

Abstract
Growing evidence indicates that sphingosine-1-P (S1P) upregulates glutamate secretion in hippocampal neurons. However, the molecular mechanisms through which S1P enhances excitatory activity remain largely undefined. The aim of this study was to identify presynaptic targets of S1P action controlling exocytosis. Confocal analysis of rat hippocampal neurons showed that S1P applied at nanomolar concentration alters the distribution of Synapsin I (SynI), a presynaptic phosphoprotein that controls the availability of synaptic vesicles for exocytosis. S1P induced SynI relocation to extrasynaptic regions of mature neurons, as well as SynI dispersion from synaptic vesicle clusters present at axonal growth cones of developing neurons. S1P-induced SynI relocation occurred in a Ca2+-independent but ERK-dependent manner, likely through the activation of S1P3 receptors, as it was prevented by the S1P3 receptor selective antagonist CAY1044 and in neurons in which S1P3 receptor was silenced. Our recent evidence indicates that microvesicles (MVs) released by microglia enhance the metabolism of endogenous sphingolipids in neurons and stimulate excitatory transmission. We therefore investigated whether MVs affect SynI distribution and whether endogenous S1P could be involved in the process. Analysis of SynI immunoreactivity showed that exposure to microglial MVs induces SynI mobilization at presynaptic sites and growth cones, whereas the use of inhibitors of sphingolipid cascade identified S1P as the sphingolipid mediating SynI redistribution. Our data represent the first demonstration that S1P induces SynI mobilization from synapses, thereby indicating the phosphoprotein as a novel target through which S1P controls exocytosis.
SIGNIFICANCE STATEMENT Growing evidence indicates that the bioactive lipid sphingosine and its metabolite sphingosine-1-P (S1P) stimulate excitatory transmission. While it has been recently clarified that sphingosine influences directly the exocytotic machinery by activating the synaptic vesicle protein VAMP2 to form SNARE fusion complexes, the molecular mechanism by which S1P promotes neurotransmission remained largely undefined. In this study, we identify Synapsin I, a presynaptic phosphoprotein involved in the control of availability of synaptic vesicles for exocytosis, as the key target of S1P action. In addition, we provide evidence that S1P can be produced at mature axon terminals as well as at immature growth cones in response to microglia-derived signals, which may be important to stabilize nascent synapses and to restore or potentiate transmission.
Keywords: microglia, neurotransmitter release, S-FTY720-vinylphosphonate, S1P3 receptor, sphingosine-1-phosphate, synapsin I
Introduction
Sphingolipids are potent bioactive lipids, which have signaling functions beyond their structural role in the nervous system (Davletov and Montecucco, 2010; García-Martínez et al., 2013). Their signaling action depends on mobilization from neuronal membranes. The sphingolipid sphingosine (Sph) is produced from plasma membrane sphingomyelin by sequential action of sphingomyelinase and ceramidase and can be converted to its polar metabolite sphingosine-1-P (S1P) by sphingosine kinase (SphK).
Growing evidence indicates that sphingolipids stimulate transmitter release from excitatory synaptic terminals. Externally added Sph and S1P stimulate depolarization-induced glutamate release from synaptosomes (Kajimoto et al., 2007; Darios et al., 2009) and increase the rate of spontaneous glutamate transmission in hippocampal slices or in cultures (Darios et al., 2009; Kanno et al., 2010). Endogenous production of Sph or S1P, triggered by sphingomyelinase activity, also activates exocytosis (Darios et al., 2009; Okada et al., 2009; Kanno and Nishizaki, 2011; Antonucci et al., 2012). Furthermore, knock-out of SphK-1, which is recruited to presynaptic terminals in response to neuronal activity (Chan and Sieburth, 2012), results in reduction of evoked transmitter and impairment of long term-potentiation, spatial learning, and memory (Kajimoto et al., 2007; Chan and Sieburth, 2012), confirming that endogenous S1P crucially regulates synaptic activity.
Recently, it has been clarified that Sph, but not S1P, influences directly the exocytotic machinery by activating the synaptic vesicle (SV) protein VAMP2 to form SNARE fusion complexes (Darios et al., 2009; Okada et al., 2009), a molecular event associated with SV tethering (García-Martínez et al., 2013). Conversely, the molecular mechanism by which S1P promotes neurotransmission remains largely undefined. Unlike Sph, S1P, formed intracellularly at presynaptic sites, may stimulate synaptic transmission by an extracellular pathway, after release into the pericellular space and binding to its cell surface receptors in paracrine or autocrine manners (Kajimoto et al., 2007). S1P receptors Type 1 (S1P1), Type 2 (S1P2), Type 3 (S1P3), and Type 5 (S1P5) have been detected by immunofluorescence analysis in neurons (Doi et al., 2013; Kempf et al., 2014). However, the specificity of their labeling by available antibodies still deserves to be assessed (Choi et al., 2011). Out of these receptor types, S1P3 likely controls glutamate release from mossy fiber terminals in the hippocampus (Kanno and Nishizaki, 2011; Choi and Chun, 2013). However, the synaptic effectors of S1P receptors that ultimately control exocytosis remain undefined. Here we identify Synapsin I (SynI), a presynaptic phosphoprotein, as a key target of extracellular S1P action, that promotes availability of SVs for exocytosis.
Materials and Methods
Animals.
All the experimental procedures followed the guidelines established by the European Legislation (Directive 2010/63/EU) and the Italian Legislation (L.D. no 26/2014).
Cell cultures.
Primary neuronal cultures were obtained from the hippocampi of 18-d -old fetal Sprague Dawley rats of either sex (Charles River), A-SMase KO or wild-type (WT) C57BL/6 mice (of either sex) as described previously (Banker and Groslin, 1998; Antonucci et al., 2012). Briefly, dissociated cells were plated onto poly-l-lysine (Sigma-Aldrich) treated coverslips and maintained in Neurobasal with 2% B27 supplement (Invitrogen), antibiotics, glutamine, and glutamate. Neurons were used for experiments at 6 or 14 DIV. Primary microglia from P1 Sprague Dawley rats were obtained and maintained as previously described (Antonucci et al., 2012).
Drugs and reagents.
Stock solutions were prepared by dissolving the following molecules: imipramine, PD98059, H89, ATP, SMase (all from Sigma-Aldrich) in Krebs-Ringer solution (KRH, 25 mm NaCl, 5 mm KCl, 1.2 mm MgSO4, 1.2 mm KH2PO, 2 mm CaCl2, 6 mm d-glucose, and 25 mm HEPES/NaOH, pH 7.4); SK inhibitor (SKI) (Echelon Biosciences), CAY1044 (Cayman Chemicals), and W146 (Avanti Polar Lipids) in DMSO; OEA (Sigma-Aldrich) in ethanol, S1P (Enzo Biochem), and S-FTY720-vinylphosphonate (kind gift from Prof. Robert Bittman) in fatty acid free BSA (4 mg/ml in PBS). Stock solutions were then diluted in fresh medium and administered to neurons.
Microvesicle (MV) isolation and characterization.
MVs shed from microglia upon exposure to 1 mm ATP for 30 min in KRH were pelleted at 10,000 × g after preclearing from cells and debris as described previously (Gabrielli et al., 2015). Routinely, protein content of MVs was determined by Micro BCA protein assay kit (Thermo Fischer Scientific), whereas the number and dimension of MVs were assessed using Nanosight LM10-HS system (Malvern Instruments) configured with a 405 nm laser and EMCDD camera (Hamamatsu Photonics). Videos were collected and analyzed using the NTA software (version 2.3), with the minimal expected particle size, minimum track length, and blur setting all set to automatic. Camera shutter speed was fixed at 15 ms, and camera gain was set to 300. Room temperature was ranging from 25°C to 28°C. MV pellets were resuspended in 400 μl of 0.1 μm-filtered sterile KRH, and five recordings of 30 s were performed for each sample. No particles were detected in 0.1 μm filtered sterile KRH. MVs were used immediately after isolation. The 1.7 × 105 neurons were incubated with 1.42 μg of MVs produced by primary microglia in a microglia-to-neuron ratio of 2:1, for 30 min at 37°C, corresponding to a concentration of 3.55 μg/ml, obtained diluting MVs in 400 μl of medium.
Immunofluorescence staining.
Neuronal cultures were fixed in PBS with 4% PFA and 4% sucrose. The following antibodies were used: mouse anti-SynI (1:100), guinea pig anti-Bassoon (1:300), rabbit anti-SV2 (1:300), rabbit anti-vesicular glutamate transporter 1 (VGLUT1) (1:1000) (all from Synaptic Systems), human anti-glutamic acid decarboxylase (1:100) (from serum of patients, kindly provided by Prof. M. Solimena, Dresden, Germany), rabbit anti-tubulin (1:80, Sigma-Aldrich), rabbit anti-S1P3 (1:100, Cayman Chemicals), and mouse anti-PSD95 (1:400, NeuroMab). Secondary antibodies were conjugated with Alexa-488, Alexa-568, Alexa-633 (Invitrogen). Images were acquired using a Leica SPE confocal microscope equipped with an ACS APO × 63/1.30 oil objective (Leica Microsystems). Fluorescence image processing was performed with ImageJ Software (National Institutes of Health).
Analysis of SynI dispersion.
Acquisition parameters (i.e., laser power, gain, and offset) were kept constant among different experimental settings. To quantify the degree of SynI dispersion (dispersion index) in growth cones, the profile of SynI fluorescence was measured along arbitrary lines in growth cones by using ImageJ software. The number of peaks per micron (peak density), which reflects the number of SynI clusters, was counted in the plot profile by putting threshold equal to 100 arbitrary units. Ratio between the mean peak density from treated and control cultures was subtracted from the value of 1 to obtain the dispersion index. For the quantification of SynI dispersion as cone fraction, excitatory cones with dispersion index >0.2 were considered as dispersed (Gelsomino et al., 2013). Unless otherwise mentioned, only quite large growth cones (≥8 × 10 μm in length/width) were analyzed. Such large growth cones belong to excitatory neurons, whereas inhibitory cones are <5 × 4 μm in size at 6 DIV. At synapses, SynI dispersion was quantified as follows: SynI and Bassoon double-positive puncta (i.e., synaptic SynI puncta) were revealed by generating a SynI/Bassoon double-positive image using the ‘and’ option of ‘image calculator’ function. Total colocalizing area was quantified using the ‘analyze particle’ function. Total SynI fluorescence area was measured in SynI fluorescence images. Finally, synaptic SynI (SynI-Bassoon colocalizing area) area was normalized to total SynI area in each field, and the ratio was considered as an index of Syn redistribution out of synaptic sites. All data are results of at least three independent experiments.
RNA interference.
The 2 DIV hippocampal neurons were incubated with S1P3-specific Accell smart pool siRNAs or nontargeting control pool siRNAs (1:500, Dharmacon, Carlo Erba Reagents) for 96 h (two additions, every 48 h). Neurons were then fixed or lysated and S1P3 protein levels evaluated by immunofluorescence or Western blotting analysis, respectively.
Ca2+ imaging.
The 6 DIV hippocampal neurons were loaded with 2 μm Fura-2 pentacetoxy methylester for 40 min at 37°C, washed and transferred to the recording chamber of an inverted microscope (Axiovert 100; Zeiss) equipped with a Ca2+ imaging unit Polychrome V (TILL Photonics) as described previously (Verderio et al., 2004). Images were collected with a CCD Imago-QE camera (TILL Photonics) and analyzed with TILLvisION 4.5.66 software. After excitation at 340 and 380 nm wavelengths, the emitted light was acquired at 505 nm at 1 Hz. [Ca2+]i was expressed as F340/F380 fluorescence ratio. The ratio values in selected region of interest corresponding to neuronal cell bodies or growth cones were calculated from sequences of images to obtain temporal analysis.
Western blotting.
Neurons were lysed in Laemmli buffer, separated by electrophoresis, blotted on nitrocellulose membrane filters, and probed using rabbit anti-ERK 1/2, anti-P-ERK 1/2 T202/Y204 (1:1000; Cell Signaling Technology), mouse anti-synapsin I (1:1000), mouse anti-SV2 (1:1000), rabbit anti-VGLUT1 (1:1000) (all from Synaptic Systems), rabbit anti-phosphosite 4,5-synapsin (1:3000 (Jovanovic et al., 1996), mouse anti-Bassoon (1:500; Stressgen), rabbit anti-S1P3 receptor (1:1000; Cayman Chemicals), mouse anti-β3 tubulin (1:4000; Promega). Photographic development was by chemiluminescence (GE Healthcare) according to the manufacturer's instructions. Western blot bands were quantified by ImageJ software.
Electrophysiology.
Whole-cell voltage-clamp recordings were performed using a MultiClamp 700A amplifier (Molecular Devices) coupled to a pCLAMP 10 Software (Molecular Devices), and using an inverted Axiovert 200 microscope (Zeiss). Experiments were performed at room temperature (20°C–25°C) in the external control solution KRH. Signals were sampled at 10 kHz and filtered to 4 kHz. Miniature EPSCs (mEPSCs) were recorded at −70 mV in the presence of 1 μm TTX (Tocris Bioscience), using the following internal solution: 130 mm potassium gluconate,10 mm KCl, 1 mm EGTA, 10 mm HEPES, 2 mm MgCl2, 4 mm MgATP, 0.3 mm Tris-GTP, and analyzed using Clampfit software. For measurements of the readily releasable pool (RRP), hypertonic solution containing 6 mm sucrose was infused with a puffer pipette for 4 s in the extracellular solution KRH. Before stimulation, mEPSCs were recorded for 1 min to evaluate the miniature quantal charge. The ratio between the charge transferred by sucrose delivery with respect to the quantal charge of miniature events indicated the number of readily releasable vesicles (Rosenmund and Stevens, 1996).
Statistical analysis.
All data are presented as mean ± SEM from the indicated number of experiments (N). Statistical analysis was performed using Graphpad Prism 6.0 software (Graphpad Software). After testing data for normal distribution, the appropriate statistical test has been used (see figure legends).
Results
S1P induces dispersion of SynI immunoreactivity in the growth cones of excitatory hippocampal neurons
Axonal growth cones of excitatory neurons represent an ideal compartment to analyze the spatial distribution of molecules controlling SV fusion upon S1P stimulation as they are quite large structures, up to 40–50 μm in size, enriched in SVs (Bonanomi et al., 2005; Schenk et al., 2005; Gelsomino et al., 2013). Among components of the exocytotic machinery, SynI is a major regulatory phosphoprotein, which controls the availability of SVs for exocytosis, by tethering them to each other and to the actin cytoskeleton (Cesca et al., 2010; Fornasiero et al., 2010). Therefore, SynI is a good candidate for mediating changes in neurotransmitter release. Confocal analysis showed that S1P administration at 50 nm for 30 min induced relocation of SynI from a clustered to a fully dispersed distribution, in axonal growth cones of developing hippocampal neurons (Fig. 1A,A′). The effects of S1P were reversible, and growth cones fully recovered a clustered SynI distribution 5 h after S1P removal (Fig. 1A,A′). SynI relocation was quantified by calculating the immunoreactivity dispersion index (Fig. 1B) and the fraction of cones with dispersed SynI distribution (Fig. 1C) in control and S1P-treated neurons (see Materials and Methods). Growth cones responding to S1P were found to be 70.642 ± 4.369% and belonged mostly to excitatory neurons, unequivocally identified by staining for the VGLUT1 (Fig. 1D). Indeed, no significant alteration of SynI distribution was induced by S1P in growth cones of GABAergic neurons, identified by glutamic acid decarboxylase staining (Fig. 1D; dispersion index: ctrl 0 ± 0.083, S1P 0.1768 ± 0.044). At 6 DIV, inhibitory cones were rather small (maximum length: 5.3 ± 0.35 μm; maximum width; 3.97 ± 0.39 μm; N = 40), and most of them lacked a flattened lamellipodium. Given their small size, GABAergic growth cones were likely excluded by all subsequent analyses, which were performed in quite large cones, larger than 8 × 10 μm in size. Relocation of SynI was paralleled by SV dispersion, as evidenced by staining for VGLUT1 (Fig. 1E,E′), suggesting dissociation of SVs from the actin cytoskeleton. Western blotting analysis revealed that SynI and VGLUT1 protein levels remained constant after S1P treatment, thus excluding that the reduction in the SynI/VGLUT1 clustered fluorescence may result from protein degradation (Fig. 1F).
Figure 1.

Low nanomolar S1P induces dispersion of SynI immunoreactivity in growth cones. A, Immunofluorescence of 6 DIV hippocampal neurons treated or not with S1P (50 nm) for 30 min and fixed immediately or 5 h after S1P treatment. Neurons are stained for SynI in green and tubulin in red. SynI is clustered in small puncta in control neurons (top), becomes dispersed after S1P exposure (middle), and reacquires a clustered distribution 5 h after S1P removal (bottom). Scale bar, 10 μm. A′, Fluorescence intensity plot (gray levels) of SynI along lines depicted in A. B, Quantification of SynI dispersion under the different experimental conditions (dispersion index, ctrl = 0.047 ± 0.03026; S1P = 0.7179 ± 0.07696; S1P wash = −0.09127 ± 0.0183, one-way ANOVA p < 0.001, Holm–Sidak's multiple-comparison test). C, Fraction of cones with dispersed SynI under the different experimental conditions (ctrl = 1 ± 0.054; S1P 2.784 ± 0.255; S1P wash = 1.086 ± 0.072, N = 3, Kruskal–Wallis one-way ANOVA p < 0.001, Dunn's multiple-comparison test). D, Fraction of cones with dispersed SynI in control and S1P-treated excitatory or inhibitory hippocampal neurons (VGLUT1+ neurons: ctrl = 1 ± 0.222; S1P = 4.866 ± 1.655; N = 3, Mann–Whitney Rank Sum Test p = 0.008; glutamic acid decarboxylase+ neurons: ctrl = 1 ± 0.108; S1P = 1.586 ± 0.242; N = 3, Mann–Whitney Rank Sum Test p = 0.343). E, E′, Immunofluorescence (E) and plot profiles of fluorescence intensity (E′) showing relocation of SynI and V-GLUT1 at growth cones after exposure to S1P. Peak fluorescence of SynI and VGLUT1 decreases and the intensity profiles of SynI and VGLUT1 fluorescence become more uniformly distributed in S1P-treated neurons. F, Western blotting analysis of developing neurons showing lack of degradation of SynI and VGLUT1 after S1P treatment. β3 tubulin is used as sample loading control. *p < 0.05. **p < 0.01. ns, Not significant.
Dispersion of SynI immunoreactivity by S1P occurs through S1P3 and ERK/MAPK pathway
To explore the involvement of S1P receptors in S1P-induced SynI dispersion, neurons were preincubated with the full antagonist of S1P1–3-5 S-FTY720-vinylphosphonate (100 nm) (Valentine et al., 2010), the selective S1P3 antagonist CAY1044 (10 μm) (Koide et al., 2002; Pyne and Pyne, 2011), or the selective S1P1 antagonist W146 (10 μm) (Di Menna et al., 2013). Both the general block of S1P receptors and the selective block of S1P3 largely prevented S1P-induced SynI dispersion at growth cones (Fig. 2A), indicating a role for S1P3 in the control of SynI location. Selective block of S1P1 produced a much less pronounced, but still significant, inhibition of SynI relocation (Fig. 2A). Antagonists alone did not have any effect on SynI distribution (data not shown). To confirm the involvement of S1P3 in SynI dispersion, we knocked down the receptor by exposing developing neurons to S1P3-specific pool siRNAs for 96 h. S1P3 downregulation was assessed by immunofluorescence staining and Western blotting (Fig. 2B,C). We found that S1P-mediated SynI redistribution was largely prevented in neurons in which S1P3 was silenced but not in neurons exposed to nontargeting control pool siRNAs (Fig. 2D).
Figure 2.

S1P-induced dispersion of SynI is mediated mainly by S1P3 via MAPK. A, Fraction of growth cones characterized by dispersed SynI in control neurons or neurons exposed to S1P upon different pharmacological treatments. Application of either the pan S1P receptors inhibitor S-FTY720-vinylphosphonate (S-FTY) or the S1P3 selective antagonist CAY10444 (CAY) fully prevents SynI relocation. The S1P1 selective antagonist W146 inhibits SynI dispersion to a lower extent (ctrl 0.985 ± 0.1077; S1P 2.534 ± 0.1069; S1P+S-FTY 1.414 ± 0.3171; S1P+CAY 1.015 ± 0.017; S1P +W146 1.822 ± 0.1228; N = 4, one-way ANOVA p < 0.001, Holm–Sidak's multiple-comparison test). B, Immunolabeling for S1P3 (red) and SynI (green) in 6 DIV hippocampal neurons. Note the decrease in S1P3 immunoreactivity in neurons treated with S1P3-specific siRNAs. C, Western blotting analysis of neurons treated with S1P3-specific siRNAs or with nontargeting siRNAs for S1P3. β3 tubulin is used as loading control (Student's t test p = 0.0051). D, Fraction of growth cones characterized by dispersed SynI under S1P treatment in control neurons, neurons treated with S1P3-specific siRNAs, or nontargeting control siRNAs (nontargeting siRNA-treated neurons: ctrl 1 ± 0.059, S1P 2.073 ± 0.070; t test p < 0.001; S1P3-siRNA-treated neurons: ctrl 1.015 ± 0.068, S1P 0.699 ± 0.083; t test p = 0.074). E, Representative temporal plot of [Ca2+]i responses (measured as variation in the F340/F380 fluorescence ratio) induced by application of 100 nm S1P, followed by depolarization with 30 mm KCl in the cell body and the axonal growth cone of a developing neuron. F, Analysis of S1P-induced SynI dispersion, expressed as normalized fraction of cones with dispersed SynI staining, in either control neurons (+Ca2+) or neurons maintained in Ca2+-free medium and loaded with the cell-permeable Ca2+ chelator BAPTA (−Ca2+). Intracellular and extracellular Ca2+ chelation does not impact on SynI dispersion induced by 50 nm S1P (ctrl+Ca+ + 0.988 ± 0.125; S1P+Ca+ + 2.136 ± 0.31; ctrl-Ca2+ 1.228 ± 0.037; S1P-Ca2+ 2.379 ± 0.152; N = 3, one-way ANOVA p = 0.0008, Holm–Sidak's for multiple-comparison test). G, Fraction of growth cones characterized by dispersed SynI in control neurons and in neurons treated with S1P in the presence of either the MEK inhibitor PD98059 or the PKA inhibitor H89. Application of PD98059 prevents SynI relocation (ctrl 1.008 ± 0.039; S1P 2.487 ± 0.364; S1P+PD98059 1.107 ± 0.150; S1P+H89 2.099 ± 0.159; N = 3, one-way ANOVA p = 0.0027, Holm–Sidak's for multiple-comparison test). H, Western blotting of 6 DIV neurons for phosphosites 4/5 Syn (P-4/5 Syn) and total SynI. Quantification shows phosphorylation of SynI at ERK sites after treatment with 50 nm S1P (one-way ANOVA p = 0.0439, Kruskal–Wallis for multiple-comparison test). *p < 0.05. **p < 0.01. ***p < 0.001. ns, Not significant.
S1P3 is coupled to several heterotrimeric G-proteins and effector kinases (Choi and Chun, 2013), including PKA, CaMKs, and ERK/MAPK, which phosphorylate SynI at multiple sites and decrease the binding of the protein to SVs and/or F-actin (Cesca et al., 2010). We first explored the involvement of Ca2+ mobilization and Ca2+-dependent kinases in SynI dispersion triggered by S1P receptors. As previously described (Giussani et al., 2007), single-cell Ca2+ imaging experiments showed the lack of Ca2+ responses to 10–100 nm S1P in growth cones or cell bodies of developing neurons, loaded with the Ca2+ dye FURA-2 (Fig. 2E; no growth cones showed Ca2+ response, N = 20; no cell body showed Ca2+ response, N = 34). Consistently, SynI dispersion still occurred in neurons loaded with the Ca2+ chelator BAPTA and challenged with S1P in Ca2+-free medium (Fig. 2F). Collectively, these findings excluded a role of Ca2+ mobilization and Ca2+-dependent kinases in SynI dispersion. We next investigated the possible involvement of ERK/MAPK and PKA pathways in the process. Western blotting of developing neurons with antibodies recognizing the phosphorylated form of ERK 1/2 (p-ERK) showed a significant increase of ERK 1/2 phosphorylation level in response to 50 nm S1P (ctrl 1.013 ± 0.01033; S1P 1.857 ± 0.1099; N = 3, t test p = 0.0044). Accordingly, S1P-induced SynI dispersion was largely prevented by the specific MEK1 inhibitor PD98059 (30 μm), but not by the PKA inhibitor H89 (10 μm), thus selectively involving ERK 1/2 signaling pathways in SynI relocation (Fig. 2G). To provide a direct demonstration that ERK-dependent phosphorylation of SynI was involved in S1P action, neurons were exposed or not to S1P and analyzed by Western blotting with specific antibodies for SynI phosphorylated by ERK at sites 4 and 5. A significant increase in SynI phosphorylation was detected 5–8 min after S1P treatment (Fig. 2H).
Endogenous S1P controls SynI distribution in axonal growth cones
We have recently shown that microglia, the immune cells of the nervous system, release extracellular MVs, which enhance sphingolipid metabolism in neurons and stimulate excitatory transmission (Antonucci et al., 2012; Gabrielli et al., 2015). Thus, we used microglia-derived MVs to stimulate sphingolipid metabolism, trigger production of Sph and S1P in neurons, and test whether endogenous S1P may regulate SynI distribution. Concentration and dimension of MVs produced by cultured microglia were assessed by Nanosight Tracking Analysis (Fig. 3A), whereas protein content of MVs was evaluated by BCA protein assay. Incubation of neurons with microglial MVs (3.55 μg protein/ml) for 30 min produced a clear dispersion of SynI in axonal growth cones (Fig. 3B,B′), as indicated by the analysis of dispersion index or by the fraction of dispersed growth cones (dispersion index: ctrl 0 ± 0.1; MVs 0.4561 ± 0.1104; N = 5, t test p = 0.0129; cone fraction: ctrl 0.145 ± 0.024; MVs 0.4082 ± 0.045; N = 14, t test p < 0.0001). SynI dispersion evoked by MVs was largely prevented by preincubation of neurons with specific inhibitors of the sphingolipid cascade: the acid sphingomyelinase inhibitor imipramine (1 μm), which blocks sphingomyelin metabolism to ceramide; the ceramidase inhibitor N-oleoylethanolamine (37 μm OEA), which inhibits ceramide metabolism to sphingosine (Strelow et al., 2000); or the sphingosine kinase inhibitor SKI-1 (2 μm), which prevents biosynthesis of S1P from Sph (Kanno et al., 2010) (Fig. 3C). Accordingly, SynI dispersion evoked by MVs did not occur in acid sphingomyelin knock-out (ASMase−/−) neurons, in which S1P biosynthesis from sphingomyelin is genetically impaired (Fig. 3D), whereas it could be induced by application of exogenous sphingomyelinase (cone fraction: ctrl 0.16 ± 0.06; A-SMase-treated 0.773 ± 0.028; N = 3, t test p = 0.0008). Similarly to exogenous S1P, endogenous S1P, produced upon neuron exposure to microglial MVs, triggered SynI dispersion by an extracellular action, acting on S1P3. Indeed, SynI redistribution was blocked by the pan S1P antagonist S-FTY720-vinylphosphonate and the S1P3 selective antagonist CAY1044, but not by the S1P1 selective antagonist W146 (Fig. 3E). SynI relocation, induced by extracellular S1P generated upon treatment with MVs, occurred in a Ca2+-independent (Fig. 3F) but ERK-dependent manner, as it was almost completely prevented by the specific ERK inhibitor PD98059 (30 μm) (Fig. 3G).
Figure 3.
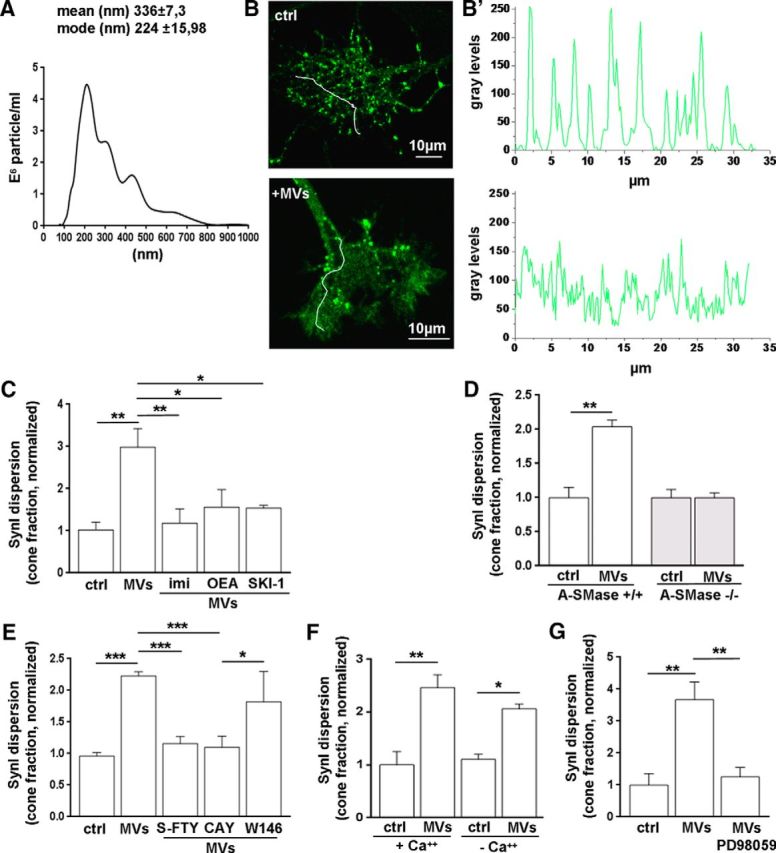
Endogenous S1P induces SynI dispersion in growth cones. A, Representative size profile of MVs isolated by differential centrifugation at 10,000 × g from the supernatant of 1 × 106 primary microglial cells after removal of cellular debris, resuspended in 400 μl of 0.1 μm-filtered sterile KRH and analyzed by Nanosight. B, B′, Fluorescence images of growth cones, from control (top) or MVs-treated (bottom) neurons, stained for SynI. The relative plot profiles of fluorescence intensity along arbitrary lines (white lines) are shown in B′. Scale bars, 10 μm. C, Quantification of SynI dispersion (cone fraction) in neurons treated with MVs in the presence or absence of the A-SMase inhibitor imipramine (imi), the ceramidase inhibitor OEA, and the sphingosine kinase inhibitor SKI-1 (ctrl 1.022 ± 0.186; MVs 2.986 ± 0.214; MVs+imi 1.183 ± 0.336; MVs+OEA 1.563 ± 0–412; MVs+SKI-1 1.538 ± 0.066; N = 3–5, one-way ANOVA p = 0.0028, Holm–Sidak's for multiple-comparison test). D, Dispersion of SynI in developing neurons established from WT or A-SMase KO mice. Data are expressed as dispersed cone fraction (WT ctrl 1 ± 0.15; WT MVs 2.04 ± 0.095; KO ctrl 1 ± 0.118; KO MVs 0.997 ± 0.072; N = 3, t test p = 0.021). E, Graphs represent the fraction of growth cones displaying dispersed SynI upon various pharmacological treatments. The pan S1P receptor antagonist (s-FTY) and the selective S1P3 antagonist (CAY), but not S1P1 antagonist (W146), fully prevent SynI relocation in response to MVs (ctrl 0.961 ± 0.055; MVs 2.231 ± 0.063; MVs+S-FTY 1.157 ± 0.111; MVs+CAY 1.099 ± 0.177; MVs+W146 1.819 ± 0.48; N = 3–7, one-way ANOVA p < 0.0001, Holm–Sidak's multiple-comparison test). F, Chelation of extracellular and intracellular Ca2+ does not influence SynI relocation evoked by MVs (ctrl+Ca2+ 1 ± 0.254; MVs+Ca2+ 2.461 ± 0.242; ctrl-Ca2+ 1.106 ± 0.1; MVs-Ca2+ 2.063 ± 0.091; N = 3; one-way ANOVA p = 0.013, Holm–Sidak's for multiple-comparison test). G, Application of the MAPK inhibitor PD98059 prevents SynI relocation evoked by MVs (ctrl 0.999 ± 0.355; MVs 3.671 ± 0.555; MVs+PD98059 1.258 ± 0.296; N = 3, one-way ANOVA p = 0.028, Holm–Sidak's for multiple-comparison test). *p < 0.05. **p < 0.01. ***p < 0.001.
S1P-induced SynI relocation occurs upstream glutamate release
Our previous evidence indicates that activation of presynaptic AMPA receptors induces ERK 1/2 phosphorylation and SynI dispersion at growth cones (Schenk et al., 2005). Thus, endogenous glutamate, released upon S1P stimulation, may induce SynI dispersion through activation of presynaptic AMPA receptors. This would imply that SynI is a target rather than a promoter of glutamate release. To address this possibility, neurons were stimulated with S1P or microglial MVs in the presence of the AMPA receptor antagonist CNQX (10 μm). In both cases, SynI dispersion was unaffected by CNQX, indicating that SynI does not redistribute as a consequence of enhanced glutamate release and presynaptic glutamate receptor stimulation (Fig. 4A,B).
Figure 4.

S1P induces SynI relocation upstream glutamate release. A, B, Graphs represent normalized fraction of growth cones displaying dispersed SynI in control neurons, neurons exposed to S1P (A) or exposed to microglia-derived MVs (B) in the presence or absence of the AMPA receptor antagonist CNQX (A: ctrl 1.00 ± 0.22; S1P 2.613 ± 0.076; S1P+CNQX/APV 2.701 ± 0.063; N = 3, one-way ANOVA p < 0.0001, Holm–Sidak's for multiple-comparison test. B, ctrl 1.001 ± 0.03; MVs 2.128 ± 0.088; MVs+CNQX/APV 1.982 ± 0.084, N = 3, one-way ANOVA p < 0.0001, Holm–Sidak's for multiple-comparison test). ***p < 0.001.
S1P affects SynI distribution at the mature synapse
We next investigated whether S1P also affects SynI distribution in the presynaptic compartment of fully developed neurons; 14–16 DIV neurons were exposed to 50 nm S1P for 30 min, fixed, and counterstained for the presynaptic active zone protein Bassoon. We observed a reduction of the typical punctuate staining of SynI upon challenge with S1P, whereas staining for Bassoon remained punctuate (Fig. 5A,A′). Accordingly, a clear decrease in the ratio between synaptic and total SynI immunoreactivity was measured in neurons exposed to S1P (Fig. 5B). Colocalization analysis for the SV integral protein SV2 (Bassoon/SynI/SV2) revealed that SynI dispersion was accompanied by SV relocation into the perisynaptic area (Fig. 5A,A′). Conversely, postsynaptic density organization was unchanged. Indeed, PSD-95 staining remained strictly punctate after S1P application (Fig. 5C), and no changes of the mean size of PSD95 clusters were observed (mean size μm2: ctrl 0.1562 ± 0.019; S1P 0.1519 ± 0.004; N = 3). The presynaptic action of S1P was reversible, prevented by the S1P3 receptor antagonist CAY1044 (Fig. 5B) and not associated with synaptic protein degradation (Fig. 5D), similarly to the S1P action at the developing growth cone.
Figure 5.

S1P-mediated SynI relocation at mature synapses. A, Confocal images showing synaptic contacts triple stained for SynI (green), the SV protein SV2 (red), and the active zone protein Bassoon (blue) in 14 DIV neurons maintained in control conditions or exposed to microglia-derived MVs or S1P. Scale bars, 10 μm. A′, Fluorescence intensity profiles of SynI, SV2, and Bassoon along the white lines depicted in A (top images) show SynI and SV relocation after S1P or MVs exposure. B, Quantification of synaptic versus total SynI area in mature neurons fixed either immediately after S1P treatment or 5 h after S1P removal (S1P wash), and in neurons pretreated with the S1P3 receptor antagonist CAY10444 (synaptic/total SynI area, ctrl 1.000; S1P 0.640 ± 0.041; S1P-wash 1.170 ± 0.106; CAY+S1P 0.944 ± 0.145, N = 3, normalized data, one-way ANOVA p < 0.001, Holm–Sidak's multiple-comparison test; for details, see Materials and Methods). C, Representative images of 14 DIV cultures stained for the postsynaptic protein PSD-95 (green) and tubulin (red). Scale bars, 5 μm. D, Western blotting analysis of mature neurons showing lack of degradation of SynI, Bassoon (Basn), VGLUT1, and SV2 under S1P treatment for 30 min. β3 tubulin is used as sample loading control (Student's t test: SynI p = 0.8497, Bassoon p = 0. 8968,VGLUT1 p = 0.9967, SV2 p = 0.6362). E, Quantification of SynI dispersion evoked by microglia-derived MVs in the presence or not of inhibitors of sphingolipid metabolism (synaptic/total SynI area, normalized to control: ctrl 0.99 ± 0.0002; MVs 0.48 ± 0.06; MVs+OEA 0.9375 ± 0.1424; MVs+SKI-I 0.8198 ± 0.062; MVs+S-FTY 0.9177 ± 0.1205; N = 3, one-way ANOVA p = 0.0063, Holm–Sidak's for multiple-comparison test). *p < 0.05. **p < 0.01.
A similar redistribution of SynI immunoreactivity was observed in response to microglial MVs (Fig. 5E), which enhance Sph/S1P metabolism from neuronal plasma membrane. SynI relocation to extrasynaptic regions evoked by S1P was largely prevented by inhibitors of the sphingolipid cascade (Fig. 5E).
S1P increases the size of ready releasable pool of SVs
The next step was to investigate how the activation of S1P receptors and SynI dispersion impact neurotransmission. Analysis of mEPSCs revealed lack of changes in either mEPSC frequency (ctrl = 2.1563 ± 0.24406 Hz vs 2.53015 ± 0.30 Hz; Fig. 6B) or amplitude (ctrl = 20.3563 ± 1.54 pA vs 22.77733 ± 1.88 pA; Fig. 6C) in neurons treated with nanomolar S1P (50 nm) for 40 min (Fig. 6A–C). However, analysis of quantal charge revealed reduced charge transfer in S1P-treated neurons with respect to the untreated cells (Fig. 6D), suggesting an effect of S1P on postsynaptic AMPA receptors. Exposure of neurons to S1P in the presence of S1P3 inhibitor CAY1044 did not alter frequency and amplitude of excitatory miniature events (Fig. 6A–C; S1P+CAY: frequency = 2.632 ± 0.249 Hz; amplitude = 20.266 ± 1.70318 pA) but produced a partial recovery of the quantal charge reduction associated to S1P treatment (Fig. 6D; ctrl = 129.154 ± 13.82 pC; S1P = 85.055 ± 14.64 pC; S1P+CAY = 107.301 ± 14.74 pC). We next estimated the size of the readily releasable pool of synaptic vesicles (RRP) in response to local applications of hypertonic sucrose solution (4-s-long; Fig. 6E,F). The total charge transferred during the sucrose-evoked EPSCs was not different between control, S1P-treated cultures, and S1P+CAY neurons (Fig. 6E,F; ctrl = 1127.54 ± 134.36 pC; S1P = 1039.46 ± 166.52 pC; S1P+CAY = 782.55 ± 103.017 pC), but a significant increase in the number of released vesicles was observed in S1P-treated neurons (Fig. 6G), which was fully normalized by application of the S1P3 antagonist CAY1044 (Fig. 6G; ctrl = 1.00 ± 0.095 vs 1.87 ± 0.197 normalized value; S1P+CAY = 1.29 ± 0.14; t test ctrl vs S1P: p = 0.038). Thus, all these data, together with immunocytochemical results, indicate that Syn-I dispersion evoked by S1P3 activation associates with an increased number of releasable SVs.
Figure 6.

Low nanomolar S1P increases the size of RRP without influencing mEPSC frequency. A–D, Quantification of mEPSC frequency, amplitude, and quantal charge of 14 DIV hippocampal neurons in control conditions or incubated for 40 min with S1P in the presence or absence of CAY10444. E, Representative sucrose-induced responses under the same conditions as in A. F, G, Quantitative analysis of charge transfer and number of released vesicles measured in hippocampal neurons after hypertonic solution stimulation. The number of vesicles corresponds to the ratio between the total charge transferred during the sucrose delivery and the quantal charge of mEPSCs recorded during the “baseline” period. *p < 0.05. **p < 0.01.
Discussion
Here we identify SynI, a phosphoprotein that controls the availability of SVs for exocytosis, as the molecular target mediating extracellular action of S1P on excitatory synapses. By quantitative immunofluorescence analysis, we show that S1P, applied at nanomolar concentrations (50 nm), activates ERK/MAPK via S1P3 receptors and influences the spatial organization of SynI, both in the growth cones and at mature axon terminals of hippocampal neurons. S1P action is relatively fast, as SynI relocation occurs within 30 min from S1P application. ERK/MAPK pathway, through phosphorylation of SynI at sites 4, 5, and 6 (Cesca et al., 2010), is known to decrease SynI interaction with actin but not with SVs (Jovanovic et al., 2001; Cesca et al., 2010). Accordingly, we show that, upon S1P application, SynI is phosphorylated and redistributes along with SV markers to the presynaptic regions of mature synapses and in the growth cones. In fully developed neurons, mobilization of SVs increases the size of RRP, estimated by measuring responses to high sucrose solution, without changing the rate of spontaneous quantal events. The selective action of S1P-induced SynI dispersion in evoked but not spontaneous transmission is in agreement with a previous study, which associated SynI dispersion, evoked by PKA, to larger RRP of SVs and to enhanced transmission under sustained stimulation but not basal condition (Menegon et al., 2006). Interestingly, the larger pool of releasable SVs following SynI dispersion counteracted synaptic depression and accelerated recovery from depression (Menegon et al., 2006), suggesting that extracellular S1P may increase synapse resistance to fatigue. Our results are also consistent with the evidence that impaired S1P production does not influence quantal release from motor terminals in C. elegans mutants, lacking Sph kinase (Chan et al., 2012). Other studies reported that application of high nanomolar or micromolar concentrations of S1P significantly increases mEPSC frequency in hippocampal slices or cultures (Kajimoto et al., 2007; Kanno et al., 2010; Kanno and Nishizaki, 2011; Antonucci et al., 2012). However, high concentrations of S1P may impact spontaneous glutamate transmission through intracellular targets rather than extracellular action, consistent with the recent identification of several proteins that bind S1P within cells (Kunkel et al., 2013).
The extracellular action of S1P on excitatory transmission differs from that of its lipid precursor Sph which, by activating VAMP-2 to form SNARE fusion complexes, upregulates not only evoked but also spontaneous glutamate transmission in hippocampal neurons and neuromuscular junctions (Darios et al., 2009; Antonucci et al., 2012). It is worth noting, however, that both Sph and S1P increase the size of SVs ready to be exocytosed during stimulation (RRP) (Darios et al., 2009; Antonucci et al., 2012; current study). Thus, RRP appears an important target downstream Sph and S1P production, supporting the idea that the two sphingolipids promote glutamate release through either distinct or codependent mechanisms (Chan et al., 2012).
Identification of the mechanism of action by which S1P promotes exocytosis is of high relevance, not least because of the extensive use of the S1P1,3,4,5 agonist Fingolimod in the treatment of multiple sclerosis (Cohen et al., 2010; Kappos et al., 2010). As the S1P parent compound diffuses into the brain, where it even accumulates (Foster et al., 2007; Hait et al., 2014), it is becoming increasingly important to better define its action on brain cells, including neurons.
On the other hand, which are the stimuli that trigger neurons to endogenously produce and secrete S1P, promoting evoked exocytosis via SynI dispersion? In rat hippocampal neurons, depolarization regulates Sph kinase-1 translocation from cytosol to presynaptic membranes, which results in S1P accumulation at the synapse (Kajimoto et al., 2007). Similarly, in C. elegans, S1P is produced in response to neuronal activity, thanks to the recruitment of Sph kinase to presynaptic regions of motor neurons following muscarinic signaling (Chan and Sieburth, 2012; Chan et al., 2012). However, we recently showed that sphingolipid metabolism can be also regulated at the synapse by non-neuronal cells. We identified microglia, the brain resident immune cells, as a source of lipid messengers, which enhance ceramide and Sph production in rodent neurons, through activation of acid sphingomyelinase (Antonucci et al., 2012). Neuroactive lipids are secreted by reactive microglia through extracellular MVs and signal to neurons upon MV contact with the neuronal surface (Antonucci et al., 2012). We here provide evidence that the sphingolipid cascade induced by microglial MVs does not stop at Sph but proceeds to S1P, which participates to presynaptic stimulation downstream Sph by selectively targeting SynI.
Stimulation of S1P production at axon terminals induced by MVs may be an important mechanism for microglia to restore or potentiate glutamate transmission. At mature synapses, microglia is kept in a resting state by “off signals” released by active neurons (Neumann, 2001; Pocock and Kettenmann, 2007) and production of MVs is low (Verderio et al., 2012). It can be speculated that deficits in neurotransmission, by triggering microglia activation and MV release, may enhance Sph and S1P production to restore synaptic activity, a possibility that will be worth exploring (Turola et al., 2012). However, during brain inflammation, S1P synthesis in response to excessive MV production from overreactive microglia may trigger an excitatory-inhibitory unbalance by selectively promoting transmitter release and resistance to fatigue at excitatory synapses (Antonucci et al., 2012; Gabrielli et al., 2015).
The impact of S1P production in response to microglia-derived MVs may be even stronger at growth cones of immature neurons, where ERK-mediated SynI dispersion has been shown to enhance SV exocytosis (Schenk et al., 2005). S1P-induced acceleration of glutamate release may influence motility of filopodia and favor stabilization of nascent synapses through stimulation of AMPA and kainate receptors (Verderio et al., 1994; Chang and De Camilli, 2001; Schenk et al., 2005; Gelsomino et al., 2013). This effect adds to the complex action by which extracellular S1P orchestrates neurite extension through other signaling pathways, including Rac and Rho, which couple various S1P receptors to actin cytoskeleton (Toman et al., 2004; Milstien et al., 2007). Relevant to this context, a clear role for microglia in favoring synaptogenesis has been recently established, in particular during the early stages of brain development, when microglia display functions, which are later on performed by astrocytes (Schlegelmilch et al., 2011). Hence, the control of S1P production by microglia-derived MVs at growth cones may provide a novel mechanism for the dynamic regulation of neural circuits during development.
Footnotes
Dedicated to the memory of Robert Bittmann, Professor of Chemistry and Biochemistry at City University and Queens College, New York, who kindly provided S-FTY720-vinylphosphonate, a fundamental molecule for experiments of this work. This work was supported in part by FISM Grant2012/R/17 and Eranet-Neuron project “Micromet” to C.V., and 2010JFYFY2-2008 to M.M., I.P. was supported by a Fondazione Umberto Veronesi Fellowship. F.A. was supported by the Italian Ministry of Research and Education program “FIRB giovani” 2010 protocol RBFR10ZBYZ. We thank Dr. Laura Cantone for the use of Nanosight (Università di Milano) and Giulia D'Arrigo (SISSA, Trieste) for help in image analysis.
The authors declare no competing financial interests.
References
- Antonucci F, Turola E, Riganti L, Caleo M, Gabrielli M, Perrotta C, Novellino L, Clementi E, Giussani P, Viani P, Matteoli M, Verderio C. Microvesicles released from microglia stimulate synaptic activity via enhanced sphingolipid metabolism. EMBO J. 2012;31:1231–1240. doi: 10.1038/emboj.2011.489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banker G, Groslin K. Culturing nerve cell. Cambridge, MA: Massachusetts Institute of Technology; 1998. [Google Scholar]
- Bonanomi D, Menegon A, Miccio A, Ferrari G, Corradi A, Kao HT, Benfenati F, Valtorta F. Phosphorylation of synapsin I by cAMP-dependent protein kinase controls synaptic vesicle dynamics in developing neurons. J Neurosci. 2005;25:7299–7308. doi: 10.1523/JNEUROSCI.1573-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cesca F, Baldelli P, Valtorta F, Benfenati F. The synapsins: key actors of synapse function and plasticity. Prog Neurobiol. 2010;91:313–348. doi: 10.1016/j.pneurobio.2010.04.006. [DOI] [PubMed] [Google Scholar]
- Chan JP, Sieburth D. Localized sphingolipid signaling at presynaptic terminals is regulated by calcium influx and promotes recruitment of priming factors. J Neurosci. 2012;32:17909–17920. doi: 10.1523/JNEUROSCI.2808-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan JP, Hu Z, Sieburth D. Recruitment of sphingosine kinase to presynaptic terminals by a conserved muscarinic signaling pathway promotes neurotransmitter release. Genes Dev. 2012;26:1070–1085. doi: 10.1101/gad.188003.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang S, De Camilli P. Glutamate regulates actin-based motility in axonal filopodia. Nat Neurosci. 2001;4:787–793. doi: 10.1038/90489. [DOI] [PubMed] [Google Scholar]
- Choi JW, Chun J. Lysophospholipids and their receptors in the central nervous system. Biochim Biophys Acta. 2013;1831:20–32. doi: 10.1016/j.bbalip.2012.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi JW, Gardell SE, Herr DR, Rivera R, Lee CW, Noguchi K, Teo ST, Yung YC, Lu M, Kennedy G, Chun J. FTY720 (fingolimod) efficacy in an animal model of multiple sclerosis requires astrocyte sphingosine 1-phosphate receptor 1 (S1P1) modulation. Proc Natl Acad Sci U S A. 2011;108:751–756. doi: 10.1073/pnas.1014154108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen JA, Barkhof F, Comi G, Hartung HP, Khatri BO, Montalban X, Pelletier J, Capra R, Gallo P, Izquierdo G, Tiel-Wilck K, de Vera A, Jin J, Stites T, Wu S, Aradhye S, Kappos L. Oral fingolimod or intramuscular interferon for relapsing multiple sclerosis. N Engl J Med. 2010;362:402–415. doi: 10.1056/NEJMoa0907839. [DOI] [PubMed] [Google Scholar]
- Darios F, Wasser C, Shakirzyanova A, Giniatullin A, Goodman K, Munoz-Bravo JL, Raingo J, Jorgacevski J, Kreft M, Zorec R, Rosa JM, Gandia L, Gutiérrez LM, Binz T, Giniatullin R, Kavalali ET, Davletov B. Sphingosine facilitates SNARE complex assembly and activates synaptic vesicle exocytosis. Neuron. 2009;62:683–694. doi: 10.1016/j.neuron.2009.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davletov B, Montecucco C. Lipid function at synapses. Curr Opin Neurobiol. 2010;20:543–549. doi: 10.1016/j.conb.2010.06.008. [DOI] [PubMed] [Google Scholar]
- Di Menna L, Molinaro G, Di Nuzzo L, Riozzi B, Zappulla C, Pozzilli C, Turrini R, Caraci F, Copani A, Battaglia G, Nicoletti F, Bruno V. Fingolimod protects cultured cortical neurons against excitotoxic death. Pharmacol Res. 2013;67:1–9. doi: 10.1016/j.phrs.2012.10.004. [DOI] [PubMed] [Google Scholar]
- Doi Y, Takeuchi H, Horiuchi H, Hanyu T, Kawanokuchi J, Jin S, Parajuli B, Sonobe Y, Mizuno T, Suzumura A. Fingolimod phosphate attenuates oligomeric amyloid β-induced neurotoxicity via increased brain-derived neurotrophic factor expression in neurons. PLoS One. 2013;8:e61988. doi: 10.1371/journal.pone.0061988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fornasiero EF, Bonanomi D, Benfenati F, Valtorta F. The role of synapsins in neuronal development. Cell Mol Life Sci. 2010;67:1383–1396. doi: 10.1007/s00018-009-0227-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster CA, Howard LM, Schweitzer A, Persohn E, Hiestand PC, Balatoni B, Reuschel R, Beerli C, Schwartz M, Billich A. Brain penetration of the oral immunomodulatory drug FTY720 and its phosphorylation in the central nervous system during experimental autoimmune encephalomyelitis: consequences for mode of action in multiple sclerosis. J Pharmacol Exp Ther. 2007;323:469–475. doi: 10.1124/jpet.107.127183. [DOI] [PubMed] [Google Scholar]
- Gabrielli M, Battista N, Riganti L, Prada I, Antonucci F, Cantone L, Matteoli M, Maccarrone M, Verderio C. Active endocannabinoids are secreted on extracellular membrane vesicles. EMBO Rep. 2015;16:213–220. doi: 10.15252/embr.201439668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Martínez V, Villanueva J, Torregrosa-Hetland CJ, Bittman R, Higdon A, Darley-Usmar VM, Davletov B, Gutiérrez LM. Lipid metabolites enhance secretion acting on SNARE microdomains and altering the extent and kinetics of single release events in bovine adrenal chromaffin cells. PLoS One. 2013;8:e75845. doi: 10.1371/journal.pone.0075845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelsomino G, Menna E, Antonucci F, Rodighiero S, Riganti L, Mulle C, Benfenati F, Valtorta F, Verderio C, Matteoli M. Kainate induces mobilization of synaptic vesicles at the growth cone through the activation of protein kinase A. Cereb Cortex. 2013;23:531–541. doi: 10.1093/cercor/bhs026. [DOI] [PubMed] [Google Scholar]
- Giussani P, Ferraretto A, Gravaghi C, Bassi R, Tettamanti G, Riboni L, Viani P. Sphingosine-1-phosphate and calcium signaling in cerebellar astrocytes and differentiated granule cells. Neurochem Res. 2007;32:27–37. doi: 10.1007/s11064-006-9219-3. [DOI] [PubMed] [Google Scholar]
- Hait NC, Wise LE, Allegood JC, O'Brien M, Avni D, Reeves TM, Knapp PE, Lu J, Luo C, Miles MF, Milstien S, Lichtman AH, Spiegel S. Active, phosphorylated fingolimod inhibits histone deacetylases and facilitates fear extinction memory. Nat Neurosci. 2014;17:971–980. doi: 10.1038/nn.3728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jovanovic JN, Benfenati F, Siow YL, Sihra TS, Sanghera JS, Pelech SL, Greengard P, Czernik AJ. Neurotrophins stimulate phosphorylation of synapsin I by MAP kinase and regulate synapsin I-actin interactions. Proc Natl Acad Sci U S A. 1996;93:3679–3683. doi: 10.1073/pnas.93.8.3679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jovanovic JN, Sihra TS, Nairn AC, Hemmings HC, Jr, Greengard P, Czernik AJ. Opposing changes in phosphorylation of specific sites in synapsin I during Ca2+-dependent glutamate release in isolated nerve terminals. J Neurosci. 2001;21:7944–7953. doi: 10.1523/JNEUROSCI.21-20-07944.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kajimoto T, Okada T, Yu H, Goparaju SK, Jahangeer S, Nakamura S. Involvement of sphingosine-1-phosphate in glutamate secretion in hippocampal neurons. Mol Cell Biol. 2007;27:3429–3440. doi: 10.1128/MCB.01465-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanno T, Nishizaki T. Endogenous sphingosine 1-phosphate regulates spontaneous glutamate release from mossy fiber terminals via S1P(3) receptors. Life Sci. 2011;89:137–140. doi: 10.1016/j.lfs.2011.05.021. [DOI] [PubMed] [Google Scholar]
- Kanno T, Nishizaki T, Proia RL, Kajimoto T, Jahangeer S, Okada T, Nakamura S. Regulation of synaptic strength by sphingosine 1-phosphate in the hippocampus. Neuroscience. 2010;171:973–980. doi: 10.1016/j.neuroscience.2010.10.021. [DOI] [PubMed] [Google Scholar]
- Kappos L, Radue EW, O'Connor P, Polman C, Hohlfeld R, Calabresi P, Selmaj K, Agoropoulou C, Leyk M, Zhang-Auberson L, Burtin P. A placebo-controlled trial of oral fingolimod in relapsing multiple sclerosis. N Engl J Med. 2010;362:387–401. doi: 10.1056/NEJMoa0909494. [DOI] [PubMed] [Google Scholar]
- Kempf A, Tews B, Arzt ME, Weinmann O, Obermair FJ, Pernet V, Zagrebelsky M, Delekate A, Iobbi C, Zemmar A, Ristic Z, Gullo M, Spies P, Dodd D, Gygax D, Korte M, Schwab ME. The sphingolipid receptor S1PR2 is a receptor for Nogo: a repressing synaptic plasticity. PLoS Biol. 2014:12. doi: 10.1371/journal.pbio.1001763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koide Y, Hasegawa T, Takahashi A, Endo A, Mochizuki N, Nakagawa M, Nishida A. Development of novel EDG3 antagonists using a 3D database search and their structure-activity relationships. J Med Chem. 2002;45:4629–4638. doi: 10.1021/jm020080c. [DOI] [PubMed] [Google Scholar]
- Kunkel GT, Maceyka M, Milstien S, Spiegel S. Targeting the sphingosine-1-phosphate axis in cancer, inflammation and beyond. Nat Rev Drug Discov. 2013;12:688–702. doi: 10.1038/nrd4099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menegon A, Bonanomi D, Albertinazzi C, Lotti F, Ferrari G, Kao HT, Benfenati F, Baldelli P, Valtorta F. Protein kinase A-mediated synapsin I phosphorylation is a central modulator of Ca2+-dependent synaptic activity. J Neurosci. 2006;26:11670–11681. doi: 10.1523/JNEUROSCI.3321-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milstien S, Gude D, Spiegel S. Sphingosine 1-phosphate in neural signalling and function. Acta Paediatr. 2007;96:40–43. doi: 10.1111/j.1651-2227.2007.00206.x. [DOI] [PubMed] [Google Scholar]
- Neumann H. Control of glial immune function by neurons. Glia. 2001;36:191–199. doi: 10.1002/glia.1108. [DOI] [PubMed] [Google Scholar]
- Okada T, Kajimoto T, Jahangeer S, Nakamura S. Sphingosine kinase/sphingosine 1-phosphate signalling in central nervous system. Cell Signal. 2009;21:7–13. doi: 10.1016/j.cellsig.2008.07.011. [DOI] [PubMed] [Google Scholar]
- Pocock JM, Kettenmann H. Neurotransmitter receptors on microglia. Trends Neurosci. 2007;30:527–535. doi: 10.1016/j.tins.2007.07.007. [DOI] [PubMed] [Google Scholar]
- Pyne NJ, Pyne S. Selectivity and specificity of sphingosine 1-phosphate receptor ligands: “off-targets” or complex pharmacology? Frontiers Pharmacol. 2011;2:26. doi: 10.3389/fphar.2011.00026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenmund C, Stevens CF. Definition of the readily releasable pool of vesicles at hippocampal synapses. Neuron. 1996;16:1197–1207. doi: 10.1016/S0896-6273(00)80146-4. [DOI] [PubMed] [Google Scholar]
- Schenk U, Menna E, Kim T, Passafaro M, Chang S, De Camilli P, Matteoli M. A novel pathway for presynaptic mitogen-activated kinase activation via AMPA receptors. J Neurosci. 2005;25:1654–1663. doi: 10.1523/JNEUROSCI.3074-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlegelmilch T, Henke K, Peri F. Microglia in the developing brain: from immunity to behaviour. Curr Opin Neurobiol. 2011;21:5–10. doi: 10.1016/j.conb.2010.08.004. [DOI] [PubMed] [Google Scholar]
- Strelow A, Bernardo K, Adam-Klages S, Linke T, Sandhoff K, Krönke M, Adam D. Overexpression of acid ceramidase protects from tumor necrosis factor-induced cell death. J Exp Med. 2000;192:601–612. doi: 10.1084/jem.192.5.601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toman RE, Payne SG, Watterson KR, Maceyka M, Lee NH, Milstien S, Bigbee JW, Spiegel S. Differential transactivation of sphingosine-1-phosphate receptors modulates NGF-induced neurite extension. J Cell Biol. 2004;166:381–392. doi: 10.1083/jcb.200402016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turola E, Furlan R, Bianco F, Matteoli M, Verderio C. Microglial microvesicle secretion and intercellular signaling. Front Physiol. 2012;3:149. doi: 10.3389/fphys.2012.00149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valentine WJ, Kiss GN, Liu J, Gotoh M, Murakami-Murofushi K, Pham TC, Baker DL, Parrill AL, Lu X, Sun C, Bittman R, Pyne NJ, Tigyi G. (S)-FTY720-vinylphosphonate, an analogue of the immunosuppressive agent FTY720, is a pan-antagonist of sphingosine 1-phosphate GPCR signaling and inhibits autotaxin activity. Cell Signal. 2010;22:1543–1553. doi: 10.1016/j.cellsig.2010.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verderio C, Coco S, Fumagalli G, Matteoli M. Spatial changes in calcium signaling during the establishment of neuronal polarity and synaptogenesis. J Cell Biol. 1994;126:1527–1536. doi: 10.1083/jcb.126.6.1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verderio C, Pozzi D, Pravettoni E, Inverardi F, Schenk U, Coco S, Proux-Gillardeaux V, Galli T, Rossetto O, Frassoni C, Matteoli M. SNAP-25 modulation of calcium dynamics underlies differences in GABAergic and glutamatergic responsiveness to depolarization. Neuron. 2004;41:599–610. doi: 10.1016/S0896-6273(04)00077-7. [DOI] [PubMed] [Google Scholar]
- Verderio C, Muzio L, Turola E, Bergami A, Novellino L, Ruffini F, Riganti L, Corradini I, Francolini M, Garzetti L, Maiorino C, Servida F, Vercelli A, Rocca M, Dalla Libera D, Martinelli V, Comi G, Martino G, Matteoli M, Furlan R. Myeloid microvesicles are a marker and therapeutic target for neuroinflammation. Ann Neurol. 2012;72:610–624. doi: 10.1002/ana.23627. [DOI] [PubMed] [Google Scholar]