

Abstract
Aberrant branch formation of granule cell axons (mossy fiber sprouting) is observed in the dentate gyrus of many patients with temporal lobe epilepsy and in animal models of epilepsy. However, the mechanisms underlying mossy fiber sprouting remain elusive. Based on the hypothesis that seizure-mediated gene expression induces abnormal mossy fiber growth, we screened activity-regulated genes in the hippocampus and found that neuritin, an extracellular protein anchored to the cell surface, was rapidly upregulated after electroconvulsive seizures. Overexpression of neuritin in the cultured rat granule cells promoted their axonal branching. Also, kainic acid-dependent axonal branching was abolished in the cultured granule cells from neuritin knock-out mice, suggesting that neuritin may be involved in activity-dependent axonal branching. Moreover, neuritin knock-out mice showed less-severe seizures in chemical kindling probably by reduced mossy fiber sprouting and/or increased seizure resistance. We found that inhibition of the fibroblast growth factor (FGF) receptor attenuated the neuritin-dependent axonal branching. FGF administration also increased branching in granule neurons, whereas neuritin knock-out mice did not show FGF-dependent axonal branching. In addition, FGF and neuritin treatment enhanced the recruitment of FGF receptors to the cell surface. These findings suggest that neuritin and FGF cooperate in inducing mossy fiber sprouting through FGF signaling. Together, these results suggest that FGF and neuritin-mediated axonal branch induction are involved in the aggravation of epilepsy.
SIGNIFICANCE STATEMENT This study reveals the molecular mechanism underlying mossy fiber sprouting. Mossy fiber sprouting is the aberrant axonal branching of granule neurons in the hippocampus, which is observed in patients with epilepsy. Excess amounts of neuritin, a protein upregulated by neural activity, promoted axonal branching in granule neurons. A deficiency of neuritin suppressed mossy fiber sprouting and resulted in mitigation of seizure severity. Neuritin and fibroblast growth factor (FGF) cooperated in stimulating FGF signaling and enhancing axonal branching. Neuritin is necessary for FGF-mediated recruitment of FGF receptors to the cell surface. The recruitment of FGF receptors would promote axonal branching. The discovery of this new mechanism should contribute to the development of novel antiepileptic drugs to inhibit axonal branching via neuritin-FGF signaling.
Keywords: axon, epilepsy, mossy fiber
Introduction
Epilepsy is a chronic neurological disorder that affects ∼1% of people worldwide. It is defined by recurrent seizures and is often accompanied by cognitive deficits and mood disorders (Devinsky, 2004; Pellock, 2004; Jones et al., 2008). Seizures may be generated by the synchronous firing of small groups of neurons called epileptic foci (Stevens, 1996; Pitkänen and Sutula, 2002). Epileptic seizures cause anatomical and functional sequelae in the brain. These neuroplastic changes, which include neuronal cell loss (Represa et al., 1995; Arzimanoglou et al., 2002), dispersion of dentate gyrus granule cells (Houser, 1990), promotion of neurogenesis (Parent et al., 1997), and activation of astrocytes in the hippocampus (Sofroniew, 2009), have been found in numerous patients with temporal lobe epilepsy and in various laboratory animal models of epilepsy.
Another anatomical change is observed in the patients with epilepsy and in the animal models of epilepsy. Axons of granule cells in the dentate gyrus (mossy fibers) form aberrant axonal branches that are directed toward the dentate gyrus molecular layer and form recurrent excitatory synapses with other granule cells (Sutula et al., 1988; Parent et al., 1997). This anatomical alteration is called mossy fiber sprouting. The functional significance of mossy fiber sprouting in epilepsy has been studied using different approaches, including animal models (Zhang et al., 2002; Koyama and Ikegaya, 2004) and neuron cultures (Routbort et al., 1999; Bausch and McNamara, 2004). These studies have shown that mossy fiber sprouting is a critical factor that contributes to the hyperexcitability of the neural circuit in the hippocampus, particularly in the dentate gyrus (Zhang et al., 2002; Bausch and McNamara, 2004; Koyama and Ikegaya, 2004; Houser et al., 2012). It has also been shown that the deletion of phosphatase and tensin homolog in granule neurons causes spontaneous mossy fiber sprouting and results in intermittent seizures (Amiri et al., 2012; Pun et al., 2012), thus supporting a correlation between mossy fiber sprouting and epileptogenesis.
Neuritin, also known as candidate plasticity gene 15 (CPG15), was first identified as one of the activity-dependent gene products in the brain (Nedivi et al., 1993). Neuritin is a small extracellular protein that is anchored to the cell surface by a glycosyl-phosphoinositide (GPI) link (Naeve et al., 1997). Several studies have explored the function of neuritin in neuronal morphology. Neuritin induces neuritogenesis, neurite arborization, and neurite outgrowth (Shimada et al., 2013). The administration of purified neuritin to cultured hippocampal and cortical neurons induces extensive neurite formation (Naeve et al., 1997; Sato et al., 2012). Overexpression of neuritin increases dendritic arbors in the optic tectal neurons of Xenopus tadpoles (Nedivi et al., 1998) and promotes increases in axonal branch formation and decreases in the branch retraction in motoneurons of Xenopus (Javaherian and Cline, 2005). Furthermore, dissected hippocampal explants cocultured with neuritin-expressing cells show significantly longer neurites than do those cocultured with control cells (Fujino et al., 2008). Although neuritin mRNA is predominantly expressed in the brain, particularly in the dentate gyrus of the hippocampus (Naeve et al., 1997), the effects of neuritin on dentate granule neurons are still unknown. Moreover, the mechanisms that explain how neuritin induces morphological changes in neurons are not well studied.
Here, we elucidate the function of neuritin in mossy fibers both in dissociated cultures and in hippocampal organotypic slice cultures. The results indicate that neuritin promotes axonal branch formation without axonal elongation. We further demonstrate that neuritin interacts with fibroblast growth factor receptor (FGFR) and that neuritin-mediated axonal branch formation is abolished by the inhibition of FGFR activation. Finally, we find that neuritin-deficient mice show less aggravation in the mossy fibers after pentylenetetrazole (PTZ) kindling. Our studies suggest that neuritin and FGF cooperatively cause seizure-induced axonal branch formation in granule cells and that this neuritin-mediated axonal branching may be involved in the exacerbation of epilepsy.
Materials and Methods
DNA constructs and reagents.
To construct expression vectors for neuritin ORF, cDNA was amplified by PCR from a rat brain cDNA library and ligated into a vector with a CAG promoter (Niwa et al., 1991). For FGFRs, cDNA from just after the signal peptide to the terminal codon was amplified by PCR from cDNA of the rat brain and then ligated into the vector with the CAG promoter, signal peptide, and HA tag. The myc tag was added to neuritin through site-directed mutagenesis, and the myc peptide was inserted just before the GPI signal peptide. The EGFP tag was added to neuritin through site-directed mutagenesis. The EcoR I-XhoI site was created just after the extracellular transport signal peptide. EGFP was amplified by PCR and ligated in the enzyme site.
Kainic acid (KA), PD173074, AZD4547, U0126, LY294002, U73122, 5-chloromethylfluorescein diacetate (CMFDA), and recombinant FGFs were obtained from Sigma-Aldrich, Cayman, Selleck, LC Laboratories, Cayman, Focus Biomolecules, AdipoGen, and PeproTech, respectively.
Mice.
Neuritin knock-out (KO) mice were obtained from The Jackson Laboratory (Nrn1tm1.2Ndiv: stock # 018402, https://www.jax.org/strain/018402). Control animals were neuritin+/+ wild-type (WT) mice that were derived from heterozygous crosses. All animal experiments were approved by the Animal Care and Use Committee of the Tokyo Metropolitan Institute of Medical Science and were performed according to their recommendations.
Cell culture.
Hippocampal granule neurons were collected from P3 Sprague Dawley rats (Nihon-Clea) or P3-P4 neuritin KO mice and their WT littermates of either sex. Isolation of granule cells was performed as described by Baba et al. (2003). In brief, hippocampi were dissected from the animals, and the subiculum and the Ammon horn were carefully removed. The remaining dentate gyrus was treated with 0.05% trypsin (Invitrogen). After gentle trituration, isolated cells were plated onto 18 mm coverslips coated with poly-l-lysine in 12-well plates at a density of 2–5 × 104/well. The cells were cultured in 10% FCS/Neurobasal medium for 3 h, and the medium was then changed to Neurobasal medium/2% B-27 supplement/0.2% GlutaMAX (Invitrogen). The cells were transfected with plasmids using Nucleofector (Amaxa, Lonza) before plating. Half of the medium was changed twice per week. Granule cells were identified by immunostaining with anti-prox 1 antibody (Millipore).
Organotypic slice culture.
Hippocampal slice cultures were prepared from P7 Sprague Dawley rats (Nihon-Clea) or P6-P7 neuritin KO mice and their WT littermates of either sex as described previously (Hayashi et al., 2000; Okamoto et al., 2004). In brief, after the brain was taken from the animal and placed in cutting buffer (2.5 mm KCl, 25.6 mm NaHCO3, 1.15 mm NaH2PO4, 11 mm glucose, 238 mm sucrose, 1 mm CaCl2, and 5 mm MgCl2), the hippocampi were removed and cut with a McIlwain tissue chopper to generate 350-μm-thick slices. The hippocampal slices were placed on tissue culture inserts (Millicell, Millipore) and maintained in 5% CO2 at 35°C. Millicells were submerged in 1 ml of culture medium (788 ml MEM, 200 ml horse serum, 4.8 g glucose, 0.49 g NaHCO3, 2 ml of 1 m CaCl2, 2 ml of 1 m MgSO4, 7.16 g HEPES, 48 μl of 25% ascorbic acid, and 50 μl of 10 mg/ml insulin; pH was adjusted to 7.3 with NaOH) in a 6-well plate or 35 mm dish. The medium was completely changed 3 times per week. After 7 d of culture, a Helios gene gun (Bio-Rad) was used to transfect DNA into granule neurons. For gene gun transfection, 25 mg of 1.0 μm gold particles (Bio-Rad) was coated with a total of 50 μg of plasmid DNA (50 μg: pCAG-EGFP, or 25 μg: pCAG-EGFP and 25 μg: pCAG-neuritin-myc/1 long tube), according to the manufacturer's protocol. The slices were treated with KA, PD173074, AZD4547 or vehicle from DIV 8 to DIV 10 and were fixed on DIV 11. For reagent treatment, reagent was applied to the medium first, and then the medium was dropped onto the slices; this was followed by the transfer of medium from the culture insert to the well.
Preparation and application of Fc-tagged proteins.
Soluble recombinant neuritin-Fc and Fc were prepared as described previously by Uemura et al. (2010). In brief, respective expression vectors were transfected into the FreeStyle 293F cells (Invitrogen). Cell culture medium containing Fc-tagged protein was collected. Protein was purified by a protein A-Sepharose bead column and eluted with 3 m MgCl2. Eluted solution was concentrated with Amicon concentrators (10 kDa cutoff), and the buffer was changed to PBS. Purified proteins were administered to the cultured neurons (1 μg/ml) at DIV 1. Medium with purified proteins was used to change the culture medium.
Immunocytochemistry and immunohistochemistry.
Cultured granule neurons were fixed with 4% PFA for 20 min at room temperature, and this was followed by 3 washes with PBS. After treatment with 0.1% Triton X-100 PBS for 10 min at room temperature for permeabilization and a brief wash with PBS, the cells were treated with 2% goat serum PBS for 1 h at room temperature. The cells were then immunostained using the following antibodies: anti-Tuj1 (Covance, 1:1000), anti-prox 1 (Millipore, 1:6000), anti-myc (Roche, 1:1000), anti-FGFR1 (CST, D8E4, 1:200), and anti-GFP (Millipore, 1:1000). For cell surface FGFR1 staining, 30 μm CMFDA was applied 180 min before fixation, and Triton X-100 treatment was not performed after fixation. The cells were then immunostained with anti-FGFR1 (Abcam, M2F12, 1:300). Secondary antibodies were conjugated with Alexa-488, Alexa-568, and Alexa-647 (Invitrogen, 1:2000). We used 50% glycerol and 50% PBS as the mounting medium.
For slice culture, the slices were fixed with 4% PFA for 2 h at room temperature; this was followed by 3 washes with PBS. After treatment with 1% Triton X-100 PBS overnight at 4°C and a brief wash with PBS, the slices were treated with 20% BSA, 0.1% Triton X-100 PBS for 1 h at room temperature. The slices were then immunostained using the same primary and secondary antibodies as those used for immunostaining the cultured cells. We used 15% poly-vinyl alcohol, 5% propyl gallate, and 30% glycerol in PBS as the mounting medium.
The fixed brain slices were washed with 0.1% Triton X-100 PBS (PBST) for 10 min 3 times before being blocked with 2% goat serum in PBST for 1 h. The slices were then immunostained using the following antibodies: anti-Tuj1 (1:1000), anti-synaptoporin (Synaptic Systems, 1:500), and anti-ZnT3 (Synaptic Systems, 1:500). Secondary antibodies were conjugated with Alexa-488 and Alexa-568 (1:1000). The slices were stained with DAPI after the secondary antibody treatment. We used 50% glycerol and 50% PBS as the mounting medium.
RNA interference (RNAi).
The sequence of the targeted region of rat FGFR1 mRNA for Stealth RNAi (Invitrogen) was 5′-CAGCUGCCAAGACGGUGAAAUUCAA-3′, which corresponds to nucleotide residues 506–530. As a control, we used a sequence (5′-AAGGAGGAAAGACCGCUGAAUCCUG-3′), which does not target any known vertebrate gene. For the rescue experiment, rat FGFR1*, which is resistant to RNAi treatment, was subcloned into the vector with the CAG promoter, signal peptide, and HA tag. The FGFR1* sequence was altered as follows: CAGCUGCCAAGACGGUUAAGUUUAA (italicized nucleotides were substituted). The neurons were transfected with RNA and plasmids using Nucleofector (Amaxa, Lonza) before plating. The 293T cells were transfected using Lipofectamine 2000.
Immunoprecipitation.
The 293T cells were transfected with Lipofectamine 2000. After 30 h of culture after transfection, the cells were lysed with lysis buffer (100 mm NaCl, 3 mm MgCl2, 5 mm EDTA, 20 mm HEPES, pH 7.2, 1% Triton X-100, with Roche protease inhibitor mixture and phosphatase inhibitor, NaF and NaV3). After a brief sonication on ice, cell lysates were centrifuged, and detergent-soluble fractions were collected. Lysates were precleaned with protein L agarose beads (Santa Cruz Biotechnology) and then incubated with anti-HA (Roche) or anti-GFP (Invitrogen) antibody for 2 h; the protein L agarose was then added. Precipitates were washed with the lysis buffer and eluted in 2× SDS sample buffer.
PTZ-mediated seizure induction.
Eight-week-old mice of either sex were injected intraperitoneally with a subconvulsive dose (35 mg/kg) of pentylenetetrazole (Sigma-Aldrich) (2 mg/ml in 0.9% NaCl) every other day. After the injection, the convulsive behavior of the mice was observed for 30 min, and the resultant seizures were classified and scored as follows: 0, normal behavior; 1, immobilization; 2, facial, forelimb, or hindlimb myoclonus; 3, continuous whole-body myoclonus; 4, rearing, tonic seizure; 5, tonic-chronic seizure; and 6, death; if an animal died, the following scores were estimated as 6 (Shimada et al., 2014). This scoring system was slightly modified from that described by Itoh et al. (2004). For the seizure score graph (see Fig. 4A), 10 injections were administered. To induce mossy fiber sprouting, 25 injections with the dose of PTZ being changed from the 11th injection were administered as follows: the starting dose at 11th injection was 32 mg/kg; if a seizure score reached 5, the dose was decreased by 4 mg/kg thereafter; if a seizure score was 3 or less for 3 consecutive injections, the dose was increased by 4 mg/kg (32 mg/kg maximum) thereafter. All experiments were performed from 9:00 A.M. to 11:00 A.M.
Figure 4.

Neuritin aggravates epileptic seizures by enhancing mossy fiber sprouting. A, Development of epileptic convulsive seizures following consecutive treatments with PTZ. KO mice (n = 10) showed a significant reduction in seizure score compared with that in WT mice (n = 10) (p = 0.041). *p < 0.05 (repeated-measured ANOVA). B, Maximally projected immunohistochemical images of the dentate gyrus and hilus of the WT and neuritin KO mice, fixed before and after kindling. Anti-synaptoporin antibody was used to visualize axons of granule neurons (mossy fibers). Scale bar, 200 μm. C, Enlarged images of the hilus, granule cell layer, and molecular layer. The pictured areas correspond to the boxed areas in B. Arrowheads indicate mossy fiber sprouting. Scale bar, 50 μm. D, Schematic illustration for the quantification of the degree of mossy fiber sprouting. The linescan intensity of synaptoporin was measured along the border of the hilus (black line). Background intensity was measured as the minimum intensity at the granule cell layer (red square), and saturated intensity was measured as the maximum intensity at the hilus (blue square). Within the linescan, the number of branches (black arrowheads) that exceeded the threshold intensity, the mean intensity of the background, and saturation were measured in each hippocampal slice. And the numbers of branches per unit length of the border of the hilus were quantified. E, Quantification of the number of synaptoporin-positive branches per 100 μm of border of the hilus in hippocampal slices (WT: p < 0.001; KO: p < 0.001; WT vs KO after kindling: p = 0.032; WT vs KO before kindling: p = 0.713). The number of animals used was 4 before kindling and 5 (WT) or 3 (KO) after kindling. Two slices were prepared from each animal, and the numbers of branches were measured in both hippocampi in every slice. *p < 0.05 (one-way ANOVA between each group). ***p < 0.005 (one-way ANOVA between each group). F, Quantification of the number of synaptoporin-positive branches per 100 μm of border of the hilus in hippocampal slices (p = 0.105). The number of animals was 3 for WT and 6 for KO. Two slices were prepared from each animal, and the numbers of branches were measured in both hippocampi in every slice. Significance was assessed with a one-way ANOVA. n.s., Not significant. G, Immunohistochemical images of the hilus, granule cell layer, and molecular layer of WT and neuritin KO mice, fixed before and after kindling (average group). Anti-ZnT3 antibody was used to visualize axons of granule neurons. Arrowheads indicate mossy fiber sprouting. Scale bar, 50 μm.
Preparing brain slices.
The mice were anesthetized and perfused transcardially with 4% PFA in 0.1 m PB, pH 7.4. The brains were rapidly removed and postfixed for >24 h with 4% PFA and then cryoprotected in 20% sucrose PBS. The brains were frozen on the stage of a microtome (Microm). The sections were cut at 30 μm and preserved at 4°C with 0.02% sodium azide PBS.
Microscopy and image processing.
Images were captured with an AxioImager Z.1 (Zeiss) with the MosaiX option. For 3D images, stack images were acquired with the interval between images set to 0.48 μm. Stack images were maximally projected by the Fiji program. For reconstruction of neuronal images, projected neuronal processes were manually traced with a pencil tool in GIMP. For image processing, brightness and contrast were adjusted with GIMP.
Quantification and statistical analysis.
Quantification of axon length and primary branch length was performed with ImageJ with the NeuronJ plug-in. The longest neurite was defined as an axon, and the sum of the length of primary branches along the given axon (secondary and tertiary branches were excluded) divided by the number of primary branches was defined as the average primary branch length. The number of primary branches divided by axonal length was defined as the number of primary branches per unit length of axon (100 μm axon). Statistical significance was determined as described in the figure legends. Statistical analysis was performed with SPSS software (IBM). Data are presented as the mean ± SEM. When the program showed p = 0.000, it is shown as p < 0.001 in this manuscript.
Results
Neuritin promotes axonal branching in hippocampal granule neurons
To investigate the function of neuritin in axonal morphogenesis of granule cells in the hippocampal dentate gyrus, we overexpressed myc-tagged neuritin in granule cells. Single cells were prepared from the dentate gyrus of P3 rats and were electroporated with an EGFP expression plasmid to visualize the axonal elongation and branching. The granule cells were plated at low density on poly-l-lysine-coated coverslips and were cultured for 4 d. The longest neurite was evaluated as an axon. We measured the length of the axon, the number of branches per 100 μm of axon, and the average length of the branches of the axon. The branching of granule cell axons was promoted by the overexpression of neuritin (Fig. 1A,B), whereas the axonal length was not altered by neuritin expression (Fig. 1A,B). We measured the same parameters at 7 DIV and 10 DIV. Axonal branching was also promoted at both DIVs (Fig. 1B). Given previous reports that neuritin induces neurite elongation (Naeve et al., 1997; Fujino et al., 2008), axonal and dendrite branching (Nedivi et al., 1998; Javaherian and Cline, 2005), and neuritogenesis (Naeve et al., 1997; Sato et al., 2012), the effects of neuritin may vary across neuronal subtypes. The hippocampal granule cells may exhibit induced axonal branching but do not exhibit elongated axons in response to neuritin.
Figure 1.
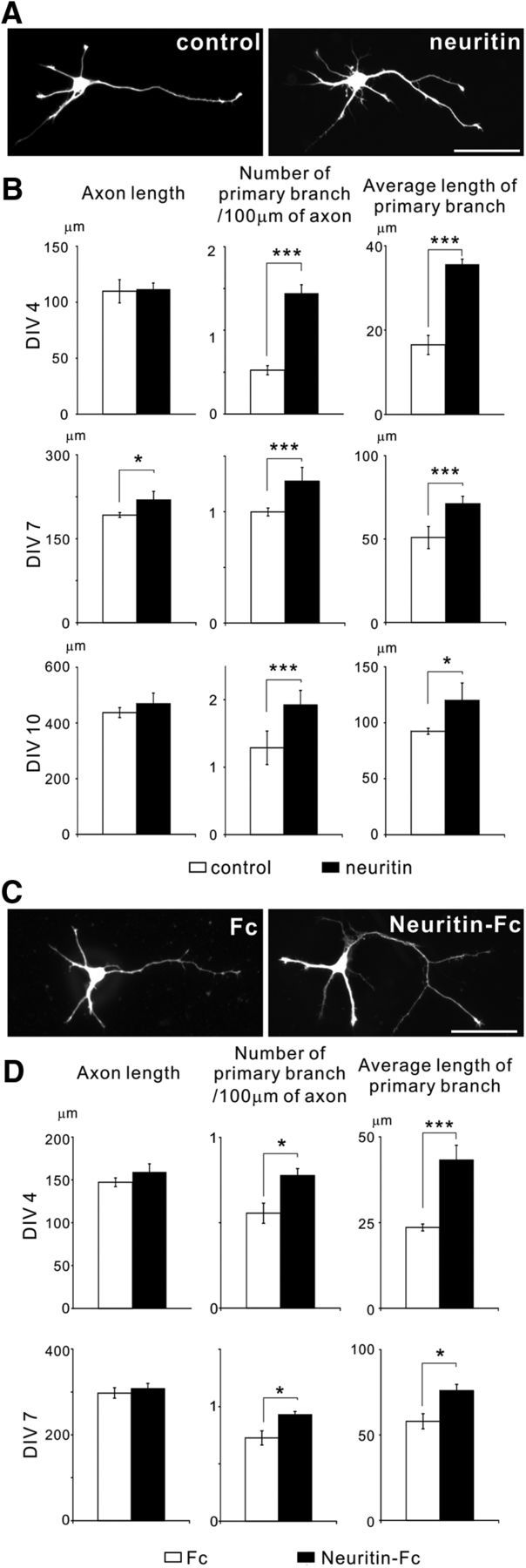
Overexpression and application of neuritin promotes axonal branching in cultured hippocampal granule cells. A, Overexpression of neuritin enhanced axonal branching. EGFP or EGFP and neuritin-transfected granule neurons were fixed and imaged at DIV 4. Representative neurons are shown. Scale bar, 50 μm. B, Quantification of axonal length (DIV 4: p = 0.736; DIV 7: p = 0.027; and DIV 10: p = 0.549), the number of primary branches per 100 μm of axon (DIV 4: p < 0.001; DIV 7: p = 0.002; and DIV 10: p = 0.003), and the average length of primary branches (DIV 4: p < 0.001; DIV 7: p < 0.001; and DIV 10: p = 0.012) from three independently performed experiments with n = 30 for each at DIV 4, 7, and 10, respectively. *p < 0.05 (one-way ANOVA). ***p < 0.005 (one-way ANOVA). C, Application of purified neuritin also enhanced axonal branching. EGFP-transfected granule neurons treated with Fc or Neuritin-Fc were fixed and imaged at DIV 4. Representative neurons are shown. Scale bar, 50 μm. D, Quantification of axonal length (DIV 4: p = 0.109; and DIV 7: p = 0.620), the number of primary branches per 100 μm of axon (DIV 4: p = 0.021; and DIV 7: p = 0.012), and the average length of primary branches (DIV 4: p < 0.001; and DIV 7: p = 0.026) from three independent experiments with n = 30 for each at DIV 4 and DIV 7, respectively. *p < 0.05 (one-way ANOVA). ***p < 0.005 (one-way ANOVA).
Neuritin is a GPI-anchored membrane protein and is solubilized by cleavage of the GPI anchor (Putz et al., 2005). The soluble form of neuritin is also functional (Putz et al., 2005; Fujino et al., 2008). To verify the possibility that soluble neuritin promotes axonal branching, we administered recombinant soluble neuritin to the culture medium of granule cells. The granule cells cultured with soluble neuritin also exhibited enhanced axonal branching at DIV 4, but control Fc protein did not promote axonal branch formation and elongation (Fig. 1C,D). Additionally, soluble neuritin did not alter the axonal length (Fig. 1C,D). Similar morphological changes were also observed at DIV 7 (Fig. 1D), indicating that neuritin promotes only axonal branch formation and elongation. Because the application of a soluble form of neuritin caused morphological effects on granule neurons comparable with those caused by the overexpression of neuritin, soluble neuritin may act as a ligand on the axonal plasma membrane to promote branching.
We then sought to investigate the branch-forming activity of neuritin in granule cells in hippocampal slice cultures. The brain slices were prepared from P7 rats. We transfected neuritin and EGFP expression plasmids into cultured slices at DIV 7 and fixed the slices at DIV 11. The overexpression of neuritin increased the number of branches and the average length of branches, whereas axonal length was not altered by neuritin expression (Fig. 2A,B). These results suggest that neuritin enhanced axonal branching, even when neurons were embedded in other brain cells.
Figure 2.

Neuritin promotes axonal branching of hippocampal granule neurons in organotypic slice cultures. A, Images and reconstructions of axons and dendrites of a granule neuron expressing EGFP or expressing EGFP and neuritin in hippocampal slice culture. Slices were fixed and imaged at DIV 11. Scale bar, 50 μm. B, Quantification of axonal length (p = 0.963), the number of primary branches per 100 μm of axon (p = 0.041), and the average length of primary branches (p = 0.005); n = 18 total for each, and the data were collected from three independent experiments. *p < 0.05 (one-way ANOVA). ***p < 0.005 (one-way ANOVA). C, Reconstructions of axons and dendrites of granule neurons expressing EGFP in hippocampal slice cultures treated with 5 μm of KA or the vehicle (PBS). Scale bar, 50 μm. D, Quantification of axonal length (p = 0.648), the number of primary branches per 100 μm of axon (p = 0.047), and the average length of primary branches (p = 0.007); n = 18 for each, and the data were collected from three independent experiments. *p < 0.05 (one-way ANOVA). ***p < 0.005 (one-way ANOVA).
Interestingly, the application of KA showed similar morphological changes to those resulting from neuritin overexpression. We transfected the EGFP expression plasmid at DIV 7 and treated slices with KA from DIV 8 to DIV 10. The slices were fixed at DIV 11, and an EGFP signal was observed. Treatment with KA also resulted in the promotion of axonal branching but did not alter axonal length (Fig. 2C,D).
Neuritin is required for activity-dependent axonal branching
Because neuritin is inducible in hippocampal neurons by KA treatment (Naeve et al., 1997), enhanced neuronal activity may induce neuritin and subsequent axonal branch elongation. In addition, neuritin KO mice show delayed axonal arbor development in retinal ganglion cells (Fujino et al., 2011), thus suggesting that neuritin may be necessary for activity-dependent axonal branch formation. To test this hypothesis, we prepared hippocampal granule neurons from WT and neuritin KO mice. We treated these granule neurons with KA from DIV 1 to DIV 4 and fixed them at DIV 4. The application of KA also promoted axonal branching in WT granule neurons but not in KO neurons. We did not observe any increase in the number of axonal branches or in the average axonal branch length in response to kainate. Interestingly, as also observed with neuritin overexpression or treatment, KA treatment did not increase axon length in either WT or KO neurons (Fig. 3A,B).
Figure 3.
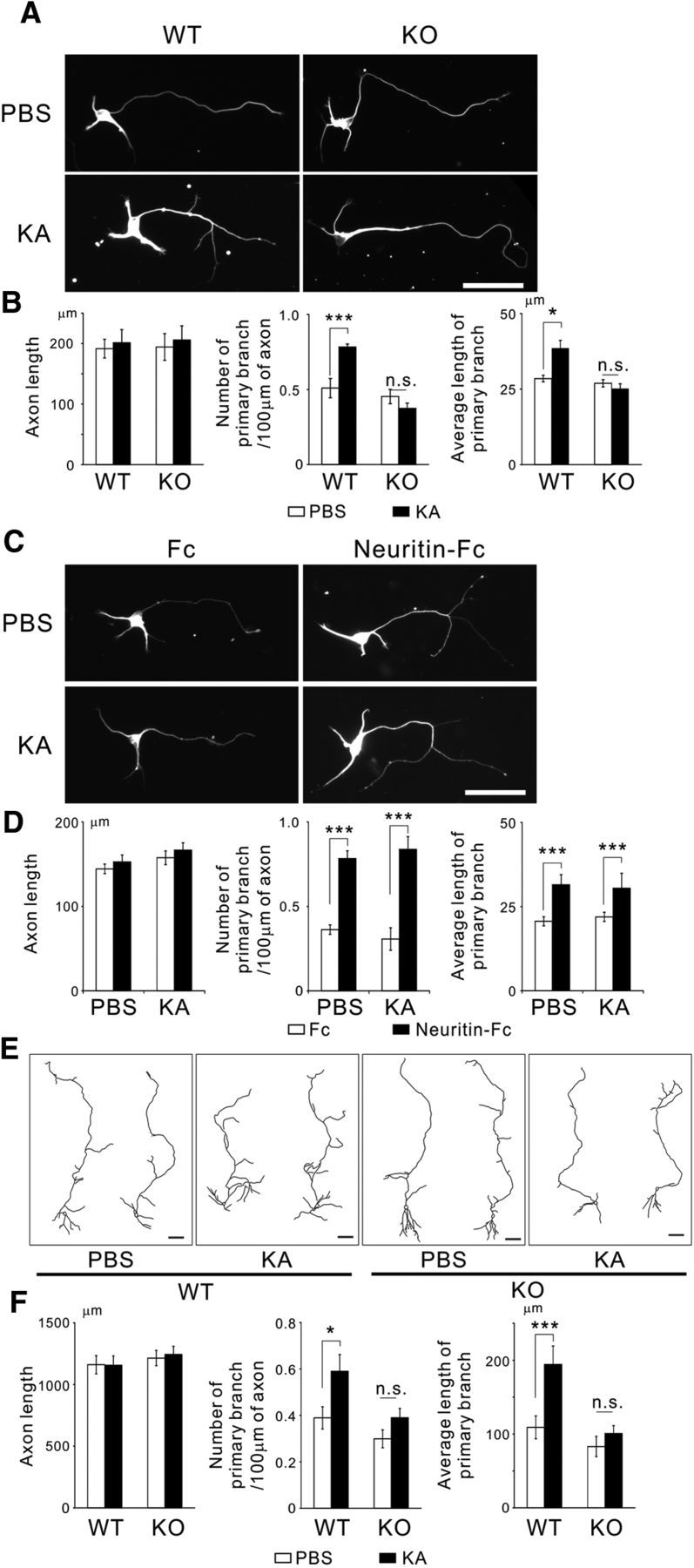
Deficiency of neuritin abolishes KA-dependent axonal branching in granule neurons. A, Neurons from neuritin KO mice did not show KA-dependent axonal branching. KA (10 μm) or the vehicle (PBS) was applied at DIV 1, and neurons were fixed at DIV 4. Representative neurons are shown. Scale bar, 50 μm. B, Quantification of axonal length (WT: p = 0.252; KO: p = 0.285), the number of primary branches per 100 μm of axon (WT: p = 0.001; KO: p = 0.338), and the average length of primary branches (WT: p = 0.014; KO: p = 0.812) from three independent experiments with n = 30 for each. *p < 0.05 (one-way ANOVA for each genotype). ***p < 0.005 (one-way ANOVA for each genotype). n.s., Not significant. C, Neuritin-Fc treatment promoted axonal branching in neuritin KO neurons. KA (10 μm) or the vehicle (PBS) and 1 μg/ml of Fc or Neuritin-Fc were applied at DIV 1, and neurons were fixed at DIV 4. Representative neurons are shown. Scale bar, 50 μm. D, Quantification of axonal length (PBS: p = 0.564; KA: p = 0.497), the number of primary branches per 100 μm of axon (PBS: p < 0.001; KA: p < 0.001), and the average length of primary branches (PBS: p < 0.001; KA: p < 0.001) from three independent experiments with n = 30 for each. ***p < 0.005 (one-way ANOVA for each treatment). n.s., Not significant. E, Granule neurons in hippocampal slice cultures from KO mice were not altered by KA treatment. Reconstructions of granule neurons expressing EGFP in hippocampal slice culture. KA (5 μm) treatment was performed from DIV 8 to DIV 10. Scale bar, 50 μm. F, Quantification of axonal length (WT: p = 0.955; KO: p = 0.808), the number of primary branches per 100 μm of axon (WT: p = 0.048; KO: p = 0.603), and the average length of primary branches (WT: p = 0.009; KO: p = 0.940); n = 20–25 total for each, and the data were collected from five independent experiments. *p < 0.05 (one-way ANOVA for each genotype). ***p < 0.005 (one-way ANOVA for each genotype). n.s., Not significant.
We performed rescue experiments to determine whether this abrogation of KA-dependent axonal branching would be recovered by treatment of recombinant neuritin. The administration of recombinant neuritin enhanced axonal branching even in neuritin KO neurons. KA treatment did not induce any additional enhancement of axonal branching (Fig. 3C,D). These results suggest that the protein level of neuritin is positively correlated with axonal branching and that KA may promote axonal branching through the induction of neuritin.
We then compared the granule neurons in organotypic slice cultures of hippocampi between WT and neuritin KO mice. EGFP-encoding plasmids were transfected into the slices at DIV 7. The slices were treated with KA from DIV 8 to DIV 10 and were fixed at DIV 11. Again, in contrast with the WT neurons, neuritin KO neurons did not show either KA-mediated axonal branch formation or branch elongation. Axonal elongation of the granule cells was not observed in either WT or KO slices (Fig. 3E,F). These results strongly suggest that neuritin is required for the activity-dependent axonal branch formation and elongation in hippocampal granule neurons.
Neuritin-mediated branch formation is involved in the aggravation of epileptic seizure
To evaluate the pathophysiological role of neuritin-mediated axonal branching in vivo, we constructed an epileptic seizure model using chemical kindling. Aberrant branch formation of granule cell axons is observed in the hippocampi of patients with temporal lobe epilepsy and in animal models of epilepsy. There is a leading hypothesis that the aberrant sprouting of mossy fibers exacerbates seizure severity in epilepsy. Epileptic seizure is caused by abnormal excessive or synchronous neuronal activity in the brain. Neural activity induces the expression of neuritin (Nedivi et al., 1993; Naeve et al., 1997). Moreover, our data suggest that upregulation of neuritin promotes axonal branching in granule cells. Thus, we investigated whether neuritin is involved in the exacerbation of epileptic seizures.
Injection of PTZ, an antagonist of GABA, induces epileptic seizures. Chronic administration of PTZ at a subconvulsive dose has been used to develop chemical kindling models in rodents. In this model, seizure severity is elevated along with the progression of PTZ injections. We injected PTZ at 35 mg/kg into WT or neuritin KO mice every other day and monitored their seizure scores for 30 min after injection (WT, n = 10 males; KO, n = 10 males). Neuritin KO mice showed significantly lower seizure scores than did WT mice over the course of 10 injections (Fig. 4A). This result suggests that neuritin-mediated axonal branching is involved in the aggravation of epilepsy. To clarify whether axonal branching is indeed observed after the kindling experiment, we performed immunohistochemical analyses of the mouse brain before and after kindling. To induce massive mossy fiber sprouting, we injected PTZ 25 times (WT, n = 19, 12 males and 7 females; KO, n = 12, 8 males and 4 females) and classified each genotype of animals by their maximum seizure score as follows: WT, severe group (maximum score = 6, n = 9 of 19); average group (maximum = 5 or 4, n = 5 of 19); and mild group (maximum 3 or less, n = 5 of 19); KO, severe group (maximum = 5, n = 6 of 12); average group (maximum = 4, n = 3 of 12); and mild group (maximum = 3 or less, n = 3 of 12). No KO animals died by seizure. Then, we stained the brain slices from animals in each average group with anti-synaptoporin antibody and anti-ZnT3 antibody. Synaptoporin and ZnT3 have been reported to be good markers for mossy fibers (Grabs et al., 1994; Wenzel et al., 1997). Aberrant synaptoporin-positive axonal branches were observed in the granule cell layer in WT mouse brains after kindling (Fig. 4B,C). Aberrant axonal branching was measured by counting the number of axons that penetrated into the granule cell layer (Fig. 4D). As shown in Figure 4E, the KO mice showed fewer axons penetrating into the dentate gyrus after kindling than did the WT mice. The numbers of sprouted axons were not different between these strains before kindling. In addition, ZnT3-positive axons also penetrated into the granule layer after kindling, and the WT mice showed more prominent axonal sprouting than did the KO mice (Fig. 4G). These results suggest that reduction in seizure-mediated axonal branching causes the slower development of seizure severity in neuritin KO mice.
Neuritin KO mice have been shown to exhibit delayed synapse formation, dendritic arborization, and axonal branching (Fujino et al., 2011). Thus, we investigated whether such axonal changes after kindling are due to a resistance to seizure activity in KO mice or a prevention of axon sprouting. We compared mossy fiber sprouting in the hippocampal slices of KO and WT mice, both showing equivalent degrees of seizure severity (WT, n = 3; KO, n = 6). The number of aberrant axonal branches was not significantly different between the WT and KO mice (Fig. 4F). These results also raise a possibility that neuritin KO mice may have a resistance to seizure, causing a slower development of mossy fiber sprouting after PTZ kindling.
FGFR is necessary for neuritin-dependent axonal branching
We next tried to clarify the downstream signaling of neuritin that induces axonal branching. Because neuritin is an extracellular or secreted protein, binding proteins could be candidates for the neuritin receptor. Furthermore, the GPI-anchored protein preferably interacts with tyrosine-kinase-coupled receptor proteins, including the glial cell line-derived neurotrophic factor receptor and C-Ret (Trupp et al., 1996), ciliary neurotrophic factor receptor and GP130 (Ip et al., 1992), and ephrin B and tyrosine receptor kinases (Marler et al., 2008). Among the tyrosine-kinase-coupled receptors, we focused on the FGFR and investigated whether FGFRs can interact with neuritin.
Four FGFRs have been identified to date (Szebenyi and Fallon, 1999), but only FGFRs 1–3 have been found in differentiated neurons (Itoh et al., 1994; Belluardo et al., 1997; Kalyani et al., 1999). We tested the interaction between neuritin and FGFRs. Immunoprecipitation analysis showed that FGFR1 was strongly associated with neuritin (Fig. 5A). FGFR3 showed weak binding, but FGFR2 failed to interact with neuritin (Fig. 5A), which indicated that neuritin can selectively associate with FGFR1.
Figure 5.

Neuritin causes axonal branch formation in granule cells through the activation of FGFRs. A, Immunoprecipitation analysis showed the interaction between neuritin and FGFR1. The 293T cells expressing HA-tagged FGFR and GFP-tagged neuritin were lysed, and subsequent immunoprecipitation of the lysates was performed with anti-HA antibody. FGFR1 showed an interaction with neuritin. B, PD173074 (50 nm) or AZD4547 (100 nm), FGFR inhibitors, was applied to rat granule neurons expressing EGFP or expressing EGFP and neuritin at DIV 1. Neurons were fixed and imaged at DIV 4. Representative neurons are shown. Scale bar, 50 μm. C, Quantification of the number of primary branches per 100 μm of axon (DMSO: p < 0.001; PD173074: p = 0.065; and AZD4547: p = 0.751) and the average length of primary branches (DMSO: p < 0.001; PD173074: p = 0.621; and AZD4547: p = 0.843) from three or four independent experiments; n = 30 for each. ***p < 0.005 (one-way ANOVA for each reagent). n.s., Not significant. D, Reconstructions of EGFP-expressing neurons or EGFP and neuritin-expressing neurons in rat hippocampal slice cultures. PD 173074 (100 nm) or AZD4547 (200 nm) was administered from DIV 8 to DIV 11, and slices were fixed at DIV 11. Scale bar, 50 μm. E, Quantification of the number of primary branches per 100 μm of axon (DMSO: p = 0.034; PD173074: p = 0.116; and AZD4547: p = 0.198) and the average length of primary branches (DMSO: p < 0.001; PD173074: p = 0.526; and AZD4547: p = 0.140); n = 19–21 total for each; the data were collected from three to five independent experiments. *p < 0.05 (one-way ANOVA for each reagent). ***p < 0.005 (one-way ANOVA for each reagent). n.s., Not significant. F, Validation of Stealth RNAi against rat FGFR1. The 293T cells were transfected with control RNA or Stealth RNAi against rat FGFR1 and a mock vector, HA-FGFR1, or HA-FGFR1* (RNAi resistant mutant), as indicated in the figure. Cells were lysed 36 h later, and immunoblotting was performed with anti-HA and anti-tubulin antibody. G, Knockdown of FGFR1 expression suppressed neuritin-dependent axonal branching. Rat granule neurons were transfected with EGFP-expressing plasmids, control RNA or Stealth RNAi against rat FGFR1, and FGFR1 or FGFR1*-expressing plasmids before being plated. Fc or Neuritin-Fc (1 μg/ml) was administered at DIV 1, and neurons were fixed at DIV 4. Representative neurons with Neuritin-Fc are shown. Scale bar, 50 μm. H, Quantification of the number of primary branches per 100 μm of axon (control RNA: p < 0.001; RNAi: p = 0.787; RNAi+FGFR1: p = 0.650; and RNAi+FGFR1*: p < 0.001) and the average length of primary branches (control RNA: p < 0.001; RNAi: p = 0.846; RNAi+FGFR1: p = 0.941; and RNAi+FGFR1*: p < 0.001) from three independent experiments with n = 30 for each. ***p < 0.005 (one-way ANOVA for each set of nucleotides). n.s., Not significant.
Next, we tested whether the interaction between neuritin and FGFR activates FGFR signaling to promote axonal branching. The cultured granule neurons were treated with the potent FGFR inhibitors PD173074 or AZD4547. These treatments completely abolished axonal branching via the overexpression of neuritin at DIV 4 (Fig. 5B,C).
We also tested the requirement of FGFR for neuritin-mediated branching in slice cultures. We prepared organotypic hippocampal slice cultures from P7 rats, transfected neuritin-encoding plasmids or control EGFP-encoding plasmids at DIV 7, and treated the slices with PD173074 or AZD4547 from DIV 8 to DIV 10. The inhibition of FGFR again abolished neuritin-dependent axonal branch formation and elongation in slice culture (Fig. 5D,E).
To further confirm that FGFR1 is involved in neuritin-dependent axonal branching, we transfected siRNA against rat FGFR1 into the cultured granule neurons and quantified axonal branching. We validated the efficiency of siRNA by immunoblotting (Fig. 5F) and immunocytochemistry (data not shown), which showed that FGFR1 protein was significantly decreased by siRNA, and RNAi-refractory FGFR1 (FGFR1*) was not affected by RNAi treatment. We administered recombinant soluble neuritin to the siRNA-transfected granule neurons. FGFR1 knockdown markedly suppressed neuritin-dependent axonal branching, and additional transfection of FGFR1* recovered the axonal branching (Fig. 5G,H). Together, these results suggest that the interaction between neuritin and FGFR1 may activate FGFR1 and its downstream signaling, which is required for neuritin-mediated axonal branching.
Canonical downstream signals of FGFR1 are required for neuritin-dependent axonal branching
We further analyzed which downstream signals of FGFR1 are involved in neuritin-dependent axonal branching. We pharmacologically inhibited representative downstream signaling cascades, including the mitogen-activated protein kinase (MAPK) cascade, phosphatidylinositol-3 kinase (PI3K)/Akt, and phospholipase C (PLC) (Katz et al., 2007). Unexpectedly, all inhibitors affected neuritin-mediated axonal branch formation and/or elongation (Fig. 6A,B). U0126, an inhibitor of MAPK cascade protein MEK1/2, abolished the increase in branch number and branch elongation; however, it did not alter the basal branching ability (Fig. 6B). LY294002, an inhibitor of PI3K, diminished basal branching activity and suppressed neuritin-mediated enhancement of axonal branching (Fig. 6B). Interestingly, U73122, an inhibitor of PLC, did not affect the increase in branch number but abolished the branch elongation (Fig. 6B). Recent studies have shown that the inhibition of PLC activity attenuates growth factor-dependent-axonal elongation (Xie et al., 2006; Hashimoto and Ishima, 2011), suggesting that PLC activity is not involved in the formation of axonal branches but is involved in the elongation of axonal branches. These results suggest that neuritin activates three signaling cascades, which promote axonal branch formation and/or elongation in various regulatory systems of axonal branching.
Figure 6.

Downstream signaling of FGFR1 is required for neuritin-mediated axonal branching. A, U0126 (10 μm), a MEK1/2 inhibitor, LY294002 (20 μm), a PI3K inhibitor, or U73122 (2 μm), a PLC inhibitor, was applied to rat granule neurons expressing EGFP or expressing EGFP and neuritin at DIV 1. Neurons were fixed and imaged at DIV 4. Representative neurons expressing neuritin are shown. Scale bar, 50 μm. B, Quantification of the number of primary branches per 100 μm of axon (control vs neuritin, DMSO: p < 0.001; U0126: p = 0.222; LY294002: p = 0.717; and U73122: p = 0.001; DMSO vs each reagent in control, U0126: p = 0.693; LY294002: p = 0.016; and U73122: p = 0.562) and the average length of primary branches (control vs neuritin, DMSO: p < 0.001; U0126: p = 0.411; LY294002: p = 0.395; and U73122: p = 0.099; DMSO vs each reagent in control, U0126: p = 0.377; LY294002: p = 0.002; and U73122: p = 0.881) from three or four independent experiments with n = 30 for each. Control versus neuritin: ***p < 0.005 (one-way ANOVA for each reagent). n.s., Not significant. DMSO versus each reagent: #p < 0.05 (a one-way ANOVA followed by Tukey's post-test for the control). ###p < 0.005 (a one-way ANOVA followed by Tukey's post-test for the control). N, Not significant, compared with DMSO.
Determination of the neuritin-interacting domain of FGFR1
We next designed a deletion mutant of FGFR1 to specify the domain that is necessary for interacting with neuritin. FGFRs contain three Ig-like domains, D1, D2, and D3 (Fig. 7A). Deletion of either D1 and D2 or D3 in FGFR1 did not disturb the interaction between FGFR and neuritin. However, deletion of both D2 and D3 abolished the binding ability of FGFR1 with neuritin (Fig. 7B). Crystal structure analysis has revealed that FGF-2 binds the ligand-binding domain (D2 and D3) of FGFR1 (Fig. 7A) (Plotnikov et al., 1999). Both neuritin and FGF bind to D2–D3 of FGFR1, thereby raising the possibility that neuritin and FGFs may cooperate in the activation of FGFR1 signalings and that those FGFs are involved in neuritin-mediated axonal branching.
Figure 7.

The FGF-binding domain in FGFRs also interacts with neuritin, and FGFs promote axonal branching. A, A schematic illustration of the domain structure of FGFR1 and its FGF binding site. D1–D3 indicate Ig-like domains 1–3, respectively. B, Schematic illustrations of the domain structure of full-length FGFR and its truncated mutants. The immunoprecipitation analysis results are shown below the illustrations. The mutant lacking both D2 and D3 did not bind to neuritin, whereas the mutants lacking either D2 or D3 were able to interact with neuritin. C, FGF-4, FGF-5, FGF-7, and FGF-8 promoted axonal branching in rat granule neurons. Neurons were cultured with 2 nm recombinant FGF from DIV 2 to DIV 4. Representative neurons are shown. Scale bar, 50 μm. D, Quantification of the number of primary branches per 100 μm of axon (FGF-1: p = 1.000; FGF-2: p = 0.063; FGF-4: p < 0.001; FGF-5: p < 0.001; FGF-7: p = 0.006; FGF-8: p = 0.007; and FGF-9: p = 1.000) and the average length of primary branches (FGF-1: p = 1.000; FGF-2: p = 0.001; FGF-4: p < 0.001; FGF-5: p < 0.001; FGF-7: p = 0.141; FGF-8: p < 0.001; and FGF-9: p = 1.000) from four independently performed experiments with n = 30 for each. **p < 0.01, compared with the control (one-way ANOVA followed by Tukey's post-test). ***p < 0.005, compared with the control (one-way ANOVA followed by Tukey's post-test). E, Evolutionary relationships within the human/mouse FGF gene family by phylogenetic analysis, modified from the picture by Itoh and Ornitz (2011). FGF-4 and FGF-5 belong to the same subfamily.
The FGF-4, FGF-7, and FGF-8 subgroups are involved in the axonal branching of granule neurons
There are 22 FGF subtypes in mammals, and they are divided into 7 subfamilies (Fig. 7E). We chose FGFs that represent these subfamilies, except the FGF-11/12/13/14 and FGF-19/21/23 subfamilies, because FGF-11/12/13/14 are not secreted and FGF-19/21/23 show weak FGFR signaling activation, at a level <1% of that in other paracrine subfamilies (Zhang et al., 2006). We added each FGF to cultured granule neurons. Interestingly, FGF-4, FGF-5, FGF-7, and FGF-8 promoted axonal branch formation in dissociated granule neurons (Fig. 7C,D). Surprisingly, FGF-2 had an inhibitory effect on axonal branching in the cultured granule neurons (Fig. 7D). FGF-2 has been reported to promote branching in various types of neurons (Szebenyi et al., 2001; Klimaschewski et al., 2004). However, recent reports showed that overexpression of FGF-2 or cell grafts enriched with FGF-2 suppressed mossy fiber sprouting in vivo (Rao et al., 2006; Paradiso et al., 2011). These findings indicate that the granule neurons may exhibit distinct responses to FGF-2, which downregulates mossy fiber branching. Overall, the FGF-4, FGF-7, and FGF-8 subfamilies promoted axonal branching in the cultured granule neurons, whereas FGF-2 decreased the branching ability.
Neuritin facilitates FGF-4-mediated recruitment of FGFR1 to the cell surface
Because both neuritin and FGFs interact with D2–D3 of FGFR1 and the inhibition of FGFR1 abolished neuritin-mediated axonal branching, we hypothesized that FGF signaling is involved in neuritin-mediated axonal branching in granule neurons. In addition, because the FGF-4, FGF-7, and FGF-8 subfamilies enhanced axonal branching in the granule neurons, the FGF signaling of these three subfamilies should be regulated by neuritin. To verify this possibility, we first investigated which FGF subfamily is involved in neuritin-mediated axonal branching. Granule cells from neuritin KO mice were cultured with the above FGFs. Interestingly, FGF-4 did not promote axonal branching in neuritin-deficient granule cells, whereas FGF-7 and FGF-8 showed branching effects, even in neuritin-deficient cells and WT neurons (Fig. 8A,B). These results indicate that neuritin is required for FGF-4-mediated axonal branch formation and elongation but that the FGF-7 and FGF-8 subfamilies promote axonal branching in a neuritin-independent manner.
Figure 8.
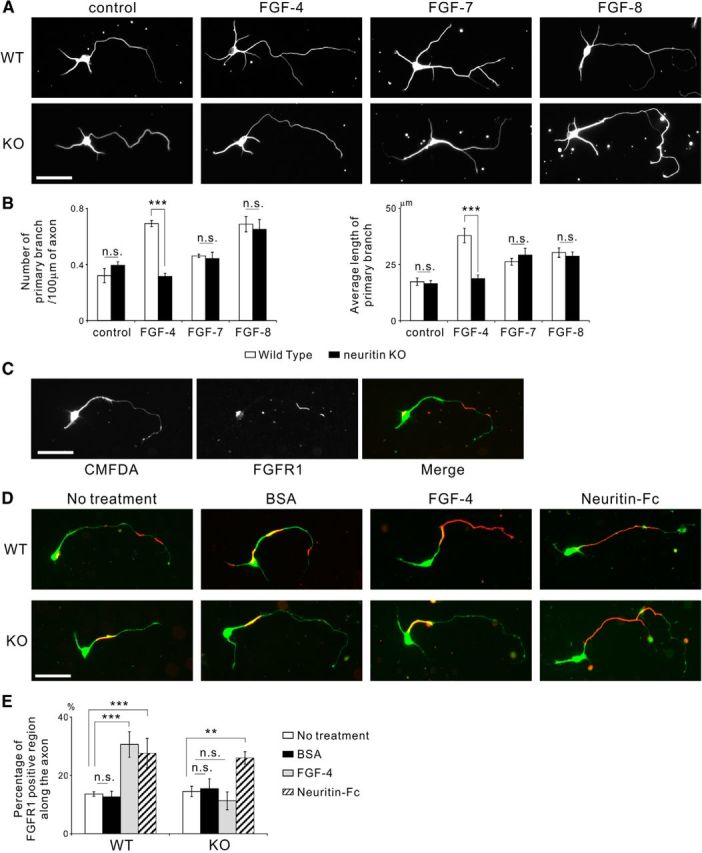
The FGF-4/5/6 subfamily is involved in neuritin-mediated axonal branching. In addition, neuritin and FGF-4/5/6 aid the recruitment of FGFR1 to the cell surface. A, FGF-4 failed to promote axonal branching in neurons from neuritin KO mice, whereas FGF-7 and FGF-8 had a branching effect on neurons from both WT and KO mice. Neurons were cultured with 2 nm recombinant FGF from DIV 2 to DIV 4. Representative neurons are shown. Scale bar, 50 μm. B, Quantification of the number of primary branches per 100 μm of axon (control: p = 0.307; FGF-4: p < 0.001; FGF-7: p = 0.818; and FGF-8: p = 0.704) and the average length of primary branches (control: p = 0.801; FGF-4: p < 0.001; FGF-7: p = 0.578; and FGF-8: p = 0.661) from three independently performed experiments; n = 30 for each. ***p < 0.005 (one-way ANOVA for each FGF). n.s., Not significant. C, FGFR1 localized to parts of the cell surface along the neurite and on the cell body. The cultured mouse granule neurons were treated with 30 μm CMFDA for 3 h before fixation at DIV 4 and were immunostained without permeabilization. CMFDA treatment visualized the whole-cell body and neurites. FGFR1 unevenly localized on the cell surface. Scale bar, 50 μm. D, FGF-4 failed to recruit FGFR1 to the axonal surface in the neurons from neuritin KO mice, whereas Neuritin-Fc increased cell surface FGFR1 along the axon in the neurons from both WT and KO mice. Neurons were cultured with 30 μm CMFDA for 3 h and 2 nm of FGF-4 or 1 μg/ml of Neuritin-Fc for 15 min before fixation at DIV 4; they were immunostained without permeabilization. Representative neurons with CMFDA (green) and cell surface FGFR1 (red) are shown. Scale bar, 50 μm. E, Quantification of the ratio of FGFR1-positive region along the axonal surface (no treatment vs each treatment, in WT, BSA: p = 0.995; FGF-4: p < 0.001; and Neuritin-Fc: p = 0.004; in KO, BSA: p = 0.993; FGF-4: p = 0.815; and Neuritin-Fc: p = 0.010). **p < 0.01, compared with no treatment (one-way ANOVA for each genotype followed by Tukey's post-test). ***p < 0.005, compared with no treatment (one-way ANOVA for each genotype followed by Tukey's post-test).
To investigate the functional role of neuritin in FGF signaling, we observed the cell surface FGFR1 in the cultured granule neurons after application of FGF-4 and recombinant neuritin. Immunostaining without permeabilization showed that FGFR1 localized on parts of the axon and the cell body surface of the cultured granule neurons (Fig. 8C). We found that the surface FGFR1 immunoreactivity was greatly increased on the axon after the administration of FGF-4 or recombinant neuritin to the WT neurons (Fig. 8D,E), suggesting that both FGF-4 and neuritin cooperatively recruit FGFR1 to the surface of granule neurons. Indeed, the absence of neuritin abolished the FGF-4-mediated recruitment of FGFR1 to the axonal surface, and the application of recombinant neuritin increased the cell surface FGFR1 (Fig. 8D,E). These results suggest that neuritin is required for FGF-4-dependent recruitment of FGFR1 to the cell surface and that excess amounts of neuritin translocate FGFR1 to the cell surface without FGF-4. The inhibition of FGFR1 endocytosis has been shown to promote FGF-dependent axonal branching (Hausott et al., 2011). Thus, FGF-4- and neuritin-mediated recruitment of FGFR1 to the axonal surface would induce axonal branch formation and elongation in granule neurons. Furthermore, a deficiency of neuritin abolished the FGF-4-dependent FGFR1 recruitment to the cell surface, resulting in a reduction in FGF-4-mediated axonal branching.
Discussion
The present findings demonstrate that the activity-dependent protein neuritin promotes axonal branching in granule neurons. Overexpression and administration of neuritin enhanced axonal branching in cultured hippocampal granule cells. KA treatment caused a similar promotion of axonal branching in WT granule cells; however, neuritin-deficient granule neurons did not show KA-dependent axonal branch formation and elongation. Pharmacological inhibition or knockdown of FGFR1 abolished neuritin-mediated axonal branching, suggesting that neuritin induces axonal branching through the activation of FGFR. In addition, neuritin is required for FGF-mediated recruitment of FGFR1 to the axonal surface. This recruitment of FGFR1 may be involved in axonal branching. Finally, neuritin KO mice exhibited slower chemical kindling development by PTZ than did WT mice. These results suggest that neuritin promotes axonal branching in hippocampal granule cells and that the activity-dependent induction of neuritin may cause the aggravation of epileptic seizures.
Interacting partner of neuritin
Previous studies have shown that neuritin promotes neuritogenesis (Naeve et al., 1997; Sato et al., 2012), neurite elongation (Naeve et al., 1997; Fujino et al., 2008), and neurite arborization (Nedivi et al., 1998; Javaherian and Cline, 2005) both in cultured neurons and in vivo. Analysis of neuritin KO mice has also shown that a deficiency of neuritin delays axonal and dendritic arbor development (Fujino et al., 2011). However, the molecular mechanism of neuritin in regulating neural morphology remains elusive. A recent study showed that neuritin activated insulin-like growth factor receptors (IGFRs) in mouse cerebellar granule neurons (Yao et al., 2012). Although the interaction between neuritin and the receptor protein was not examined in that study, an inhibitory reagent against IGFRs suppressed neuritin function (Yao et al., 2012). FGFRs and IGFRs are both tyrosine-kinase-coupled receptors. Therefore, neuritin may interact with both receptors. Neuritin regulates postsynaptic potassium channel insertion into dendrites through IGFRs (Yao et al., 2012). Moreover, neuritin KO mice show delayed synaptic maturation and decreased spine stabilization (Fujino et al., 2011); therefore, neuritin may play a crucial role in postsynaptic development through IGFRs. In contrast, our findings indicate that neuritin affects axonal morphology through FGFRs. These data suggest that an interaction partner of neuritin may vary by cellular location. IGFRs act along the dendrites, whereas FGFRs are required for axonal morphogenesis. Another possibility could be that neuritin uses different receptor proteins in a cell type-specific manner. That is, neuritin might act on cerebellar and hippocampal neurons via IGFRs and FGFRs, respectively.
Neuritin facilitates the FGF-mediated recruitment of FGFRs to the cell surface, thus promoting axonal branching
We found that neuritin strongly bound to FGFR1 and that FGF-4 did not enhance axonal branching in neuritin-deficient neurons. We also found that neuritin and FGF bound to the common domain of FGFRs, D2–D3, and that an absence of neuritin suppressed the FGF-4-mediated FGFR1 translocation to the cell surface along the axon. A recent study has revealed that an inhibition of FGFR1 endocytosis promotes FGF-dependent axonal branching (Hausott et al., 2011), indicating that an increase in surface FGFR1 enhances axonal branching. These findings suggest that FGF-4 and neuritin cooperate to recruit FGFR1 to the surface of axons, resulting in activation of FGFR1 signaling, thus promoting axonal branching. Our findings also suggest that excess amounts of neuritin recruit FGFR1 to the cell surface without FGF treatment, indicating that neuritin promotes axonal branching by activating FGF signaling without FGF. However, we cannot exclude the possibility that a basal level of FGF may be sufficient to enhance axonal branching with overexpression of neuritin. Another study showed that FGF-5 and FGFR1 were also upregulated in the hippocampus by neural activity (Gómez-Pinilla et al., 1995), as was neuritin. This finding raises the possibility that the FGF-4/5/6 subgroup and neuritin are also upregulated by seizure and that they synergize, together promoting activity-dependent axonal branch formation through the recruitment of FGFR1 to the axonal surface.
Further downstream signaling
Pharmacological inhibition and knockdown of FGFRs abolished neuritin-mediated axonal branching. We investigated whether three canonical downstream signaling pathways of FGFR1, namely, PI3K-Akt (Eswarakumar et al., 2005), MAPK (Simon, 2000), and PLC signaling, are involved in neuritin-dependent axonal branching. Direct activation of PI3K elicits the formation of axonal filopodia and branches in sensory neurons (Ketschek and Gallo, 2010). Akt is a major downstream effector of PI3K signaling. Expression of constitutively active Akt promotes branching of sensory axons (Markus et al., 2002; Grider et al., 2009). Thus, the activation of PI3K and Akt along axons is one of the earliest steps in axonal branch formation (Gallo and Letourneau, 1998; Gallo, 2011). Our findings showed that inhibition of PI3K resulted in a suppression of basal and neuritin-mediated axonal branching. This indicates that PI3K signaling has a pivotal role in the formation and elongation of axonal branches.
MAPK signaling inhibits the activity of the actin-capping protein Eps8 (Menna et al., 2009). In hippocampal neurons, KO of Eps8 results in increased numbers of axonal filopodia (Menna et al., 2009), which suggests that Eps8 is involved in the suppression of protrusive activity along the axon. Thus, MAPK signaling may regulate the emergence of filopodia from actin patches through the regulation of Eps8. Axonal branches may be initiated as axonal filopodial protrusions (Bastmeyer and O'Leary, 1996; Gallo and Letourneau, 1998, 1999; Portera-Cailliau et al., 2005; Cohen-Cory et al., 2010). Our findings revealed that inhibition of the MAPK cascade abolished neuritin-mediated axonal branching but did not affect basal branching ability. This result suggests that the MAPK cascade is involved in extracellular signaling-dependent axonal branch formation and elongation.
PLC activity has been shown to promote the dynamics of growth cone and axonal elongation (Zhang et al., 2012). Activated PLC cleaves phosphatidylinositol 4,5-bisphosphate into diacylglycerol and inositol 1,4,5-trisphosphate, and inositol 1,4,5-trisphosphate stimulates the release of Ca2+ from internal stores (Rebecchi and Pentyala, 2000). Calcineurin is enriched in growth cones and has been implicated in Ca2+-dependent regulation of neurite extension and filopodium dynamics (Arie et al., 2009). Inhibiting PLC activity has been shown to suppress NGF- or Netrin-mediated axonal elongation (Xie et al., 2006; Hashimoto and Ishima, 2011). Thus, PLC activity would not control branch formation but would regulate branch elongation. These results are consistent with our finding that pharmacological inhibition of PLC did not alter neuritin-mediated branch formation but suppressed the extension of axonal branches.
Neuritin-mediated axonal branching in the aggravation of epilepsy
Mossy fiber sprouting is one of the pathological features of temporal lobe epilepsy. Although there is still some controversy, the “mossy fiber sprouting hypothesis” may explain the physiological role of this abnormal axonal branching in granule cells in the hippocampus. Aberrant axonal branches enter the dentate gyrus molecular layer and can form recurrent circuits, thus promoting epileptogenesis. Our data suggest that neuritin is a good candidate to promote mossy fiber sprouting in epileptic seizure-mediated conditions. As noted above, neuritin may be involved in the activity-dependent axonal branching of granule cells in vitro. Moreover, neuritin KO mice showed slower increases in seizure severity in chemical kindling and decreases in mossy fiber sprouting after kindling. These results positively support the mossy fiber sprouting hypothesis because the inhibition of mossy fiber sprouting mitigated epileptic seizures induced by chemical kindling.
However, we cannot exclude the possibility that mossy fiber sprouting has an inhibitory effect on epileptogenesis. Deficiency of neuritin might abrogate PTZ-induced seizure in an axonal branching-independent mechanism, and mild seizures in neuritin KO mice may result in the slower development of mossy fiber sprouting. As another mechanism, neuritin KO mice may have a resistance to seizure activity. That is, KO mice may have revealed less severe seizures by the resistance to seizure activity, resulting in poor induction of mossy fiber sprouting. Neuritin KO mice have been shown to exhibit delayed synapse formation, dendrite arborization, and axonal branching (Fujino et al., 2011). Such delayed development may result in an inefficient formation of neuronal circuits and may fail to propagate seizure activity. Further analyses are required to determine the causal relationship between mossy fiber sprouting and the exacerbation of epilepsy.
In conclusion, this study suggests that neuritin mediates activity-dependent axonal branching in granule cells in the hippocampus through the activation of FGFR signaling in vitro. Moreover, neuritin is involved in mossy fiber sprouting and in the aggravation of epilepsy in vivo. A pharmacological approach to inhibit neuritin/FGFR signaling may shed new light on the development of new antiepileptic drugs.
Footnotes
This work was supported in part by Japan Society for the Promotion of Science KAKENHI Grants 24700349, 24659093, and 25293239 and MEXT KAKENHI Grants 25110737 and 23110525. We thank Dr. Yasuo Ohashi (Chairman of Board of Directors, Statcom Co., Ltd.) for expert advice in statistics.
The authors declare no competing financial interests.
References
- Amiri A, Cho W, Zhou J, Birnbaum SG, Sinton CM, McKay RM, Parada LF. Pten deletion in adult hippocampal neural stem/progenitor cells causes cellular abnormalities and alters neurogenesis. J Neurosci. 2012;32:5880–5890. doi: 10.1523/JNEUROSCI.5462-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arie Y, Iketani M, Takamatsu K, Mikoshiba K, Goshima Y, Takei K. Developmental changes in the regulation of calcium-dependent neurite outgrowth. Biochem Biophys Res Commun. 2009;379:11–15. doi: 10.1016/j.bbrc.2008.11.128. [DOI] [PubMed] [Google Scholar]
- Arzimanoglou A, Hirsch E, Nehlig A, Castelnau P, Gressens P, Pereira de Vasconcelos A. Epilepsy and neuroprotection: an illustrated review. Epileptic Disord. 2002;4:173–182. [PubMed] [Google Scholar]
- Baba A, Yasui T, Fujisawa S, Yamada RX, Yamada MK, Nishiyama N, Matsuki N, Ikegaya Y. Activity-evoked capacitative Ca2+ entry: implications in synaptic plasticity. J Neurosci. 2003;23:7737–7741. doi: 10.1523/JNEUROSCI.23-21-07737.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastmeyer M, O'Leary DD. Dynamics of target recognition by interstitial axon branching along developing cortical axons. J Neurosci. 1996;16:1450–1459. doi: 10.1523/JNEUROSCI.16-04-01450.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bausch SB, McNamara JO. Contributions of mossy fiber and CA1 pyramidal cell sprouting to dentate granule cell hyperexcitability in kainic acid-treated hippocampal slice cultures. J Neurophysiol. 2004;92:3582–3595. doi: 10.1152/jn.01028.2003. [DOI] [PubMed] [Google Scholar]
- Belluardo N, Wu G, Mudo G, Hansson AC, Pettersson R, Fuxe K. Comparative localization of fibroblast growth factor receptor-1, -2, and -3 mRNAs in the rat brain: in situ hybridization analysis. J Comp Neurol. 1997;379:226–246. doi: 10.1002/(SICI)1096-9861(19970310)379:2<226::AID-CNE5>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- Cohen-Cory S, Kidane AH, Shirkey NJ, Marshak S. Brain-derived neurotrophic factor and the development of structural neuronal connectivity. Dev Neurobiol. 2010;70:271–288. doi: 10.1002/dneu.20774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devinsky O. Effects of seizures on autonomic and cardiovascular function. Epilepsy Curr. 2004;4:43–46. doi: 10.1111/j.1535-7597.2004.42001.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eswarakumar VP, Lax I, Schlessinger J. Cellular signaling by fibroblast growth factor receptors. Cytokine Growth Factor Rev. 2005;16:139–149. doi: 10.1016/j.cytogfr.2005.01.001. [DOI] [PubMed] [Google Scholar]
- Fujino T, Wu Z, Lin WC, Phillips MA, Nedivi E. cpg15 and cpg15-2 constitute a family of activity-regulated ligands expressed differentially in the nervous system to promote neurite growth and neuronal survival. J Comp Neurol. 2008;507:1831–1845. doi: 10.1002/cne.21649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujino T, Leslie JH, Eavri R, Chen JL, Lin WC, Flanders GH, Borok E, Horvath TL, Nedivi E. CPG15 regulates synapse stability in the developing and adult brain. Genes Dev. 2011;25:2674–2685. doi: 10.1101/gad.176172.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallo G. The cytoskeletal and signaling mechanisms of axon collateral branching. Dev Neurobiol. 2011;71:201–220. doi: 10.1002/dneu.20852. [DOI] [PubMed] [Google Scholar]
- Gallo G, Letourneau PC. Localized sources of neurotrophins initiate axon collateral sprouting. J Neurosci. 1998;18:5403–5414. doi: 10.1523/JNEUROSCI.18-14-05403.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallo G, Letourneau PC. Different contributions of microtubule dynamics and transport to the growth of axons and collateral sprouts. J Neurosci. 1999;19:3860–3873. doi: 10.1523/JNEUROSCI.19-10-03860.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gómez-Pinilla F, van der Wal EA, Cotman CW. Possible coordinated gene expressions for FGF receptor, FGF-5, and FGF-2 following seizures. Exp Neurol. 1995;133:164–174. doi: 10.1006/exnr.1995.1019. [DOI] [PubMed] [Google Scholar]
- Grabs D, Bergmann M, Schuster T, Fox PA, Brich M, Gratz M. Differential expression of synaptophysin and synaptoporin during pre- and postnatal development of the rat hippocampal network. Eur J Neurosci. 1994;6:1765–1771. doi: 10.1111/j.1460-9568.1994.tb00569.x. [DOI] [PubMed] [Google Scholar]
- Grider MH, Park D, Spencer DM, Shine HD. Lipid raft-targeted Akt promotes axonal branching and growth cone expansion via mTOR and Rac1, respectively. J Neurosci Res. 2009;87:3033–3042. doi: 10.1002/jnr.22140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto K, Ishima T. Neurite outgrowth mediated by translation elongation factor eEF1A1: a target for antiplatelet agent cilostazol. PLoS One. 2011;6:e17431. doi: 10.1371/journal.pone.0017431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hausott B, Rietzler A, Vallant N, Auer M, Haller I, Perkhofer S, Klimaschewski L. Inhibition of fibroblast growth factor receptor 1 endocytosis promotes axonal branching of adult sensory neurons. Neuroscience. 2011;188:13–22. doi: 10.1016/j.neuroscience.2011.04.064. [DOI] [PubMed] [Google Scholar]
- Hayashi Y, Shi SH, Esteban JA, Piccini A, Poncer JC, Malinow R. Driving AMPA receptors into synapses by LTP and CaMKII: requirement for GluR1 and PDZ domain interaction. Science. 2000;287:2262–2267. doi: 10.1126/science.287.5461.2262. [DOI] [PubMed] [Google Scholar]
- Houser CR. Granule cell dispersion in the dentate gyrus of humans with temporal lobe epilepsy. Brain Res. 1990;535:195–204. doi: 10.1016/0006-8993(90)91601-C. [DOI] [PubMed] [Google Scholar]
- Houser CR, Zhang N, Peng Z, Huang CS, Cetina Y. Neuroanatomical clues to altered neuronal activity in epilepsy: from ultrastructure to signaling pathways of dentate granule cells. Epilepsia. 2012;53(Suppl 1):67–77. doi: 10.1111/j.1528-1167.2012.03477.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ip NY, Nye SH, Boulton TG, Davis S, Taga T, Li Y, Birren SJ, Yasukawa K, Kishimoto T, Anderson DJ. CNTF and LIF act on neuronal cells via shared signaling pathways that involve the IL-6 signal transducing receptor component gp130. Cell. 1992;69:1121–1132. doi: 10.1016/0092-8674(92)90634-O. [DOI] [PubMed] [Google Scholar]
- Itoh K, Watanabe M, Yoshikawa K, Kanaho Y, Berliner LJ, Fujii H. Magnetic resonance and biochemical studies during pentylenetetrazole-kindling development: the relationship between nitric oxide, neuronal nitric oxide synthase and seizures. Neuroscience. 2004;129:757–766. doi: 10.1016/j.neuroscience.2004.09.025. [DOI] [PubMed] [Google Scholar]
- Itoh N, Ornitz DM. Fibroblast growth factors: from molecular evolution to roles in development, metabolism and disease. J Biochem. 2011;149:121–130. doi: 10.1093/jb/mvq121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh N, Yazaki N, Tagashira S, Miyake A, Ozaki K, Minami M, Satoh M, Ohta M, Kawasaki T. Rat FGF receptor-4 mRNA in the brain is expressed preferentially in the medial habenular nucleus. Brain Res Mol Brain Res. 1994;21:344–348. doi: 10.1016/0169-328X(94)90265-8. [DOI] [PubMed] [Google Scholar]
- Javaherian A, Cline HT. Coordinated motor neuron axon growth and neuromuscular synaptogenesis are promoted by CPG15 in vivo. Neuron. 2005;45:505–512. doi: 10.1016/j.neuron.2004.12.051. [DOI] [PubMed] [Google Scholar]
- Jones JE, Austin JK, Caplan R, Dunn D, Plioplys S, Salpekar JA. Psychiatric disorders in children and adolescents who have epilepsy. Pediatr Rev. 2008;29:e9–e14. doi: 10.1542/pir.29-2-e9. [DOI] [PubMed] [Google Scholar]
- Kalyani AJ, Mujtaba T, Rao MS. Expression of EGF receptor and FGF receptor isoforms during neuroepithelial stem cell differentiation. J Neurobiol. 1999;38:207–224. doi: 10.1002/(SICI)1097-4695(19990205)38:2<207::AID-NEU4>3.3.CO;2-7. [DOI] [PubMed] [Google Scholar]
- Katz M, Amit I, Yarden Y. Regulation of MAPKs by growth factors and receptor tyrosine kinases. Biochim Biophys Acta. 2007;1773:1161–1176. doi: 10.1016/j.bbamcr.2007.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ketschek A, Gallo G. Nerve growth factor induces axonal filopodia through localized microdomains of phosphoinositide 3-kinase activity that drive the formation of cytoskeletal precursors to filopodia. J Neurosci. 2010;30:12185–12197. doi: 10.1523/JNEUROSCI.1740-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klimaschewski L, Nindl W, Feurle J, Kavakebi P, Kostron H. Basic fibroblast growth factor isoforms promote axonal elongation and branching of adult sensory neurons in vitro. Neuroscience. 2004;126:347–353. doi: 10.1016/j.neuroscience.2004.04.004. [DOI] [PubMed] [Google Scholar]
- Koyama R, Ikegaya Y. Mossy fiber sprouting as a potential therapeutic target for epilepsy. Curr Neurovasc Res. 2004;1:3–10. doi: 10.2174/1567202043480242. [DOI] [PubMed] [Google Scholar]
- Markus A, Zhong J, Snider WD. Raf and akt mediate distinct aspects of sensory axon growth. Neuron. 2002;35:65–76. doi: 10.1016/S0896-6273(02)00752-3. [DOI] [PubMed] [Google Scholar]
- Marler KJ, Becker-Barroso E, Martínez A, Llovera M, Wentzel C, Poopalasundaram S, Hindges R, Soriano E, Comella J, Drescher U. A TrkB/EphrinA interaction controls retinal axon branching and synaptogenesis. J Neurosci. 2008;28:12700–12712. doi: 10.1523/JNEUROSCI.1915-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menna E, Disanza A, Cagnoli C, Schenk U, Gelsomino G, Frittoli E, Hertzog M, Offenhauser N, Sawallisch C, Kreienkamp HJ, Gertler FB, Di Fiore PP, Scita G, Matteoli M. Eps8 regulates axonal filopodia in hippocampal neurons in response to brain-derived neurotrophic factor (BDNF) PLoS Biol. 2009;7:e1000138. doi: 10.1371/journal.pbio.1000138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naeve GS, Ramakrishnan M, Kramer R, Hevroni D, Citri Y, Theill LE. Neuritin: a gene induced by neural activity and neurotrophins that promotes neuritogenesis. Proc Natl Acad Sci U S A. 1997;94:2648–2653. doi: 10.1073/pnas.94.6.2648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nedivi E, Hevroni D, Naot D, Israeli D, Citri Y. Numerous candidate plasticity-related genes revealed by differential cDNA cloning. Nature. 1993;363:718–722. doi: 10.1038/363718a0. [DOI] [PubMed] [Google Scholar]
- Nedivi E, Wu GY, Cline HT. Promotion of dendritic growth by CPG15, an activity-induced signaling molecule. Science. 1998;281:1863–1866. doi: 10.1126/science.281.5384.1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niwa H, Yamamura K, Miyazaki J. Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene. 1991;108:193–199. doi: 10.1016/0378-1119(91)90434-D. [DOI] [PubMed] [Google Scholar]
- Okamoto K, Nagai T, Miyawaki A, Hayashi Y. Rapid and persistent modulation of actin dynamics regulates postsynaptic reorganization underlying bidirectional plasticity. Nat Neurosci. 2004;7:1104–1112. doi: 10.1038/nn1311. [DOI] [PubMed] [Google Scholar]
- Paradiso B, Zucchini S, Su T, Bovolenta R, Berto E, Marconi P, Marzola A, Navarro Mora G, Fabene PF, Simonato M. Localized overexpression of FGF-2 and BDNF in hippocampus reduces mossy fiber sprouting and spontaneous seizures up to 4 weeks after pilocarpine-induced status epilepticus. Epilepsia. 2011;52:572–578. doi: 10.1111/j.1528-1167.2010.02930.x. [DOI] [PubMed] [Google Scholar]
- Parent JM, Yu TW, Leibowitz RT, Geschwind DH, Sloviter RS, Lowenstein DH. Dentate granule cell neurogenesis is increased by seizures and contributes to aberrant network reorganization in the adult rat hippocampus. J Neurosci. 1997;17:3727–3738. doi: 10.1523/JNEUROSCI.17-10-03727.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellock JM. Understanding co-morbidities affecting children with epilepsy. Neurology. 2004;62(Suppl 2):S17–S23. doi: 10.1212/WNL.62.5_suppl_2.S17. [DOI] [PubMed] [Google Scholar]
- Pitkänen A, Sutula TP. Is epilepsy a progressive disorder? Prospects for new therapeutic approaches in temporal-lobe epilepsy. Lancet Neurol. 2002;1:173–181. doi: 10.1016/S1474-4422(02)00073-X. [DOI] [PubMed] [Google Scholar]
- Plotnikov AN, Schlessinger J, Hubbard SR, Mohammadi M. Structural basis for FGF receptor dimerization and activation. Cell. 1999;98:641–650. doi: 10.1016/S0092-8674(00)80051-3. [DOI] [PubMed] [Google Scholar]
- Portera-Cailliau C, Weimer RM, De Paola V, Caroni P, Svoboda K. Diverse modes of axon elaboration in the developing neocortex. PLoS Biol. 2005;3:e272. doi: 10.1371/journal.pbio.0030272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pun RY, Rolle IJ, Lasarge CL, Hosford BE, Rosen JM, Uhl JD, Schmeltzer SN, Faulkner C, Bronson SL, Murphy BL, Richards DA, Holland KD, Danzer SC. Excessive activation of mTOR in postnatally generated granule cells is sufficient to cause epilepsy. Neuron. 2012;75:1022–1034. doi: 10.1016/j.neuron.2012.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putz U, Harwell C, Nedivi E. Soluble CPG15 expressed during early development rescues cortical progenitors from apoptosis. Nat Neurosci. 2005;8:322–331. doi: 10.1038/nn1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao MS, Hattiangady B, Shetty AK. Fetal hippocampal CA3 cell grafts enriched with FGF-2 and BDNF exhibit robust long-term survival and integration and suppress aberrant mossy fiber sprouting in the injured middle-aged hippocampus. Neurobiol Dis. 2006;21:276–290. doi: 10.1016/j.nbd.2005.07.009. [DOI] [PubMed] [Google Scholar]
- Rebecchi MJ, Pentyala SN. Structure, function, and control of phosphoinositide-specific phospholipase C. Physiol Rev. 2000;80:1291–1335. doi: 10.1152/physrev.2000.80.4.1291. [DOI] [PubMed] [Google Scholar]
- Represa A, Niquet J, Pollard H, Ben-Ari Y. Cell death, gliosis, and synaptic remodeling in the hippocampus of epileptic rats. J Neurobiol. 1995;26:413–425. doi: 10.1002/neu.480260313. [DOI] [PubMed] [Google Scholar]
- Routbort MJ, Bausch SB, McNamara JO. Seizures, cell death, and mossy fiber sprouting in kainic acid-treated organotypic hippocampal cultures. Neuroscience. 1999;94:755–765. doi: 10.1016/S0306-4522(99)00358-9. [DOI] [PubMed] [Google Scholar]
- Sato H, Fukutani Y, Yamamoto Y, Tatara E, Takemoto M, Shimamura K, Yamamoto N. Thalamus-derived molecules promote survival and dendritic growth of developing cortical neurons. J Neurosci. 2012;32:15388–15402. doi: 10.1523/JNEUROSCI.0293-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimada T, Sugiura H, Yamagata K. Neuritin: a therapeutic candidate for promoting axonal regeneration. World J Neurol. 2013;3:138. doi: 10.5316/wjn.v3.i4.138. [DOI] [Google Scholar]
- Shimada T, Takemiya T, Sugiura H, Yamagata K. Role of inflammatory mediators in the pathogenesis of epilepsy. Mediators Inflamm. 2014;2014:1–8. doi: 10.1155/2014/901902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon MA. Receptor tyrosine kinases: specific outcomes from general signals. Cell. 2000;103:13–15. doi: 10.1016/S0092-8674(00)00100-8. [DOI] [PubMed] [Google Scholar]
- Sofroniew MV. Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci. 2009;32:638–647. doi: 10.1016/j.tins.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens JM. Epilepsy: structural or functional? AJNR Am J Neuroradiol. 1996;17:243. [PMC free article] [PubMed] [Google Scholar]
- Sutula T, He XX, Cavazos J, Scott G. Synaptic reorganization in the hippocampus induced by abnormal functional activity. Science. 1988;239:1147–1150. doi: 10.1126/science.2449733. [DOI] [PubMed] [Google Scholar]
- Szebenyi G, Fallon JF. Fibroblast growth factors as multifunctional signaling factors. Int Rev Cytol. 1999;185:45–106. doi: 10.1016/s0074-7696(08)60149-7. [DOI] [PubMed] [Google Scholar]
- Szebenyi G, Dent EW, Callaway JL, Seys C, Lueth H, Kalil K. Fibroblast growth factor-2 promotes axon branching of cortical neurons by influencing morphology and behavior of the primary growth cone. J Neurosci. 2001;21:3932–3941. doi: 10.1523/JNEUROSCI.21-11-03932.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trupp M, Arenas E, Fainzilber M, Nilsson AS, Sieber BA, Grigoriou M, Kilkenny C, Salazar-Grueso E, Pachnis V, Arumäe U. Functional receptor for GDNF encoded by the c-ret proto-oncogene. Nature. 1996;381:785–789. doi: 10.1038/381785a0. [DOI] [PubMed] [Google Scholar]
- Uemura T, Lee SJ, Yasumura M, Takeuchi T, Yoshida T, Ra M, Taguchi R, Sakimura K, Mishina M. Trans-synaptic interaction of GluRδ2 and Neurexin through Cbln1 mediates synapse formation in the cerebellum. Cell. 2010;141:1068–1079. doi: 10.1016/j.cell.2010.04.035. [DOI] [PubMed] [Google Scholar]
- Wenzel HJ, Cole TB, Born DE, Schwartzkroin PA, Palmiter RD. Ultrastructural localization of zinc transporter-3 (ZnT-3) to synaptic vesicle membranes within mossy fiber boutons in the hippocampus of mouse and monkey. Proc Natl Acad Sci U S A. 1997;94:12676–12681. doi: 10.1073/pnas.94.23.12676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Y, Hong Y, Ma XY, Ren XR, Ackerman S, Mei L, Xiong WC. DCC-dependent phospholipase C signaling in netrin-1-induced neurite elongation. J Biol Chem. 2006;281:2605–2611. doi: 10.1074/jbc.M512767200. [DOI] [PubMed] [Google Scholar]
- Yao JJ, Gao XF, Chow CW, Zhan XQ, Hu CL, Mei YA. Neuritin activates insulin receptor pathway to up-regulate Kv4.2-mediated transient outward K+ current in rat cerebellar granule neurons. J Biol Chem. 2012;287:41534–41545. doi: 10.1074/jbc.M112.390260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang XF, Hyland C, Van Goor D, Forscher P. Calcineurin-dependent cofilin activation and increased retrograde actin flow drive 5-HT-dependent neurite outgrowth in Aplysia bag cell neurons. Mol Biol Cell. 2012;23:4833–4848. doi: 10.1091/mbc.E12-10-0715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Cui SS, Wallace AE, Hannesson DK, Schmued LC, Saucier DM, Honer WG, Corcoran ME. Relations between brain pathology and temporal lobe epilepsy. J Neurosci. 2002;22:6052–6061. doi: 10.1523/JNEUROSCI.22-14-06052.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Ibrahimi OA, Olsen SK, Umemori H, Mohammadi M, Ornitz DM. Receptor specificity of the fibroblast growth factor family: the complete mammalian FGF family. J Biol Chem. 2006;281:15694–15700. doi: 10.1074/jbc.M601252200. [DOI] [PMC free article] [PubMed] [Google Scholar]