonicated using Sonifer 450 (Branson) until the DNA fragments were 200–500 bp in length. This chromatin sample was diluted 1:10 with dilution buffer (1.1% Triton X-100, 0.11% NaDOC, 50 mm Tris-HCl, pH 8.1, 167 mm NaCl), and 10% of the total volume was stored as input at −20°C until use. Immunoprecipitation was performed at 4°C overnight with 2 μg of an antibody against histone H3-Lys27 trimethylation (H3K27me3) or histone H3-Lys4 trimethylation (H3K4me3). Immunocomplexes were then incubated with 20 μl of anti-mouse IgG Dynabeads magnetic beads for at least 6 h. The beads were washed successively with the following four buffers: low-salt buffer (0.1% SDS, 1% Triton X-100, 0.1% NaDOC,1 mm EDTA, 50 mm Tris-HCl, pH 8.1, 167 mm NaCl), high-salt buffer (0.1% SDS, 1% Triton X-100, 0.1% NaDOC, 1 mm EDTA, 50 mm Tris-HCl, pH 8.1, 500 mm NaCl), LiCl buffer (0.25 m LiCl, 0.5% NP-40, 0.5% NaDOC, 1 mm EDTA, 10 mm Tris-HCl, pH 8.1), and twice with TE buffer (10 mm Tris-HCl, 1 mm EDTA, pH 8.0). The cross-linking in immunocomplexes and input samples was reversed with direct elution buffer (0.5% SDS, 5 mm EDTA, 10 mm Tris-HCl, pH 8.1, 300 mm NaCl) at 65°C overnight. The DNA was further treated with RNase at 37°C for 30 min, followed by incubation with proteinase K (Nacalai Tesque) at 65°C for 1 h. The DNA was purified by phenol-chloroform extraction followed by ethanol precipitation. The DNA pellet was dissolved in 20 μl of H2O and used as a template for PCR or quantitative PCR. Primer sequences for PCRs are available upon request.
Statistical analysis.
At least three mice per group were analyzed. Statistical analyses were performed using either Student's t test (for comparisons between two groups) or one-way ANOVA with Tukey's multiple comparison test (for multiple groups comparison). All experiments were independently replicated at least three times. Differences were considered statistically significant at p < 0.05.
Results
DNMT1 is expressed in NSCs during development of the DG
Hippocampus development begins at ∼E11–E12. NSCs in the ventricular zone of the telencephalon begin to produce pyramidal neurons of the cornu ammonis region and terminate their production ∼E18 (Angevine, 1965; Caviness, 1973). On the other hand, production of precursors for granule neurons in the DG begins at a later embryonic stage (∼E15), and the majority of granule neurons are generated in the postnatal stage (Altman and Bayer, 1990a). In a first step of the development of the DG, immature granule neurons originating from the dentate neuroepithelium near the fimbria migrate into the incipient dentate area ∼E15 (Fig. 1A) (Altman and Bayer, 1990a; Seki et al., 2014). We first examined the expression of DNMT1 in this initial step of DG development and found that DNMT1 was abundant in the ventricular zone (VZ) (Fig. 1B,C). Coimmunostaining of DNMT1 with several cell type markers showed that DNMT1 expression was highly enriched in cells positive for the proliferation marker Ki67 and the NSC markers Sox2 and Nestin (Fig. 1C1–C3). On the other hand, DNMT1 expression was much weaker in the cortical plate, which was labeled with the neuronal marker DCX, and in NeuroD+ immature neurons (Fig. 1D,E). These data indicate that DNMT1 is highly expressed in NSCs in the developing DG.
Figure 1.

DNMT1 is highly expressed in proliferating cells in the developing DG. A, Schematic illustration of the DG at E15. NSCs localize in the VZ and start to migrate into the FDJ. F, Fimbria. B, Expression of DNMT1 (red), Ki67 (green), and Sox2 (cyan) in the DG at E15. Inset, Hoechst staining. Scale bar, 200 μm. C, Representative immunofluorescence images of DNMT1 (red) and Ki67 (green) (C1), Sox2 (cyan) (C2), or Nestin (green) (C3) in the DG at E15. Scale bar, 100 μm. DNMT1 was detected in the proliferating NSCs. D, E, Expression of DNMT1 (red) and either NeuroD (green) (D) or DCX (green) (E) in the DG at E15. Scale bar, 100 μm. DNMT1 expression is weaker in neurons than in NSCs.
Loss of Dnmt1 in NSCs impairs DG development, resulting in a reduced GCL volume
To investigate the role of Dnmt1 in the development of the DG, we ablated Dnmt1 in NSCs at the beginning of DG development using the TAM-inducible Cre recombinase (CreERT2) system (Imayoshi et al., 2006). Dnmt1flox/flox mice were crossed with Nestin-CreERT2 mice to achieve TAM-inducible deletion of Dnmt1 in NSCs (Fig. 2A). After TAM administration at E15, we checked DNMT1 expression in cKO embryonic brain at E18. In the control DG, DNMT1 expression was observed in Ki67+ and Sox2+ proliferating NSCs (Fig. 2B) in the VZ. However, in the cKO mice, DNMT1 expression was efficiently abolished in Ki67+ and Sox2+ proliferating NSCs. These Dnmt1 cKO mice develop to adults (8–10 weeks) without displaying any obvious abnormality in brain weight or morphology (data not shown). We next investigated the structure of the hippocampus. The cornu ammonis regions in the hippocampus were indistinguishable between control and cKO mice, as indicated by immnostaining with NeuN, a marker for mature neurons (Fig. 2C). However, Dnmt1 cKO mice displayed a smaller GCL in the DG compared with controls (Fig. 2C,D). These results show that DNMT1 expression in late-gestational NSCs is necessary for proper development of the DG.
Figure 2.
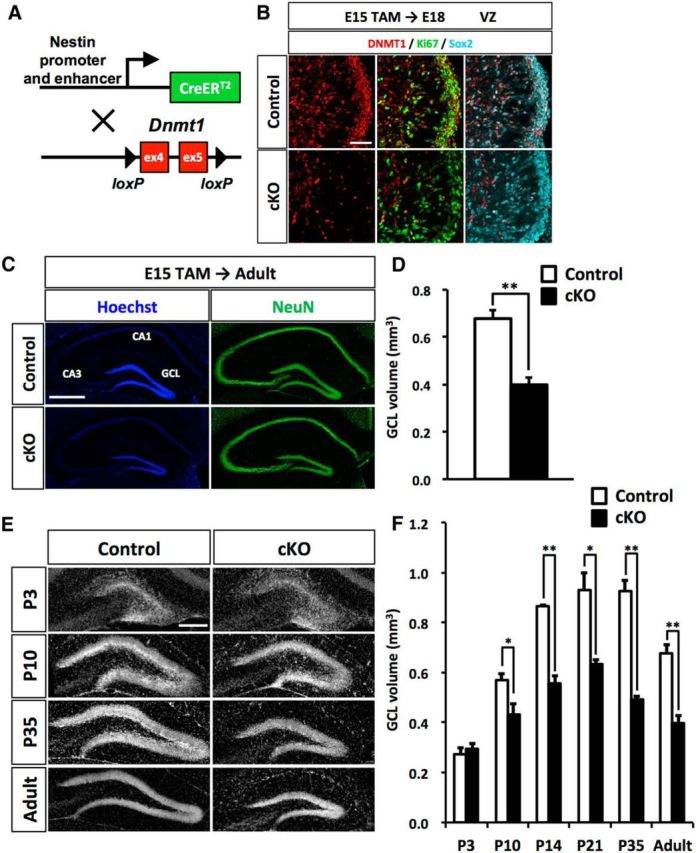
Loss of Dnmt1 impairs development of the DG and results in a reduced GCL. A, Diagram of the Dnmt1 conditional knock-out strategy in NSCs at the beginning of DG development. Dnmt1flox/flox mice were crossed with Nestin-CreERT2 mice, yielding progeny in which TAM administration results in the deletion of Dnmt1 (exons 4 and 5) in Nestin-expressing NSCs. Pregnant mice were injected with TAM at E15. B, Expression of DNMT1 (red) in Ki67+ (green) and Sox2+ (cyan) proliferating NSCs in the DGs of control and Dnmt1 cKO mice at E18. DNMT1 expression is diminished in proliferating NSCs in Dnmt1 cKO mice. Scale bar, 100 μm. C, Representative immunofluorescence images for NeuN (green) in the DGs of control and Dnmt1 cKO mice at the adult stage after prenatal deletion of Dnmt1. Hoechst (blue) staining shows nuclei. Scale bar, 500 μm. D, Bar graph represents the volume of the GCL in the DGs of control and Dnmt1 cKO mice. Prenatal deletion of Dnmt1 results in a reduction of the GCL volume in the adult stage. E, Representative Hoechst staining in the DGs of control and Dnmt1 cKO mice at the indicated developmental stages after TAM injection at E15. Scale bar, 200 μm. F, Bar graph represents the volume of GCL of DGs in control and Dnmt1 cKO mice at each time point. Prenatal deletion of Dnmt1 compromises development of the DG in the postnatal stage. Data are mean ± SEM. *p < 0.05 (Student's t test). **p < 0.01 (Student's t test).
Development of the DG occurs through multiple processes in both embryonic and postnatal stages (Li and Pleasure, 2005). Thus, we next asked when the reduction of GCL volume in Dnmt1 cKO DGs is first observable during DG development. After TAM administration at E15, mice were fixed at several time points, and the volume of the GCL was measured (Fig. 2E,F). The majority of granule neurons are produced in postnatal stages, and GCL structure develops in the first 2 postnatal weeks (Angevine, 1965; Altman and Bayer, 1990a). We observed that the GCL volume dramatically increased from P3 to P14 in the control DG, consistent with previous reports. In contrast, Dnmt1 cKO mice showed markedly weaker expansion of the GCL (Fig. 2E,F). Although the GCL volume in cKO DGs was indistinguishable from controls at P3, Dnmt1 cKO mice started to display a significantly smaller GCL volume from P10. Moreover, the difference in GCL volume between control and cKO mice widened from P10 to P14. These data suggest that deletion of Dnmt1 affects the developmental process of DGs during postnatal rather than embryonic stages.
NSCs fail to localize in the SGZ or to develop proper secondary radial glial scaffold in Dnmt1 cKO mice
NSCs and NPs migrate toward the hilus of DG and form a proliferative zone, called the SGZ, at the border between the hilus and the GCL in the second postnatal week (Fig. 3A) (Li and Pleasure, 2005; Li et al., 2009). Thereafter, NSCs settle in the SGZ by adulthood, contributing to persistent neurogenesis in the SGZ throughout life. During this migration, NSCs produce NPs and granule neurons that are integrated into the GCL. This increases the volume of the GCL during postnatal stages. Given that Dnmt1 cKO mice showed impaired DG development in postnatal stages, we investigated whether Dnmt1 deletion affects the migration of NSCs into the SGZ of the DG. Immunohistochemical analysis for the NSC marker Sox2 revealed that Sox2+ NSCs started to localize in the inner region of the GCL at P7 in control mice (Fig. 3B). As development progressed, Sox2+ cells in control mice were arranged mainly in the vicinity of the border between the hilus and the GCL by P10 and formed the SGZ. In contrast, NSCs in Dnmt1 cKO DGs showed scattered localization throughout entire GCL. Quantifying the distribution of Sox2+ cells in the GCL showed an increased population of Sox2+ cells on the outer side of the GCL in Dnmt1 cKO DG compared with controls (Fig. 3C). Even after P14, NSCs in Dnmt1 cKO DGs had failed to attain residence in the inner region of the GCL (Fig. 3D). Collectively, these results indicate that deletion of Dnmt1 impairs the migration of NSCs to their final destination during postnatal development.
Figure 3.
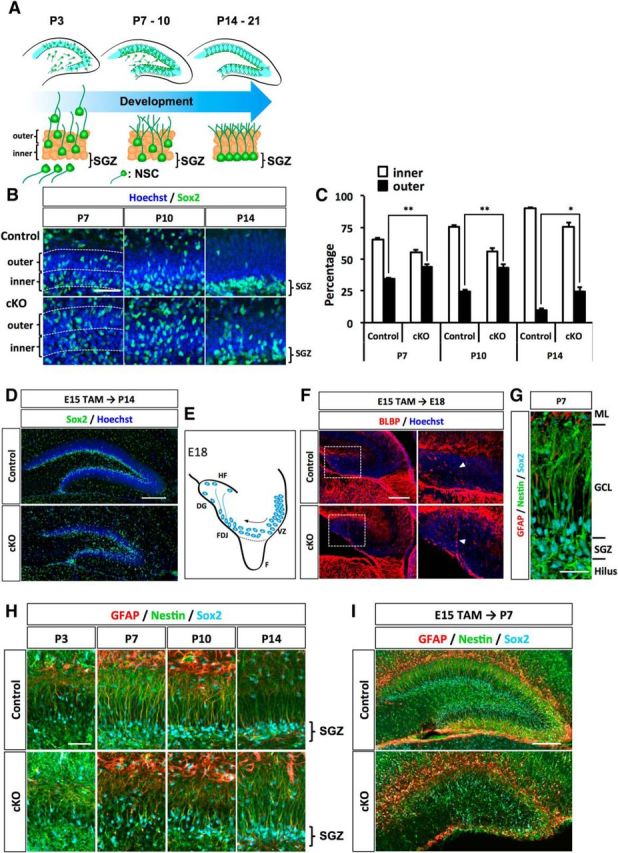
In the absence of Dnmt1, NSCs display disorganized localization and secondary radial glial scaffolds at the postnatal stage. A, Schematic illustration of migration and morphological change of NSCs during DG development. B, Representative immunofluorescence images for Sox2 (green) in the DGs of control and Dnmt1 cKO mice at the indicated developmental stages. Hoechst staining shows nuclei. Scale bar, 100 μm. C, Quantification of the positions of NSCs in the DG. Bar graph represents the percentage of Sox2+ cells in either the inner or the outer half of the dentate blade. D, Position of NSCs shown by Sox2 (green) staining in the DGs at P14. NSCs are disorganized in the Dnmt1 cKO DG. E, Schematic illustration of the DG at E18. NSCs localized in the FDJ establish a primordial radial glial scaffold, which extends to the hippocampal fissure (HF). F, Fimbria. F, BLBP+ processes were observed in DGs of both control and Dnmt1 cKO mice at E18. Scale bar, 200 μm. Higher magnification of the white boxes in the left images (right) showed that BLBP+ processes of NSCs were indistinguishable between control and Dnmt1 cKO mice. G, Representative 3D immunofluorescence image of secondary radial glial scaffolds of NSCs in DGs at P7. NSCs and their processes were stained with GFAP (red), Nestin (green), and Sox2 (cyan). Scale bar, 50 μm. H, I, Representative immunofluorescence images for GFAP (red), Nestin (green), and Sox2 (cyan) in the DGs of control and Dnmt1 cKO mice at various time points after TAM injection at E15. Deletion of Dnmt1 impairs the development of the secondary radial glial scaffold. Scale bars: H, 100 μm; I, 200 μm. Data are mean ± SEM. *p < 0.05 (Student's t test). **p < 0.01 (Student's t test).
While they are migrating into the SGZ, NSCs alter their morphology. In the late embryonic stage, NSCs residing in the fimbriodentate junction (FDJ) extend a process, termed the primordial radial glial scaffold, to the pial surface of the DG (Fig. 3E) (Li et al., 2009). In subsequent postnatal stages, the primordial radial glial scaffold disappears. Instead, as NSCs reach the SGZ, they establish a process that extends to the ML across the GCL, called the secondary radial scaffold (Bignami and Dahl, 1974; Rickmann et al., 1987; Sievers et al., 1992). Previous studies have demonstrated that the secondary radial scaffold is important for the migration of NPs and granule neurons to their final positions in the GCL (Tian et al., 2012; Brunne et al., 2013). We therefore also analyzed the morphology of NSCs at each developmental stage by immunohistochemistry. At E18, NSCs developed the primordial radial glial scaffold to the pial surface of the DG, as indicated by the radial glial marker BLBP (Fig. 3F). In Dnmt1 cKO mice, NSCs established BLBP+ processes that were indistinguishable from those in controls (Fig. 3F), indicating that the loss of Dnmt1 in NSCs at the late gestational stage does not affect the formation of the primordial radial glial scaffold. We then examined the development of the secondary radial glial scaffold in control and Dnmt1 cKO mice. The secondary radial glial scaffold of NSCs is established in the first postnatal week and is fully developed ∼P10-P14 (Brunne et al., 2010). We assessed the processes of NSCs at four time points of the postnatal stage: P3, P7, P10, and P14. By performing immunostaining with antibodies against the NSC marker Nestin and the glial marker GFAP, we found that the secondary radial scaffold became clearly visible by P7 in control mice, and was fully elaborated in the control DG by P10-P14, when NSCs are located in the SGZ (Fig. 3G,H). On the other hand, Nestin+ and GFAP+ scaffolds of NSCs in Dnmt1 cKO DGs were shorter than those of control NSCs and disoriented even at P10 (Fig. 3H,I). We could not observe correctly directed processes of NSCs until P14 in Dnmt1 cKO mice. Together, these results suggest that deletion of Dnmt1 in NSCs at the late gestational stage impairs both the localization of NSCs to the SGZ and the morphological development of NSCs.
Deletion of Dnmt1 leads to mispositioning of NPs in the ML
The secondary radial glial scaffold has been assumed to help NPs and granule neurons to migrate to their final positions in the GCL during DG development (Brunne et al., 2013). Indeed, we found that NPs positive for the specific marker Tbr2, attached to the secondary radial glial scaffold labeled with GFAP (Fig. 4A). NSCs in Dnmt1 cKO mice showed misregulated localization in the DG and morphological abnormality in the secondary radial glial scaffold. These defects may also influence the localization of NPs. Thus, we next asked whether deletion of Dnmt1 impairs the production and positioning of NPs in the postnatal stage. To this end, we analyzed the number and distribution of NPs at each developmental stage by immunostaining with anti-Tbr2 antibody.
Figure 4.

Deletion of Dnmt1 leads to aberrant localization of NPs in the ML of the DG. A, Tbr2+ NPs attach to the GFAP+ processes of NSCs, suggesting that the radial processes of NSCs function as scaffolds for NPs. B, Schematic illustration of the DG. C, D, Representative immunofluorescence images for Tbr2 (green) in the DGs of control and Dnmt1 cKO mice at various developmental stages after TAM injection at E15. DNA is stained with Hoechst (blue). The asterisk indicates fewer Tbr2+ NPs in the hilus of Dnmt1 cKO mice (C). Scale bar, 200 μm. E, Quantification of the positions of NPs in DGs. Bar graph represents the percentage of Tbr2+ cells in the ML. Deletion of Dnmt1 increases the population of NPs in the ML. Data are mean ± SEM. *p < 0.05 (Student's t test). **p < 0.01 (Student's t test).
From P7 to P21, we observed no obvious difference in the total number of Tbr2+ cells in the DG between control and Dnmt1 cKO mice (data not shown), suggesting that the loss of Dnmt1 in late-gestational NSCs does not impair the differentiation of NSCs into NPs. However, while the majority of Tbr2+ cells in control mice were distributed in the hilus and ML of the DG at P7, these cells in Dnmt1 cKO DG were more abundant in the ML than in the hilus (Fig. 4B,C). In control mice, NPs in the ML decreased as development progressed, as indicated by the ratio of Tbr2+ cells in the ML to total Tbr2+ cells. In Dnmt1 cKO DGs, however, although the percentage of Tbr2+ cells in the ML also tended to decrease with development, 12.9% of Tbr2+ cells remained in the ML of Dnmt1 cKO DGs at P21, which was in sharp contrast to only 2% in the control DGs (Fig. 4D,E). These results indicate that Dnmt1 deficiency leads to mispositioning of NPs during DG development.
Loss of Dnmt1 leads to aberrant neuronal production in the ML and fewer mature neurons in the GCL
We hypothesized that mispositioned NPs in the ML fail to produce granule neurons in the GCL, resulting in the smaller GCL in Dnmt1 cKO mice. Hence, we next asked whether mispositioned NPs in the ML could differentiate into neurons, and whether such neurons could contribute to GCL formation. To address these questions, we administered BrdU to trace the fate of proliferating NPs at P10, when Dnmt1 cKO DGs displayed the greatest significant difference in the percentage of mispositioned NPs in the ML compared with control DGs. After BrdU injection, brain sections for immunohistochemistry were prepared at different time points: 2 h, 4 d (P14), and 11 d (P21) (Fig. 5A). Two hours after the BrdU injection, we observed that ∼50% of BrdU+ cells expressed the NP marker Tbr2 in both control and Dnmt1 cKO DGs (Fig. 5B,C). Quantification data showed no significant difference in the number of BrdU+ cells, or the percentage of BrdU+ cells among Tbr2+ cells, between control and Dnmt1 cKO DGs (Fig. 5D,E), suggesting that deletion of Dnmt1 does not affect the proliferation of these cells, at least in early postnatal stages. Reminiscent of the mispositioned NPs in Dnmt1 cKO DG, we observed significantly more BrdU+ cells in the ML of Dnmt1 cKO DGs than of control DGs (Fig. 5F).
Figure 5.
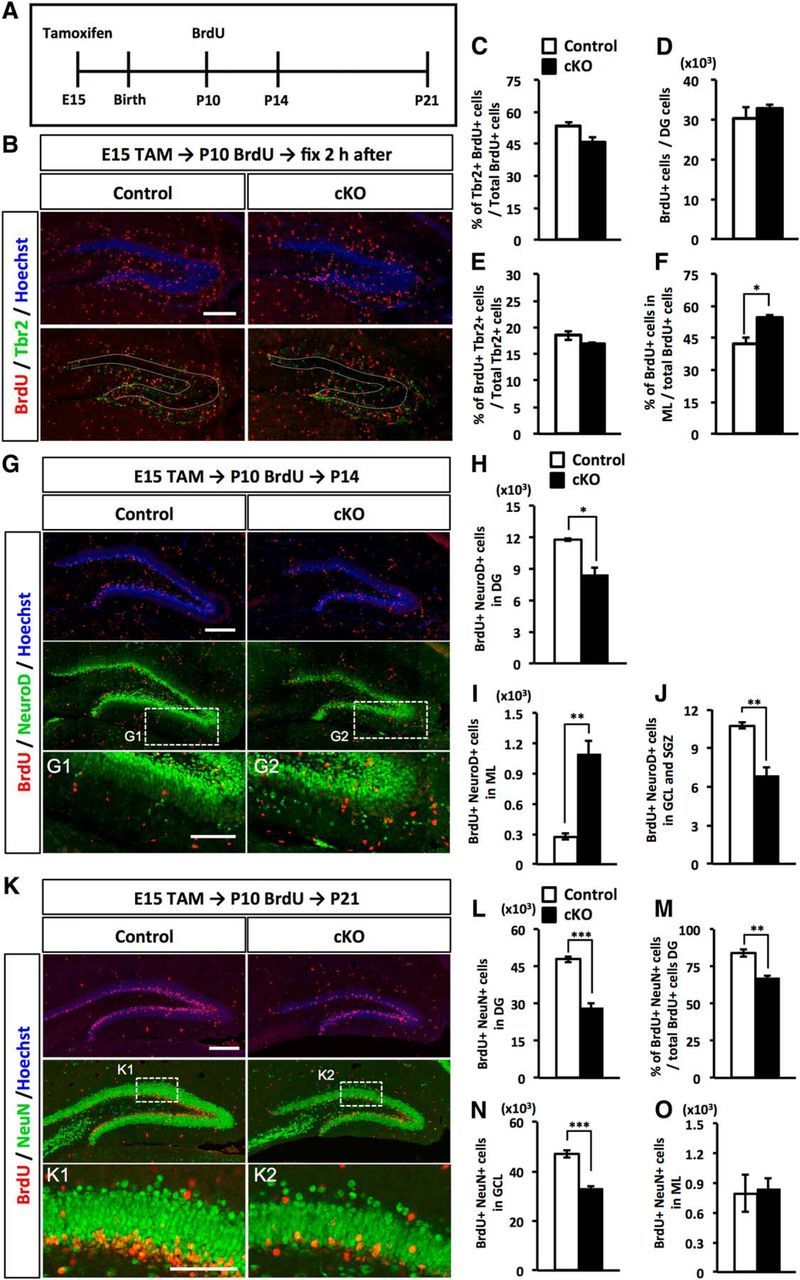
Deletion of Dnmt1 leads to aberrant neuronal production in the ML and a subsequent reduction of mature granule neurons in the GCL. A, Experimental scheme of TAM and BrdU injection for fate-tracing analysis. Pups, treated prenatally with TAM at E15, were injected with BrdU at P10 and fixed at 2 h, 4 d (P14), or 11 d (P21) after BrdU injection. B, Representative immunofluorescence images for BrdU (red) and Tbr2 (green) in the DGs of control and Dnmt1 cKO mice at 2 h after BrdU injection at P10. DNA is stained with Hoechst (blue). Scale bar, 200 μm. Dotted line indicates the GCL region. C, Bar graph represents populations of Tbr2+ cells among BrdU+ cells. D, Quantification of the number of BrdU+ cells in the DGs. E, Quantification of the proliferating population of Tbr2+ cells. Bar graph represents the percentage of BrdU+ cells among Tbr2+ cells. Dnmt1 deficiency did not affect the production or proliferation of NPs in the early postnatal stage. F, Quantification of BrdU+ cells in the ML. Proliferating cells in Dnmt1 cKO DGs were more numerous in the ML, reflecting the increased population of Tbr2+ cells in the ML. G, Representative immunofluorescence images for BrdU (red) and NeuroD (green) in the DGs of control and Dnmt1 cKO mice at P14, 4 d after BrdU injection at P10. DNA is stained with Hoechst (blue). Scale bar, 200 μm. G1, G2, Magnified images of the white dashed line boxes. Scale bar, 100 μm. H, I, Quantification of BrdU+/NeuroD+ cells in the DG (H) and ML (I). J, Quantification of BrdU+/NeuroD+ cells in the GCL and SGZ. Immature neurons were increased in the ML of Dnmt1 cKO mice but decreased in the GCL and SGZ. K, Representative immunofluorescence images for BrdU (red) and NeuN (green) in the DGs of control and Dnmt1 cKO mice 11 d after BrdU injection at P10. DNA is stained with Hoechst (blue). Scale bar, 200 μm. K1, K2, Magnified images of the white dashed line boxes. Scale bar, 100 μm. L, Quantification of BrdU+/NeuN+ cells in DGs. M, Bar graph represents the percentage of NeuN+ cells among BrdU+ cells. N, O, Quantification of BrdU+/NeuN+ cells in the GCL (N) and ML (O). The number of mature neurons was significantly reduced in the GCL of Dnmt1 cKO mice. Data are mean ± SEM. *p < 0.05 (Student's t test). **p < 0.01 (Student's t test). ***p < 0.001 (Student's t test).
Next, to investigate the potential of Tbr2+ cells to differentiate into immature neurons, BrdU-labeled cells were coimmunostained with NeuroD, a marker of immature neurons, at P14, 4 d after BrdU injection. In Dnmt1 cKO DGs, the number of BrdU+/NeuroD+ cells was significantly reduced compared with control DGs (Fig. 5G,H). Given that Dnmt1 cKO and control DGs showed comparable numbers of BrdU+ and BrdU+/ Tbr2+ cells at P10 (Fig. 5C,D), the decreased number of BrdU+/NeuroD+ cells in Dnmt1 cKO DGs suggests that deletion of Dnmt1 impairs the production of granule neurons from NPs. We further compared the number of BrdU+/NeuroD+ cells in each region of the DG. Interestingly, the number of BrdU+/NeuroD+ immature neurons in the ML of Dnmt1 cKO DG was approximately 4 times higher than that in the control (Fig. 5I). On the other hand, the number of BrdU+/NeuroD+ immature neurons in the GCL and SGZ was significantly lower in the Dnmt1 cKO mice (Fig. 5J). These data indicate that the loss of Dnmt1 in NSCs at the late embryonic stage causes the abnormal production of immature neurons in the ML, which is probably attributable to the mislocalization of NPs.
We further examined whether these immature mispositioned neurons in the ML are later integrated into the GCL. Eleven days after BrdU injection at P10 (P21), most BrdU+ cells were located in both control and Dnmt1 KO GCLs (Fig. 5K). In the Dnmt1 cKO DG, however, the number of BrdU-labeled NeuN+ mature neurons in GCL was markedly lower than that in control DGs (Fig. 5L). In addition, the percentage of BrdU+/NeuN+ cells among total BrdU+ cells was also significantly reduced in Dnmt1 cKO DGs (Fig. 5M), and Dnmt1 cKO DGs displayed a significant decrease of BrdU+/NeuN+ cells in the GCL (Fig. 5N). These results further strengthen the idea that Dnmt1 deletion disturbs neuronal production. Surprisingly, despite the abnormal accumulation of immature neurons in the ML at P14 in Dnmt1 cKO DGs (Fig. 5I), the number of BrdU+/NeuN+ mature neurons in the ML at P21 was comparable between control and cKO (Fig. 5O). These results suggest that, although mispositioned NPs in the ML of Dnmt1 cKO DGs can differentiate into immature neurons, these neurons fail to become integrated into the GCL and are eliminated from the DG. This may partially explain the reduced volume of the GCL in Dnmt1 cKO DGs.
Dnmt1 ablation induces apoptotic cell death in late postnatal stage
We hypothesized that the reduction in GCL volume and granule neurons in Dnmt1 cKO was due to increased cell death. To test this possibility, we assessed apoptotic cell death during development of the DG. The number of Active caspase 3+ apoptotic cells in Dnmt1 cKO DGs was similar to that in control DGs during the early postnatal stage (P3-P7). However, Dnmt1 cKO DGs displayed a remarkable increase of apoptotic cells in the later postnatal stage (P10-P21), consistent with the period when the reduction of granule neurons and GCL volume was observed in Dnmt1 cKO mice (Fig. 6A,B). Because Dnmt1 cKO DGs showed abnormal accumulation of immature neurons in the ML at P14, and these neurons subsequently diminished (Fig. 5I,O), it is conceivable that they were eliminated by apoptotic cell death as development progressed. Indeed, we were able to detect NeuN+/Active caspase 3+ cells in Dnmt1 cKO DGs at P14 (Fig. 6C). Furthermore, we found that some apoptotic cells were detectable in the ML, where abnormally localized immature neurons were observed in Dnmt1 cKO DGs (Fig. 6C, arrow), supporting the idea that mislocalized immature neurons fail to integrate properly into the GCL and are eliminated by apoptotic cell death. Collectively, these data suggest that induced cell death at the late postnatal stage accounts for the reduction of GCL volume and granule neuron numbers in the DGs of Dnmt1 cKO mice.
Figure 6.

Deletion of Dnmt1 increases apoptotic cell death. A, Representative immunofluorescence images for Active caspase 3 (red) in the DGs of control and Dnmt1 cKO mice at postnatal stages P3 and P21. DNA is stained with Hoechst (blue). Scale bar, 100 μm. B, Quantification of Active caspase 3+ cells in DGs at the indicated developmental stages. C, Representative immunofluorescence images for active caspase 3 (red) and NeuN (green) in the DG of control and Dnmt1 cKO mice at P14. Scale bar, 50 μm. White arrowheads indicate Active caspase 3+/NeuN+ cells. White arrow indicates an Active caspase 3+/NeuN+ cell in the ML. Data are mean ± SEM. *p < 0.05 (Student's t test). **p < 0.01 (Student's t test).
Loss of Dnmt1 promotes differentiation of NSCs into astrocytes in late postnatal stage
It has been reported that Dnmt1 deletion in telencephalic neural progenitor cells during cortical development triggers precocious astrocyte differentiation (Fan et al., 2005; Hutnick et al., 2009). We therefore also investigated whether the loss of Dnmt1 induces astrocyte differentiation during DG development. In the DG, the majority of astrocytes localize in the ML. During DG development, NSCs in the SGZ are a major source of astrocytes in the ML and start to transform into astrocytes at the late postnatal stage (Brunne et al., 2010). As the astrocytic transformation progresses, astroglially committed NSCs migrate toward the ML through the GCL, and they gradually display astrocytic morphology by losing their radial glial fibers, increasing the branching of their processes, and acquiring multipolar processes. When the cells reach the ML, they lose expression of the radial glial markers Nestin and BLBP and instead start to express the mature astrocyte marker S100β (Brunne et al., 2010). Indeed, we could observe S100β+ cells that localized around the border between the GCL and ML in P21 DGs (Fig. 7A, white arrowheads). Next, after inducing Dnmt1 deletion at E15, we injected BrdU at P10 and assessed astrocytic differentiation of BrdU-labeled cells at P21. Coimmunostaining for BrdU and the mature astrocyte marker S100β showed an increased percentage of S100β+ cells among BrdU+ cells in Dnmt1 cKO mice (Fig. 7A,B). Consistent with the migration route of astroglially committed cells into the ML during astrocyte differentiation, the majority of S100β+/BrdU+ cells were observed in the ML of DGs in control mice (Fig. 7A,D). In Dnmt1 cKO mice, however, many S100β+ cells were detected in the inner region of the GCL and especially in the SGZ of Dnmt1-deficient DGs, whereas these cells were rarely seen in the corresponding areas of controls, consistent with a previous study (Brunne et al., 2010). These data indicate that the loss of Dnmt1 leads to aberrant production of astrocytes in the GCL. Astrocytes normally have multiple processes, and the morphology of astrocytes in the ML was indistinguishable between control and Dnmt1 cKO DGs (Fig. 7D). Nevertheless, when we carefully analyzed these S100β+ cells in the GCL and SGZ of Dnmt1 cKO DGs, some of them had a single and less-branched process, reminiscent of the radial glial fiber of NSCs (Fig. 7A, yellow arrowheads), and the projections of such S100β+ cells displayed merged GFAP and Nestin immunostaining (Fig. 7E). These results suggest that deletion of Dnmt1 increases the production of astrocytes in the GCL and generates NSC-like astrocytes, which express both the mature astrocyte marker S100β and the NSC markers Nestin and GFAP in the postnatal DG.
Figure 7.
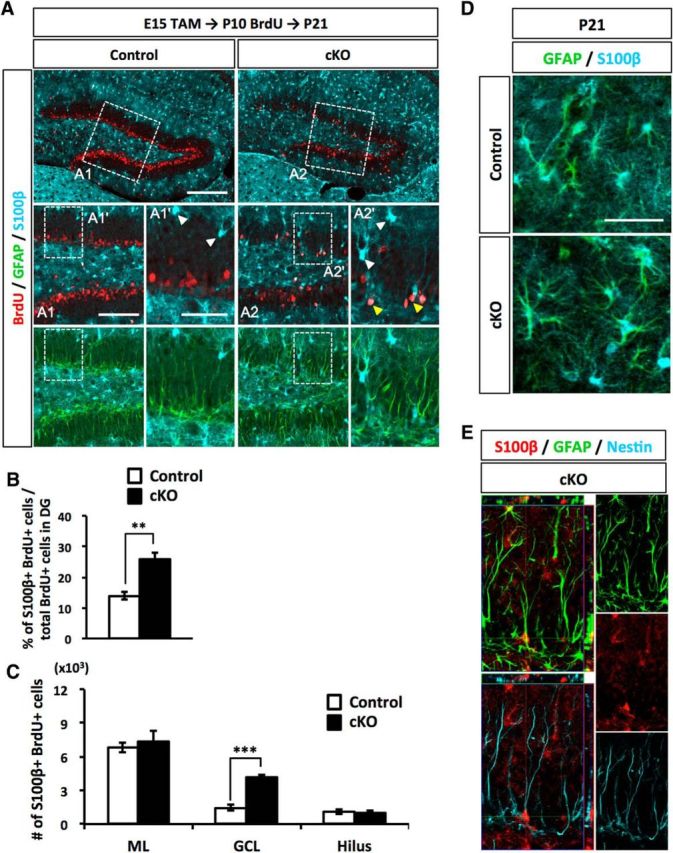
Loss of Dnmt1 increases astrocyte differentiation in the late postnatal stage. A, Representative immunofluorescence images for BrdU (red), GFAP (green), and S100β (cyan) in the DGs of control and Dnmt1 cKO mice at 11 d after BrdU injection at P10. Scale bar, 200 μm. Successively magnified images of the white dashed line boxes are shown (first) (A1, A2) and (second) (A1′, A2′). Scale bars: A1, A2, 100 μm; A1′, A2′, 50 μm. White arrowheads indicate the astrocytes located in the border between GCL and ML. Yellow arrowheads indicate BrdU+/S100β+ cells having the radial glial fiber. B, Bar graph represents the percentage of S100β+ cells among BrdU+ cells in DGs. C, Quantification of S100β+/BrdU+ cells in the indicated regions of DGs. Dnmt1 cKO increases astrocyte numbers in the SGZ. D, Representative immunofluorescence images for GFAP (green) and S100β (cyan) in the DGs of control and Dnmt1 cKO mice at P21. Scale bar, 50 μm. E, Orthogonal analysis of a representative cell expressing GFAP, Nestin, and S100β in the DG of a Dnmt1 cKO mouse. Data are mean ± SEM. **p < 0.01 (Student's t test). ***p < 0.001 (Student's t test).
Prenatal deletion of Dnmt1 leads to NSC loss in adulthood and impairs the neurogenic ability of NSCs
For proper neurogenesis to occur in the adult DG, NSCs must be maintained in the SGZ into adulthood. Our results showed that Dnmt1 cKO leads to mispositioning of NSCs in the SGZ and disturbed development of the secondary radial glial scaffold (Fig. 3). Moreover, deletion of Dnmt1 produces more astrocytes in the SGZ compared with the number in controls, and some of them have NSC-like morphology (Fig. 7). These abnormalities in DG development of Dnmt1 cKO mice raise the questions of whether prenatal deletion of Dnmt1 impairs lifelong maintenance of the NSC pool and/or impairs neurogenesis. In the adult SGZ, radial NSCs, defined as Type 1 cells, express GFAP, Nestin, and Sox2 (Ming and Song, 2011). Type 1 cells extend their radial processes across the GCL and toward the ML, as shown in a control DG (Fig. 8A). However, Type1 cells in Dnmt1 cKO DG displayed disoriented radial processes, similar to those seen in P7-P14 (Fig. 3H). This result indicates that Dnmt1 deficiency at the late gestational stage continuously has a sustained effect on the morphology of NSCs. Moreover, when we quantified the number of Type 1 cells in the SGZ, we found significantly fewer in Dnmt1 cKO DGs than in control DGs (Fig. 8B). These results demonstrate that Dnmt1 in late-gestational NSCs is important for the proper maintenance of NSCs into adulthood.
Figure 8.

Prenatal deletion of Dnmt1 impairs adult neurogenesis and proliferation of NSCs. A, Representative immunofluorescence images for GFAP (red), Nestin (green), and Sox2 (cyan) in the DGs of control and Dnmt1 cKO mice at the adult stage (8–10 weeks) after TAM administration at E15. Scale bar, 200 μm. Inset, Hoechst staining for DNA. A1, A2, Magnified images of the white dashed line boxes. Scale bars: A1, A2, 100 μm. B, Quantification of GFAP+/Nestin+/Sox2+ Type 1 cells NSCs in the DGs of control and cKO mice. C, Representative immunofluorescence images for DCX (red). DNA is stained with Hoechst (blue). D, Quantification of DCX+ cells in the DGs of control and cKO mice. E, Bar graph represents the ratio of DCX+ cells to Type 1 cells in DGs. Neurogenesis is impaired in Dnmt1 cKO mice. F, Representative immunofluorescence images for Sox2 (red) and Ki67 (green) in the DGs of control and Dnmt1 cKO mice. G, Quantification of Ki67+ cells in the DGs of control and Dnmt1 cKO mice. H, Bar graph represents the ratio of Ki67+ cells to Sox2+ NSCs in DGs. I, Representative immunofluorescence images for pHH3 (green) in the DGs of control and Dnmt1 cKO mice at P3 and P7. DNA is stained with Hoechst (blue). J, Quantification of pHH3+ cells in the DGs of control and cKO mice. K, Representative immunofluorescence images for Ki67 (green) in the DGs of control and Dnmt1 cKO mice at P14 and P21. DNA is stained with Hoechst (blue). L, Quantification of Ki67+ cells in the DGs of control and cKO mice. Data are mean ± SEM. *p < 0.05 (Student's t test). **p < 0.01 (Student's t test). ***p < 0.001 (Student's t test).
We next investigated the effect of prenatal Dnmt1 deletion on adult neurogenesis by immunostaining for DCX, a marker of newborn neurons, and found that DCX+ cells were significantly decreased in Dnmt1 cKO DGs (Fig. 8C,D). Speculating that the reduced number of newborn neurons in Dnmt1 cKO could result from the reduction of NSCs, we also calculated the ratio of the number of newborn neurons to the number of NSCs to assess the neurogenic ability of NSCs. Dnmt1 cKO DGs displayed a ratio of newborn neurons of approximately half that in control DGs (Fig. 8E), indicating that the neurogenic ability of NSCs was impaired in Dnmt1 cKO DGs.
Adult NSCs are relatively quiescent and enter the cell cycle as they generate transit amplifying NPs. Next, we assessed proliferation of NSCs in Dnmt1 cKO DGs and found that cells positive for the proliferation marker Ki67 were markedly reduced in the SGZ of Dnmt1 cKO DGs (Fig. 8F,G). Quiescent NSCs are mostly in the G0 phase of the cell cycle and thus negative for Ki67. To assess the quiescent state of NSCs, we calculated the ratio of Ki67+ cells among Sox2+ cells. In Dnmt1 cKO DGs, the percentage of Ki67+ cells among Sox2+ cells was significantly reduced (Fig. 8H), implying that more NSCs in Dnmt1 cKO DGs than in control DGs remain in the quiescent state. These data suggest that prenatal deletion of Dnmt1 leads to the impaired proliferation of NSCs in the adult DG. However, data presented above showed no difference in the number of BrdU+ cells 2 h after BrdU injection at P10 (Fig. 5D), indicating that deletion of Dnmt1 does not impair the proliferation of NSCs until at least P10. Thus, we next investigated when the effects of Dnmt1 deletion on the proliferation of NSCs appear during development. Consistent with previous data using BrdU, immunostaining for a cell proliferation-specific marker, phosphorylated histone H3 (pHH3) showed that there were a comparable number of pHH3+ cells in the DGs of control and Dnmt1 cKO mice at P3 and P7. However, we found that the number of Ki67+ cells was significantly reduced in the DGs of Dnmt1 cKO mice at P14 and P21. These data suggest that prenatal deletion of Dnmt1 does not impair the proliferation of NSCs during early postnatal stages (prior to P10), but does so significantly in the later stages of DG development, which may lead to an increased quiescent population in the adult stage. Together, these data demonstrate that deletion of Dnmt1 in NSCs at the prenatal stage impairs the neurogenic ability and proliferation of NSCs in adult stages, resulting in impaired lifelong maintenance of the NSC pool and impaired adult neurogenesis.
Dnmt1 deletion attenuates the expression of key genes involved in Reelin signaling
Our results demonstrated that the loss of Dnmt1 leads to impaired development of the secondary radial glial scaffold as well as impaired neuronal differentiation of NSCs (Figs. 3, 4), resulting in a reduced volume of the GCL. Next, we tried to gain a molecular insight into how the loss of Dnmt1 leads to these defects in development. Previous studies using mutant mice have indicated the crucial signal pathways in the establishment of the secondary radial glial scaffold during development of the DG, such as Shh and Reelin signaling (Förster et al., 2002; Weiss et al., 2003; Breunig et al., 2008; Han et al., 2008; Brunne et al., 2013). We investigated whether Dnmt1 deletion induces dysregulation of these signaling pathways. We collected mRNA from the DGs of Dnmt1 cKO and control mice at P7 and analyzed the expression of Shh and Reelin signaling pathway components (Fig. 9A), but we could not detect any difference in the expression of three key transcription factors modulating Shh signaling, GLI Family Zinc Finger (Gli)1, Gli2, and Gli3 (Fig. 9B). However, we did find significant downregulation in the Reelin signaling transducer Disabled 1 (Dab1), and in the Reelin receptors Apolipoprotein E receptor 2 (ApoER2), and very low density lipoprotein receptor (Vldlr), in Dnmt1 cKO DGs compared with control DG (Fig. 9C). These data suggest that the loss of Dnmt1 interferes with Reelin signaling during DG development.
Figure 9.

Loss of Dnmt1 decreases the expression of the Reelin signaling components in developing DGs. A, Experimental scheme for assessing the expression of Shh and Reelin signaling components in Dnmt1 cKO DGs. After TAM administration at E15, DGs were collected at P7 for expression analysis. B, qRT-PCR analyses of Shh signaling component expression in Dnmt1 cKO DGs. C, qRT-PCR analyses of Reelin signaling component expression in Dnmt1 cKO DGs. Expression of Reelin, but not Shh, signaling components is decreased in Dnmt1 cKO DGs. D, Experimental scheme for assessing the expression of Reelin signaling components in Dnmt1 KD NSCs. P1 DG-derived NSCs were infected with lentivirus expressing Dnmt1-targeting shRNA and subjected to qRT-PCR analysis at 7 d after infection. E, qRT-PCR analyses of Reelin signaling component expression in Dnmt1 KD NSCs. Data are mean ± SD. *p < 0.05 (Student's t test). **p < 0.01 (Student's t test). ***p < 0.001 (Student's t test). F, Experimental scheme for assessing the expression of Reelin signaling components in RG108-treated NSCs. P1 DG-derived NSCs were treated with the DNA methyltransferase inhibitor RG108 for 5 d before qRT-PCR analyses. G, qRT-PCR analyses of Reelin signaling component expression in RG108-treated NSCs. Expression of the Reelin receptor ApoER2 is reduced by inhibition of DNA methyltransferase activity. Data are mean ± SD. *p < 0.05 (ANOVA with Tukey post hoc tests).
As well as NSCs, the DGs of P7 mice contain other cell types, such as NPs, granule neurons, and astrocytes. Thus, we next asked whether the decreased expression of Reelin signaling components occurs specifically in NSCs following the loss of Dnmt1. To address this, we isolated NSCs from developing DGs at P1 and induced knockdown (KD) of Dnmt1 by shRNA using lentivirus (Fig. 9D). Six days after the induction of Dnmt1 KD, we tested whether Dnmt1 KD in isolated NSCs also decreases the expression of Reelin signaling components, and found that the expression of ApoER2 was significantly lower, whereas that of Vldlr and Dab1 was unaffected. We also found decreased expression of ApoER2, but not of Vldlr or Dab1, when we treated wild-type NSCs with the DNA methyltransferase inhibitor RG108 (Fig. 9F,G). These data suggest that Dnmt1 modulates ApoER2 expression in NSCs via DNA methylation. In contrast to ApoER2 expression, we could not observe the decreased expression of Vldlr and Dab1 that we found in P7 DGs of Dnmt1 cKO mice. Astrocytes, NPs, and granule neurons also exist in the DG of P7 mice, and these cell types are generated from NSCs during DG development. Because it is likely that prenatal deletion of Dnmt1 in NSCs also affects gene expression in these differentiated cells, the decreased expression of Vldlr and Dab1 that we found in P7 DGs was probably caused by changes in expression that occurred in one or more of the differentiated cell populations. Nonetheless, our findings suggest that Dnmt1 contributes to the development of the DG, at least in part, by controlling the Reelin signaling pathway.
Loss of Dnmt1 increases the expression of the cell cyclin-dependent kinase inhibitors p21 and p57, leading to decreased proliferation of NSCs
Our data showed that prenatal deletion of Dnmt1 impairs proliferation of NSCs in adult stages. Next, we tried to elucidate the molecular mechanism whereby Dnmt1 regulates this NSC proliferation. The cyclin-dependent kinase (CDK) inhibitors p21 and p57 have been reported to be crucial factors in regulating the proliferation and quiescence status of adult NSCs (Kippin et al., 2005; Qiu et al., 2009; Furutachi et al., 2013). Using isolated NSCs from P1 DGs, we examined whether the loss of Dnmt1 affects the expression of three CDK inhibitors, p16, p21, and p57 (Fig. 10A), and found that expression of p21 and p57 was significantly increased in Dnmt1 KD NSCs (Fig. 10B). Furthermore, p21 and p57 expression in Dnmt1 cKO DGs was elevated relative to that in the control DG. These data indicate that prenatal deletion of Dnmt1 increases the expression of p21 and p57 in NSCs, which could account for the impaired proliferation of NSCs in the adult Dnmt1 cKO DG. To address this possibility, we inhibited the expression of p21 and p57 in Dnmt1 KD NSCs by cointroducing shRNA for p21 or p57 together with that for Dnmt1 (Fig. 10D). The elevated expression of p21 and p57 in Dnmt1 KD NSCs was indeed suppressed by induction of p21 and p57 mRNA-targeting shRNA. Dnmt1 KD significantly decreased the proliferation of NSCs, whereas this defect was partially overcome by simultaneous KD of p21 and p57 in Dnmt1 KD NSCs (Fig. 10E,F). These findings suggest that the Dnmt1 KD-mediated decreased proliferation of NSCs is attributable to upregulation of p21 and p57 expression. Because we have previously shown that deletion of Dnmt1 in the adult stage does not impair the proliferation of NSCs (Noguchi et al., 2015), we also checked whether Dnmt1 KD affects the expression of p21 and p57 in adult mice DG-derived NSCs (adult NSCs). In contrast to the case of prenatal mice DG-derived NSCs, we found that there was no difference in the expression of p21 and p57 between control and Dnmt1 KD adult NSCs (Fig. 10G,H). Together, these findings suggest that Dnmt1 regulates the proliferation of NSCs in prenatal stages, but not in adult stage by controlling the expression of p21 and p57.
Figure 10.
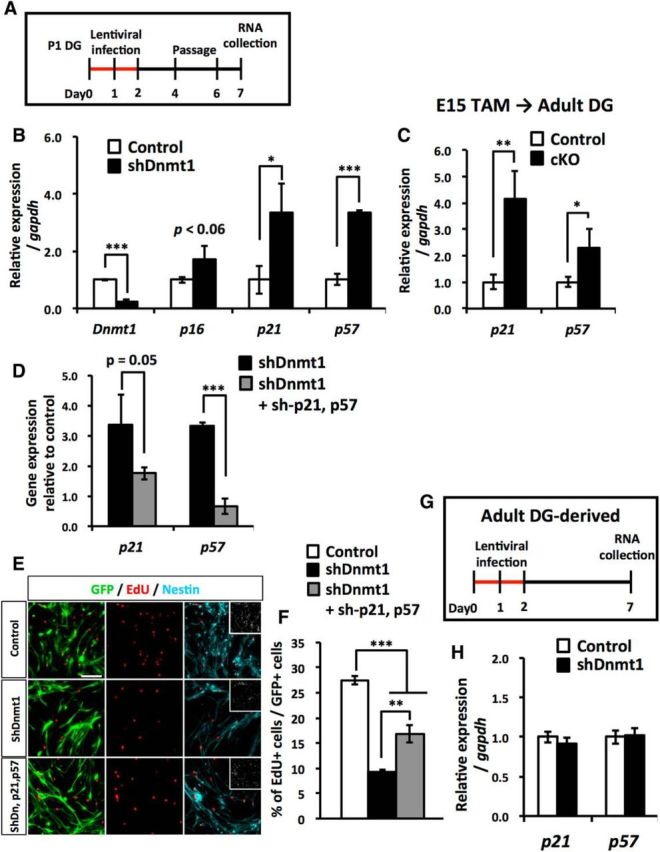
Loss of Dnmt1 increases the expression of the cyclin-dependent kinase inhibitors p21 and p57. A, Experimental scheme for assessing the expression of CDK inhibitors in Dnmt1 KD. P1 DG-derived NSCs were infected with lentivirus expressing Dnmt1-targeting shRNA. B, qRT-PCR analyses of CDK inhibitor expression in Dnmt1 KD NSCs. C, qRT-PCR analyses of CDK inhibitor expression in adult DGs (8–10 weeks) of Dnmt1 cKO mice. Prenatal deletion of Dnmt1 increases the expression of p21 and p57 in adult DGs. D, qRT-PCR analyses of p21 and p57 expression in NSCs infected with lentivirus expressing either p21- or p57-targeting shRNA. Data are mean ± SD. *p < 0.05 (Student's t test). **p < 0.01 (Student's t test). ***p < 0.001 (Student's t test). E, Proliferation assay of Dnmt1 KD NSCs after induction with both p21- and p57-targeting shRNA. NSCs were treated with EdU for 30 min before fixing. Representative immunofluorescence images for GFP (green), EdU (red), and Nestin (cyan). F, Bar graph represents the percentage of EdU+ cells among GFP+ cells. Loss of Dnmt1 impairs the proliferation of NSCs, and induction of p21- and p57-targeting shRNA partially rescues this impaired proliferation. Data are mean ± SEM. **p < 0.01 (ANOVA with Tukey post hoc tests). ***p < 0.001 (ANOVA with Tukey post hoc tests). G, Experimental scheme for assessing the expression of CDK inhibitors in Dnmt1 KD adult NSCs. Adult mouse DG-derived NSCs were infected with lentivirus expressing Dnmt1-targeting shRNA for 2 d, and RNA was collected 6 d after infection. H, qRT-PCR analyses of CDK inhibitor expression in Dnmt1 KD adult NSCs. Data are mean ± SD Student's t test.
Dnmt1 affects histone modifications in the p21 and p57 promoters and modulates the gene's expression in DNA methylation-independent manner
We assessed the importance of the DNA methyltransferase activity of DNMT1 for the regulation of p21 and p57 expression by treating wild-type NSCs with RG108 (Fig. 11A). Contrary to our expectation, we found no upregulation of p21 or p57 in NSCs treated with RG108 (Fig. 11B). This implies that DNMT1 inhibits the expression of these genes in a DNA methylation-independent manner. Recent functional analyses of DNMT1 have provided evidence that DNMT1 interacts with histone modification enzymes (Fuks et al., 2000; Qin et al., 2011; Clements et al., 2012) and raised the possibility of histone modification-mediated gene regulation by Dnmt1. Finally, therefore, we assessed whether the loss of Dnmt1 affects histone modifications in p21 and p57 gene regulatory regions, namely the promoter and transcription start site (TSS), by ChIP assay (Fig. 11C). We found that Dnmt1 KD in NSCs increased the active histone modification mark H3K4me3 and decreased the repressive histone modification mark H3K27me3 in the p21 gene promoter and TSS, respectively (Fig. 11D). Similarly H3K27me3 in both the p57 gene promoter and TSS was significantly decreased in Dnmt1 KD NSCs (Fig. 11E). These data suggest that loss of Dnmt1 decreases repressive histone modification marks in both p21 and p57 gene regulatory regions, which may account for the mechanism by which Dnmt1 modulates the expression of p21 and p57 in a DNA methylation-independent manner.
Figure 11.

Loss of Dnmt1 decreases repressive histone modification mark from p21 and p57 gene expression regulatory regions. A, Experimental scheme for assessing the expression of CDK inhibitors in RG108-treated NSCs. P1 DG-derived NSCs were treated with RG108 for 5 d. B, qRT-PCR analyses of p21 and p57 expression in RG108-treated NSCs. p21 and p57 expression is not affected by RG108 treatment. Data are mean ± SD. n.s., Not significant (p > 0.05; ANOVA with Tukey post hoc tests). C, Experimental scheme for assessing histone modification in Dnmt1 KD NSCs. P1 DG-derived NSCs were infected with lentivirus expressing Dnmt1-targeting shRNA for 2 d, and DNA was collected 7 d after infection. D, E, Diagram of the p21 (D) and p57 (E) gene promoter and TSS. The lower horizontal lines demarcate the gene promoter (left) and TSS (right) regions analyzed by ChIP assay. Bar graphs indicate ChIP-qPCR for H3K4me3 and H3K27me3 enrichment in the p21 (D) and p57 (E) gene promoter and TSS in Dnmt1 KD NSCs. Data are represented as the percentage of bound DNA to input DNA (Input %). Data are mean ± SEM. *p < 0.05 (Student's t test). **p < 0.01 (Student's t test).
Discussion
We investigated the role of Dnmt1 in DG development and revealed that Dnmt1 is required for proper DG development to occur. Our data show that deletion of Dnmt1 in NSCs at the beginning of DG development impacts on multiple steps in the development, eventually resulting in a smaller GCL in the adult DG; this diminution is associated with impaired proliferation and neurogenic ability of NSCs (Fig. 12). These abnormalities in the DGs of adult Dnmt1 cKO mice are likely to be attributable to the disruption of multiple steps in DG development, beginning with impaired formation of the secondary radial glial scaffolds at the postnatal stage.
Figure 12.

Schematic diagram summarizing the effects of Dnmt1 knock-out on development of the DG. In the absence of Dnmt1, NSCs fail to localize in the SGZ or to establish secondary radial glial scaffolds in the early postnatal stage, resulting in aberrant neuronal production in the ML of the DG and fewer granule neurons in the GCL in the late postnatal stage. Loss of Dnmt1 reduces the expression of ApoER2, a key component of Reelin signaling, which may account for the developmental defects in Dnmt1 cKO DGs. Additionally, prenatal deletion of Dnmt1 interferes with the proliferative and neurogenic ability of NSCs in adult DGs. Prenatal deletion of Dnmt1 also results in the increased expression of the cyclin-dependent kinase inhibitors p21 and p57 in adult DGs, leading to impaired proliferation and impaired neurogenic ability of NSCs.
Dnmt1-deficient NSCs failed to establish proper secondary radial glial scaffolds but could establish normal primordial radial glial scaffolds, suggesting that Dnmt1 is specifically involved in the former scaffold formation (Fig. 3). Analysis of mice deficient for Reelin signaling has indicated that development of the secondary radial glial scaffold depends on the activity of Reelin signaling (Weiss et al., 2003). Reeler and scrambler mice, which lack Reelin and Dab1, an intracellular adaptor protein in Reelin signaling, respectively, show severe defects in the establishment of secondary radial glial scaffolds (Förster et al., 2002; Weiss et al., 2003). In contrast, single-knock-out mice for one of the two receptors for Reelin show mild defects. In these mice, NSCs belatedly but eventually established radial processes in the late postnatal stage. In the present study, we have shown that deletion of Dnmt1 leads to impaired development of the secondary radial glial scaffold, which is reminiscent of what was observed in Reelin signaling-deficient mice (Weiss et al., 2003). In addition, we found that deletion or inhibition of Dnmt1 decreased the expression of ApoER2 in NSCs (Fig. 9C,E,G). These findings support our idea that Dnmt1 contributes to the development of the secondary radial glial scaffold by regulating the Reelin signaling pathway.
Impaired development of the secondary radial glial scaffold leads to further impairments of DG development. Previous reports have demonstrated that the secondary radial glial scaffold contributes to the migration of NPs and granule neurons to their final positions. In reeler mice, the secondary radial glial scaffold is disorganized, and Tbr2+ cells accumulate in the ML (Förster et al., 2002; Li et al., 2009). This suggests that the abnormal accumulation of Tbr2+ cells that we observed in the ML in Dnmt1 cKO mice reflects disturbed migration of NPs as a result of hypoplasia of the secondary radial glial scaffold. In addition, mismigration of NSCs probably also contributed to the increased number of NPs in the ML. As NSCs transit the from the ML to the SGZ, neurogenic regions are confined to the SGZ in the first 2 postnatal weeks (Li et al., 2009) (Figs. 3B, 4C,D). However, our data demonstrated that deletion of Dnmt1 leads to the impairment of NSC migration into the SGZ, resulting in the overproduction of Tbr2+ cells in the ML. reeler mice also show a defect in the transition of NSC positioning from the ML to the SGZ. This further strengthens our idea that developmental defects in Dnmt1 cKO mice result from attenuated Reelin signaling.
Because migration of Dnmt1-deficient NPs to the SGZ is impaired, these cells accumulate in the ML, where they produce more neurons than controls. However, increased cell death and decreased mature neuron numbers were observed in Dnmt1 cKO DGs, suggesting that such mispositioned neurons are eliminated from the ML over time. This supports earlier findings that DNA hypomethylation caused by the loss of Dnmt1 in embryonic cortical NSCs induces cell death of differentiated neurons (Fan et al., 2001; Hutnick et al., 2009). We have also previously reported that Dnmt1 deletion in adult NSCs, but not in neurons, impairs the survival of newly generated neurons (Noguchi et al., 2015).
In the present study, we further showed that deletion of Dnmt1 increased astrocyte differentiation of NSCs in the SGZ. However, these S100β+ astrocytes in the SGZ of Dnmt1 cKO DGs retained some features of NSCs (i.e., a single radial process and expression of the NSC marker Nestin). In response to inflammatory signals, astrocytes become reactive astrocytes, which express Nestin. Thus, it is possible that these abnormally produced astrocytes are reactive astrocytes rather than cells that had failed to fully differentiate into mature astrocytes. We found increased cell death in Dnmt1 cKO DGs, and dead cells may have triggered inflammation to make the astrocytes become the reactive type.
We also found that prenatal deletion of Dnmt1 impaired the proliferation of NSCs and increased their quiescent population in the adult DG. Loss of Dnmt1 increased expression of the cell cycle inhibitor genes p21 and p57, suggesting that Dnmt1 modulates the proliferation and quiescent status of NSCs via the expression of p21 and p57. In particular, p57 expression is enriched in quiescent NSCs in the adult DG, and recent studies have indicated the relevance of p57 to the quiescent state of NSCs (Furutachi et al., 2013, 2015). Increased expression of p57 and the abundance of a quiescent population of NSCs in Dnmt1 cKO DGs suggest that Dnmt1 plays a key role in regulating the quiescent state of NSCs by regulating p57 expression. However, we have previously revealed that deleting Dnmt1 in NSCs at adult stages does not impair the proliferation or the number of quiescent NSCs (Noguchi et al., 2015). Moreover, we have shown in the current study that KD of Dnmt1 in adult NSCs, but not prenatal NSCs, does not affect expression of p21 or p57 (Fig. 10H). These findings suggest that Dnmt1 is not involved in regulating the proliferation or the quiescent state of NSCs in the adult stages but rather plays a crucial role in such regulation during developmental stages. The embryonic brain has relatively dormant NSCs, and it has been demonstrated that adult NSCs in the SVZ originate from these cells (Furutachi et al., 2015). This subpopulation of NSCs expresses a high level of p57, and gain- and loss-of-function experiments revealed that p57 contributes to the emergence of adult NSCs in the SVZ. These findings suggest that, although Dnmt1 participates in mechanisms that establish the quiescent state of NSCs by regulating p57 expression during DG development, it does not do so in the adult stage. The detailed mechanisms by which embryonic NSCs acquire the properties of adult NSCs must await future investigation, but we believe that our data provide the important idea that Dnmt1-mediated epigenetic regulation contributes to this transition.
We have shown that the loss of Dnmt1 results in reduced expression of Reelin signaling components (Fig. 9C). Inhibition of DNMT1 activity by RG108 decreased the expression of ApoER2, suggesting that DNMT1 regulates ApoER2 expression through DNA methylation. However, because DNA methylation at gene promoters is generally associated with gene repression (Klose and Bird, 2006), Dnmt1 deletion or inhibition of its enzymatic activity should increase its target genes' expression. Considering that the loss of Dnmt1 decreased expression of ApoER2, Dnmt1 may indirectly regulate ApoER2 expression through DNA methylation-mediated suppression of a negative regulator for ApoER2 expression. On the other hand, at least for p21 and p57 expression, Dnmt1 does not require DNA methyltransferase activity because RG108 treatment did not affect the expression of these genes (Fig. 11B). Dnmt1 has been shown to interact with histone modification enzymes and to suppress gene expression via histone modification (Fuks et al., 2000; Qin et al., 2011; Clements et al., 2012). Thus, it is likely that Dnmt1 inhibits p21 and p57 expression through such histone modification. In this context, recent studies have indicated that enzymes for histone deacetylation and methylation are involved in silencing p21 and p57 expression, respectively (Hsieh et al., 2004; Yang et al., 2009). Supporting these observations, we found that the loss of Dnmt1 decreased H3K27me3 in the p21 and p57 promoters (Fig. 11D,E), suggesting that Dnmt1 represses the expression of p21 and p57 in a DNA methylation-independent manner through interactions with enzymes, responsible for the H3K27me3, such as the enhancer of zeste 2 polycomb repressive complex 2 subunit.
In the present study, we have shown that Dnmt1 plays critical roles in the development of the DG that are apparently both DNA methylation-dependent and -independent. Our findings should stimulate efforts to further understand the role of epigenetic regulation in the development of nervous systems.
Footnotes
This work was supported by the Japan Agency for Medical Research and Development, Core Research for Evolutional Science and Technology to K.N., Challenging Exploratory Research Grant-in-Aid 15K14452 to K.N., Sasakawa Scientific Research Grant to H.N., and Japan Society for the Promotion of Science Fellows Grant-in-Aid 13J09870 to H.N. We thank Y. Bessho, T. Matsui, Y. Nakahata, T. Imamura, and S. Katada for valuable discussions; M.E. Greenberg and Z. Zhou for sharing reagents; Dr. R. Jaenisch for Dnmt1flox mice; Dr. R. Kageyama for Nestin-CreERT2 mice; members of our laboratories, in particular T. Sanosaka and B. Juliandi, for technical help and suggestions; A.M.D. Adefuin and M.C. Sanosaka for helping to write the manuscript; I. Smith for helpful comments and grammatical corrections on the manuscript; M. Tano and Y. Nakagawa for their excellent secretarial assistance; and the Research Support Center, Research Center for Human Disease Modeling, Kyushu University Graduate School of Medical Sciences for technical assistance.
The authors declare no competing financial interests.
References
- Altman J, Bayer SA. Migration and distribution of two populations of hippocampal granule cell precursors during the perinatal and postnatal periods. J Comp Neurol. 1990a;301:365–381. doi: 10.1002/cne.903010304. [DOI] [PubMed] [Google Scholar]
- Altman J, Bayer SA. Mosaic organization of the hippocampal neuroepithelium and the multiple germinal sources of dentate granule cells. J Comp Neurol. 1990b;301:325–342. doi: 10.1002/cne.903010302. [DOI] [PubMed] [Google Scholar]
- Altman J, Das GD. Autoradiographic and histological evidence of postnatal hippocampal neurogenesis in rats. J Comp Neurol. 1965;124:319–335. doi: 10.1002/cne.901240303. [DOI] [PubMed] [Google Scholar]
- Angevine JB., Jr Time of neuron origin in the hippocampal region: an autoradiographic study in the mouse. Exp Neurol Suppl. 1965;2:1–70. [PubMed] [Google Scholar]
- Ansorg A, Witte OW, Urbach A. Age-dependent kinetics of dentate gyrus neurogenesis in the absence of cyclin D2. BMC Neurosci. 2012;13:46. doi: 10.1186/1471-2202-13-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asano H, Aonuma M, Sanosaka T, Kohyama J, Namihira M, Nakashima K. Astrocyte differentiation of neural precursor cells is enhanced by retinoic acid through a change in epigenetic modification. Stem Cells. 2009;27:2744–2752. doi: 10.1002/stem.176. [DOI] [PubMed] [Google Scholar]
- Bignami A, Dahl D. Astrocyte-specific protein and neuroglial differentiation: an immunofluorescence study with antibodies to the glial fibrillary acidic protein. J Comp Neurol. 1974;153:27–38. doi: 10.1002/cne.901530104. [DOI] [PubMed] [Google Scholar]
- Breunig JJ, Sarkisian MR, Arellano JI, Morozov YM, Ayoub AE, Sojitra S, Wang B, Flavell RA, Rakic P, Town T. Primary cilia regulate hippocampal neurogenesis by mediating sonic hedgehog signaling. Proc Natl Acad Sci U S A. 2008;105:13127–13132. doi: 10.1073/pnas.0804558105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunne B, Zhao S, Derouiche A, Herz J, May P, Frotscher M, Bock HH. Origin, maturation, and astroglial transformation of secondary radial glial cells in the developing dentate gyrus. Glia. 2010;58:1553–1569. doi: 10.1002/glia.21029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunne B, Franco S, Bouché E, Herz J, Howell BW, Pahle J, Müller U, May P, Frotscher M, Bock HH. Role of the postnatal radial glial scaffold for the development of the dentate gyrus as revealed by Reelin signaling mutant mice. Glia. 2013;61:1347–1363. doi: 10.1002/glia.22519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caviness VS., Jr Time of neuron origin in the hippocampus and dentate gyrus of normal and reeler mutant mice: an autoradiographic analysis. J Comp Neurol. 1973;151:113–120. doi: 10.1002/cne.901510203. [DOI] [PubMed] [Google Scholar]
- Clements EG, Mohammad HP, Leadem BR, Easwaran H, Cai Y, Van Neste L, Baylin SB. DNMT1 modulates gene expression without its catalytic activity partially through its interactions with histone-modifying enzymes. Nucleic Acids Res. 2012;40:4334–4346. doi: 10.1093/nar/gks031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng W, Aimone JB, Gage FH. New neurons and new memories: how does adult hippocampal neurogenesis affect learning and memory? Nat Rev Neurosci. 2010;11:339–350. doi: 10.1038/nrn2822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eriksson PS, Perfilieva E, Björk-Eriksson T, Alborn AM, Nordborg C, Peterson DA, Gage FH. Neurogenesis in the adult human hippocampus. Nat Med. 1998;4:1313–1317. doi: 10.1038/3305. [DOI] [PubMed] [Google Scholar]
- Fan G, Beard C, Chen RZ, Csankovszki G, Sun Y, Siniaia M, Biniszkiewicz D, Bates B, Lee PP, Kuhn R, Trumpp A, Poon C, Wilson CB, Jaenisch R. DNA hypomethylation perturbs the function and survival of CNS neurons in postnatal animals. J Neurosci. 2001;21:788–797. doi: 10.1523/JNEUROSCI.21-03-00788.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan G, Martinowich K, Chin MH, He F, Fouse SD, Hutnick L, Hattori D, Ge W, Shen Y, Wu H, ten Hoeve J, Shuai K, Sun YE. DNA methylation controls the timing of astrogliogenesis through regulation of JAK-STAT signaling. Development. 2005;132:3345–3356. doi: 10.1242/dev.01912. [DOI] [PubMed] [Google Scholar]
- Fasano CA, Dimos JT, Ivanova NB, Lowry N, Lemischka IR, Temple S. shRNA knockdown of Bmi-1 reveals a critical role for p21-Rb pathway in NSC self-renewal during development. Cell Stem Cell. 2007;1:87–99. doi: 10.1016/j.stem.2007.04.001. [DOI] [PubMed] [Google Scholar]
- Förster E, Tielsch A, Saum B, Weiss KH, Johanssen C, Graus-Porta D, Müller U, Frotscher M. Reelin, Disabled 1, and 1 integrins are required for the formation of the radial glial scaffold in the hippocampus. Proc Natl Acad Sci U S A. 2002;99:13178–13183. doi: 10.1073/pnas.202035899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuks F, Burgers WA, Brehm A, Hughes-Davies L, Kouzarides T. DNA methyltransferase Dnmt1 associates with histone deacetylase activity. Nat Genet. 2000;24:88–91. doi: 10.1038/71750. [DOI] [PubMed] [Google Scholar]
- Furutachi S, Matsumoto A, Nakayama KI, Gotoh Y. p57 controls adult neural stem cell quiescence and modulates the pace of lifelong neurogenesis. EMBO J. 2013;32:970–981. doi: 10.1038/emboj.2013.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furutachi S, Miya H, Watanabe T, Kawai H, Yamasaki N, Harada Y, Imayoshi I, Nelson M, Nakayama KI, Hirabayashi Y, Gotoh Y. Slowly dividing neural progenitors are an embryonic origin of adult neural stem cells. Nat Neurosci. 2015;18:657–665. doi: 10.1038/nn.3989. [DOI] [PubMed] [Google Scholar]
- Han YG, Spassky N, Romaguera-Ros M, Garcia-Verdugo JM, Aguilar A, Schneider-Maunoury S, Alvarez-Buylla A. Hedgehog signaling and primary cilia are required for the formation of adult neural stem cells. Nat Neurosci. 2008;11:277–284. doi: 10.1038/nn2059. [DOI] [PubMed] [Google Scholar]
- Hirabayashi Y, Gotoh Y. Epigenetic control of neural precursor cell fate during development. Nat Rev Neurosci. 2010;11:377–388. doi: 10.1038/nrn2810. [DOI] [PubMed] [Google Scholar]
- Hirabayashi Y, Suzki N, Tsuboi M, Endo TA, Toyoda T, Shinga J, Koseki H, Vidal M, Gotoh Y. Polycomb limits the neurogenic competence of neural precursor cells to promote astrogenic fate transition. Neuron. 2009;63:600–613. doi: 10.1016/j.neuron.2009.08.021. [DOI] [PubMed] [Google Scholar]
- Hodge RD, Nelson BR, Kahoud RJ, Yang R, Mussar KE, Reiner SL, Hevner RF. Tbr2 is essential for hippocampal lineage progression from neural stem cells to intermediate progenitors and neurons. J Neurosci. 2012;32:6275–6287. doi: 10.1523/JNEUROSCI.0532-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh J, Nakashima K, Kuwabara T, Mejia E, Gage FH. Histone deacetylase inhibition-mediated neuronal differentiation of multipotent adult neural progenitor cells. Proc Natl Acad Sci U S A. 2004;101:16659–16664. doi: 10.1073/pnas.0407643101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutnick LK, Golshani P, Namihira M, Xue Z, Matynia A, Yang XW, Silva AJ, Schweizer FE, Fan G. DNA hypomethylation restricted to the murine forebrain induces cortical degeneration and impairs postnatal neuronal maturation. Hum Mol Genet. 2009;18:2875–2888. doi: 10.1093/hmg/ddp222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imayoshi I, Ohtsuka T, Metzger D, Chambon P, Kageyama R. Temporal regulation of Cre recombinase activity in neural stem cells. Genesis. 2006;44:233–238. doi: 10.1002/dvg.20212. [DOI] [PubMed] [Google Scholar]
- Imayoshi I, Sakamoto M, Ohtsuka T, Takao K, Miyakawa T, Yamaguchi M, Mori K, Ikeda T, Itohara S, Kageyama R. Roles of continuous neurogenesis in the structural and functional integrity of the adult forebrain. Nat Neurosci. 2008;11:1153–1161. doi: 10.1038/nn.2185. [DOI] [PubMed] [Google Scholar]
- Kippin TE, Martens DJ, van der Kooy D. p21 loss compromises the relative quiescence of forebrain stem cell proliferation leading to exhaustion of their proliferation capacity. Genes Dev. 2005;19:756–767. doi: 10.1101/gad.1272305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klose RJ, Bird AP. Genomic DNA methylation: the mark and its mediators. Trends Biochem Sci. 2006;31:89–97. doi: 10.1016/j.tibs.2005.12.008. [DOI] [PubMed] [Google Scholar]
- Kriegstein A, Alvarez-Buylla A. The glial nature of embryonic and adult neural stem cells. Annu Rev Neurosci. 2009;32:149–184. doi: 10.1146/annurev.neuro.051508.135600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kronenberg G, Reuter K, Steiner B, Brandt MD, Jessberger S, Yamaguchi M, Kempermann G. Subpopulations of proliferating cells of the adult hippocampus respond differently to physiologic neurogenic stimuli. J Comp Neurol. 2003;467:455–463. doi: 10.1002/cne.10945. [DOI] [PubMed] [Google Scholar]
- Kuhn HG, Dickinson-Anson H, Gage FH. Neurogenesis in the dentate gyrus of the adult rat: age-related decrease of neuronal progenitor proliferation. J Neurosci. 1996;16:2027–2033. doi: 10.1523/JNEUROSCI.16-06-02027.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G, Pleasure SJ. Morphogenesis of the dentate gyrus: what we are learning from mouse mutants. Dev Neurosci. 2005;27:93–99. doi: 10.1159/000085980. [DOI] [PubMed] [Google Scholar]
- Li G, Kataoka H, Coughlin SR, Pleasure SJ. Identification of a transient subpial neurogenic zone in the developing dentate gyrus and its regulation by Cxcl12 and reelin signaling. Development. 2009;136:327–335. doi: 10.1242/dev.025742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lois C, Alvarez-Buylla A. Proliferating subventricular zone cells in the adult mammalian forebrain can differentiate into neurons and glia. Proc Natl Acad Sci U S A. 1993;90:2074–2077. doi: 10.1073/pnas.90.5.2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lois C, Hong EJ, Pease S, Brown EJ, Baltimore D. Germline transmission and tissue-specific expression of transgenes delivered by lentiviral vectors. Science. 2002;295:868–872. doi: 10.1126/science.1067081. [DOI] [PubMed] [Google Scholar]
- Meehan RR. DNA methylation in animal development. Semin Cell Dev Biol. 2003;14:53–65. doi: 10.1016/S1084-9521(02)00137-4. [DOI] [PubMed] [Google Scholar]
- Ming GL, Song H. Adult neurogenesis in the mammalian brain: significant answers and significant questions. Neuron. 2011;70:687–702. doi: 10.1016/j.neuron.2011.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murao N, Noguchi H, Nakashima K. Epigenetic regulation of neural stem cell property from embryo to adult. Neuroepigenetics. 2016;5:1–10. doi: 10.1016/j.nepig.2016.01.001. [DOI] [Google Scholar]
- Namihira M, Kohyama J, Semi K, Sanosaka T, Deneen B, Taga T, Nakashima K. Committed neuronal precursors confer astrocytic potential on residual neural precursor cells. Dev Cell. 2009;16:245–255. doi: 10.1016/j.devcel.2008.12.014. [DOI] [PubMed] [Google Scholar]
- Noguchi H, Kimura A, Murao N, Matsuda T, Namihira M, Nakashima K. Expression of DNMT1 in neural stem/precursor cells is critical for survival of newly generated neurons in the adult hippocampus. Neurosci Res. 2015;95:1–11. doi: 10.1016/j.neures.2015.01.014. [DOI] [PubMed] [Google Scholar]
- Oldekamp J, Kraemer N, Alvarez-Bolado G, Skutella T. bHLH gene expression in the Emx2-deficient dentate gyrus reveals defective granule cells and absence of migrating precursors. Cereb Cortex. 2004;14:1045–1058. doi: 10.1093/cercor/bhh064. [DOI] [PubMed] [Google Scholar]
- Qin W, Leonhardt H, Pichler G. Regulation of DNA methyltransferase 1 by interactions and modifications. Nucleus. 2011;2:392–402. doi: 10.4161/nucl.2.5.17928. [DOI] [PubMed] [Google Scholar]
- Qiu J, Takagi Y, Harada J, Topalkara K, Wang Y, Sims JR, Zheng G, Huang P, Ling Y, Scadden DT, Moskowitz MA, Cheng T. p27Kip1 constrains proliferation of neural progenitor cells in adult brain under homeostatic and ischemic conditions. Stem Cells. 2009;27:920–927. doi: 10.1002/stem.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rickmann M, Amaral DG, Cowan WM. Organization of radial glial cells during the development of the rat dentate gyrus. J Comp Neurol. 1987;264:449–479. doi: 10.1002/cne.902640403. [DOI] [PubMed] [Google Scholar]
- Seki T, Sato T, Toda K, Osumi N, Imura T, Shioda S. Distinctive population of Gfap-expressing neural progenitors arising around the dentate notch migrate and form the granule cell layer in the developing hippocampus. J Comp Neurol. 2014;522:261–283. doi: 10.1002/cne.23460. [DOI] [PubMed] [Google Scholar]
- Shen L, Nam HS, Song P, Moore H, Anderson SA. FoxG1 haploinsufficiency results in impaired neurogenesis in the postnatal hippocampus and contextual memory deficits. Hippocampus. 2006;16:875–890. doi: 10.1002/hipo.20218. [DOI] [PubMed] [Google Scholar]
- Sievers J, Hartmann D, Pehlemann FW, Berry M. Development of astroglial cells in the proliferative matrices, the granule cell layer, and the hippocampal fissure of the hamster dentate gyrus. J Comp Neurol. 1992;320:1–32. doi: 10.1002/cne.903200102. [DOI] [PubMed] [Google Scholar]
- Steiner B, Klempin F, Wang L, Kott M, Kettenmann H, Kempermann G. Type-2 cells as link between glial and neuronal lineage in adult hippocampal neurogenesis. Glia. 2006;54:805–814. doi: 10.1002/glia.20407. [DOI] [PubMed] [Google Scholar]
- Takizawa T, Nakashima K, Namihira M, Ochiai W, Uemura A, Yanagisawa M, Fujita N, Nakao M, Taga T. DNA methylation is a critical cell-intrinsic determinant of astrocyte differentiation in the fetal brain. Dev Cell. 2001;1:749–758. doi: 10.1016/S1534-5807(01)00101-0. [DOI] [PubMed] [Google Scholar]
- Theil T, Aydin S, Koch S, Grotewold L, Rüther U. Wnt and Bmp signalling cooperatively regulate graded Emx2 expression in the dorsal telencephalon. Development. 2002;129:3045–3054. doi: 10.1242/dev.129.13.3045. [DOI] [PubMed] [Google Scholar]
- Tian C, Gong Y, Yang Y, Shen W, Wang K, Liu J, Xu B, Zhao J, Zhao C. Foxg1 has an essential role in postnatal development of the dentate gyrus. J Neurosci. 2012;32:2931–2949. doi: 10.1523/JNEUROSCI.5240-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss KH, Johanssen C, Tielsch A, Herz J, Deller T, Frotscher M, Förster E. Malformation of the radial glial scaffold in the dentate gyrus of reeler mice, scrambler mice, and ApoER2/VLDLR-deficient mice. J Comp Neurol. 2003;460:56–65. doi: 10.1002/cne.10644. [DOI] [PubMed] [Google Scholar]
- Yang X, Karuturi RK, Sun F, Aau M, Yu K, Shao R, Miller LD, Tan PB, Yu Q. CDKN1C (p57) is a direct target of EZH2 and suppressed by multiple epigenetic mechanisms in breast cancer cells. PLoS One. 2009;4:e5011. doi: 10.1371/journal.pone.0005011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, Hong EJ, Cohen S, Zhao WN, Ho HY, Schmidt L, Chen WG, Lin Y, Savner E, Griffith EC, Hu L, Steen JA, Weitz CJ, Greenberg ME. Brain-specific phosphorylation of MeCP2 regulates activity-dependent Bdnf transcription, dendritic growth, and spine maturation. Neuron. 2006;52:255–269. doi: 10.1016/j.neuron.2006.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou P, Yoshihara H, Hosokawa K, Tai I, Shinmyozu K, Tsukahara F, Maru Y, Nakayama K, Nakayama KI, Suda T. p57(Kip2) and p27(Kip1) cooperate to maintain hematopoietic stem cell quiescence through interactions with Hsc70. Cell Stem Cell. 2011;9:247–261. doi: 10.1016/j.stem.2011.07.003. [DOI] [PubMed] [Google Scholar]