

Abstract
ARALAR/AGC1/Slc25a12, the aspartate-glutamate carrier from brain mitochondria, is the regulatory step in the malate-aspartate NADH shuttle, MAS. MAS is used to oxidize cytosolic NADH in mitochondria, a process required to maintain oxidative glucose utilization. The role of ARALAR was analyzed in two paradigms of glutamate-induced excitotoxicity in cortical neurons: glucose deprivation and acute glutamate stimulation. ARALAR deficiency did not aggravate glutamate-induced neuronal death in vitro, although glutamate-stimulated respiration was impaired. In contrast, the presence of l-lactate as an additional source protected against glutamate-induced neuronal death in control, but not ARALAR-deficient neurons. l-Lactate supplementation increased glutamate-stimulated respiration partially prevented the decrease in the cytosolic ATP/ADP ratio induced by glutamate and substantially diminished mitochondrial accumulation of 8-oxoguanosine, a marker of reactive oxygen species production, only in the presence, but not the absence, of ARALAR. In addition, l-lactate potentiated glutamate-induced increase in cytosolic Ca2+, in a way independent of the presence of ARALAR. Interestingly, in vivo, the loss of half-a-dose of ARALAR in aralar+/− mice enhanced kainic acid-induced seizures and neuronal damage with respect to control animals, in a model of excitotoxicity in which increased l-lactate levels and l-lactate consumption have been previously proven. These results suggest that, in vivo, an inefficient operation of the shuttle in the aralar hemizygous mice prevents the protective role of l-lactate on glutamate excitotoxiciy and that the entry and oxidation of l-lactate through ARALAR-MAS pathway is required for its neuroprotective function.
SIGNIFICANCE STATEMENT Lactate now stands as a metabolite necessary for multiple functions in the brain and is an alternative energy source during excitotoxic brain injury. Here we find that the absence of a functional malate-aspartate NADH shuttle caused by aralar/AGC1 disruption causes a block in lactate utilization by neurons, which prevents the protective role of lactate on excitotoxicity, but not glutamate excitotoxicity itself. Thus, failure to use lactate is detrimental and is possibly responsible for the exacerbated in vivo excitotoxicity in aralar+/− mice.
Keywords: ARALAR/AGC1, glutamate excitotoxicity, kainic acid, l-lactate, malate-aspartate NADH shuttle, neuroprotection
Introduction
l-Glutamate, the major excitatory neurotransmitter in the CNS, plays an important role in neuronal differentiation, migration, and survival in the developing brain, largely through facilitating Ca2+ entry (Hack and Balázs, 1994; Yano et al., 1998).
Glutamate activates postsynaptic receptors, such as ionotropic AMPA or NMDA and metabotropic receptors (Farooqui and Farooqui, 2009), allowing the influx of Ca2+ and Na+ and the release of Ca2+ from intracellular compartments. Increased neuronal activity imposed by glutamate has been shown to upregulate respiration in neurons (Jekabsons and Nicholls, 2004; Gleichmann et al., 2009). Excessive activation of glutamate receptors can also evoke neuronal dysfunction, damage, or death through glutamate excitotoxicity (Choi, 1988; Wang and Qin, 2010), which is involved in acute or chronic neurodegenerative processes.
Mitochondrial Ca2+ plays a key role in glutamate excitotoxicity, and preventing Ca2+ uptake by mitochondria protects against neuronal death (Stout et al., 1998; Qiu et al., 2013). Another important player is poly(ADP ribose)polymerase-1 (PARP-1), which is activated by DNA strand breaks, and its detrimental role in excitotoxicity has been attributed to PARP1-mediated NAD+ loss and PAR synthesis, which blocks glycolysis and leads to ATP loss (Zhang et al., 1994; Alano et al., 2010). Other sources of NAD+ depletion, including SARM1 protein, an essential mediator of axonal degeneration (Gerdts et al., 2015), may also play a role.
These two types of glutamate actions, physiological and excitotoxic, may be influenced by the nutrients used by neurons. Glucose is the principal energy source of the adult brain (Sokoloff, 1992). It can be stored as glycogen mainly by astrocytes or broken down to pyruvate in glycolysis. l-Lactate is a common metabolite between glycolysis and oxidative phosphorylation, and it is considered a preferred fuel for brain metabolism (Smith et al., 2003). Neural activity is also accompanied by an acute rise in tissue l-lactate (Prichard et al., 1991; Hu and Wilson, 1997; Barros, 2013). l-Lactate has been shown to rescue from neuronal cell death during ischemic-related processes both in vitro (Schurr et al., 1997a, b, 2001a; Maus et al., 1999; Berthet et al., 2009) and in vivo (Ros et al., 2001; Schurr et al., 2001b; Berthet et al., 2009). Whether and when neurons produce or consume l-lactate during neural activity remains a controversial issue (San Martín et al., 2013). Some effects of l-lactate cannot be interpreted as a fuel, which suggests that l-lactate has intercellular signaling functions. G-protein-coupled l-lactate receptor, GPR81, was found in the mammalian brain and downregulates cAMP levels (Lauritzen et al., 2014). Indeed, the neuroprotective effect of lactate in vivo probably occurs both through a receptor-mediated signal transduction and also by the classic metabolic pathway as an alternative source of energy (Castillo et al., 2015).
In neurons, glucose oxidation or intracellular use of l-lactate by mitochondria requires an NADH shuttle system. The malate-aspartate NADH shuttle (MAS) is the main shuttle in brain (Satrústegui et al., 2007), and its regulatory component is ARALAR/AGC1/Slc25a12, the mitochondrial transporter of aspartate–glutamate. Activation of ARALAR-MAS by extramitochondrial Ca2+ in the nm range results in an increase in NADH production in neuronal mitochondria (Pardo et al., 2006). Ca2+ activation of ARALAR-MAS will function to “push” pyruvate into mitochondria by increasing: (1) the cytosolic redox state of NAD/NADH, (2) the conversion of lactate into pyruvate, and (3) glucose oxidation (Fig. 1).
Figure 1.

Schematic representation of the malate-aspartate NADH shuttle involvement in glucose oxidation and l-lactate utilization. Extramitochondrial Ca2+ activation of ARALAR-MAS will function to “push” pyruvate into mitochondria by increasing the cytosolic redox state of NAD/NADH both by promoting glucose oxidation and the conversion of lactate into pyruvate. Aralar/AGC, mitochondrial aspartate/glutamate carrier; OGC, αKG/malate carrier; PyrC, mitochondrial pyruvate carrier. cMDH, mMDH, cytosolic and mitochondrial malate dehydrogenases; cAAT, mAAT, cytosolic and mitochondrial aspartate amino transferases.
The expression of aralar is regulated by active, phosphorylated CREB protein and by signal pathways that modify the activity of CREB itself, including intracellular Ca2+ levels (Menga et al., 2015). Ca2+ activation of ARALAR increases pyruvate formation and supply into mitochondria, which increases mitochondrial matrix NADH production and respiration. Moreover, the ARALAR-MAS pathway is required to upregulate respiration under conditions of increased workload in neurons using glucose (Llorente-Folch et al., 2013; Rueda et al., 2014).
Both glucose and l-lactate require ARALAR-MAS for oxidation. Glutamate imposes a strong workload in neurons, suggesting that ARALAR-MAS may be required for the physiological response to glutamate and also in response to excitotoxicity. In the present work, we have addressed the requirement of the shuttle in physiological and excitotoxic conditions triggered by glutamate with different nutrients and using aralar KO mice.
Materials and Methods
Animals.
Male SVJ129 × C57BL/6 mice carrying a deficiency for aralar expression (aralar−/−, aralar +/−, and aralar +/+) obtained from Lexicon Pharmaceuticals were used (Jalil et al., 2005). The mice were housed in a humidity- and temperature-controlled room on a 12 h light/dark cycle, receiving water and food ad libitum. All animal procedures were approved by the corresponding institutional ethical committee (Centro de Biología Molecular Severo Ochoa) and were performed in accordance with Spanish regulations (BOE 67/8509-12, 1988) and European regulations (EU directive 86/609, EU decree 2001-486). All efforts were made to minimize animal suffering. Reporting followed the ARRIVE Guidelines.
Neuronal cell culture.
Cortical neuronal cultures were prepared from E15-E16 mouse embryos as described previously (Ramos et al., 2003; Pardo et al., 2006). Embryos were obtained from crosses between C57BL/6SV129 aralar+/− mice, and nonbrain tissue was used for determination of DNA genotype as previously described for aralar (Jalil et al., 2005; Pardo et al., 2006). Neurons represented >80% of the total cell population (Ramos et al., 2003; Pardo et al., 2006).
Viability assay.
In glucose deprivation assays, cellular viability was evaluated 24 h after maintaining cultures for 6 h in conditions of: normoglycemia (5.0 mm glucose), glucose deprivation (nominally 0.0 mm glucose), or 10 mm l-lactate. In glutamate administration assays, 5–50 μm was added in MEM for 5 or 30 min. Cultures were maintained for 24 h in fresh serum-free B27-supplemented Neurobasal medium until testing the cellular viability. When used, l-lactate (10 mm) was added 30 min before the experiment and was present during glutamate stimulation and in the subsequent fresh media. CL/PI treatment was performed as previously described (Mattson et al., 1995).
Measurement of glutamate release.
The concentration of l-glutamic acid in cell-free supernatants was measured by an enzymatic assay (Lund and Bergmeyer, 1986). Briefly, cell supernatants from each condition were collected and used after deproteinization (5% PCA)-neutralization. Each media sample was mixed with Tris/hydrazine buffer (Tris 0.1 m, EDTA 2 mm, hydrazine 0.63 m, pH 9). The enzymatic reaction was started by addition of glutamic dehydrogenase (14 IU/ml). l-Glutamate suffers an oxidative deamination to 2-oxoglutarate in the presence of NAD+ and glutamic dehydrogenase, with the corresponding NADH formation. The increase in NADH concentration, measured by the change in absorbance at 340 nm by microplate reader (FLUOstar OPTIMA, BMG Lab Tech), is proportional to the amount of glutamate. Concentrations of glutamate in the experimental samples were calculated according to the optical densities obtained from standards of glutamate solution. The amount of glutamate produced by cells was calculated after subtracting the values obtained from wells containing distilled water.
Cytosolic Na+ and Ca2+ imaging in primary neuronal cultures.
Single-cell measurements of cytosolic Ca2+ and Na+ were performed as previously described (Llorente-Folch et al., 2013) using neurons loaded with fura-2 AM as Ca2+ indicator (Ruiz et al., 1998; Pardo et al., 2006) and SBFI as Na+ indicator (Rose and Ransom, 1997) at 37°C. Experiments were performed in HEPES-control salt solution (HCSS) containing 15 mm glucose with 2 mm CaCl2. Image acquisition was performed with the Aquacosmos 2.5 software (Hamamatsu), and data analysis was done with Origin software (Origin-Lab).
Measurement of cytosolic pH.
Cytosolic pH was measured with the fluorescent probe 2′, 7′-bis (2-carboxyethyl)-5(6)-carboxy-fluorescein (BCECF; Invitrogen). Neurons growing on poly-lysine-coated coverslips were incubated in HCSS with 15 mm glucose and 2 mm CaCl2 containing 0.12 μm BCECF-AM and 0.025% pluronic F.127 (Invitrogen) for 30 min at 37°C, and rinsed in HCSS for 20 min before use. Then coverslips were mounted on the microscope stage equipped with a 40× objective as described previously (Ruiz et al., 1998), and BCECF fluorescence was imaged ratiometrically using alternate excitation at 450 and 490 nm, and a 530 nm emission filter with a Neofluar 40×/0.75 objective in an Axiovert 75M microscope (Zeiss). Additions were made as a bolus. For single-cell analysis of pHi, the ratio of fluorescence intensity at 450 nm (F(450)) and 490 nm (F(490)), (F(450)/F(490)), was obtained. Image acquisition was performed with the Aquacosmos 2.5 software (Hamamatsu), and data analysis was done with Origin software (Origin-Lab).
Imaging of mitochondrial Ca2+ and cytosolic ATP/ADP ratio in primary neuronal cultures.
Single-cell measurements of cytosolic ATP/ADP ratio and mitochondrial Ca2+ were performed in neurons transfected using calcium phosphate protocol 24 or 48 h before the experiments either with the plasmid coding for cytosolic targeted ratiometric Perceval-HR (Tantama et al., 2013) or with the plasmid genetically encoded chameleon probe specifically targeted into mitochondria 4mitD3cpv (Palmer and Tsien, 2006). Neurons were seeded in chambered coverglass 4-well plates at a density of 105 cell/cm2 and at 8–9 DIV were transfected. Experiments were performed in HCSS, 15 mm glucose with 2 mm CaCl2. When used, 2 mm l-lactate was added immediately before starting the experiment.
Briefly, cells were excited for 100 ms alternatively at 426-44 (Venus) and 472-27 (GFP) for Perceval-HR measurements and for 40 ms at 440 ± 10 nm (440AF21, Omega Optical) for mit4D3cpv. The emitted fluorescence was collected at 520/35 (GFP) for Perceval HR probe and through 480 at 535 nm for mit4D3cpv. Images were collected every 5 s using a filter wheel (Lambda 10-2, Sutter Instruments, all filters purchased from Chroma) and recorded by a Hamamatsu C9100-02 camera mounted on an Axiovert 200M inverted microscope equipped with a 40×/1.3 Plan-Neofluar objective.
Perceval-HR emission ratio was GFP/CFP, whereas mit4D3cpv emission ratio was YFP/CFP, reflecting cytosolic ATP/ADP ratio and mitochondrial Ca2+, respectively. Single-cell fluorescence recordings were analyzed using ImageJ (National Institutes of Health) or MetaMorph (Universal Imaging).
Measurement of cellular oxygen consumption.
Cellular oxygen consumption rate (OCR) was measured using a Seahorse XF24 Extracellular Flux Analyzer (Seahorse Bioscience) (Qian and Van Houten, 2010) as previously described (Llorente-Folch et al., 2013). Cells were equilibrated with bicarbonate-free low-buffered DMEM (without pyruvate, l-lactate, glucose, glutamine, and Ca2+) supplemented with 2.5 mm glucose and 2 mm CaCl2 for 1 h before XF assay. Additional substrates apart from glucose (i.e., 2 mm l-lactate and 2 mm pyruvate) were prepared in the experimental buffer and were incubated with the neurons 30 min before the XF assay and maintained throughout the experiment.
Mitochondrial function in neurons was determined through sequential addition of 6 μm oligomycin (Oli), 0.5 mm 2,4-dinitrophenol (DNP), and 1 μm antimycin/1 μm rotenone. This allowed determination of basal oxygen consumption, oxygen consumption linked to ATP synthesis (ATP), non-ATP linked oxygen consumption (leak), mitochondrial uncoupled respiration, and nonmitochondrial oxygen consumption (Qian and Van Houten, 2010) for review see (Brand and Nicholls, 2011).
Membrane potential.
Cells were loaded with 10 μg/ml Rhod-123 (Invitrogen) for ∼10–15 min at room temperature. Rhod-123 fluorescence was excited at 488 nm and measured at 530 nm (Emaus et al., 1986; Duchen et al., 1990). Mitochondrial potential (Δψmit) was evaluated by measuring Rhod-123 fluorescence quench using the FLUOStart OPTIMA (BGM Labtech) spectrofluorometer. Mitochondrial depolarization increases the fluorescence signal, and we analyzed the effect of 100 μm glutamate. Uncoupling agents (0.5 mm DNP) increased fluorescence, whereas fluorescence was reduced by blockade of the Fo proton channel of the mitochondrial ATP synthase complex (6 μm oligomycin). Experiments were performed in the presence of 2 mm Ca2+ or in the absence of Ca2+ with 100 μm EGTA. We have presented all data as a ratio F/Fo referring the changes in fluorescence to the baseline level before glutamate addition.
Immunodetection of 8-hydroxy-2′-deoxiguanosine (8-OHdG).
Immunodetection assays of 8-OHdG were performed as described previously (Soultanakis et al., 2000). Four different groups were established. Control group (CTR) only suffered the replacement of the media like the others, in l-lactate group (Lac) neurons were incubated with 10 mm l-lactate for 30 min, the glutamate group (Glu) was stimulated with 100 μm glutamate for 30 min in MEM and the glutamate + l-lactate group (Glu+Lac), where cells were preincubated with l-lactate before the excitotoxic stimulus. Anti-8-oxo-dG immunostaining assay was performed according to the manufacturer's protocol from Trevigen.
Confocal scanning laser microscopy was performed in a confocal Zeiss microscope with 63× oil-immersion objective. The analysis was performed in individual cells, and intensity thresholds were set that highlighted the areas to correspond to nucleus and mitochondria (from TOPRO-3 and Mn-SOD labeling). With these thresholds (nuclear 500-1999, and mitochondrial 2000-16383), we have analyzed α-8-OHdG fluorescence measuring the average intensity (considering constant nuclear area) and integrated intensity (considering the perinuclear area occupied by mitochondria). Image analysis was performed with MetaMorph 5.0r7 (Universal Imaging).
Kainic acid (KA) treatment, seizure induction, and quantification of degenerating neurons.
KA (22.5 mg/kg; Sigma-Aldrich), freshly prepared, was administered intraperitoneally to 3-month-old male wild-type (WT) and aralar hemizygous (HT) mice (n = 13 mice for each genotype). An initial treatment of 15 mg/kg KA was reinforced with half of the initial dose 1 h later. This sublethal dose of KA induced continuous tonic clonic seizures and at least one episode of Level 4 (Table 1). Seizure severity was quantified in a blind manner using the previously established 6 point seizure scale (Schauwecker and Steward, 1997) adapted from a 5 point scale for rat (Racine, 1972) (Table 1).
Table 1.
KA administration scheme and seizure scoresa
| Seizure score level | Characteristic behavior |
|---|---|
| 1 | Unmoving and crouched in a corner |
| 2 | Stretched body out, tail becomes straight and rigid, ears laid back, bulging eyes |
| 3 | Repetitive head bobbing, rears into a sitting position with forepaws resting on belly |
| 4 | Rearing and falling clonic seizures broken by periods of total stillness, jumping clonus, running clonus |
| 5 | Continuous level 4 seizures |
| 6 | Body in clonus, no longer using limbs to maintain posture, tonic-clonic seizures, usually precursor to death |
| 7 | Death |
aMice were given an initial intraperitoneal injection of 15 mg/kg KA, which was reinforced 60 min later with a supplemental dose that was half the initial one. Seizure recording was performed over 3 h. A 7 point scale was used to rate a subject visually following systemic administration of KA (22.5 mg/kg final dose) (Schauwecker and Steward, 1997).
One week after the treatment, animals were intracardially perfused with saline buffer (0.9% NaCl in PBS) and 4% PFA (in 0.1 m phosphate buffer, pH 7.4). Brains were removed, postfixed for 24 h in 4% PFA at 4°C, and transferred into sucrose (30% sucrose in 0.1 m phosphate buffer) 48 h at 4°C. Coronal sections were cut with a freezing cryotome (free-floating sections) and kept in cryoprotectant solution (25% glycerol, 25% ethylene glycol in 50 mm PB) at −20°C until processing. Fluoro-Jade B dye (Millipore) was used for the detection of neuronal degeneration as previously described (Schmued et al., 1997). Sections were examined with a confocal Zeiss microscope with a 10× objective.
Image analysis was performed with the ImageJ program. The analysis shows the total number of degenerating (Fluoro Jade B-positive) neurons in a subfield referred to the area of the corresponding hippocampal subfield. Each tissue section was also examined for neurodegeneration in other brain regions, including neocortex, striatum, thalamus, and amygdala, to obtain a qualitative assessment of a strain's pattern of neuronal cell death throughout the brain (Table 2).
Table 2.
Qualitative analysis of damaged regions in wild-type and Aralar HT micea
| aralar+/+ | aralar+/− | |
|---|---|---|
| Hipoccampus | 3/13 | 9/12 |
| Cerebral cortex | 0/13 | 3/12 |
| Septal nucleus | 3/13 | 9/12 |
| Thalamus | 0/13 | 5/12 |
| Hypothalamus | 0/13 | 1/12 |
| Endopiriform nucleus | 0/13 | 3/12 |
| Ectorhinal, endorhinal, perirhinal cortex | 0/13 | 4/12 |
| Amygdala nucleus | 2/13 | 3/12 |
| Cerebellum | 0/13 | 0/12 |
aOne week after the KA treatment, brains from WT (n = 13) and aralar HT (n = 12) mice were analyzed. Fluoro-Jade B immunostaining was used for the detection of neuronal degeneration in several regions as indicated.
Results
Stimulation of respiration in response to glutamate is impaired in ARALAR-deficient neurons
Glutamate (100 μm) activates ionotropic NMDA and AMPA receptors, and metabotropic receptors, and induced a pronounced rise in cytosolic Ca2+ in cerebral cortex neurons (Fig. 2A–C), which was prevented in Ca2+-free media (data not shown). It also induced a sustained rise in mitochondrial Ca2+ levels (Fig. 2D), as reported by mit4D3cpv, a genetically encoded Ca2+ sensor expressed in the mitochondrial matrix (Jean-Quartier et al., 2012), which did not vary with ARALAR deficiency (Fig. 2E,F) and a prominent loss of mitochondrial membrane potential as estimated from rhodamine 123 fluorescence, which was blocked in Ca2+-free media (Fig. 2G). NMDA receptors also transport Na+; and even though a typical NMDA receptor may have a 10:1 selectivity for Ca2+ over Na+ (Mayer and Westbrook, 1987; Nicholls, 2008), the higher concentration of the latter leads to a Na+ flux that can greatly exceed that of Ca2+. The increase in cytoplasmic Na+ concentration caused by glutamate was measured in neurons loaded with the Na+ indicator SBFI-AM (Rose and Ransom, 1997) and was not affected by ARALAR deficiency (Fig. 2H). Thus, except for a slightly smaller glutamate-induced cytosolic calcium peak (Fig. 2C), ionic fluxes induced by glutamate were the same in control and ARALAR-deficient neurons.
Figure 2.
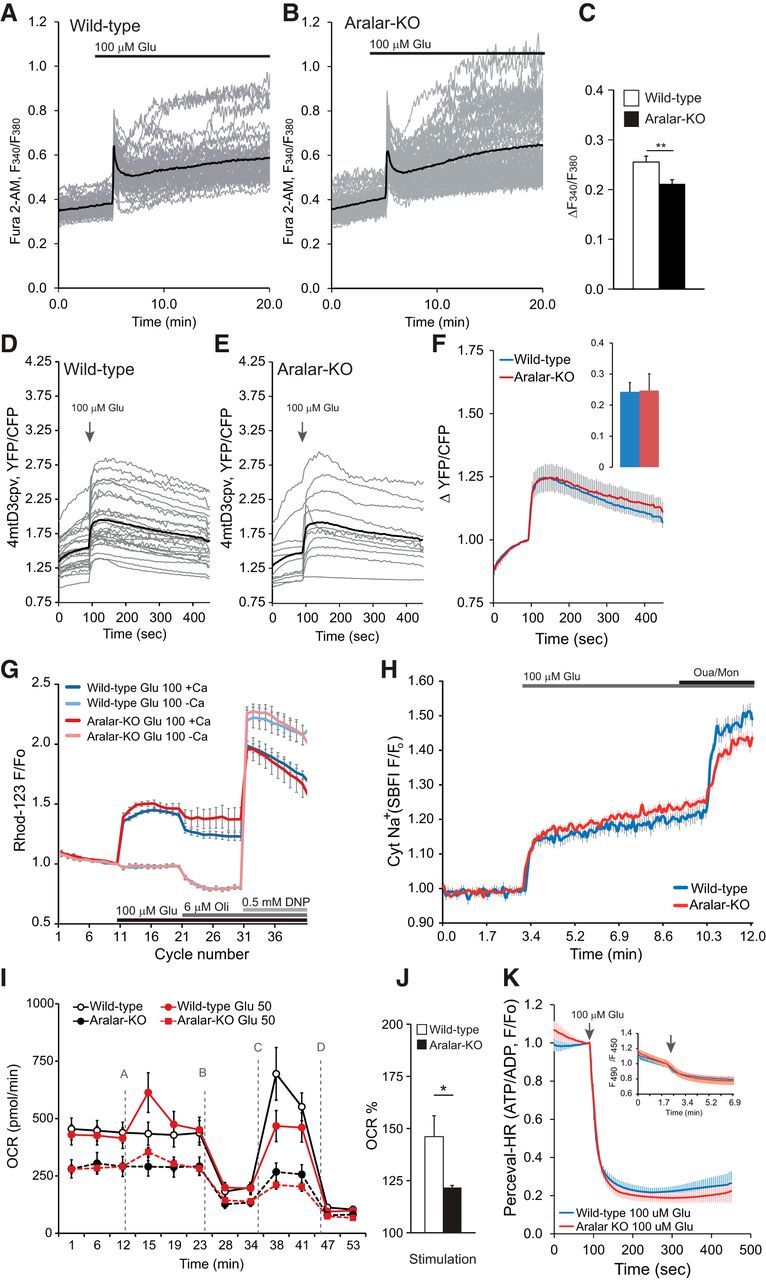
Stimulation of respiration in response to glutamate is impaired in ARALAR-deficient neurons. A–C, Change in cytosolic Ca2+ in fura-2 AM-loaded neurons obtained by stimulation with 100 μm glutamate (100 μm Glu) in 15 mm glucose and 2 mm Ca2+ media. Fluorescence F340/F380 ratio is represented from >50 neurons from two independent experiments in WT cultures and >70 neurons in four independent experiments in aralar-KO cultures. Individual cell recordings (gray) and average (thick black trace) are shown. D–F, Corresponding data in neurons transfected with 4mtD3cpv probe to determine changes in mitochondrial Ca2+ in 2.5 mm glucose and 2 mm Ca2+ media. Recording from 26 and 11 transfected cells, in 15 and 7 independent experiments, in WT and aralar-KO primary neuronal cultures, respectively. Individual cell recordings (gray) and average (thick black trace) are shown. G, Mitochondrial membrane potential was analyzed using the single-wavelength fluorescent indicator Rhodamine 123 (Rhod-123) in the quench mode. Mitochondrial depolarization is observed as an increase in fluorescence. The 100 μm glutamate in the presence or the absence of 2 mm Ca2+ in 2.5 mm glucose media was added as indicated. Data correspond to ∼10 independent experiments in WT and aralar-KO primary neuronal cultures. Glu, Glutamate; Oli, oligomycin. H, Changes in cytosolic Na+ in SBFI-loaded neurons by stimulation with 100 μm glutamate (100 μm Glu) in 15 mm glucose and 2 mm Ca2+ media. Normalized ratio is represented. Data correspond to 28 neurons from two independent experiments (blue line, WT) and 75 neurons in four independent experiments (red line, aralar-KO), Ouabain (Oua; 0.1 μm), and monensin (Mon; 10 mm) were added for equilibration of extracellular and intracellular [Na+] at the end of the experiments. I, Stimulation of OCR upon 50 μm glutamate (Glu 50) in WT and aralar-KO neurons in 2.5 mm glucose and 2 mm Ca2+ media. A, 50 μm glutamate. B, 6 μm oligomycin. C, 0.5 mm DNP. D, 1 μm/1 μm antimycin/rotenone. n = 33–42 from 11 to 18 independent experiments in WT and n = 25–34 from 9 to 16 aralar-KO primary neuronal cultures. J, Stimulation of respiration (as percentage of basal values). K, Cytosolic ATP/ADP ratio (Perceval-HR occupancy, GFP/Venus ratio fluorescence) was measured in WT and aralar-KO neurons 24 h after Perceval-HR transfection. Neurons were stimulated with 100 μm glutamate in 15 mm glucose and 2 mm Ca2+ medium. Data are mean ± SEM (5–16 neurons and independent platings). Inset, Variations in intracellular pH (mean ± SEM) determined in BCECF-loaded neurons in WT and KO neurons, under the same conditions of the Perceval-HR experiments. *p ≤ 0.05 (Student's t test). **p ≤ 0.01 (Student's t test).
The glutamate-induced ionic unbalance results in a net increase in ATP consumption to extrude Ca2+ and Na+ and in stimulation of mitochondrial respiration to match the increased ATP demand (Nicholls et al., 2003; Jekabsons and Nicholls, 2004). As previously described (Llorente-Folch et al., 2013), basal OCR in aralar-KO neuronal cultures was severely reduced (Fig. 2I), indicating that the lack of MAS prevents adequate glucose-derived substrate supply to mitochondria under basal conditions. Moreover, glutamate-induced increase in OCR in cortical neurons was clearly reduced in ARALAR-deficient cells (146.12 ± 7.64% and 122.71 ± 0.99%, p ≤ 0.05, in WT and aralar-KO neurons, respectively; Fig. 2I,J). Using NMDA (25 μm), a nonmetabolized glutamate agonist, a similar decrease in OCR stimulation was found in aralar-KO neurons (156 ± 5% vs 127 ± 3% in WT and KO neurons, respectively, p < 0.001).
The increase in respiration was oligomycin-sensitive. To test whether this increase in respiration was able to compensate for the ATP breakdown promoted by glutamate, we performed single-cell measurements of cytosolic ATP/ADP ratio in neurons transfected with the plasmid coding for cytosolic targeted ratiometric Perceval-HR (Tantama et al., 2013). As glutamate induces an intracellular acidification (Wang et al., 1994) and Perceval-HR fluorescence is pH-sensitive, the variations in neuronal pH were investigated in BCECF- loaded neurons. Glutamate induced the same acidification in wt and KO neurons (Fig. 2K, inset) and caused a similar fall in ATP/ADP ratio in both genotypes (Fig. 2K), suggesting that the increase in respiration does not compensate for glutamate-induced ATP breakdown.
Together, these results indicate that the lack of ARALAR does not cause major changes in ionic fluxes due to glutamate stimulation in cortical neurons but limits upregulation of mitochondrial function in response to the high workload imposed by glutamate. We have recently observed that the rapid activation of respiration in response to NMDA appears to be critical to prevent delayed excitotoxicity (Rueda et al., 2015). Therefore, this suggested that ARALAR deficiency would impact on the delayed response to glutamate. To explore this possibility, we have studied the role of ARALAR in the excitotoxic paradigm in vivo and in vitro.
Aralar hemizygote mice display an increased susceptibility to KA-induced seizures and neuronal damage
To study the in vivo response to KA excitotoxicity, we used aralar HT mice as aralar-KO mice only live up to 20 d. Ben-Ari et al. (1979a, b) showed that excessive neuronal activity following KA intraperitoneal administration, linked to epileptiform activity (Olney et al., 1979), is closely associated with neuronal damage. Thus, we analyzed the epileptic activity and the subsequent neurodegenerative effects induced by KA in WT and aralar HT mice.
Mouse strain background determines both KA-evoked seizures as well as the severity of seizure-induced damage and excitotoxic cell death (Schauwecker, 2011). For the SVJ129 × C57BL6 hybrid background of the aralar mice, we used a final dose of 22.5 mg/kg KA (see administration protocol in Materials and Methods), and the time course of the seizures was followed for 3 h after injection. Brain histological analysis was performed 1 week after the treatment. At this dose, the administration protocol guaranteed the achievement of at least one seizure scored Level 4 for each animal (Table 1) while maintaining overall viability (one aralar HT animal died few days after the test).
A cohort of 26 adult animals (WT n = 13, aralar HT n = 13) were used. Neither control nor aralar HT mice showed hyperexcitability or spontaneous seizures during standard housing. However, after KA administration, aralar HT mice showed an enhanced seizure profile (Fig. 3A). WT mice showed a predominance of seizure score Level 2 events (Table 1; Fig. 3A) and latency to first seizure score Level 4 event of 88.39 ± 15.71 min and only 46.54 ± 12.43 min for aralar HT mice (p ≤ 0.05) (Fig. 3B). In addition to the early burst of seizures, aralar HT mice underwent a sustained seizure profile with seizures lasting several minutes, reaching an average crisis duration of ∼34 min well above WT mice (∼9 min) (Fig. 3C). WT mice never showed tonic-clonic seizure score Level 6 events and fewer number of seizure score Level 4 and 5 than aralar HT mice (Fig. 3A).
Figure 3.
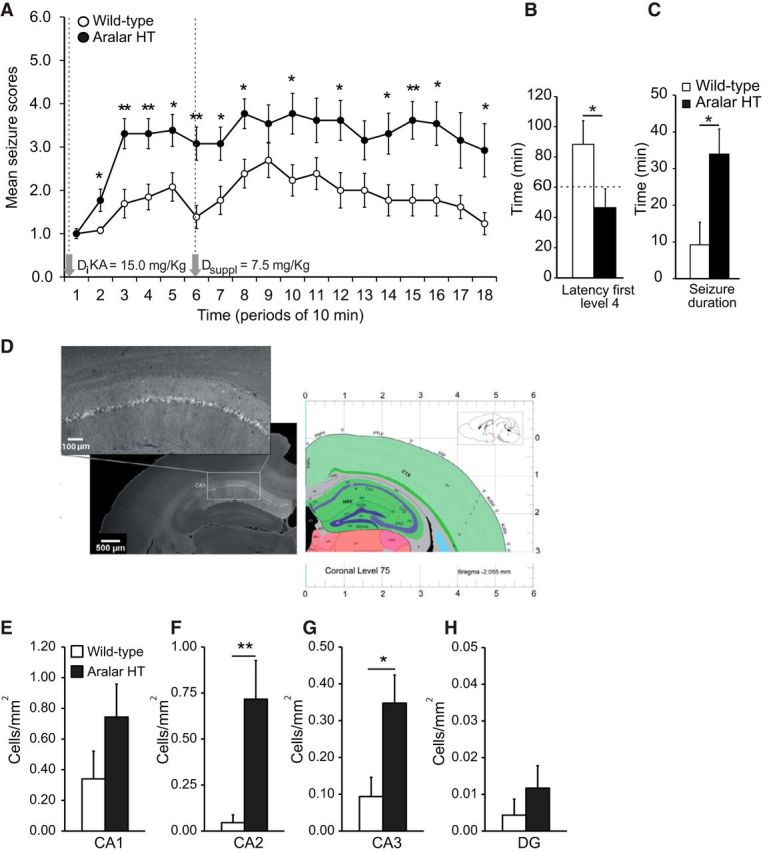
Increased susceptibility to KA and higher neuronal degeneration in aralar HT mice. A, Progression of behavioral changes after systemic KA administration (22.5 mg/kg i.p., final dose) in WT and aralar HT following the score Table 1 over 3 h. B, Latency to the first Level 4 event calculated from the time of KA initial administration. C, Total time performing seizures during the complete experiment comparing WT versus aralar HT mice. Data are mean seizure scores or recorded time from 13 WT (empty cycles or bars) and 12 aralar HT (filled cycles or black bars) mice. Variance homogeneity and normality of data were tested by means of Levene and Shapiro–Wilk tests, respectively. Data did meet specifications for parametric analysis, so factorial ANOVA was used: *p ≤ 0.05; **p ≤ 0.01. D, Fluoro-Jade B immunostaining in a representative aralar HT brain. E–H, Degenerating Fluoro JadeB-positive neurons were counted using ImageJ and referred to area of the hippocampal subfields in square millimeters. Data are mean ± SEM. *p ≤ 0.05 (Student's t test). **p ≤ 0.01 (Student's t test).
Analysis of neuronal death showed damaged (Fluoro-Jade B)-positive cells in different regions. Table 2 summarizes the results obtained and regions analyzed in WT and aralar HT. Importantly, and as shown previously (Lothman and Collins, 1981), the hippocampus was one of the most affected areas in the brain. Representative image of hippocampus is shown in Figure 3D. Quantitative analysis performed in the hippocampal region showed that the number of degenerating neurons in CA1, CA2, CA3, and DG per square millimeters was much higher in aralar HT than in control mice (Fig. 3E–H). These results indicate that the lack of an aralar allele increases susceptibility to KA seizures and KA-induced neuronal damage.
Glutamate-induced cell death in hypoglycemic conditions and upon external glutamate administration is independent of ARALAR
We next studied the influence of ARALAR on glutamate excitotoxicity in vitro using primary cortical neuronal cultures from WT and aralar-KO embryos. We focused on two conditions: hypoglycemia-induced and glutamate-induced excitotoxicity. As shown previously, glucose deprivation leads to glutamate release to culture media, which induces neuronal death through the action on NMDA receptors (Kauppinen et al., 1989). Glutamate release was the same in WT and aralar-KO cultures (1.72 ± 0.10 and 1.38 ± 0.08 nmol/105cell in WT and aralar-KO cultures, respectively, Fig. 4A) and neuronal death occurred in both to a similar extent (Fig. 4B). Administration of 5 μm (+)-MK-801 hydrogen maleate (MK-801), a noncompetitive inhibitor of the NMDA receptor (Wong et al., 1986), completely blocked neurotoxicity and glutamate release to the culture media induced in hypoglycemic conditions (data not shown), verifying the involvement of excitotoxicity in this paradigm.
Figure 4.
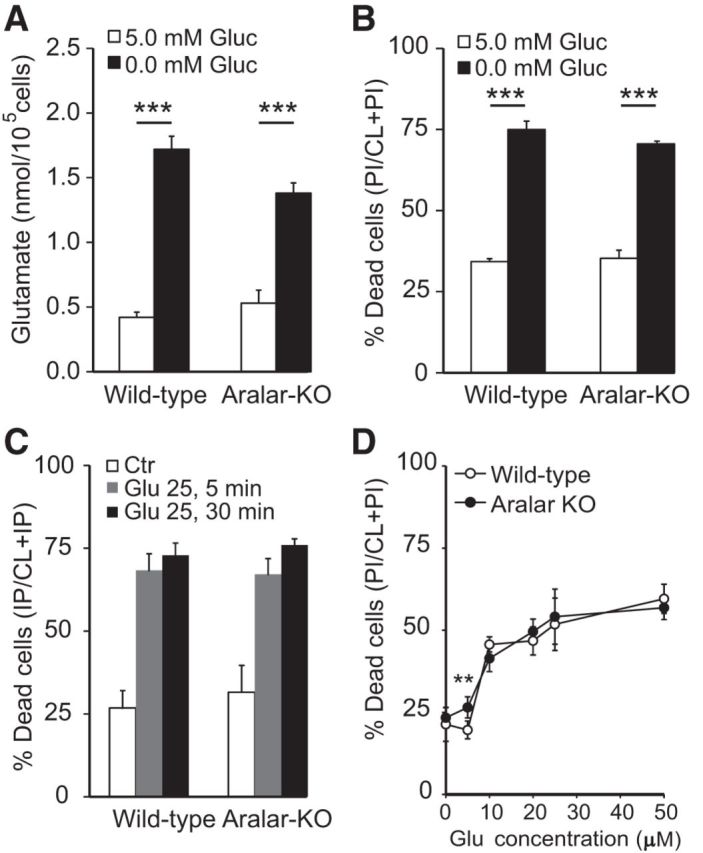
Glutamate-induced cell death in hypoglycemic conditions and upon external glutamate administration is independent of ARALAR. A, Glutamate released to the neuronal culture media 24 h after treatment in conditions of normoglycemia (5.0 mm glucose) and glucose deprivation (nominally 0.0 mm glucose) in WT and aralar-KO cultures. B, Cellular viability was evaluated 24 h after maintaining cultures for 6 h in the previously mentioned conditions. C, Neuronal cultures were treated with 25 μm glutamate 5 or 30 min and then maintained for 24 h testing the cellular sensitivity. Data are calculated as percentage of dead as PI/(CL + PI) by using the calcein-AM/propidium iodide method. Genetic deletion of aralar in cortical neuronal cultures had no effect on basal glutamate-mediated cell death under the present experimental conditions. D, Dose–response curve to glutamate (present during 30 min) in WT and aralar-KO neurons. Data are mean ± SEM from three independent experiments. **p ≤ 0.01 (one-way ANOVA followed by Student-Newman-Keuls t test). ***p ≤ 0.001 (one-way ANOVA followed by Student-Newman-Keuls t test).
As hypoglycemia-induced excitotoxicity was not dependent on the presence of ARALAR, we hypothesized that acute exposure to 25 μm glutamate in normoglycemia would reveal ARALAR-dependent differences in excitotoxicity 24 h later. Surprisingly, neuronal survival 24 h after 5 or 30 min exposure to 25 μm glutamate did not differ between genotypes (Fig. 4C). Basal neuronal survival in vitro was also the same in aralar-KO than in control cultures (Fig. 4C). Moreover, neuronal survival in response to increasing glutamate concentrations (5–50 μm) was the same for both genotypes, except for a small difference at 5 μm glutamate (Fig. 4D). The lack of influence of ARALAR on these various forms of in vitro glutamate excitotoxicity is in striking contrast with the results observed in vivo. We reasoned that this paradoxical result may be due to the utilization of alternative energy sources in vivo, different from glucose, the only exogenous substrate present in vitro conditions.
A major event in glutamate excitotoxicity is the activation of the nuclear enzyme PARP1, which uses cytosolic, but not mitochondrial, NAD+, and thereby inhibits glycolysis at the glyceraldehyde 3-phosphate dehydrogenase (GA3PDH) step (Alano et al., 2010; Kim et al., 2011; Duchen, 2012). In a scenario of block in GA3PDH and limited pool of cytosolic pyridine nucleotides, MAS would be dispensable, and this may explain why the presence of ARALAR does not protect against in vitro excitotoxicity. In addition, a direct action of PAR polymers in mitochondria-associated hexokinase1 (Andrabi et al., 2014; Fouquerel et al., 2014) would also lead to a limitation in substrate supply. Although the extent to which this happens in neurons is yet unknown, together, these findings support the notion that ARALAR is not protective in in vitro excitotoxicity on glucose as only energy source, due to the block of glycolysis caused by glutamate, which makes MAS dispensable. Therefore, we have considered that, during in vivo kainate administration, neurons may use other energy sources whose utilization may be hampered in the absence of ARALAR, particularly l-lactate.
Limited aerobic l-lactate utilization in ARALAR-deficient neurons
Glucose is the obligate energy substrate for the brain. Abundant experimental evidence, however, suggests that l-lactate is also an important brain energy substrate in hypoxia or during high brain activity, not a mere waste byproduct issued from anaerobic metabolism.
Therefore, we have studied the role of ARALAR on lactate utilization during excitotoxicity. We first studied the effect of l-lactate on glutamate-induced Ca2+ signaling. A 30 min preincubation in the presence of 15 mm glucose and 2 mm l-lactate resulted in an increase in glutamate-induced Ca2+ peak both in WT and ARALAR-deficient neurons (Fig. 5A,B; compare with Fig. 2A,B). Glutamate-induced increase in mitochondrial Ca2+ was not modified by l-lactate preincubation and was the same in both genotypes (Fig. 5D–F; compare with Fig. 2D–F). This result is consistent with those of Yang et al. (2014) who found that high concentrations of l-lactate (≥2.5 mm) potentiate over time the effects of glutamate, greatly amplifying glutamate-induced increase in cytosolic Ca2+. These effects required the uptake of l-lactate, as they were blocked by an inhibitor of the l-lactate transporter (UK5099). Whether this effect of l-lactate involves l-lactate signaling through a G-protein-coupled receptor, GPR81/HCA1 (Lauritzen et al., 2014; Mosienko et al., 2015), to downregulate cell cAMP levels is yet unknown.
Figure 5.
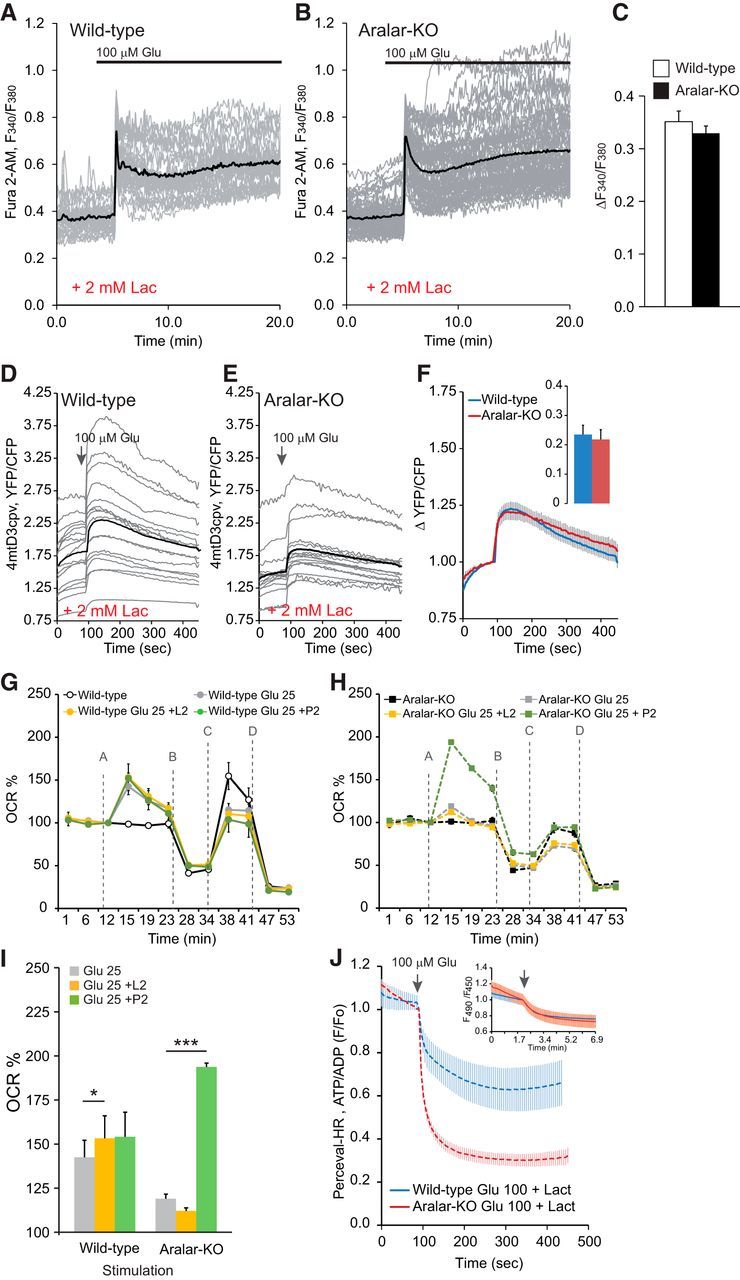
l-Lactate on glutamate-induced Ca2+ signaling. Limited aerobic l-lactate utilization in ARALAR-deficient neurons. A–C, Change in cytosolic Ca2+ in fura-2 AM-loaded neurons obtained by stimulation with 100 μm glutamate (100 μm Glu) in 15 mm glucose and 2 mm Ca2+ media in the presence of 2 mm l-lactate. Fluorescence F340/F380 ratio is represented from 30 neurons form two independent experiments in WT cultures and 59 neurons in four independent experiments in aralar-KO neurons. Individual cell recordings (gray) and average (thick black trace) are shown. Horizontal lines indicate cytosolic Ca2+ increase in the absence of l-lactate for each experimental condition. No differences were observed comparing genotypes. D–F, Corresponding data in neurons transfected with 4mtD3cpv probe to determine changes in mitochondrial Ca2+ in 2.5 mm glucose and 2 mm Ca2+ media in the presence of 2 mm l-lactate. Recording from 16 and 17 transfected cells, in 11 and 9 independent experiments, in WT and aralar-KO primary neuronal cultures, respectively, for mitochondrial Ca2+ imaging. Individual cell recordings (gray) and average (thick black trace) are shown. G, H, Effects of additional substrates, 2 mm l-lactate (L2) and 2 mm pyruvate (P2), in the presence of 2.5 mm glucose, on glutamate-induced stimulation of mitochondrial respiration using upon 25 μm glutamate (Glu 25). n = 4–6 from two or three independent experiments from control and aralar-KO primary neuronal cultures.*p ≤ 0.05 (Student's t test). ***p ≤ 0.001 (Student's t test). Mitochondrial function in neurons was determined through sequential addition of (A) vehicle/glutamate, (B) 6 μm oligomycin (Oli), (C) 0.5 mm DNP, and (D) 1 μm antimycin/1 μm rotenone (A/R) where indicated by the dashed line using the XF24 extracellular flux analyzer. I, Stimulation of respiration (as percentage of basal values). J, Cytosolic ATP/ADP ratio (Perceval-HR occupancy, GFP/Venus ratio fluorescence) was measured in WT and aralar-KO neurons 24 h after Perceval-HR transfection, and stimulated with 100 μm glutamate in 15 mm glucose and 2 mm Ca2+ medium in the presence of 2 mm l-lactate. Quantification 300 s after glutamate is shown. Data are from 5 to 12 neurons and two or three independent platings. *p ≤ 0.05 (Student's t test). Inset, Variations in intracellular pH, determined in BCECF-loaded neurons in WT and KO neurons, under the same conditions of the Perceval-HR experiments.
The effects of l-lactate on glutamate-induced increase in OCR were next studied in WT (Fig. 5G) and aralar-KO (Fig. 5H) neurons incubated in the presence of 2.5 mm glucose alone, or supplemented with 2 mm l-lactate. Pyruvate (2 mm) was used as a substrate independent of ARALAR-MAS activity. The presence of these additional substrates did not change the basal respiratory rate or apparent respiratory control ratio, which reflects the contribution of ATP synthesis and proton leak to the OCR (results not shown) but the presence of pyruvate increased mitochondrial uncoupled respiration in both genotypes (results not shown). As shown before, aralar-KO neurons had a decreased OCR in the presence of glucose as only substrate, which remained lower than control neurons also when l-lactate and pyruvate were supplied (data not shown).
d-Lactate or pyruvate supply in the presence of 2.5 mm glucose enhanced the initial glutamate-stimulation of OCR in WT cultures (Fig. 5G,I). This was matched by an increase in ATP production, as indicated by the smaller decrease in the ATP/ADP ratio upon glutamate addition when 2 mm l-lactate was supplied (Fig. 5J). However, l-lactate had a much smaller effect on the fall of the ATP/ADP ratio in aralar-KO neurons (Fig. 5J), even though intracellular pH acidification was the same in both genotypes (Fig. 5J, inset), and no effect on glutamate-stimulated respiration (Fig. 5H,I), which remained much smaller than in control neurons. The inability of l-lactate to sustain glutamate-stimulated respiration strikingly contrasts with the effect of pyruvate supplementation, which greatly increased glutamate-stimulated respiration in aralar-KO cultures (Fig. 5H,I).
These results indicate that, in this high-energy demanding state, glycolysis and l-lactate utilization by mitochondria, both of which require MAS, are unable to meet the demand for substrates in mitochondria from aralar-KO neurons, whereas the pyruvate supply does. They also confirm that l-lactate utilization by mitochondria is completely dependent on MAS and ARALAR. Together, the results indicate that most of the energy effects of l-lactate are lost in aralar-KO neurons, particularly those involving mitochondrial function. However, l-lactate potentiation of glutamate-induced increase in cytosolic calcium is maintained in aralar-KO neurons, suggesting that it does not require l-lactate effects on mitochondrial metabolism.
Effects of l-lactate on survival and oxidative stress of ARALAR-deficient primary neuronal cultures challenged with exogenous glutamate
To test whether the exogenous l-lactate confers resistance to glutamate excitotoxicity through a mechanism requiring ARALAR-MAS, we have studied the effect of l-lactate (and pyruvate) in addition to glucose on in vitro glutamate excitotoxicity. Figure 6 shows that the presence of 10 mm l-lactate in hypoglycemic conditions (Fig. 6A,B) or during exposure to 25 μm glutamate (Fig. 6C) confers a marked resistance to cell death in control, but not in aralar-KO neurons (Fig. 6B,C), whereas 2 mm pyruvate, whose utilization does not require MAS, protects both types of neurons (Fig. 6C).
Figure 6.
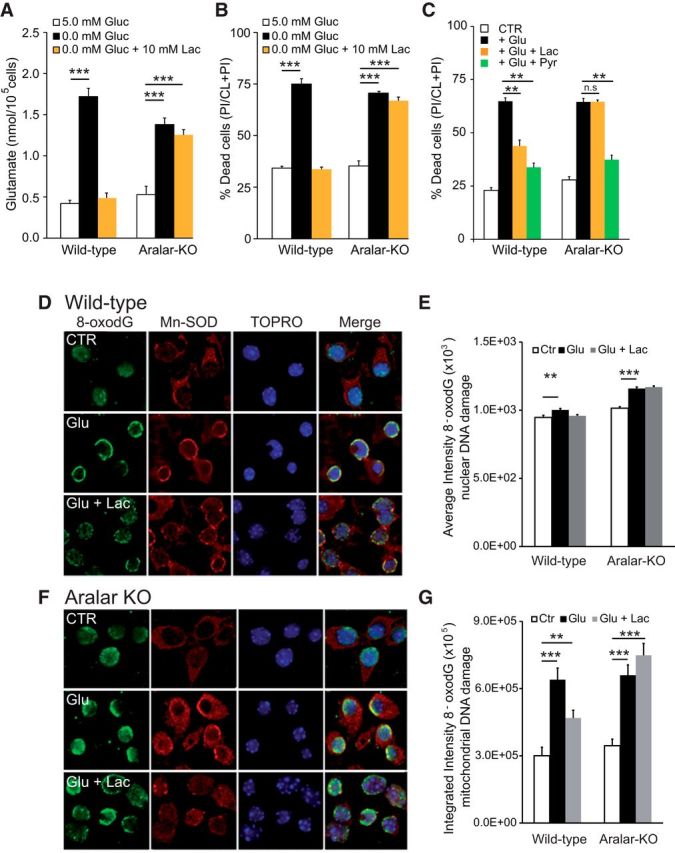
Effects of l-lactate on survival and oxidative stress of ARALAR-deficient primary neuronal cultures challenged with exogenous glutamate. A, Increase in glutamate released to the neuronal culture media, 24 h after treatment in conditions of 10 mm l-lactate under glucose deprivation in WT and aralar-KO cultures. B, Cellular viability was evaluated 24 h after maintaining cultures for 6 h in the previously mentioned condition. Data are calculated as an increase in percentage of dead cells, compared with control condition with 5.0 mm glucose using the calcein-AM/propidium iodide method. C, Cortical cell cultures were pretreated with 10 mm l-lactate or 2 mm pyruvate 30 min before 5 min exposure to 25 μm glutamate and maintained for 24 h in the presence of l-lactate or pyruvate. Data are expressed as percentage of dead cells, PI/(CL+PI), under control treatment (Ctr), glutamate exposure (Glu 25), glutamate plus l-lactate (Glu 25 + Lac), or glutamate plus pyruvate (Glu25 + pyr). Data are mean ± SEM from three independent experiments. *p ≤ 0.05 (one-way ANOVA followed by Student-Newman-Keuls t test). **p ≤ 0.01 (one-way ANOVA followed by Student-Newman-Keuls t test). D–G, Detection of 8-OHdG immunostaining in mitochondrial and nuclear DNA induced by 100 μm glutamate exposure for 30 min alone or in the presence of 10 mm l-lactate. Photomicrographs representative for control (CTR), glutamate (Glu), or glutamate plus l-lactate (Glu + Lac) conditions are shown in control (D) and aralar-KO (F). Coimmunostaining was performed using TOPRO-3 (nuclear marker) or Mn-SOD (mitochondrial marker) and quantified in E and G, respectively, to elucidate the primary DNA damage induced in our experimental conditions. Integrated and average fluorescence intensity was calculated using the MetaMorph program. n = 177–349, or 218–304 cells per condition, in WT and aralar-KO cultures, respectively. *p ≤ 0.05 (ANOVA followed by followed by Student-Newman-Keuls t test). **p ≤ 0.01 (ANOVA followed by followed by Student-Newman-Keuls t test). ***p ≤ 0.001 (ANOVA followed by followed by Student-Newman-Keuls t test). n.s, Not significant.
In vitro, any l-lactate generated by cultured neurons rapidly diffuses out into the cell culture medium promoting a negligible intracellular to extracellular volume ratio. Moreover, in our cellular cultures, astrocytes are quite scarce so the supply of lactate from astrocytes to neurons is inconsiderable. Thus, in normoglycemic conditions, it may be assumed that glucose is the sole substrate, and endogenous lactate has an irrelevant metabolic impact.
Glutamate induces the production of superoxide and other reactive oxygen species (ROS) through different routes (Nicholls, 2008; Brennan-Minnella et al., 2013), and ROS scavengers protect against excitotoxic cell death (Vergun et al., 2001). Oxidative stress causes the appearance of oxidized nucleosides, particularly 8-OHdG in nuclear and mitochondrial DNA, which can be detected by 8-OHdG antibodies. Immunofluorescence and confocal microscopy were used to recognize 8-OHdG using selective mitochondria versus nucleus labeling. Thirty minute 100 μm glutamate-exposure induced an early appearance of 8-OHdG in mitochondria (assessed by colocalization with anti Mn-SOD antibodies) in both WT and aralar-KO neurons (Fig. 6D,F). Glutamate induced 8-OHdG staining in nuclei (Fig. 6E) as judged from colabeling with TOPRO-3 (Fig. 6D,F) was clearly weaker than that in mitochondria (Fig. 6G). This could reflect a specific increase in ROS levels in neuronal mitochondrial during glutamate exposure together with an enhanced vulnerability of mitochondrial versus nuclear DNA to oxidative damage (Barja and Herrero, 2000).
Mitochondrial 8-OHdG staining was increased after glutamate exposure both in WT in aralar-KO neurons (Fig. 6D–F), suggesting that, even though stimulation of OCR is lower in aralar-KO neurons, no difference in mitochondrial ROS-mediated damage to DNA occurs after glutamate exposure. Interestingly, 10 mm l-lactate significantly protected from glutamate-induced 8-OHdG labeling of mitochondrial DNA in WT but not in aralar-KO neurons (Fig. 6D,F,G). We have previously shown that the striatum, a particularly oxidative stress-sensitive brain region, undergoes a prominent rise in the GSSG/GSH ratio in aralar KO mice, which was attributed to a limited NADH formation in mitochondria, and subsequent reduced activity of the detoxifying system (NADH-NADP transhydrogenase, GSH reductase, and peroxidase) (Llorente-Folch et al., 2013). Similarly, deficiency in l-lactate utilization through ARALAR-MAS activity might limit mitochondrial NADH formation and jeopardize the ROS detoxifying capacity as previously observed in striatum in aralar-KO brain (Llorente-Folch et al., 2013).
Discussion
Glutamate, the main excitatory neurotransmitter in brain, imposes a high workload in cerebral cortical neurons, mostly due to Na+ entry. Neuronal respiration is stimulated in response to this workload and stimulation requires the activity of MAS.
In the present work, we report that the lack of ARALAR does not lead to major differences in ionic fluxes due to glutamate stimulation in cortical neurons, but it results in a substantial failure to activate glutamate-stimulated respiration. Indeed, the presence of ARALAR allows a larger response to glutamate in terms of upregulation of respiration. However, contrary to our initial expectations, ARALAR deficiency does not aggravate excitotoxic neuronal death under hypoglycemia, when glucose metabolism must be stopped, or in the presence of glucose, but it does when l-lactate is present as an additional source. As the role of ARALAR-MAS is to oxidize cytosolic NADH in mitochondria and maintain oxidative glucose utilization, halting glucose use in hypoglycemia explains why the lack of ARALAR has no influence on glutamate release or glutamate excitotoxicity. But it does not explain why it has no influence when glucose is present.
A possible explanation for these findings lies on the mechanism of neuronal death triggered by glutamate, which ends up in energetic failure (Nicholls et al., 2007; Duchen, 2012; Rodriguez-Rodriguez et al., 2013). It involves the early activation of the nuclear PARP1 (Zhang et al., 1994), which cleaves NAD+ to nicotinamide and ADP-ribose and couples one or more ADP-ribose units to acceptor proteins. Loss of NAD+ is one of the reasons why ARALAR-MAS may become dispensable in glutamate excitotoxicity. Cytosolic NAD+ loss may lead to a block of glycolysis at the level of the only enzyme of the pathway, which requires NAD+ and the operation of MAS, GA3PDH1 (Alano et al., 2010; Kim et al., 2011; Duchen, 2012), making MAS dispensable under that condition. In axons, NAD+ destruction may be also caused by SARM1 activation (Gerdts et al., 2015).
PAR polymers are rapidly released to the cytosol and other cellular compartments and have a direct action on mitochondria leading to a fall in mitochondrial and cytosolic ATP and a decrease in OCR (Rueda et al., 2015). The fall in mitochondrial ATP is partly counteracted by the entry of adenine nucleotides in mitochondria through the mitochondrial carrier of ATP-Mg/Pi, SCaMC3 (Rueda et al., 2015). PAR also inhibits mitochondria-associated hexokinase1, leading to a block in glycolysis (Andrabi et al., 2014; Fouquerel et al., 2014) and, consequently, substrate supply.
Another explanation for the transient nature of glutamate-stimulated respiration lies on the counteracting effects of the increase in matrix Ca2+ caused by glutamate. Matrix Ca2+ increases respiration by activation of mitochondrial dehydrogenases but also lowers matrix α-ketoglutarate (αKG) levels (due to Ca2+-dependent drop of the Km for αKG of αKG dehydrogenase) and thus the activity of the OGC, the second member of MAS, causing an inhibition of MAS (Pardo et al., 2006; Contreras and Satrústegui, 2009; Satrústegui and Bak, 2015) opposing the increase in respiration.
The combined and not mutually exclusive effects of NAD+ loss, matrix Ca2+-dependent inhibition of OGC-MAS, limited respiratory response, and reduced glucose phosphorylation and substrate supply may result in a metabolic limitation in which ARALAR-MAS is probably dispensable.
d-Lactate is an alternative energy substrate for the brain, which may be used by neurons when glucose use is impaired, as in glutamate excitotoxicity. It can be a preferential substrate particularly in conditions of hypoxia (Schurr et al., 1997a) and during periods of high activity (Tildon et al., 1993; Pellerin et al., 1998; Kasischke et al., 2004; Pellerin and Magistretti, 2004), as confirmed in vivo (Smith et al., 2003; Serres et al., 2004; Boumezbeur et al., 2010); and it can spare cerebral glucose in the injured human brain (Bouzat et al., 2014).
In the brain, astrocytes are the main source of l-lactate, and glutamate stimulation enhances glucose transport (Loaiza et al., 2003), utilization (Pellerin and Magistretti, 1994, 1996; Porras et al., 2008), and lactate production in astrocytes. Together with the activation of astrocytic glycogen degradation by glutamate (Shulman et al., 2001), this may explain the large increase in local extracellular lactate that follows cortical activation (Hu and Wilson, 1997) and subconvulsive kainate administration (Walls et al., 2014). l-Lactate acts as a buffer between glycolysis and oxidative metabolism and it is exchanged as a fuel between cells and tissues with different glycolytic and oxidative rates. As l-lactate utilization by mitochondria in neurons requires cytosolic NADH oxidation through MAS, ARALAR-deficient neurons may be unable to use it, which may lead to cell demise.
Our results clearly show that l-lactate was able to rescue from glutamate excitotoxicity control, but not ARALAR-deficient neurons (Fig. 6). Both l-lactate and pyruvate potentiated the respiratory response to glutamate in control neurons, but only pyruvate, and not l-lactate, supplementation increased glutamate-stimulated respiration of aralar-KO neurons to the level of control neurons, consistent with an energetic failure at the level of pyruvate supply to mitochondria in these neurons. l-Lactate metabolism bypasses the PARP-1 block of the hexokinase1 step; therefore, substrate supply to mitochondria will be maintained, even if other events downstream of PARP-1 activation remain unchanged. This explains the effect of l-lactate supply in preventing the large decrease in the ATP/ADP ratio caused by glutamate in controls, but not in ARALAR-deficient neurons.
We have also found that l-lactate supply markedly reduces glutamate-induced mitochondrial accumulation of 8-oxoguanosine, a marker of ROS production in control, but not in aralar-KO neurons. l-Lactate metabolism supplies redox equivalents in the cytosol and, through the action of ARALAR-MAS, also to the mitochondrial matrix. Glutathione is more oxidized in the striatum of aralar-KO mice than in that of controls, possibly due to the inability to transfer redox equivalents to the mitochondrial matrix in ARALAR deficiency (Llorente-Folch et al., 2013). In the paradigm of glutamate excitotoxicity, failure to transfer l-lactate-derived reducing equivalents to mitochondria also explains lack of protection by l-lactate in aralar-KO neurons.
We also noted that l-lactate potentiated glutamate signaling, resulting in a larger glutamate-stimulated [Ca2+]i increase. l-Lactate has been shown to potentiate NMDA receptor activity and its downstream signaling cascade leading to the expression of plasticity related genes, such as Arc, c-Fos, and Zif268 (Yang et al., 2014), and l-lactate uptake into neurons is required for these effects. However, as the increase in glutamate-induced [Ca2+]i signals by l-lactate were observed both on control and aralar-KO neurons, we conclude that they do not require l-lactate metabolism in mitochondria, and that they are probably unrelated to the protection by l-lactate against excitotoxicity.
Finally, we report that, in vivo, the loss of a single aralar allele of in the aralar+/− mouse increases KA-induced seizures and neuronal damage. As there are abundant data indicating that l-lactate levels increase during seizures and l-lactate consumption by neurons also increases (Ros et al., 2001), these results are probably due to inefficient l-lactate utilization in the aralar HT mice. This would entail that, in these mice, ARALAR levels limit the operation of the shuttle in vivo. Indeed, there is evidence that this is the case: (1) brain aspartate levels, which depend on ARALAR activity, are significantly lower than controls at 20 d postnatal (Jalil et al., 2005); (2) brain N-acetyl-aspartate peak determined in vivo by 1H-NMR spectroscopy is also reduced with respect to control (unpublished results); (3) respiration of brain mitochondria on malate plus glutamate and MAS activity reconstituted in skeletal muscle, and brain mitochondria is halved with respect to control (Jalil et al., 2005); and (4) N-acetyl-aspartate levels in cultured neurons are reduced with respect to control (Jalil et al., 2005). Although other explanations cannot be ruled out, the results suggest that l-lactate utilization during seizures requires maximal ARALAR activity to efficiently transfer cytosolic reducing equivalents from NADH to mitochondria. On these grounds, it may be suggested that other situations requiring l-lactate metabolism by neurons will be also affected by partial ARALAR deficiency.
Footnotes
This work was supported by Ministerio de Economía Grant BFU2011-30456 to J.S. and Grant SAF2014-56929R to J.S. and B.P., Centro de Investigación Biomédica en Red de Enfermedades Raras (an initiative of the Instituto de Salud Carlos III), and Comunidad de Madrid Grant S2010/BMD-2402 MITOLAB-CM to J.S., I.P.-L. is a recipient of a fellowship from the Ministerio de Economia y Competitividad. We thank Isabel Manso and Barbara Sesé for technical support.
The authors declare no competing financial interests.
References
- Alano CC, Garnier P, Ying W, Higashi Y, Kauppinen TM, Swanson RA. NAD+ depletion is necessary and sufficient for poly(ADP-ribose) polymerase-1-mediated neuronal death. J Neurosci. 2010;30:2967–2978. doi: 10.1523/JNEUROSCI.5552-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrabi SA, Umanah GK, Chang C, Stevens DA, Karuppagounder SS, Gagné JP, Poirier GG, Dawson VL, Dawson TM. Poly(ADP-ribose) polymerase-dependent energy depletion occurs through inhibition of glycolysis. Proc Natl Acad Sci U S A. 2014;111:10209–10214. doi: 10.1073/pnas.1405158111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barja G, Herrero A. Oxidative damage to mitochondrial DNA is inversely related to maximum life span in the heart and brain of mammals. FASEB J. 2000;14:312–318. doi: 10.1096/fasebj.14.2.312. [DOI] [PubMed] [Google Scholar]
- Barros LF. Metabolic signaling by lactate in the brain. Trends Neurosci. 2013;36:396–404. doi: 10.1016/j.tins.2013.04.002. [DOI] [PubMed] [Google Scholar]
- Ben-Ari Y, Tremblay E, Ottersen OP. [Primary and secondary cerebral lesions produced by kainic acid injections in the rat] Comptes rendus des seances de l'Academie des sciences Serie D, Sciences naturelles. 1979a;288:991–994. [PubMed] [Google Scholar]
- Ben-Ari Y, Lagowska J, Tremblay E, Le Gal La Salle G. A new model of focal status epilepticus: intra-amygdaloid application of kainic acid elicits repetitive secondarily generalized convulsive seizures. Brain Res. 1979b;163:176–179. doi: 10.1016/0006-8993(79)90163-X. [DOI] [PubMed] [Google Scholar]
- Berthet C, Lei H, Thevenet J, Gruetter R, Magistretti PJ, Hirt L. Neuroprotective role of lactate after cerebral ischemia. J Cereb Blood Flow Metab. 2009;29:1780–1789. doi: 10.1038/jcbfm.2009.97. [DOI] [PubMed] [Google Scholar]
- Boumezbeur F, Petersen KF, Cline GW, Mason GF, Behar KL, Shulman GI, Rothman DL. The contribution of blood lactate to brain energy metabolism in humans measured by dynamic 13C nuclear magnetic resonance spectroscopy. J Neurosci. 2010;30:13983–13991. doi: 10.1523/JNEUROSCI.2040-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouzat P, Magistretti PJ, Oddo M. Hypertonic lactate and the injured brain: facts and the potential for positive clinical implications. Intensive Care Med. 2014;40:920–921. doi: 10.1007/s00134-014-3312-x. [DOI] [PubMed] [Google Scholar]
- Brand MD, Nicholls DG. Assessing mitochondrial dysfunction in cells. Biochem J. 2011;435:297–312. doi: 10.1042/BJ20110162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan-Minnella AM, Shen Y, El-Benna J, Swanson RA. Phosphoinositide 3-kinase couples NMDA receptors to superoxide release in excitotoxic neuronal death. Cell Death Dis. 2013;4:e580. doi: 10.1038/cddis.2013.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castillo X, Rosafio K, Wyss MT, Drandarov K, Buck A, Pellerin L, Weber B, Hirt L. A probable dual mode of action for both l-and d-lactate neuroprotection in cerebral ischemia. J Cereb Blood Flow Metab. 2015;35:1561–1569. doi: 10.1038/jcbfm.2015.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi DW. Glutamate neurotoxicity and diseases of the nervous system. Neuron. 1988;1:623–634. doi: 10.1016/0896-6273(88)90162-6. [DOI] [PubMed] [Google Scholar]
- Contreras L, Satrústegui J. Calcium signaling in brain mitochondria: interplay of malate aspartate NADH shuttle and calcium uniporter/mitochondrial dehydrogenase pathways. J Biol Chem. 2009;284:7091–7099. doi: 10.1074/jbc.M808066200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duchen MR. Mitochondria, calcium-dependent neuronal death and neurodegenerative disease. Pflugers Arch. 2012;464:111–121. doi: 10.1007/s00424-012-1112-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duchen MR, Pearce RJ, Biscoe TJ. Use of microfluorimetry to monitor mitochondrial membrane potential in single dissociated mammalian cells. J Physiol. 1990;426:2P. [Google Scholar]
- Emaus RK, Grunwald R, Lemasters JJ. Rhodamine 123 as a probe of transmembrane potential in isolated rat-liver mitochondria: spectral and metabolic properties. Biochim Biophys Acta. 1986;850:436–448. doi: 10.1016/0005-2728(86)90112-X. [DOI] [PubMed] [Google Scholar]
- Farooqui T, Farooqui AA. Aging: an ant factor for the pathogenesis of neurodegenerative diseases. Mech Ageing Dev. 2009;130:203–215. doi: 10.1016/j.mad.2008.11.006. [DOI] [PubMed] [Google Scholar]
- Fouquerel E, Goellner EM, Yu Z, Gagné JP, Barbi de Moura M, Feinstein T, Wheeler D, Redpath P, Li J, Romero G, Migaud M, Van Houten B, Poirier GG, Sobol RW. ARTD1/PARP1 negatively regulates glycolysis by inhibiting hexokinase 1 independent of NAD+ depletion. Cell Rep. 2014;8:1819–1831. doi: 10.1016/j.celrep.2014.08.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerdts J, Brace EJ, Sasaki Y, DiAntonio A, Milbrandt J. SARM1 activation triggers axon degeneration locally via NAD(+) destruction. Science. 2015;348:453–457. doi: 10.1126/science.1258366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gleichmann M, Collis LP, Smith PJ, Mattson MP. Simultaneous single neuron recording of O2 consumption, [Ca2+]i and mitochondrial membrane potential in glutamate toxicity. J Neurochem. 2009;109:644–655. doi: 10.1111/j.1471-4159.2009.05997.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hack N, Balázs R. Selective stimulation of excitatory amino acid receptor subtypes and the survival of granule cells in culture: effect of quisqualate and AMPA. Neurochem Int. 1994;25:235–241. doi: 10.1016/0197-0186(94)90067-1. [DOI] [PubMed] [Google Scholar]
- Hu Y, Wilson GS. A temporary local energy pool coupled to neuronal activity: fluctuations of extracellular lactate levels in rat brain monitored with rapid-response enzyme-based sensor. J Neurochem. 1997;69:1484–1490. doi: 10.1046/j.1471-4159.1997.69041484.x. [DOI] [PubMed] [Google Scholar]
- Jalil MA, Begum L, Contreras L, Pardo B, Iijima M, Li MX, Ramos M, Marmol P, Horiuchi M, Shimotsu K, Nakagawa S, Okubo A, Sameshima M, Isashiki Y, Del Arco A, Kobayashi K, Satrústegui J, Saheki T. Reduced N-acetylaspartate levels in mice lacking Aralar, a brain- and muscle-type mitochondrial aspartate-glutamate carrier. J Biol Chem. 2005;280:31333–31339. doi: 10.1074/jbc.M505286200. [DOI] [PubMed] [Google Scholar]
- Jean-Quartier C, Bondarenko AI, Alam MR, Trenker M, Waldeck-Weiermair M, Malli R, Graier WF. Studying mitochondrial Ca(2+) uptake: a revisit. Mol Cell Endocrinol. 2012;353:114–127. doi: 10.1016/j.mce.2011.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jekabsons MB, Nicholls DG. In situ respiration and bioenergetic status of mitochondria in primary cerebellar granule neuronal cultures exposed continuously to glutamate. J Biol Chem. 2004;279:32989–33000. doi: 10.1074/jbc.M401540200. [DOI] [PubMed] [Google Scholar]
- Kasischke KA, Vishwasrao HD, Fisher PJ, Zipfel WR, Webb WW. Neural activity triggers neuronal oxidative metabolism followed by astrocytic glycolysis. Science. 2004;305:99–103. doi: 10.1126/science.1096485. [DOI] [PubMed] [Google Scholar]
- Kauppinen RA, Taipale HT, Komulainen H. Interrelationships between glucose metabolism, energy state, and the cytosolic free calcium concentration in cortical synaptosomes from the guinea pig. J Neurochem. 1989;53:766–771. doi: 10.1111/j.1471-4159.1989.tb11771.x. [DOI] [PubMed] [Google Scholar]
- Kim SH, Lu HF, Alano CC. Neuronal Sirt3 protects against excitotoxic injury in mouse cortical neuron culture. PLoS One. 2011;6:e14731. doi: 10.1371/journal.pone.0014731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauritzen KH, Morland C, Puchades M, Holm-Hansen S, Hagelin EM, Lauritzen F, Attramadal H, Storm-Mathisen J, Gjedde A, Bergersen LH. Lactate receptor sites link neurotransmission, neurovascular coupling, and brain energy metabolism. Cereb Cortex. 2014;24:2784–2795. doi: 10.1093/cercor/bht136. [DOI] [PubMed] [Google Scholar]
- Llorente-Folch I, Rueda CB, Amigo I, del Arco A, Saheki T, Pardo B, Satrústegui J. Calcium-regulation of mitochondrial respiration maintains ATP homeostasis and requires ARALAR/AGC1-malate aspartate shuttle in intact cortical neurons. J Neurosci. 2013;33:13957–13971. doi: 10.1523/JNEUROSCI.0929-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loaiza A, Porras OH, Barros LF. Glutamate triggers rapid glucose transport stimulation in astrocytes as evidenced by real-time confocal microscopy. J Neurosci. 2003;23:7337–7342. doi: 10.1523/JNEUROSCI.23-19-07337.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lothman EW, Collins RC. Kainic acid induced limbic seizures: metabolic, behavioral, electroencephalographic and neuropathological correlates. Brain Res. 1981;218:299–318. doi: 10.1016/0006-8993(81)91308-1. [DOI] [PubMed] [Google Scholar]
- Lund P, Bergmeyer HU. Methods of enzymatic analysis. Vol. 8. Weinheim: Verlagsgesellschaft; 1986. Glutamine and l-glutamate: UV-method with glutaminase and glutamate dehydrogenase; pp. 357–363. [Google Scholar]
- Mattson MP, Barger SW, Begley JG, Mark RJ. Calcium, free radicals, and excitotoxic neuronal death in primary cell culture. Methods Cell Biol. 1995;46:187–216. doi: 10.1016/S0091-679X(08)61930-5. [DOI] [PubMed] [Google Scholar]
- Maus M, Marin P, Israël M, Glowinski J, Prémont J. Pyruvate and lactate protect striatal neurons against N-methyl-d-aspartate-induced neurotoxicity. Eur J Neurosci. 1999;11:3215–3224. doi: 10.1046/j.1460-9568.1999.00745.x. [DOI] [PubMed] [Google Scholar]
- Mayer ML, Westbrook GL. Permeation and block of N-methyl-d-aspartic acid receptor channels by divalent cations in mouse cultured central neurones. J Physiol. 1987;394:501–527. doi: 10.1113/jphysiol.1987.sp016883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menga A, Iacobazzi V, Infantino V, Avantaggiati ML, Palmieri F. The mitochondrial aspartate/glutamate carrier isoform 1 gene expression is regulated by CREB in neuronal cells. Int J Biochem Cell Biol. 2015;60:157–166. doi: 10.1016/j.biocel.2015.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosienko V, Teschemacher AG, Kasparov S. Is l-lactate a novel signaling molecule in the brain? J Cereb Blood Flow Metab. 2015;35:1069–1075. doi: 10.1038/jcbfm.2015.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholls DG. Oxidative stress and energy crises in neuronal dysfunction. Ann N Y Acad Sci. 2008;1147:53–60. doi: 10.1196/annals.1427.002. [DOI] [PubMed] [Google Scholar]
- Nicholls DG, Vesce S, Kirk L, Chalmers S. Interactions between mitochondrial bioenergetics and cytoplasmic calcium in cultured cerebellar granule cells. Cell Calcium. 2003;34:407–424. doi: 10.1016/S0143-4160(03)00144-1. [DOI] [PubMed] [Google Scholar]
- Nicholls DG, Johnson-Cadwell L, Vesce S, Jekabsons M, Yadava N. Bioenergetics of mitochondria in cultured neurons and their role in glutamate excitotoxicity. J Neurosci Res. 2007;85:3206–3212. doi: 10.1002/jnr.21290. [DOI] [PubMed] [Google Scholar]
- Olney JW, Fuller T, de Gubareff T. Acute dendrotoxic changes in the hippocampus of kainate treated rats. Brain Res. 1979;176:91–100. doi: 10.1016/0006-8993(79)90872-2. [DOI] [PubMed] [Google Scholar]
- Palmer AE, Tsien RY. Measuring calcium signaling using genetically targetable fluorescent indicators. Nat Protoc. 2006;1:1057–1065. doi: 10.1038/nprot.2006.172. [DOI] [PubMed] [Google Scholar]
- Pardo B, Contreras L, Serrano A, Ramos M, Kobayashi K, Iijima M, Saheki T, Satrústegui J. Essential role of Aralar in the transduction of small Ca2+ signals to neuronal mitochondria. J Biol Chem. 2006;281:1039–1047. doi: 10.1074/jbc.M507270200. [DOI] [PubMed] [Google Scholar]
- Pellerin L, Magistretti PJ. Glutamate uptake into astrocytes stimulates aerobic glycolysis: a mechanism coupling neuronal activity to glucose utilization. Proc Natl Acad Sci U S A. 1994;91:10625–10629. doi: 10.1073/pnas.91.22.10625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellerin L, Magistretti PJ. Excitatory amino acids stimulate aerobic glycolysis in astrocytes via an activation of the Na+/K+ ATPase. Dev Neurosci. 1996;18:336–342. doi: 10.1159/000111426. [DOI] [PubMed] [Google Scholar]
- Pellerin L, Magistretti PJ. Neuroscience: let there be (NADH) light. Science. 2004;305:50–52. doi: 10.1126/science.1100428. [DOI] [PubMed] [Google Scholar]
- Pellerin L, Pellegri G, Bittar PG, Charnay Y, Bouras C, Martin JL, Stella N, Magistretti PJ. Evidence supporting the existence of an activity-dependent astrocyte-neuron lactate shuttle. Dev Neurosci. 1998;20:291–299. doi: 10.1159/000017324. [DOI] [PubMed] [Google Scholar]
- Porras OH, Ruminot I, Loaiza A, Barros LF. Na(+)-Ca(2+) cosignaling in the stimulation of the glucose transporter GLUT1 in cultured astrocytes. Glia. 2008;56:59–68. doi: 10.1002/glia.20589. [DOI] [PubMed] [Google Scholar]
- Prichard J, Rothman D, Novotny E, Petroff O, Kuwabara T, Avison M, Howseman A, Hanstock C, Shulman R. Lactate rise detected by 1H NMR in human visual cortex during physiologic stimulation. Proc Natl Acad Sci U S A. 1991;88:5829–5831. doi: 10.1073/pnas.88.13.5829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian W, Van Houten B. Alterations in bioenergetics due to changes in mitochondrial DNA copy number. Methods. 2010;51:452–457. doi: 10.1016/j.ymeth.2010.03.006. [DOI] [PubMed] [Google Scholar]
- Qiu J, Tan YW, Hagenston AM, Martel MA, Kneisel N, Skehel PA, Wyllie DJ, Bading H, Hardingham GE. Mitochondrial calcium uniporter Mcu controls excitotoxicity and is transcriptionally repressed by neuroprotective nuclear calcium signals. Nat Commun. 2013;4:2034. doi: 10.1038/ncomms3034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Racine RJ. Modification of seizure activity by electrical stimulation: II. Motor seizure. Electroencephalogr Clin Neurophysiol. 1972;32:281–294. doi: 10.1016/0013-4694(72)90177-0. [DOI] [PubMed] [Google Scholar]
- Ramos M, del Arco A, Pardo B, Martínez-Serrano A, Martínez-Morales JR, Kobayashi K, Yasuda T, Bogónez E, Bovolenta P, Saheki T, Satrústegui J. Developmental changes in the Ca2+-regulated mitochondrial aspartate-glutamate carrier Aralar1 in brain and prominent expression in the spinal cord. Brain Res Dev Brain Res. 2003;143:33–46. doi: 10.1016/S0165-3806(03)00097-X. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Rodriguez P, Almeida A, Bolaños JP. Brain energy metabolism in glutamate-receptor activation and excitotoxicity: role for APC/C-Cdh1 in the balance glycolysis/pentose phosphate pathway. Neurochem Int. 2013;62:750–756. doi: 10.1016/j.neuint.2013.02.005. [DOI] [PubMed] [Google Scholar]
- Ros J, Pecinska N, Alessandri B, Landolt H, Fillenz M. Lactate reduces glutamate-induced neurotoxicity in rat cortex. J Neurosci Res. 2001;66:790–794. doi: 10.1002/jnr.10043. [DOI] [PubMed] [Google Scholar]
- Rose CR, Ransom BR. Regulation of intracellular sodium in cultured rat hippocampal neurones. J Physiol. 1997;499:573–587. doi: 10.1113/jphysiol.1997.sp021951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rueda CB, Llorente-Folch I, Amigo I, Contreras L, González-Sánchez P, Martínez-Valero P, Juaristi I, Pardo B, del Arco A, Satrústegui J. Ca(2+) regulation of mitochondrial function in neurons. Biochim Biophys Acta. 2014;1837:1617–1624. doi: 10.1016/j.bbabio.2014.04.010. [DOI] [PubMed] [Google Scholar]
- Rueda CB, Traba J, Amigo I, Llorente-Folch I, González-Sánchez P, Pardo B, Esteban JA, del Arco A, Satrústegui J. Mitochondrial ATP-Mg/Pi carrier SCaMC-3/Slc25a23 counteracts PARP-1-dependent fall in mitochondrial ATP caused by excitotoxic insults in neurons. J Neurosci. 2015;35:3566–3581. doi: 10.1523/JNEUROSCI.2702-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz F, Alvarez G, Pereira R, Hernández M, Villalba M, Cruz F, Cerdán S, Bogónez E, Satrústegui J. Protection by pyruvate and malate against glutamate-mediated neurotoxicity. Neuroreport. 1998;9:1277–1282. [PubMed] [Google Scholar]
- San Martín A, Ceballo S, Ruminot I, Lerchundi R, Frommer WB, Barros LF. A genetically encoded FRET lactate sensor and its use to detect the Warburg effect in single cancer cells. PLoS One. 2013;8:e57712. doi: 10.1371/journal.pone.0057712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satrústegui J, Bak LK. Fluctuations in cytosolic calcium regulate the neuronal malate-aspartate NADH shuttle: implications for neuronal energy metabolism. Neurochem Res. 2015;40:2425–2430. doi: 10.1007/s11064-015-1652-8. [DOI] [PubMed] [Google Scholar]
- Satrústegui J, Pardo B, Del Arco A. Mitochondrial transporters as novel targets for intracellular calcium signaling. Physiol Rev. 2007;87:29–67. doi: 10.1152/physrev.00005.2006. [DOI] [PubMed] [Google Scholar]
- Schauwecker PE. The relevance of individual genetic background and its role in animal models of epilepsy. Epilepsy Res. 2011;97:1–11. doi: 10.1016/j.eplepsyres.2011.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schauwecker PE, Steward O. Genetic determinants of susceptibility to excitotoxic cell death: implications for gene targeting approaches. Proc Natl Acad Sci U S A. 1997;94:4103–4108. doi: 10.1073/pnas.94.8.4103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmued LC, Albertson C, Slikker W., Jr Fluoro-Jade: a novel fluorochrome for the sensitive and reliable histochemical localization of neuronal degeneration. Brain Res. 1997;751:37–46. doi: 10.1016/S0006-8993(96)01387-X. [DOI] [PubMed] [Google Scholar]
- Schurr A, Payne RS, Miller JJ, Rigor BM. Brain lactate, not glucose, fuels the recovery of synaptic function from hypoxia upon reoxygenation: an in vitro study. Brain Res. 1997a;744:105–111. doi: 10.1016/S0006-8993(96)01106-7. [DOI] [PubMed] [Google Scholar]
- Schurr A, Payne RS, Miller JJ, Rigor BM. Brain lactate is an obligatory aerobic energy substrate for functional recovery after hypoxia: further in vitro validation. J Neurochem. 1997b;69:423–426. doi: 10.1046/j.1471-4159.1997.69010423.x. [DOI] [PubMed] [Google Scholar]
- Schurr A, Payne RS, Tseng MT, Gozal E, Gozal D. Excitotoxic preconditioning elicited by both glutamate and hypoxia and abolished by lactate transport inhibition in rat hippocampal slices. Neurosci Lett. 2001a;307:151–154. doi: 10.1016/S0304-3940(01)01937-1. [DOI] [PubMed] [Google Scholar]
- Schurr A, Payne RS, Miller JJ, Tseng MT, Rigor BM. Blockade of lactate transport exacerbates delayed neuronal damage in a rat model of cerebral ischemia. Brain Res. 2001b;895:268–272. doi: 10.1016/S0006-8993(01)02082-0. [DOI] [PubMed] [Google Scholar]
- Serres S, Bezancon E, Franconi JM, Merle M. Ex vivo analysis of lactate and glucose metabolism in the rat brain under different states of depressed activity. J Biol Chem. 2004;279:47881–47889. doi: 10.1074/jbc.M409429200. [DOI] [PubMed] [Google Scholar]
- Shulman RG, Hyder F, Rothman DL. Cerebral energetics and the glycogen shunt: neurochemical basis of functional imaging. Proc Natl Acad Sci U S A. 2001;98:6417–6422. doi: 10.1073/pnas.101129298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith D, Pernet A, Hallett WA, Bingham E, Marsden PK, Amiel SA. Lactate: a preferred fuel for human brain metabolism in vivo. J Cereb Blood Flow Metab. 2003;23:658–664. doi: 10.1097/01.WCB.0000063991.19746.11. [DOI] [PubMed] [Google Scholar]
- Sokoloff L. The brain as a chemical machine. Prog Brain Res. 1992;94:19–33. doi: 10.1016/S0079-6123(08)61736-7. [DOI] [PubMed] [Google Scholar]
- Soultanakis RP, Melamede RJ, Bespalov IA, Wallace SS, Beckman KB, Ames BN, Taatjes DJ, Janssen-Heininger YM. Fluorescence detection of 8-oxoguanine in nuclear and mitochondrial DNA of cultured cells using a recombinant Fab and confocal scanning laser microscopy. Free Radic Biol Med. 2000;28:987–998. doi: 10.1016/S0891-5849(00)00185-4. [DOI] [PubMed] [Google Scholar]
- Stout AK, Raphael HM, Kanterewicz BI, Klann E, Reynolds IJ. Glutamate-induced neuron death requires mitochondrial calcium uptake. Nat Neurosci. 1998;1:366–373. doi: 10.1038/1577. [DOI] [PubMed] [Google Scholar]
- Tantama M, Martínez-Francois JR, Mongeon R, Yellen G. Imaging energy status in live cells with a fluorescent biosensor of the intracellular ATP-to-ADP ratio. Nat Commun. 2013;4:2550. doi: 10.1038/ncomms3550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tildon JT, McKenna MC, Stevenson J, Couto R. Transport of l-lactate by cultured rat brain astrocytes. Neurochem Res. 1993;18:177–184. doi: 10.1007/BF01474682. [DOI] [PubMed] [Google Scholar]
- Vergun O, Sobolevsky AI, Yelshansky MV, Keelan J, Khodorov BI, Duchen MR. Exploration of the role of reactive oxygen species in glutamate neurotoxicity in rat hippocampal neurones in culture. J Physiol. 2001;531:147–163. doi: 10.1111/j.1469-7793.2001.0147j.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walls AB, Eyjolfsson EM, Schousboe A, Sonnewald U, Waagepetersen HS. A subconvulsive dose of kainate selectively compromises astrocytic metabolism in the mouse brain in vivo. J Cereb Blood Flow Metab. 2014;34:1340–1346. doi: 10.1038/jcbfm.2014.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang GJ, Randall RD, Thayer SA. Glutamate-induced intracellular acidification of cultured hippocampal neurons demonstrates altered energy metabolism resulting from Ca2+ loads. J Neurophysiol. 1994;72:2563–2569. doi: 10.1152/jn.1994.72.6.2563. [DOI] [PubMed] [Google Scholar]
- Wang Y, Qin ZH. Molecular and cellular mechanisms of excitotoxic neuronal death. Apoptosis. 2010;15:1382–1402. doi: 10.1007/s10495-010-0481-0. [DOI] [PubMed] [Google Scholar]
- Wong EH, Kemp JA, Priestley T, Knight AR, Woodruff GN, Iversen LL. The anticonvulsant MK-801 is a potent N-methyl-d-aspartate antagonist. Proc Natl Acad Sci U S A. 1986;83:7104–7108. doi: 10.1073/pnas.83.18.7104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Ruchti E, Petit JM, Jourdain P, Grenningloh G, Allaman I, Magistretti PJ. Lactate promotes plasticity gene expression by potentiating NMDA signaling in neurons. Proc Natl Acad Sci U S A. 2014;111:12228–12233. doi: 10.1073/pnas.1322912111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yano S, Tokumitsu H, Soderling TR. Calcium promotes cell survival through CaM-K kinase activation of the protein-kinase-B pathway. Nature. 1998;396:584–587. doi: 10.1038/25147. [DOI] [PubMed] [Google Scholar]
- Zhang J, Dawson VL, Dawson TM, Snyder SH. Nitric oxide activation of poly(ADP-ribose) synthetase in neurotoxicity. Science. 1994;263:687–689. doi: 10.1126/science.8080500. [DOI] [PubMed] [Google Scholar]