

Abstract
Glaucoma, the leading cause of irreversible blindness worldwide, is characterized by the selective death of retinal ganglion cells (RGCs). Ocular hypertension is the most significant known risk factor for developing the disease, but the mechanism by which elevated pressure damages RGCs is currently unknown. The axonal-enriched microtubule-associated protein tau is a key mediator of neurotoxicity in Alzheimer's disease and other tauopathies. Using a well characterized in vivo rat glaucoma model, we show an age-related increase in endogenous retinal tau that was markedly exacerbated by ocular hypertension. Early alterations in tau phosphorylation, characterized by epitope-dependent hyperphosphorylation and hypophosphorylation, correlated with the appearance of tau oligomers in glaucomatous retinas. Our data demonstrate the mislocalization of tau in the somatodendritic compartment of RGCs subjected to high intraocular pressure. In contrast, tau was depleted from RGC axons in the optic nerve of glaucomatous eyes. Importantly, intraocular administration of short interfering RNA against tau effectively reduced retinal tau accumulation and promoted robust survival of RGC somas and axons, supporting a critical role for tau alterations in ocular hypertension-induced neuronal damage. Our study reveals that glaucoma displays signature pathological features of tauopathies, including tau accumulation, altered phosphorylation, and missorting; and identifies tau as a novel target to counter RGC neurodegeneration in glaucoma and prevalent optic neuropathies.
SIGNIFICANCE STATEMENT In this study, we investigated the role of tau in retinal ganglion cell (RGC) damage in glaucoma. We demonstrate that high intraocular pressure leads to a rapid increase in endogenous retinal tau with altered phosphorylation profile and the formation of tau oligomers. Tau accumulation was primarily observed in RGC dendrites, while tau in RGC axons within the optic nerve was depleted. Attenuation of endogenous retinal tau using a targeted siRNA led to striking protection of RGC somas and axons from hypertension-induced damage. Our study identifies novel and substantial alterations of endogenous tau protein in glaucoma, including abnormal subcellular distribution, an altered phosphorylation profile, and neurotoxicity.
Keywords: Alzheimer's disease, axon degeneration, glaucoma, neurotoxicity, retinal ganglion cell, tau
Introduction
Retinal ganglion cells (RGCs) are prototypical CNS neurons with somatodendritic compartments localized in the innermost part of the retina and axons organized as bundles forming the optic nerve. RGCs are the sole neurons that convey information from the retina to the brain; therefore, their structural and functional integrity is critical for vision. RGC death is the cause of visual deficits in prevalent optic neuropathies including glaucoma, the leading cause of irreversible blindness worldwide (Tham et al., 2014). During glaucoma, axons in the optic nerve head are injured, leading to progressive RGC neurodegeneration (Calkins, 2012; Nickells et al., 2012). High intraocular pressure is the most significant known risk factor for development of the disease (Sit, 2014; Doucette et al., 2015), but the mechanism by which elevated pressure promotes RGC damage is currently unknown. A better understanding of the neurodegenerative processes triggered by ocular hypertension (OHT) is, therefore, essential to develop effective therapeutic agents for the treatment of glaucoma.
It is increasingly recognized that glaucoma shares a number of pathological features with Alzheimer's disease (AD), the most common cognitive disorder. AD is characterized by the following two neuropathological lesions: senile plaques composed of amyloid β (Aβ) peptides; and neurofibrillary tangles made of insoluble aggregated tau, a microtubule-associated protein enriched in the axon (Binder et al., 1985; Drubin and Kirschner, 1986; Kosik and Finch, 1987). Substantial visual deficits, including a preferential loss of RGCs, are found in individuals affected by AD, which may account for the impaired contrast sensitivity and motion perception often observed in these patients (Hinton et al., 1986; Blanks et al., 1989; Katz and Rimmer, 1989; Jackson and Owsley, 2003; Lee and Martin, 2004). Likewise, there is a high occurrence of glaucoma in subjects with AD, and Aβ and hyperphosphorylated tau have been detected in ocular or CSF samples from glaucoma patients (Yoneda et al., 2005; Gupta et al., 2008; Nucci et al., 2011).
Recent work supports the idea that neuronal loss in AD and glaucoma is mediated by common neurodegenerative pathways. For example, Aβ accumulation has been observed in experimental glaucoma (McKinnon et al., 2002; Goldblum et al., 2007; Kipfer-Kauer et al., 2010; Ito et al., 2012), and blockade of the Aβ pathway reduced RGC loss (Guo et al., 2007; Salt et al., 2014). It is increasingly recognized that tau is a key mediator of Aβ toxicity (Rapoport et al., 2002; Santacruz et al., 2005; Roberson et al., 2007). Recently, transgenic mice carrying the human P301S tau mutant have been shown to contain tau aggregates in the retina (Schön et al., 2012), notably in RGCs (Gasparini et al., 2011), which was correlated with impaired anterograde and retrograde axonal transport as well as increased susceptibility to excitotoxic damage (Bull et al., 2012). These findings suggest an association between tau aggregation and RGC dysfunction. However, tau mutant models have several limitations, including differences in exogenous tau expression compared with endogenous tau expression in disease (Noble et al., 2010). As such, the role of endogenous tau in RGC damage and whether it contributes to neuronal death in glaucoma is currently unknown.
To address this question, we used a well characterized rat model of ocular hypertension glaucoma that results in selective loss of RGC somas and axons. Our data demonstrate that high intraocular pressure leads to a rapid increase in retinal tau with altered phosphorylation profile and the formation of tau oligomers. Tau accumulation was primarily observed in RGC dendrites, while tau levels in RGC axons within the optic nerve markedly decreased. Importantly, tau knockdown using a targeted short interfering RNA (siRNA) led to striking protection of RGC somas and axons from hypertension-induced damage. Our study identifies novel and substantial alterations of endogenous tau protein in experimental glaucoma, which are consistent with the cardinal features of a tauopathy, including abnormal subcellular distribution, altered phosphorylation profile, and neurotoxicity.
Materials and Methods
Experimental animals.
All procedures were performed in male Brown Norway rats aged 2 months (160–200 g) or 8 months (300–400 g; Charles River Laboratories International) in compliance with the guidelines of the Canadian Council on Animal Care for the Use of Experimental Animals (http://www.ccac.ca). Tau knock-out mice (strain Mapt-tm1[EGFP]Klt/J) were purchased (The Jackson Laboratory) and maintained in our animal facility until needed. The number of animals used in each experiment is indicated in the corresponding figure legend and table.
Induction of ocular hypertension.
Unilateral elevation of intraocular pressure was induced by the injection of hypertonic saline solution into an episcleral vein, as previously described (Morrison et al., 1997; Almasieh et al., 2010). Briefly, a plastic ring was applied to the ocular equator to confine the injection to the limbal plexus, and a microneedle was used to inject 50 μl of sterile 1.85 m NaCl through an episcleral vein. Following injection, the plastic ring was removed, and the eyes were examined to assess the extent to which the saline solution traversed the limbal microvasculature. Polysporin ophthalmic ointment (Pfizer Canada) was applied to the operated eye, and the animal was allowed to recover. Eye pressure measurements were taken from awake animals after corneal application of one drop of proparacaine hydrochloride (0.5%; Alcon Laboratories) using a calibrated tonometer (TonoPen XL, Medtronic Solan). The tonometer was held perpendicular to the corneal surface, and 10 consecutive readings per eye were taken and averaged. Intraocular pressure was measured every other day, at the same time, for the entire duration of the experiment. The mean and peak (maximum) intraocular pressure values for each eye were calculated and used to estimate the mean and peak pressure for experimental and control groups.
Western blot analyses.
Whole retinas were isolated and homogenized in the following ice-cold lysis buffer: 50 mm Tris, pH 7.4, 1 mm EDTA, 150 mm NaCl, 1% NP-40, 5 mm Na fluoride, 0.25% Na deoxycholate, and 2 mm NaVO3, supplemented with protease and phosphatase inhibitors. Protein homogenates were centrifuged at 18,000 × g for 5 min, and the supernatants were removed and resedimented to yield soluble extracts. Samples in Laemmli buffer were boiled for 5 min (except for visualization of tau oligomers), resolved in 7.5% SDS polyacrylamide gels, and transferred to nitrocellulose membranes (Bio-Rad). Blots were incubated in SuperBlock T20 blocking solution (Thermo Scientific) for 1 h at room temperature, followed by overnight incubation at 4°C with each of the following primary antibodies: total tau (K9JA, 1 μg/ml, Dako North America); phospho-tau S396-S404 (PHF1; 1:100; gift of P. Davies, Albert Einstein College of Medicine, Bronx, NY); phospho-tau S199 (PS199; 1 μg/ml; Invitrogen); phospho-tau S202-T205 (AT8; 0.8 μg/ml; Thermo Scientific); oligomeric tau (T22; 1:200; gift of R. Kayed, University of Texas, Austin, TX); or β-actin (0.5 μg/ml; Sigma-Aldrich). Membranes were washed and incubated in peroxidase-linked anti-mouse or anti-rabbit secondary antibodies (0.5 μg/ml; GE Healthcare). Blots were developed with a chemiluminescence reagent (ECL, GE Healthcare Life Sciences) and exposed to X-OMAT imaging film (Eastman Kodak). Densitometry was performed using ImageJ software on scanned autoradiographic films obtained from at least three independent Western blots each, using retinal samples from different groups.
Retina and optic nerve immunohistochemistry.
Animals were perfused with 4% paraformaldehyde (PFA), and the eyes and optic nerves were rapidly dissected. Tissue was embedded in optimal cutting temperature compound (Tissue-Tek, Miles Laboratories), and retinal (16 μm) or optic nerve (12 μm) cryosections were collected onto gelatin-coated slides, as described previously (Pernet and Di Polo, 2006; Wilson et al., 2013). Some eyes were embedded in paraffin for the generation of thin retinal cross sections (4 μm) using a microtome (Leica Biosystems). The following primary antibodies were added to retinal or optic nerve sections in blocking solution and incubated overnight at 4°C, as described previously (Planel et al., 2004; Wilson et al., 2014): total tau (2–10 μg/ml; K9JA, Dako); total tau (1:1000; Tau 46, New England BioLabs); choline acetyltransferase (ChAT; 1 μg/ml; Millipore); calbindin (1:1000; Swant); protein kinase C α (PKCα; 3 μg/ml; Enzo Life Sciences); neurofilament H (NF-H; 10 μg/ml; Sternberger Monoclonals); and/or tubulin isoform βIII (TUJ1; 2.5 μg/ml; Sigma-Aldrich). Sections were washed and incubated with the following secondary antibodies: donkey anti-rabbit or anti-mouse Alexa Fluor 594 and 488 (2 μg/ml; Life Technologies). Fluorescent labeling was observed using an Axioskop 2 Plus Microscope (Carl Zeiss) or a confocal microscope (Leica Microsystems).
Reverse transcription and quantitative real time PCR.
Total RNA was isolated from individual retinas using the RNEasy Mini Kit (Qiagen). cDNAs were generated from 1 μg of total RNA using the QuantiTect Reverse Transcription Kit (Qiagen). Real-time PCR was performed using TaqMan probes and primers that target exon 5, conserved among all known tau isoforms (pan-tau; catalog #Rn01495715), exon 4a specific to rat big tau (catalog #Rn01495711, Applied Biosystems), or 18S ribosomal RNA as control (catalog #4308329, Applied Biosystems). Amplification was performed using the 7900HT Fast Real-Time PCR System (Applied Biosystems) with the following cycle conditions: 95°C for 15 s, 60°C for 1 min, and 72°C for 1 min. Reactions were run in triplicate for each sample, and the 2-ΔΔCt formula was used for the calculation of differential gene expression.
Short interfering RNA.
The following siRNA sequences against tau (siTau) were purchased from Dharmacon (ON-TARGET plus Smartpool, Thermo Scientific; sense strands): (1) 5′-GCAUGUGACUCAAGCUCGA-3′; (2) 5′-AGUUAGGGACGAUGCGGUA-3′; (3) 5′-GAUAG AGUCCAGUCGAAGA-3′; and (4) 5′-GGACAGGAAAUGACGAGAA-3′. An siRNA against green fluorescent protein (siGFP) was used as a control. Each siRNA (2 μg/μl; total volume, 5 μl) was injected into the vitreous chamber using a Hamilton syringe fitted with a 32-gauge glass microneedle. The sclera was exposed, and the tip of the needle inserted into the superior ocular hemisphere at a 45° angle through the sclera and retina into the vitreous space using a posterior approach. This route of administration avoids injury to the iris or lens, which can promote RGC survival (Mansour-Robaey et al., 1994; Leon et al., 2000). After the injection, the needle was left in the eye for 1 min to allow the solution to diffuse into the vitreous chamber and to prevent backflow. Surgical glue (Indermil, Tyco Healthcare) was immediately applied to seal the injection site.
Quantification of RGC somas and axons.
Quantification of RGC somas and axons was performed in duplicate by an observer masked to the experimental conditions. Rats were killed by transcardial perfusion with 4% PFA, and retinas were dissected out and fixed for an additional 15 min. Brn3a immunodetection on whole-mounted retinas was performed as described previously (Nadal-Nicolas et al., 2009). Briefly, whole retinas were permeabilized in PBS containing 0.5% Triton X-100 (ThermoFisher Scientific) by freezing them at −80°C for 15 min, rinsed, and incubated overnight at 4°C with goat-anti-Brn3a (0.27 μg/ml; C-20, Santa Cruz Biotechnology) in blocking buffer (PBS, 2% normal donkey serum, 2% Triton X-100). Retinas were washed and incubated for 2 h at room temperature with Alexa Fluor donkey anti-goat IgG (1 μg/ml; Jackson ImmunoResearch Laboratories). Retinas were then rinsed, mounted with the vitreal side up, and covered with anti-fade solution (SlowFade, Invitrogen). Brn3a-labeled neurons were counted within three square areas at distances of 1, 2, and 3 mm from the rat optic disc in each of the four retinal quadrants for a total of 12 retinal areas. Fluorescent staining was examined with an Axioskop 2 Plus Microscope (Carl Zeiss Canada). Images were captured with a CCD video camera (Retiga, Qimaging) and analyzed with Northern Eclipse software (Empix Imaging).
For axon counts, animals received a transcardial injection of heparin (1000 U/kg) and sodium nitroprusside (10 mg/kg) followed by perfusion with 2% PFA and 2.5% glutaraldehyde in 0.1 m PBS. Optic nerves were dissected, fixed in 2% osmium tetroxide, and embedded in epon resin. Semithin sections (0.7 μm) were cut on a microtome (Reichert) and stained with 1% Toluidine Blue. Images were acquired using an oil-immersion 63× objective (numerical aperture, 1.4). RGC axons were counted at 1 mm from the optic nerve head in five nonoverlapping areas of each optic nerve section, encompassing a total area of 5500 μm2/nerve. The five optic nerve areas analyzed included the following: one in the center of the nerve; and two peripheral dorsal and two peripheral ventral regions. The total area per optic nerve cross section was measured using Northern Eclipse image analysis software (Empix Imaging), and this value was used to estimate the total number of axons per optic nerve.
Statistical analyses.
Data and statistical analyses were performed using Instat software (GraphPad Software Inc.) by a one-way ANOVA, followed by a Bonferroni post hoc test or a Student's t test, as indicated in the figure legends.
Results
Age modulates the expression and phosphorylation profile of retinal tau
Glaucoma, like AD, is an age-related neurodegenerative disorder (Leske et al., 2003). Tau is a highly soluble, natively unfolded protein with an expression profile that varies with age (Maeda et al., 2006; Morawe et al., 2012). Therefore, we first asked whether age influences the levels of endogenous retinal tau. For this purpose, retinal protein samples from 2- and 8-month-old rats without any ocular injury (Intact) were compared. Eight-month-old animals were selected because this is the typical age of retired breeder Brown Norway rats used to develop the ocular hypertension model used in this study (Morrison et al., 1997; Almasieh et al., 2010). Western blot analysis of soluble retinal extracts using an antibody against total tau (K9JA), which recognizes the C-terminus microtubule binding region regardless of its phosphorylation state (Biernat et al., 2002; Plouffe et al., 2012), demonstrated the presence of three predominant tau isoforms of 50, 55, and 100 kDa (Fig. 1A). Densitometric analysis showed 1.7-fold and 2.8-fold increases in the 50 and 100 kDa tau variants, respectively, in samples from 8-month-old animals compared with younger animals, while no significant change was detected for the 55 kDa form (Fig. 1B). To validate the specificity of the total tau antibody K9JA, retinas from wild-type (Tau+/+) and tau-deficient (Tau−/−) mice were analyzed. All tau isoforms were observed in Tau+/+ mouse retinas, whereas no signal was detected in Tau −/− murine retinal samples, confirming that K9JA selectively recognizes tau (Fig. 1C, D).
Figure 1.

Age modulates the expression and phosphorylation profile of retinal tau. A, Representative Western blots of soluble retinal extracts from 2- and 8-month-old Brown Norway rats probed with an antibody against total tau (K9JA) demonstrated the presence of three predominant tau isoforms of 50, 55, and 100 kDa. B, Densitometric analysis showed a 1.7-fold and 2.8-fold increase in the 50 and 100 kDa tau variants, respectively, in samples from 8-month-old animals (N = 6) compared with younger animals (N = 3; Student's t test, *p < 0.05). C, D, All tau isoforms were observed in Tau+/+ mouse retinas (N = 3), whereas no signal was detected in Tau −/− murine retinal samples (N = 3), validating the specificity of the antibody. E–J, Analysis of phospho-specific tau epitopes demonstrated age-dependent changes in phosphorylation with PHF1 (Ser396, Ser404) and PS199 (Ser199), while no change was detected with AT8 (Ser202, Thr205; 8 month-old, N = 6; 2 month-old, N = 3; Student's t test, *p < 0.05). Vertical lines represent nonconsecutive samples from the same gel.
Next, we examined whether aging affected tau phosphorylation using antibodies that selectively recognize the phospho-tau epitopes PHF1 (Ser396, Ser404), PS199 (Ser199), and AT8 (Ser202, Thr205; Otvos et al., 1994; Petry et al., 2014). These epitopes were selected because they are located in distinct domains of the tau protein and are readily recognized by highly specific antibodies (Petry et al., 2014). Visualization of blots probed with PHF1 and PS199 revealed an age-dependent increase in phosphorylation at these sites (Fig. 1E, F). However, densitometric analysis of phospho-tau signals with respect to total tau, which increased with age, revealed a relative decrease in tau phosphorylation at epitopes recognized by PHF1 (50 and 100 kDa) and PS199 (50 kDa) in retinal samples from 8-month-old rats (Fig. 1H, I). No significant changes were detected with AT8 (Fig. 1G, J). Collectively, these data indicate that the level of retinal tau protein increases with age and undergoes alterations in its phosphorylation profile. To exclude any confounding effect of age, all subsequent experiments were performed using age-matched controls.
Ocular hypertension promotes retinal tau upregulation and epitope-dependent changes in phosphorylation
Unilateral ocular hypertension was induced in 8-month-old Brown Norway rats by a single injection of hypertonic saline into an episcleral vein, as described previously (Morrison et al., 1997; Almasieh et al., 2010; Cueva Vargas et al., 2015). This procedure leads to the blockade of aqueous humor outflow, a gradual increase in eye pressure, and a selective loss of RGC somas and axons. Using this model system, we first asked whether there are variations in endogenous tau levels following glaucoma induction. Our analysis focused on events at 3 weeks after saline solution injection because increased intraocular pressure is stable at this time point, and neuronal degeneration has started but is not yet overt, as it is in later stages of the disease (Cueva Vargas et al., 2015). Figure 2A shows the intraocular pressure for individual animals over the 3 week period. A mean increase in pressure of 38 mmHg in glaucomatous eyes was observed, compared with 22 mmHg in noninjured (Intact) eyes, which is well within the range of pressure elevation reported in this model (Almasieh et al., 2013; Cueva Vargas et al., 2015; Morrison et al., 2015).
Figure 2.

Ocular hypertension (OHT) promotes retinal tau upregulation and epitope-dependent changes in phosphorylation. A, Intraocular pressure (IOP) measurements for individual rats during a 3 week period following injection of hypertonic saline solution into an episcleral vein (Intact, N = 10; OHT, N = 14). B, Western blot analysis showed a substantial increase in all tau isoforms in retinas with OHT compared with Intact controls. Two examples of retinas with OHT are shown, using different samples and exposure times, to better demonstrate the independent increase of the 50 and 55 kDa bands. C, Densitometry demonstrated a 3.2-fold, 3.4-fold, and 4.9-fold increase in the 50, 55, and 100 kDa bands, respectively, in glaucomatous retinas compared with Intact controls (OHT, N = 10; Intact, N = 6; Student's t test, *p < 0.05, ***p < 0.001). D, Real-time qPCR analysis demonstrated no significant change in retinal tau gene expression (OHT, N = 6; Intact, N = 6). E–G, Western blot analysis of phospho-specific epitopes revealed alterations in tau phosphorylation at PHF1 and PS199, while no change was detected at AT8. H–J, Densitometric analysis demonstrated a significant increase in phosphorylation on S396 and S404 (PHF1), accompanied by reduced phosphorylation on S199 (PS199) relative to total tau, while no change was detected on AT8 (OHT, N = 9–10/group; Intact, N = 6; Student's t test, *p < 0.05, **p < 0.01). Vertical lines represent nonconsecutive samples from the same gel.
Western blot analysis showed a substantial increase of tau in retinas with high intraocular pressure compared with Intact controls (Fig. 2B). Densitometry demonstrated 3.2-fold, 3.4-fold, and 4.9-fold increases in the 50, 55, and 100 kDa bands, respectively (Fig. 2C). Real-time quantitative PCR (qPCR) analysis was performed using primers that recognize pan-tau or big tau, a high-molecular-weight isoform (110 kDa) that has been detected in the peripheral nervous system, spinal cord, and retina (Oblinger et al., 1991; Goedert et al., 1992; Boyne et al., 1995). No changes in tau mRNA levels were detected in glaucomatous retinas compared with controls (Fig. 2D), suggesting that tau protein upregulation is not the result of increased gene expression.
Marked changes in retinal tau phosphorylation on epitopes PHF1 and PS199 were observed in ocular hypertensive eyes (Fig. 2E, F). Densitometric analysis of phospho-tau signals relative to total tau showed that all forms of tau underwent increased phosphorylation on S396 and S404 residues recognized by PHF1 in glaucomatous retinas compared with Intact controls (Fig. 2H). In contrast, reduced phosphorylation of S199 was detected relative to that in total tau (Fig. 2I). Changes in S202 and Thr205 phosphorylation using AT8 were not statistically significant (Fig. 2G, J). These results demonstrate that ocular hypertension increases the levels of retinal tau protein and, intriguingly, alters its phosphorylation profile toward epitope-dependent hyperphosphorylation or hypophosphorylation.
Altered phosphorylation drives tau accumulation and oligomerization in glaucomatous retinas
Accumulating evidence indicates that tau oligomers, an intermediate species between native tau monomers and aggregated tau found in neurofibrillary tangles, are highly toxic and promote neuronal deficits and death (Gerson and Kayed, 2013). Thus, we asked whether the increase in the 100 kDa tau form observed in glaucomatous retinas was the result of dimerization of 50/55 kDa tau monomers. Analysis of 100 kDa/total tau versus total tau demonstrated a positive correlation between the levels of the 50 kDa species and accumulation of the 100 kDa form (Fig. 3A). In contrast, no significant correlation was observed between the levels of the 55 kDa and 100 kDa tau species (Fig. 3B), suggesting that the 55 kDa form does not contribute to tau dimerization. Abnormal phosphorylation of tau monomers has been shown to promote accumulation and, ultimately, oligomerization (Sahara et al., 2007; Tepper et al., 2014). Therefore, we also analyzed the correlation between phosphorylated tau/total tau and total tau in glaucomatous retinas. Phosphorylation on the PHF1 epitope (S396, S404) correlated positively with 50 kDa tau accumulation (Fig. 3C), while no significant correlation was found for 55 kDa tau (Fig. 3D). A negative correlation was found between S199 phosphorylation on 50 and 55 kDa tau and total protein level (Fig. 3E, F), suggesting that the loss of phosphorylation at this residue favors accumulation and subsequent oligomerization.
Figure 3.

Altered phosphorylation drives tau accumulation and oligomerization in glaucomatous retinas. A, Pearson correlation analysis of 100 kDa/total tau vs total tau demonstrated a positive association between the levels of the 50 kDa species and the 100 kDa/50 kDa ratio. B, No significant correlation was observed between the levels of the 55 kDa species and the 100 kDa/55 kDa ratio. C, D, Analysis of phosphorylated tau/total tau vs total tau showed that phosphorylation of the PHF1 epitope (S396, S404) correlated positively with 50 kDa tau accumulation, while no significant correlation was found for 55 kDa tau. E, F, A negative correlation was found between S199 phosphorylation on 50 and 55 kDa tau and their levels. R, correlation coefficient. N = 9/group. G, H, Western blot analysis of retinal extracts in nondenaturing conditions using T22, an antibody that recognizes tau oligomers, confirmed the appearance of tau oligomeric complexes in glaucomatous retinas (OHT, N = 5; Intact, N = 3; Student's t test, *p < 0.05, ***p < 0.001). Vertical lines represent nonconsecutive samples from the same gel.
To further investigate whether ocular hypertension leads to retinal tau oligomerization, we performed Western blot analysis of retinal samples under nondenaturing conditions using T22, an antibody that selectively recognizes tau oligomers at the exclusion of monomers (Lasagna-Reeves et al., 2010; Lasagna-Reeves et al., 2012). Our data show a marked increase in T22-positive 100 kDa tau (Fig. 3G, H), supporting the possibility that this tau form results from the oligomerization of monomeric 50/55 kDa tau. In addition, novel 250 and 200 kDa tau species were detected with T22 in hypertensive retinas (Fig. 3G, H), suggesting that tau monomers can aggregate to form larger multimeric complexes. Together, these data suggest that altered phosphorylation and accumulation of monomers promote tau oligomerization in glaucoma.
Tau accumulates in the RGC somatodendritic compartment in glaucoma
To establish the cellular localization of tau, we first performed immunohistochemical analysis using antibodies against total tau. Low levels of endogenous tau, primarily in the synaptic outer plexiform layer (OPL) and inner plexiform layer (IPL), were detected in control noninjured retinas (Fig. 4A). In glaucomatous retinas, tau protein levels increased substantially (Fig. 4B), confirming our biochemical findings. A marked tau increase was observed in the IPL, where RGC dendrites are located, with a modest increase in the ganglion cell layer (GCL), which is the location of RGC somas (Fig. 4B). In addition to RGCs, displaced amacrine cells and astrocytes populate the GCL (Perry, 1981); hence, we performed experiments using an antibody against TUJ1, which selectively labels RGC somas and dendrites in the retina (Cui et al., 2003; Yin et al., 2003). Colabeling of glaucomatous retinas with K9JA and TUJ1 demonstrated a tau increase in RGC somas and dendrites (Fig. 4C–E). Confocal microscopy analysis validated tau protein expression in TUJ1-positive RGCs, both in the cytoplasm and in dendritic branches (Fig. 4F–H). Some tau-positive cells were also detected in the inner nuclear layer (INL) and were identified as calbindin-positive horizontal cells (Fig. 4I–K), as previously described in human glaucomatous retinas (Gupta et al., 2008), ChAT-positive amacrine cells (Fig. 4L–N), and PKCα-positive bipolar cells (Fig. 4O–Q). We conclude that tau protein accumulates in glaucomatous retinas, most notably in the somatodendritic compartment of RGCs, but also in some retinal interneurons.
Figure 4.
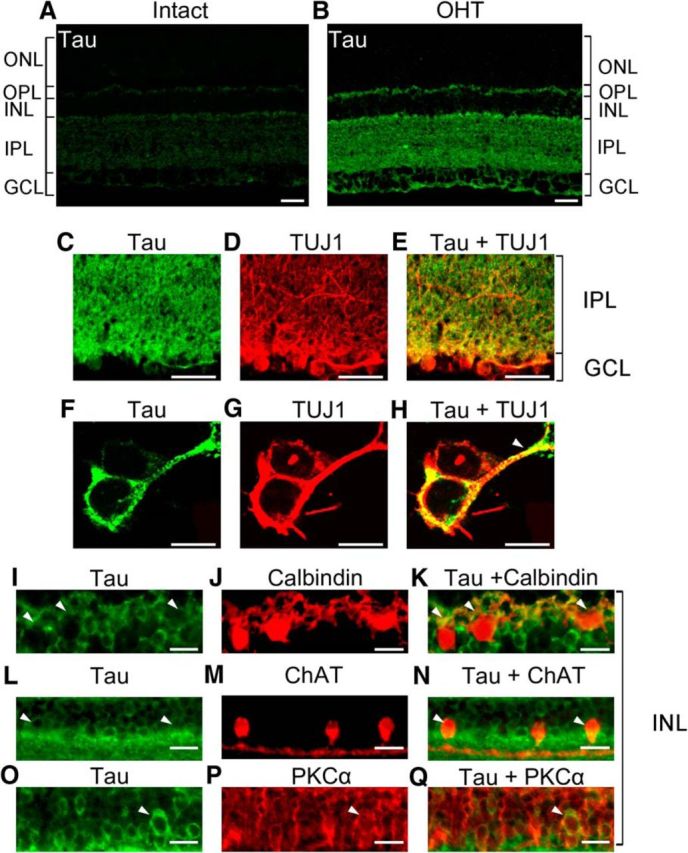
Tau accumulates in the RGC somatodendritic compartment in glaucoma. A, B, Retinal immunohistochemistry using antibodies against total tau (K9JA) revealed marked tau upregulation in glaucomatous eyes compared with Intact controls. C–E, Colabeling of hypertensive retinas with total tau and TUJ1, an RGC-specific marker, demonstrated tau upregulation in RGC soma in the GCL and dendrites in the IPL. F–H, Confocal microscopy analysis validated tau protein expression in RGC somas and dendrites (arrowhead). I–Q, In the INL, cells expressing tau were identified as calbindin-positive horizontal cells, ChAT-positive amacrine cells, and PKCα-positive bipolar cells (white arrowheads). Scale bars: A–E, 25 μm; F–H, 5 μm; I–Q, 12.5 μm. ONL, Outer nuclear layer.
Tau is markedly downregulated in optic nerve RGC axons in glaucoma
In physiological conditions, tau protein is predominantly localized to axons, with only low levels found in the cytoplasm and dendrites (Migheli et al., 1988). In AD, tau detaches from axonal microtubules and accumulates in the somatodendritic compartment of affected neurons (Ballatore et al., 2007). RGC axons have been identified as a major site of injury in glaucoma; therefore, we examined tau expression in the optic nerve at 3 weeks after the induction of ocular hypertension. In control normotensive eyes, tau was enriched in RGC axons visualized with the axonal marker NF-H (Fig. 5A–F). In contrast, optic nerves from glaucomatous eyes displayed a striking reduction in tau protein levels (Fig. 5G). Although many RGC axons are lost at 3 weeks of ocular hypertension, low tau levels could not be solely attributed to axonal death because colabeling with NF-H established that RGC axons were still abundant at this time point (Fig. 5H, I). Indeed, confocal microscopy analysis of fascicular axon bundles confirmed intense NF-H labeling in axons that lacked or had very low levels of tau (Fig. 5J–L), indicating a striking loss of tau in surviving axons. Consistent with this, Western blot analysis of optic nerve homogenates showed a substantial decrease in total tau; most notably the 50 and 55 kDa forms, in nerves from glaucomatous eyes compared with Intact controls (Fig. 5M, N). To investigate the presence of tau in intraretinal, nonmyelinated RGC axons, immunolabeling of whole-mounted retinas with tau and NF-H antibodies was performed. Our results show low levels of tau in RGC intraretinal axons of naive retinas that increased visibly with ocular hypertension (Fig. 5O–T). These results indicate that tau is markedly reduced in RGC axons within the optic nerve, while it accumulates in the RGC somatodendritic compartment and in intraretinal axons, suggesting abnormalities in tau transport and/or localization in glaucoma.
Figure 5.
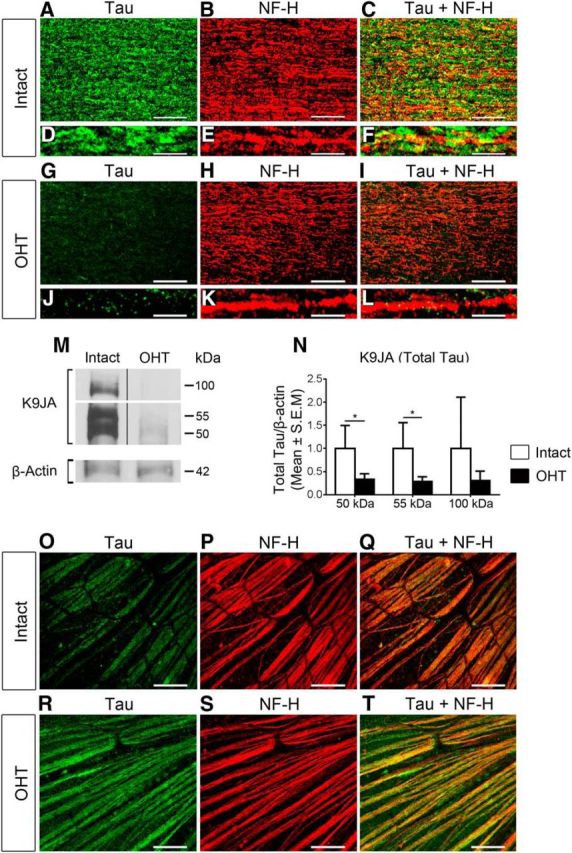
Tau is downregulated in optic nerve RGC axons in glaucoma. A–F, Tau is abundantly expressed in RGC axons, visualized with the axonal-enriched marker NF-H, in control normotensive eyes. G–L, In contrast, optic nerves with OHT displayed a striking reduction in tau protein expression. Tau downregulation was not due to axonal loss because colabeling with NF-H confirmed that RGC axons were still abundant at this time point. Scale bars: A–C, G–I, 25 μm; D–F, J–L, 5 μm. M, N, Western blot analysis of optic nerve homogenates showed a substantial decrease in total tau in nerves from glaucomatous eyes compared with Intact controls. OHT, N = 3; Intact, N = 3; Student's t test, *p < 0.05. Vertical lines represent nonconsecutive samples from the same gel. O–T, Immunolabeling of whole-mounted retinas with tau and NF-H antibodies shows low levels of tau in RGC intraretinal axons of naive retinas that increased markedly with OHT. Scale bars: O–T, 25 μm.
Selective tau knockdown promotes the survival of RGC somas and axons
To establish whether alterations in tau mediate the loss of RGCs in glaucoma, we sought to reduce tau expression by using siRNA followed by analysis of RGC survival. First, we assessed the ability of a targeted siTau to reduce retinal tau protein levels. We previously demonstrated that siRNA delivered by intravitreal injection is rapidly taken up by RGCs (Wilson et al., 2013, 2014; Morquette et al., 2015). Western blot analysis of retinal homogenates from eyes that received siTau 1 week after the induction of ocular hypertension showed a significant reduction in Tau protein, most notably the 50 and 100 kDa forms, while a control siGFP had no effect (Fig. 6A, B). The findings of immunohistochemistry of glaucomatous retinas confirmed that tau downregulation occurred primarily in the IPL and GCL following siTau administration, while no change was observed with control siGFP (Fig. 6C, D). Next, we investigated whether siRNA-mediated tau knockdown resulted in RGC rescue. For this purpose, siTau was injected intraocularly at 1 and 2 weeks after the induction of ocular hypertension, and RGC survival was evaluated a week later (3 weeks after injury onset). Cell soma and axon quantification was performed blinded to treatment. Flat-mounted retinas from eyes treated with siTau showed higher densities of cells labeled with Brn3a, an RGC-specific marker (Nadal-Nicolás et al., 2009), compared with retinas treated with control siGFP (Fig. 7A–C). Quantitative analysis demonstrated that siTau promoted significant RGC survival (92%; mean ± SEM, 1966 ± 39 RGCs/mm2; N = 10) with respect to eyes that received siGFP (75%; 1599 ± 40 RGCs/mm2; N = 9; ANOVA, p < 0.05; Fig. 7G; Table 1).
Figure 6.

Intravitreal delivery of siRNA against tau downregulates retinal tau protein levels. A, B, Western blot analysis of retinal homogenates from eyes that received siRNA at 1 week after the induction of OHT. Eyes injected with siTau showed a significant reduction in tau protein, while a control siGFP had no effect (siGFP, N = 4; siTau, N = 3; Student's t test, *p < 0.05). Vertical lines represent nonconsecutive samples from the same gel. C, D, Results of immunohistochemistry of glaucomatous retinas confirmed that tau downregulation occurred primarily in the IPL and GCL following siTau administration. No change was observed with control siGFP. Scale bars, 25 μm. ONL, Outer nuclear layer.
Figure 7.

Selective tau knockdown promotes RGC survival. A–C, Flat-mounted retinas from eyes treated with siTau displayed higher RGC densities, visualized with the RGC-specific marker Brn3a, compared with control retinas treated with vehicle at 3 weeks after OHT. Scale bars, 25 μm. D–F, Optic nerves from siTau-treated eyes contained more RGC axon fibers with normal morphology, visualized in Toluidine Blue-stained cross sections, compared with control eyes. Scale bars, 5 μm. G, H, Quantitative analysis confirmed that siTau (black bars) promoted significant survival of RGC somas (N = 10) and axons (N = 6) compared with control eyes treated with siGFP (gray bars; somas, N = 9; axons, N = 6). The densities of RGC somas and axons in Intact, nonglaucomatous Brown Norway rat retinas are shown as reference (white bars; 100% survival, N = 6). Intraocular injection of siTau did not significantly alter the density of RGC somas or axons in naive animals (light gray bars, N = 4). Values are expressed as the mean ± SEM. Overall ANOVA for RGC somas, p < 0.001; Bonferroni post hoc test, nontreated Intact vs Intact + siTau, p > 0.05; nontreated Intact vs OHT + siGFP, ***p < 0.001; OHT + siGFP vs OHT + siTau, ***p < 0.001. Overall ANOVA for RGC axons, p < 0.001; Bonferroni post hoc test, nontreated Intact vs Intact + siTau, p > 0.05; nontreated Intact vs OHT + siGFP, ***p < 0.001; OHT + siGFP vs OHT + siTau, *p < 0.05. n.s., Not significant.
Table 1.
Role of Tau on RGC survival in glaucoma
| Treatment | RGC soma/mm2 |
RGC axons/optic nerve |
||||
|---|---|---|---|---|---|---|
| N | Mean ± SEM | % | N | Mean ± SEM | % | |
| Intact | 6 | 2127 ± 81 | 100 | 6 | 102,783 ± 3623 | 100 |
| Anti-GFP siRNA | 9 | 1599 ± 40 | 75 | 6 | 58,083 ± 6246 | 57 |
| Anti-Tau siRNA | 10 | 1966 ± 39 | 92 | 6 | 79,701 ± 4606 | 78 |
Tau knockdown promotes RGC soma and axon survival. Intact control retinas or optic nerves are used as a reference (100%).
A hallmark of glaucoma is the degeneration of RGC axons in the optic nerve posterior to the lamina cribrosa; therefore, we also investigated the capacity of siTau to protect axons. Analysis of optic nerve cross sections showed a substantially larger number of RGC axon fibers with normal morphology in siTau-treated eyes compared with siGFP-treated controls (Fig. 7D–F). The latter displayed extensive axon degeneration, including the disarray of fascicular organization and the degradation of myelin sheaths. Quantitative analysis confirmed that siTau promoted marked protection of RGC axons (78%; mean ± SEM, 79,701 ± 4606 axons/nerve; N = 6) compared with control siRNA treatment (57%; 58,083 ± 6246 axons/nerve; N = 6; ANOVA, p < 0.05; Fig. 7H; Table 1). Intraocular injection of siTau in naive eyes following the same regimen used for glaucomatous eyes did not alter the density of RGC somas or axons (Fig. 7G, H), suggesting that tau downregulation does not affect RGC survival in physiological conditions over the 3 week period studied. The mean pressure elevation among siTau-treated and siGFP-treated groups was similar, thus ruling out pressure reduction by siTau as the cause of neuroprotection. In summary, our results demonstrate that the attenuation of retinal tau levels protects both RGC somas and axons from ocular hypertension-induced damage, suggesting a prominent pathological role of tau in glaucomatous RGC death.
Discussion
Data presented here using a well characterized rat glaucoma model reveal that the dysregulation of endogenous tau plays a significant role in RGC neurodegeneration. First, we demonstrate that tau accumulates with age and ocular hypertension, and undergoes epitope-dependent changes in phosphorylation. Second, we show that tau accumulation drives the formation of oligomers in the glaucomatous retina. Third, our data indicate that tau buildup in RGCs occurs primarily in dendrites, whereas it is nearly depleted from RGC axons. Last, our results demonstrate that tau knockdown promotes robust RGC survival, supporting a critical role for tau alterations in ocular hypertension-induced neuronal damage. Collectively, this study reveals that ocular hypertension glaucoma displays signature pathological features of tauopathies, including tau accumulation, altered phosphorylation, and missorting leading to neurodegeneration.
Tau protein binds to and stabilizes microtubules; therefore, it plays a crucial role in the maintenance and function of axons. The expression of the tau gene (MAPT) is intricately regulated by alternative splicing to generate multiple isoforms, six of which have been identified in the adult human brain (Andreadis, 2005; Caillet-Boudin et al., 2015). Tau splicing depends on developmental stage, tissue type, and species. In the adult rat retina, we detected three predominant tau forms weighing 50, 55, and 100 kDa. Our data show that retinal tau levels increase with age, which is consistent with tau accumulation in the retinas of elderly individuals (Leger et al., 2011). Age is a major risk factor for developing AD and glaucoma, and age-dependent progression of tau pathology, including behavioral phenotypes and memory deficits, is found in most tauopathies (Ghoshal et al., 2002; Ashe and Zahs, 2010). Therefore, the accumulation of retinal tau with age may enhance neuronal susceptibility to stress-related damage in glaucoma.
Our data show a marked increase in all retinal tau species accompanied by changes in phosphorylation in glaucomatous eyes compared with age-matched controls. Intriguingly, we found that while tau residues S396 and S404 were hyperphosphorylated, there was a net decrease in phospho-S199 relative to total tau levels. Phosphorylation is a critical post-translational modification of tau during development and in pathological conditions (Hanger et al., 2009). Inclusions of highly phosphorylated tau are found in most tauopathies, including AD, and correlate with severity of disease (Augustinack et al., 2002; Sergeant et al., 2008). Specifically, phosphorylation on residues S396 and S404 has been described in the brains of tauopathy patients, and both residues are known to impair microtubule binding and facilitate tau aggregation (Fontaine et al., 2015). Although S199 phosphorylation is also found in tauopathies and promotes tau accumulation (Hanger et al., 2009), we report a relative loss of S199 phosphorylation in experimental glaucoma. Accumulating evidence indicates that some stress signals, including oxidative stress, excitotoxicity, and starvation, do not induce hyperphosphorylation but rather hypophosphorylation of tau (Davis et al., 1997; Kuszczyk et al., 2009; Mohamed et al., 2014). Furthermore, tau dephosphorylation has been reported following ischemia, hypoxia, and glucose deprivation in in vivo models and in human brain tissue (Shackelford and Nelson, 1996; Burkhart et al., 1998; Shackelford and Yeh, 1998; Mailliot et al., 2000). Our findings indicate that ocular hypertension-induced changes in tau phosphorylation are complex and epitope specific, and suggest that both tau hyperphosphorylation and hypophosphorylation might regulate neurodegenerative changes.
Tau neurofibrillary tangles have been historically considered a hallmark of tauopathies; however, accumulating evidence indicates that they are not the most toxic tau species. For example, neuronal loss occurs before neurofibrillary tangle formation in AD (Gómez-Isla et al., 1997; Vogt et al., 1998; Terry, 2000; van de Nes et al., 2008), and synaptic dysfunction, cell death, and behavioral deficits in tauopathy models occur in the absence of tangles (Wittmann et al., 2001; Santacruz et al., 2005; Spires et al., 2006; Yoshiyama et al., 2007). Tau oligomers, an intermediate species between tau monomers and neurofibrillary tangles, are highly toxic and have emerged as crucial agents in the onset and propagation of disease (Maeda et al., 2006; Patterson et al., 2011; Lasagna-Reeves et al., 2012; Gerson et al., 2014). Our data demonstrate a correlation between tau monomer increase and the accumulation of 100 kDa tau. This could not be attributed to transcriptional upregulation of big tau, a 110 kDa isoform found in retinal and peripheral neurons (Boyne et al., 1995), because there was no change in mRNA levels in glaucomatous versus control eyes. Furthermore, higher-molecular-weight tau multimeric complexes (250 kDa) were only detected in ocular hypertensive retinas. These findings suggest that endogenous retinal tau monomers are converted to oligomers in glaucoma, which can be potentially toxic to susceptible RGCs.
In AD and other tauopathies, abnormal tau localization to compartments other than the axon, including somas, dendrites, and spines, strongly correlates with neuronal loss and cognitive decline (Zempel and Mandelkow, 2014). We demonstrate robust tau accumulation in RGC dendrites and intraretinal axons in experimental glaucoma. Gene expression in the GCL is rapidly altered following intraocular pressure elevation in rats (Guo et al., 2011); hence, a simple explanation would be that ocular hypertension upregulates retinal tau gene transcription. However, lack of changes in tau mRNA levels rules out transcriptional regulation as a mechanism for tau accumulation in glaucomatous retinas. Impaired axonal transport along RGCs, with early anterograde transport deficits, has been well documented in animal models and in human glaucoma (Crish et al., 2010; Fahy et al., 2015). Since tau protein is sorted from RGC somas to axons (Scholz and Mandelkow, 2014), anterograde transport impairment can potentially result in tau accumulation in RGC dendrites. Missorted tau in dendrites has been shown to target the postsynaptic protein Fyn and consequently to increase Aβ-mediated excitotoxicity in mice (Ittner et al., 2010; Roberson et al., 2011). Microtubule breakdown accompanied by spine and mitochondrial loss occurs in dendrites invaded by missorted tau, a process mediated by the microtubule-severing enzyme spastin (Lacroix et al., 2010; Zempel et al., 2013). Of interest, emerging data indicate that ocular hypertension or acute optic nerve damage triggers rapid dendritic pathology in RGCs, which can lead to synaptic rearrangements, functional deficits, and death (Agostinone and Di Polo, 2015). Future work is needed to establish whether tau accumulation in RGC dendrites is linked to dendritic pathology and neuronal death in glaucoma.
The increase of tau in other retinal layers together with our finding that there is no change in retinal tau mRNA prompt us to put forward the hypothesis that tau accumulates in non-RGC cells by the following two potential mechanisms: (1) impaired tau degradation due to defective autophagy or proteasomal pathways (Wang and Mandelkow, 2012; Lee et al., 2013); and/or (2) cell-to-cell spreading. Indeed, tau protein can spread along anatomically connected neurons, from one cell to another, presumably through synaptic release or secretion into the extracellular milieu and uptake by neighboring cells (Liu et al., 2012; Pooler et al., 2013; Medina and Avila, 2014). In Alzheimer's disease, tau propagates in a stereotypical manner from the entorhinal cortex to the hippocampus and neocortex by neuron-to-neuron spreading of tau aggregates (Clavaguera et al., 2015). Similarly, accruing evidence supports the notion that neurons other than RGCs are affected in glaucoma and undergo some form of dysfunction. High intraocular pressure dramatically alters retinal function before irreversible structural damage in the optic nerve occurs, suggesting early synaptic defects in interneurons (Frankfort et al., 2013; Pang et al., 2015). Other studies have demonstrated alterations in outer retinal neurons in experimental glaucoma (Fernández-Sánchez et al., 2014; Ortín-Martínez et al., 2015; Vidal-Sanz et al., 2015). It will be of interest to experimentally test the mechanisms of tau accumulation in retinal interneurons and, more importantly, to ascertain whether this affects visual function and RGC susceptibility in glaucoma.
We also report marked depletion of tau from RGC axons in glaucoma. Loss of axonal tau could not be solely ascribed to axon death because a significant number of axons in the optic nerve were devoid of tau despite their well preserved structure. As tau protein moves along RGC axons in physiological conditions (Scholz and Mandelkow, 2014), impaired anterograde transport may account for some loss of axonal tau. Alternatively, changes in tau phosphorylation might reduce its affinity for axonal microtubules and increase it for dendritic microtubules, as shown in cultured spinal cord neurons (Kanai and Hirokawa, 1995). Our observation that tau accumulates in the retina at the expense of tau in axons without gene expression changes suggests that increased retinal tau protein might be axonally derived. Loss of tau leads to destabilization and breakdown of the axonal microtubule network, disruption of axonal transport, and, potentially, neuronal death (Feinstein and Wilson, 2005; Levy et al., 2005). Thus, it is possible that the loss of tau in RGC axons reported here compromises axonal stability and function, thus contributing to neurodegeneration.
To test whether tau accumulation in the retina is toxic for RGCs, we used siRNA administered intraocularly to reduce tau levels in glaucomatous eyes. This siRNA-based strategy was chosen based on the capacity to selectively attenuate tau, without completely inhibiting it, and our observation that siRNAs are readily taken up by RGCs when injected into the vitreous space (Ahmed et al., 2011; Wilson and Di Polo, 2012; Morquette et al., 2015). The reason for the apparent preference of intravitreally delivered siRNA for RGCs is unclear, but it may result from the strategic position of these neurons, directly exposed to the vitreous humor, thus allowing rapid siRNA uptake. The onset of siRNA treatment at 1 week after induction of ocular hypertension was selected to more closely model a common clinical situation in which people with glaucoma do not experience noticeable symptoms and do not seek medical help until vision loss has begun, a time when there is already tangible RGC death (Quigley, 2011). Our data show that the administration of siTau successfully reduced retinal tau levels, and promoted robust RGC soma and axon survival in glaucomatous eyes. The observation that siTau effectively rescued RGCs, without altering intraocular pressure, provides strong proof of principle for a detrimental gain-of-function role of tau in glaucoma. The altered pattern of tau phosphorylation we report here suggests a potential contribution in the formation of tau oligomers and RGC death. Therefore, by reducing the total amount of retinal tau protein available for phosphorylation, siTau is likely to attenuate tau aggregation and neuronal death. Of interest, although this siRNA approach only decreased tau levels in the retina, we observed substantial survival of both RGC somas and axons. The decrease of tau burden in the retina appears to have a widespread beneficial effect on the overall health of RGCs, leading to improvements in axonal transport and functionality. Previous studies have demonstrated the benefits of modulating tau levels and activity by kinase inhibitors and phosphatase activators, immunotherapies, inhibitors of protein aggregation, and microtubule-stabilizing agents (Himmelstein et al., 2012). The use of siRNA is now added to the arsenal of strategies to limit the toxic effects of tau while re-establishing cellular homeostasis.
In summary, our data demonstrate a number of important alterations in endogenous tau induced by ocular hypertension, including phosphorylation, oligomerization, and accumulation in RGC dendrites. These pathological changes contribute to neurodegeneration because reducing tau burden promoted substantial protection of RGC somas and axons from glaucomatous damage. These results reveal that glaucoma shares key common features with tauopathies, and identify tau as a novel therapeutic target to potentially counter RGC neurodegeneration in glaucoma and other optic neuropathies.
Footnotes
This work was supported by grants from the Canadian Institutes for Health Research (CIHR) and the Alzheimer's Society of Canada. C.V.V. is a CIHR New Investigator. A.D.P. is a Chercheur National of Fonds de recherche Québec-Santé. We thank Dr. Timothy Kennedy (McGill University, Montréal, QC, Canada) for helpful comments on the manuscript, Dr. Rakez Kayed (University of Texas, Austin, TX) for providing the T22 antibody, Dr. Peter Davies (Albert Einstein University, Bronx, NY) for providing the PHF1 antibody, Dr. Emmanuel Planel (Université Laval, Quebec, QC, Canada) for providing tau-null mice, and Dara O'Connor and Florence Dotigny for technical assistance.
The authors declare no competing financial interests.
References
- Agostinone J, Di Polo A. Retinal ganglion cell dendrite pathology and synapse loss: implications for glaucoma. Prog Brain Res. 2015;220:199–216. doi: 10.1016/bs.pbr.2015.04.012. [DOI] [PubMed] [Google Scholar]
- Ahmed Z, Kalinski H, Berry M, Almasieh M, Ashush H, Slager N, Brafman A, Spivak I, Prasad N, Mett I, Shalom E, Alpert E, Di Polo A, Feinstein E, Logan A. Ocular neuroprotection by siRNA targeting caspase-2. Cell Death Dis. 2011;2:e173. doi: 10.1038/cddis.2011.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almasieh M, Zhou Y, Kelly MEM, Casanova C, Di Polo A. Structural and functional neuroprotection in glaucoma: role of galantamine-mediated activation of muscarinic acetylcholine receptors. Cell Death Dis. 2010;1:e27. doi: 10.1038/cddis.2009.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almasieh M, MacIntyre JN, Pouliot M, Casanova C, Vaucher E, Kelly ME, Di Polo A. Acetylcholinesterase inhibition promotes retinal vasoprotection and increases ocular blood flow in experimental glaucoma. Invest Ophthalmol Vis Sci. 2013;54:3171–3183. doi: 10.1167/iovs.12-11481. [DOI] [PubMed] [Google Scholar]
- Andreadis A. Tau gene alternative splicing: expression patterns, regulation and modulation of function in normal brain and neurodegenerative diseases. Biochim Biophys Acta. 2005;1739:91–103. doi: 10.1016/j.bbadis.2004.08.010. [DOI] [PubMed] [Google Scholar]
- Ashe KH, Zahs KR. Probing the biology of Alzheimer's disease in mice. Neuron. 2010;66:631–645. doi: 10.1016/j.neuron.2010.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Augustinack J, Schneider A, Mandelkow EM, Hyman BT. Specific tau phosphorylation sites correlate with severity of neuronal cytopathology in Alzheimer's disease. Acta Neuropathol. 2002;103:26–35. doi: 10.1007/s004010100423. [DOI] [PubMed] [Google Scholar]
- Ballatore C, Lee VM, Trojanowski JQ. Tau-mediated neurodegeneration in Alzheimer's disease and related disorders. Nat Rev Neurosci. 2007;8:663–672. doi: 10.1038/nrn2194. [DOI] [PubMed] [Google Scholar]
- Biernat J, Wu YZ, Timm T, Zheng-Fischhöfer Q, Mandelkow E, Meijer L, Mandelkow EM. Protein kinase MARK/PAR-1 is required for neurite outgrowth and establishment of neuronal polarity. Mol Biol Cell. 2002;13:4013–4028. doi: 10.1091/mbc.02-03-0046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binder LI, Frankfurter A, Rebhun LI. The distribution of tau in the mammalian central nervous system. J Cell Biol. 1985;101:1371–1378. doi: 10.1083/jcb.101.4.1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanks JC, Hinton DR, Sadun AA, Miller CA. Retinal ganglion cell degeneration in Alzheimer's disease. Brain Res. 1989;501:364–372. doi: 10.1016/0006-8993(89)90653-7. [DOI] [PubMed] [Google Scholar]
- Boyne LJ, Tessler A, Murray M, Fischer I. Distribution of Big tau in the central nervous system of the adult and developing rat. J Comp Neurol. 1995;358:279–293. doi: 10.1002/cne.903580209. [DOI] [PubMed] [Google Scholar]
- Bull ND, Guidi A, Goedert M, Martin KR, Spillantini MG. Reduced axonal transport and increased excitotoxic retinal ganglion cell degeneration in mice transgenic for human mutant P301S tau. PLoS One. 2012;7:e34724. doi: 10.1371/journal.pone.0034724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkhart KK, Beard DC, Lehman RA, Billingsley ML. Alterations in tau phosphorylation in rat and human neocortical brain slices following hypoxia and glucose deprivation. Exp Neurol. 1998;154:464–472. doi: 10.1006/exnr.1998.6899. [DOI] [PubMed] [Google Scholar]
- Caillet-Boudin ML, Buée L, Sergeant N, Lefebvre B. Regulation of human MAPT gene expression. Mol Neurodegener. 2015;10:28. doi: 10.1186/s13024-015-0025-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calkins DJ. Critical pathogenic events underlying progression of neurodegeneration in glaucoma. Prog Ret Eye Res. 2012;31:702–719. doi: 10.1016/j.preteyeres.2012.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clavaguera F, Hench J, Goedert M, Tolnay M. Prion-like transmission and spreading of tau pathology. Neuropathol Appl Neurobiol. 2015;41:47–58. doi: 10.1111/nan.12197. [DOI] [PubMed] [Google Scholar]
- Crish SD, Sappington RM, Inman DM, Horner PJ, Calkins DJ. Distal axonopathy with structural persistence in glaucomatous neurodegeneration. Proc Natl Acad Sci U S A. 2010;107:5196–5201. doi: 10.1073/pnas.0913141107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cueva Vargas JL, Osswald IK, Unsain N, Aurousseau MR, Barker PA, Bowie D, Di Polo A. Soluble tumor necrosis factor alpha promotes retinal ganglion cell death in glaucoma via calcium-permeable AMPA receptor activation. J Neurosci. 2015;35:12088–12102. doi: 10.1523/JNEUROSCI.1273-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui Q, Yip HK, Zhao RC, So KF, Harvey AR. Intraocular elevation of cyclic AMP potentiates ciliary neurotrophic factor-induced regeneration of adult rat retinal ganglion cell axons. Mol Cell Neurosci. 2003;22:49–61. doi: 10.1016/S1044-7431(02)00037-4. [DOI] [PubMed] [Google Scholar]
- Davis DR, Anderton BH, Brion JP, Reynolds CH, Hanger DP. Oxidative stress induces dephosphorylation of τ in rat brain primary neuronal cultures. J Neurochem. 1997;68:1590–1597. doi: 10.1046/j.1471-4159.1997.68041590.x. [DOI] [PubMed] [Google Scholar]
- Doucette LP, Rasnitsyn A, Seifi M, Walter MA. The interactions of genes, age, and environment in glaucoma pathogenesis. Surv Ophthalmol. 2015;60:310–326. doi: 10.1016/j.survophthal.2015.01.004. [DOI] [PubMed] [Google Scholar]
- Drubin DG, Kirschner MW. Tau protein function in living cells. J Cell Biol. 1986;103:2739–2746. doi: 10.1083/jcb.103.6.2739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahy ET, Chrysostomou V, Crowston JG. Impaired axonal transport and glaucoma. Curr Eye Res. 2015;19:1–11. doi: 10.3109/02713683.2015.1037924. [DOI] [PubMed] [Google Scholar]
- Feinstein SC, Wilson L. Inability of tau to properly regulate neuronal microtubule dynamics: a loss-of-function mechanism by which tau might mediate neuronal cell death. Biochim Biophys Acta. 2005;1739:268–279. doi: 10.1016/j.bbadis.2004.07.002. [DOI] [PubMed] [Google Scholar]
- Fernández-Sánchez L, de Sevilla Müller LP, Brecha NC, Cuenca N. Loss of outer retinal neurons and circuitry alterations in the DBA/2J mouse. Invest Ophthalmol Vis Sci. 2014;55:6059–6072. doi: 10.1167/iovs.14-14421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontaine SN, Sabbagh JJ, Baker J, Martinez-Licha CR, Darling A, Dickey CA. Cellular factors modulating the mechanism of tau protein aggregation. Cell Mol Life Sci. 2015;72:1863–1879. doi: 10.1007/s00018-015-1839-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frankfort BJ, Khan AK, Tse DY, Chung I, Pang JJ, Yang Z, Gross RL, Wu SM. Elevated intraocular pressure causes inner retinal dysfunction before cell loss in a mouse model of experimental glaucoma. Invest Ophthalmol Vis Sci. 2013;54:762–770. doi: 10.1167/iovs.12-10581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasparini L, Crowther RA, Martin KR, Berg N, Coleman M, Goedert M, Spillantini MG. Tau inclusions in retinal ganglion cells of human P301S tau transgenic mice: effects on axonal viability. Neurobiol Aging. 2011;32:419–433. doi: 10.1016/j.neurobiolaging.2009.03.002. [DOI] [PubMed] [Google Scholar]
- Gerson JE, Kayed R. Formation and propagation of tau oligomeric seeds. Front Neurol. 2013;4:93. doi: 10.3389/fneur.2013.00093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerson J, Sengupta U, Lasagna-Reeves C, Guerrero-Munoz M, Troncoso J, Kayed R. Characterization of tau oligomeric seeds in progressive supranuclear palsy. Acta Neuropathol Comm. 2014;2:73. doi: 10.1186/2051-5960-2-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghoshal N, García-Sierra F, Wuu J, Leurgans S, Bennett DA, Berry RW, Binder LI. Tau conformational changes correspond to impairments of episodic memory in mild cognitive impairment and Alzheimer's disease. Exp Neurol. 2002;177:475–493. doi: 10.1006/exnr.2002.8014. [DOI] [PubMed] [Google Scholar]
- Goedert M, Spillantini MG, Crowther RA. Cloning of a big tau microtubule-associated protein characteristic of the peripheral nervous system. Proc Natl Acad Sci U S A. 1992;89:1983–1987. doi: 10.1073/pnas.89.5.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldblum D, Kipfer-Kauer A, Sarra GM, Wolf S, Frueh BE. Distribution of amyloid precursor protein and amyloid-beta immunoreactivity in DBA/2J glaucomatous mouse retinas. Invest Ophthalmol Vis Sci. 2007;48:5085–5090. doi: 10.1167/iovs.06-1249. [DOI] [PubMed] [Google Scholar]
- Gómez-Isla T, Hollister R, West H, Mui S, Growdon JH, Petersen RC, Parisi JE, Hyman BT. Neuronal loss correlates with but exceeds neurofibrillary tangles in Alzheimer's disease. Ann Neurol. 1997;41:17–24. doi: 10.1002/ana.410410106. [DOI] [PubMed] [Google Scholar]
- Guo L, Salt TE, Luong V, Wood N, Cheung W, Maass A, Ferrari G, Russo-Marie F, Sillito AM, Cheetham ME, Moss SE, Fitzke FW, Cordeiro MF. Targeting amyloid-beta in glaucoma treatment. Proc Natl Acad Sci U S A. 2007;104:13444–13449. doi: 10.1073/pnas.0703707104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y, Johnson EC, Cepurna WO, Dyck JA, Doser T, Morrison JC. Early gene expression changes in the retinal ganglion cell layer of a rat glaucoma model. Invest Ophthalmol Vis Sci. 2011;52:1460–1473. doi: 10.1167/iovs.10-5930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta N, Fong J, Ang LC, Yücel YH. Retinal tau pathology in human glaucomas. Can J Ophthalmol. 2008;43:53–60. doi: 10.3129/i07-185. [DOI] [PubMed] [Google Scholar]
- Hanger DP, Anderton BH, Noble W. Tau phosphorylation: the therapeutic challenge for neurodegenerative disease. Trends Mol Med. 2009;15:112–119. doi: 10.1016/j.molmed.2009.01.003. [DOI] [PubMed] [Google Scholar]
- Himmelstein DS, Ward SM, Lancia JK, Patterson KR, Binder LI. Tau as a therapeutic target in neurodegenerative disease. Pharmacol Ther. 2012;136:8–22. doi: 10.1016/j.pharmthera.2012.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinton DR, Sadun AA, Blanks JC, Miller CA. Optic-nerve degeneration in Alzheimer's disease. N Engl J Med. 1986;315:485–487. doi: 10.1056/NEJM198608213150804. [DOI] [PubMed] [Google Scholar]
- Ito Y, Shimazawa M, Tsuruma K, Mayama C, Ishii K, Onoe H, Aihara M, Araie M, Hara H. Induction of amyloid-β(1–42) in the retina and optic nerve head of chronic ocular hypertensive monkeys. Mol Vis. 2012;18:2647–2657. [PMC free article] [PubMed] [Google Scholar]
- Ittner LM, Ke YD, Delerue F, Bi M, Gladbach A, van Eersel J, Wölfing H, Chieng BC, Christie MJ, Napier IA, Eckert A, Staufenbiel M, Hardeman E, Götz J. Dendritic function of tau mediates amyloid-β toxicity in Alzheimer's disease mouse models. Cell. 2010;142:387–397. doi: 10.1016/j.cell.2010.06.036. [DOI] [PubMed] [Google Scholar]
- Jackson GR, Owsley C. Visual dysfunction, neurodegenerative diseases, and aging. Neurol Clin. 2003;21:709–728. doi: 10.1016/S0733-8619(02)00107-X. [DOI] [PubMed] [Google Scholar]
- Kanai Y, Hirokawa N. Sorting mechanisms of Tau and MAP2 in neurons: suppressed axonal transit of MAP2 and locally regulated microtubule binding. Neuron. 1995;14:421–432. doi: 10.1016/0896-6273(95)90298-8. [DOI] [PubMed] [Google Scholar]
- Katz B, Rimmer S. Ophthalmologic manifestations of Alzheimer's disease. Surv Ophthalmol. 1989;34:31–43. doi: 10.1016/0039-6257(89)90127-6. [DOI] [PubMed] [Google Scholar]
- Kipfer-Kauer A, McKinnon SJ, Frueh BE, Goldblum D. Distribution of amyloid precursor protein and amyloid-β in ocular hypertensive C57BL/6 mouse eyes. Curr Eye Res. 2010;35:828–834. doi: 10.3109/02713683.2010.494240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosik KS, Finch EA. MAP2 and tau segregate into dendritic and axonal domains after the elaboration of morphologically distinct neurites: an immunocytochemical study of cultured rat cerebrum. J Neurosci. 1987;7:3142–3153. doi: 10.1523/JNEUROSCI.07-10-03142.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuszczyk M, Gordon-Krajcer W, Lazarewicz JW. Homocysteine-induced acute excitotoxicity in cerebellar granule cells in vitro is accompanied by PP2A-mediated dephosphorylation of tau. Neurochem Int. 2009;55:174–180. doi: 10.1016/j.neuint.2009.02.010. [DOI] [PubMed] [Google Scholar]
- Lacroix B, van Dijk J, Gold ND, Guizetti J, Aldrian-Herrada G, Rogowski K, Gerlich DW, Janke C. Tubulin polyglutamylation stimulates spastin-mediated microtubule severing. J Cell Biol. 2010;189:945–954. doi: 10.1083/jcb.201001024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasagna-Reeves CA, Castillo-Carranza DL, Guerrero-Muoz MJ, Jackson GR, Kayed R. Preparation and characterization of neurotoxic tau oligomers. Biochemistry. 2010;49:10039–10041. doi: 10.1021/bi1016233. [DOI] [PubMed] [Google Scholar]
- Lasagna-Reeves CA, Castillo-Carranza DL, Sengupta U, Sarmiento J, Troncoso J, Jackson GR, Kayed R. Identification of oligomers at early stages of tau aggregation in Alzheimer's disease. FASEB J. 2012;26:1946–1959. doi: 10.1096/fj.11-199851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee AG, Martin CO. Neuro-ophthalmic findings in the visual variant of Alzheimer's disease. Ophthalmology. 2004;111:376–380. doi: 10.1016/S0161-6420(03)00732-2. [DOI] [PubMed] [Google Scholar]
- Lee MJ, Lee JH, Rubinsztein DC. Tau degradation: the ubiquitin–proteasome system versus the autophagy-lysosome system. Prog Neurobiol. 2013;105:49–59. doi: 10.1016/j.pneurobio.2013.03.001. [DOI] [PubMed] [Google Scholar]
- Leger F, Fernagut PO, Canron MH, Léoni S, Vital C, Tison F, Bezard E, Vital A. Protein aggregation in the aging retina. J Neuropathol Exp Neurol. 2011;70:63–68. doi: 10.1097/NEN.0b013e31820376cc. [DOI] [PubMed] [Google Scholar]
- Leon S, Yin Y, Nguyen J, Irwin N, Benowitz LI. Lens injury stimulates axon regeneration in the mature rat optic nerve. J Neurosci. 2000;20:4615–4626. doi: 10.1523/JNEUROSCI.20-12-04615.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leske MC, Heijl A, Hussein M, Bengtsson B, Hyman L, Komaroff E. Factors for glaucoma progression and the effect of treatment: the early manifest glaucoma trial. Arch Ophthalmol. 2003;121:48–56. doi: 10.1001/archopht.121.1.48. [DOI] [PubMed] [Google Scholar]
- Levy SF, Leboeuf AC, Massie MR, Jordan MA, Wilson L, Feinstein SC. Three- and four-repeat tau regulate the dynamic instability of two distinct microtubule subpopulations in qualitatively different manners: implications for neurodegeneration. J Biol Chem. 2005;280:13520–13528. doi: 10.1074/jbc.M413490200. [DOI] [PubMed] [Google Scholar]
- Liu L, Drouet V, Wu JW, Witter MP, Small SA, Clelland C, Duff K. Trans-synaptic spread of tau pathology in vivo. PLoS One. 2012;7:e31302. doi: 10.1371/journal.pone.0031302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda S, Sahara N, Saito Y, Murayama S, Ikai A, Takashima A. Increased levels of granular tau oligomers: an early sign of brain aging and Alzheimer's disease. Neurosci Res. 2006;54:197–201. doi: 10.1016/j.neures.2005.11.009. [DOI] [PubMed] [Google Scholar]
- Mailliot C, Podevin-Dimster V, Rosenthal RE, Sergeant N, Delacourte A, Fiskum G, Buée L. Rapid tau protein dephosphorylation and differential rephosphorylation during cardiac arrest-induced cerebral ischemia and reperfusion. J Cereb Blood Flow Metab. 2000;20:543–549. doi: 10.1097/00004647-200003000-00013. [DOI] [PubMed] [Google Scholar]
- Mansour-Robaey S, Clarke DB, Wang YC, Bray GM, Aguayo AJ. Effects of ocular injury and administration of brain-derived neurotrophic factor on survival and regrowth of axotomized retinal ganglion cells. Proc Natl Acad Sci U S A. 1994;91:1632–1636. doi: 10.1073/pnas.91.5.1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinnon SJ, Lehman DM, Kerrigan-Baumrind LA, Merges CA, Pease ME, Kerrigan DF, Ransom NL, Tahzib NG, Reitsamer HA, Levkovitch-Verbin H, Quigley HA, Zack DJ. Caspase activation and amyloid precursor protein cleavage in rat ocular hypertension. Invest Ophthalmol Vis Sci. 2002;43:1077–1087. [PubMed] [Google Scholar]
- Medina M, Avila J. The role of extracellular Tau in the spreading of neurofibrillary pathology. Front Cell Neurosci. 2014;8:113. doi: 10.3389/fncel.2014.00113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Migheli A, Butler M, Brown K, Shelanski ML. Light and electron microscope localization of the microtubule-associated tau protein in rat brain. J Neurosci. 1988;8:1846–1851. doi: 10.1523/JNEUROSCI.08-06-01846.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohamed NV, Plouffe V, Rémillard-Labrosse G, Planel E, Leclerc N. Starvation and inhibition of lysosomal function increased tau secretion by primary cortical neurons. Sci Rep. 2014;4:5715. doi: 10.1038/srep05715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morawe T, Hiebel C, Kern A, Behl C. Protein homeostasis, aging and Alzheimer's disease. Mol Neurobiol. 2012;46:41–54. doi: 10.1007/s12035-012-8246-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morquette B, Morquette P, Agostinone J, Feinstein E, McKinney RA, Kolta A, Di Polo A. REDD2-mediated inhibition of mTOR promotes dendrite retraction induced by axonal injury. Cell Death Differ. 2015;22:612–625. doi: 10.1038/cdd.2014.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison JC, Moore CG, Deppmeier LM, Gold BG, Meshul CK, Johnson EC. A rat model of chronic pressure-induced optic nerve damage. Exp Eye Res. 1997;64:85–96. doi: 10.1006/exer.1996.0184. [DOI] [PubMed] [Google Scholar]
- Morrison JC, Cepurna WO, Johnson EC. Modeling glaucoma in rats by sclerosing aqueous outflow pathways to elevate intraocular pressure. Exp Eye Res. 2015;141:23–32. doi: 10.1016/j.exer.2015.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadal-Nicolás FM, Jiménez-López M, Sobrado-Calvo P, Nieto-López L, Cánovas-Martínez I, Salinas-Navarro M, Vidal-Sanz M, Agudo M. Brn3a as a marker of retinal ganglion cells: qualitative and quantitative time course studies in naive and optic nerve injured retinas. Invest Ophthalmol Vis Sci. 2009;50:3860–3868. doi: 10.1167/iovs.08-3267. [DOI] [PubMed] [Google Scholar]
- Nickells RW, Howell GR, Soto I, John SW. Under pressure: cellular and molecular responses during glaucoma, a common neurodegeneration with axonopathy. Annu Rev Neurosci. 2012;35:153–179. doi: 10.1146/annurev.neuro.051508.135728. [DOI] [PubMed] [Google Scholar]
- Noble W, Hanger DP, Gallo JM. Transgenic mouse models of tauopathy in drug discovery. CNS Neurol Disord Drug Targets. 2010;9:403–428. doi: 10.2174/187152710791556131. [DOI] [PubMed] [Google Scholar]
- Nucci C, Martucci A, Martorana A, Sancesario GM, Cerulli L. Glaucoma progression associated with altered cerebral spinal fluid levels of amyloid beta and tau proteins. Clin Experiment Ophthalmol. 2011;39:279–281. doi: 10.1111/j.1442-9071.2010.02452.x. [DOI] [PubMed] [Google Scholar]
- Oblinger MM, Argasinski A, Wong J, Kosik KS. Tau gene expression in rat sensory neurons during development and regeneration. J Neurosci. 1991;11:2453–2459. doi: 10.1523/JNEUROSCI.11-08-02453.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortín-Martínez A, Salinas-Navarro M, Nadal-Nicolás FM, Jiménez-López M, Valiente-Soriano FJ, García-Ayuso D, Bernal-Garro JM, Avilés-Trigueros M, Agudo-Barriuso M, Villegas-Pérez MP, Vidal-Sanz M. Laser-induced ocular hypertension in adult rats does not affect non-RGC neurons in the ganglion cell layer but results in protracted severe loss of cone-photoreceptors. Exp Eye Res. 2015;132:17–33. doi: 10.1016/j.exer.2015.01.006. [DOI] [PubMed] [Google Scholar]
- Otvos L, Jr, Feiner L, Lang E, Szendrei GI, Goedert M, Lee VM. Monoclonal antibody PHF-1 recognizes tau protein phosphorylated at serine residues 396 and 404. J Neurosci Res. 1994;39:669–673. doi: 10.1002/jnr.490390607. [DOI] [PubMed] [Google Scholar]
- Pang JJ, Frankfort BJ, Gross RL, Wu SM. Elevated intraocular pressure decreases response sensitivity of inner retinal neurons in experimental glaucoma mice. Proc Natl Acad Sci U S A. 2015;112:2593–2598. doi: 10.1073/pnas.1419921112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson KR, Remmers C, Fu Y, Brooker S, Kanaan NM, Vana L, Ward S, Reyes JF, Philibert K, Glucksman MJ, Binder LI. Characterization of prefibrillar tau oligomers in vitro and in Alzheimer disease. J Biol Chem. 2011;286:23063–23076. doi: 10.1074/jbc.M111.237974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pernet V, Di Polo A. Synergistic action of brain-derived neurotrophic factor and lens injury promotes retinal ganglion cell survival, but leads to optic nerve dystrophy in vivo. Brain. 2006;129:1014–1026. doi: 10.1093/brain/awl015. [DOI] [PubMed] [Google Scholar]
- Perry VH. Evidence for an amacrine cell system in the ganglion cell layer of the rat retina. Neuroscience. 1981;6:931–944. doi: 10.1016/0306-4522(81)90174-3. [DOI] [PubMed] [Google Scholar]
- Petry FR, Pelletier J, Bretteville A, Morin F, Calon F, Hébert SS, Whittington RA, Planel E. Specificity of anti-tau antibodies when analyzing mice models of Alzheimer's disease: problems and solutions. PLoS One. 2014;9:e94251. doi: 10.1371/journal.pone.0094251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Planel E, Miyasaka T, Launey T, Chui DH, Tanemura K, Sato S, Murayama O, Ishiguro K, Tatebayashi Y, Takashima A. Alterations in glucose metabolism induce hypothermia leading to tau hyperphosphorylation through differential inhibition of kinase and phosphatase activities: implications for Alzheimer's disease. J Neurosci. 2004;24:2401–2411. doi: 10.1523/JNEUROSCI.5561-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plouffe V, Mohamed NV, Rivest-McGraw J, Bertrand J, Lauzon M, Leclerc N. Hyperphosphorylation and cleavage at D421 enhance tau secretion. PLoS One. 2012;7:e36873. doi: 10.1371/journal.pone.0036873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pooler AM, Phillips EC, Lau DH, Noble W, Hanger DP. Physiological release of endogenous tau is stimulated by neuronal activity. EMBO Rep. 2013;14:389–394. doi: 10.1038/embor.2013.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quigley HA. Glaucoma. Lancet. 2011;377:1367–1377. doi: 10.1016/S0140-6736(10)61423-7. [DOI] [PubMed] [Google Scholar]
- Rapoport M, Dawson HN, Binder LI, Vitek MP, Ferreira A. Tau is essential to β-amyloid-induced neurotoxicity. Proc Natl Acad Sci U S A. 2002;99:6364–6369. doi: 10.1073/pnas.092136199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberson ED, Scearce-Levie K, Palop JJ, Yan F, Cheng IH, Wu T, Gerstein H, Yu GQ, Mucke L. Reducing endogenous tau ameliorates amyloid ß-induced deficits in an Alzheimer's disease mouse model. Science. 2007;316:750–754. doi: 10.1126/science.1141736. [DOI] [PubMed] [Google Scholar]
- Roberson ED, Halabisky B, Yoo JW, Yao J, Chin J, Yan F, Wu T, Hamto P, Devidze N, Yu GQ, Palop JJ, Noebels JL, Mucke L. Amyloid-β/Fyn-induced synaptic, network, and cognitive impairments depend on tau levels in multiple mouse models of Alzheimer's disease. J Neurosci. 2011;31:700–711. doi: 10.1523/JNEUROSCI.4152-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahara N, Maeda S, Murayama M, Suzuki T, Dohmae N, Yen SH, Takashima A. Assembly of two distinct dimers and higher-order oligomers from full-length tau. Eur J Neurosci. 2007;25:3020–3029. doi: 10.1111/j.1460-9568.2007.05555.x. [DOI] [PubMed] [Google Scholar]
- Salt TE, Nizari S, Cordeiro MF, Russ H, Danysz W. Effect of the Aβ aggregation modulator MRZ-99030 on retinal damage in an animal model of glaucoma. Neurotox Res. 2014;26:440–446. doi: 10.1007/s12640-014-9488-6. [DOI] [PubMed] [Google Scholar]
- Santacruz K, Lewis J, Spires T, Paulson J, Kotilinek L, Ingelsson M, Guimaraes A, DeTure M, Ramsden M, McGowan E, Forster C, Yue M, Orne J, Janus C, Mariash A, Kuskowski M, Hyman B, Hutton M, Ashe KH. Tau suppression in a neurodegenerative mouse model improves memory function. Science. 2005;309:476–481. doi: 10.1126/science.1113694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholz T, Mandelkow E. Transport and diffusion of tau protein in neurons. Cell Mol Life Sci. 2014;71:3139–3150. doi: 10.1007/s00018-014-1610-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schön C, Hoffmann NA, Ochs SM, Burgold S, Filser S, Steinbach S, Seeliger MW, Arzberger T, Goedert M, Kretzschmar HA, Schmidt B, Herms J. Long-term in vivo imaging of fibrillar tau in the retina of P301S transgenic mice. PLoS One. 2012;7:e53547. doi: 10.1371/journal.pone.0053547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sergeant N, Bretteville A, Hamdane M, Caillet-Boudin ML, Grognet P, Bombois S, Blum D, Delacourte A, Pasquier F, Vanmechelen E, Schraen-Maschke S, Buée L. Biochemistry of tau in Alzheimer's disease and related neurological disorders. Expert Rev Proteomics. 2008;5:207–224. doi: 10.1586/14789450.5.2.207. [DOI] [PubMed] [Google Scholar]
- Shackelford DA, Nelson KE. Changes in phosphorylation of τ during ischemia and reperfusion in the rabbit spinal cord. J Neurochem. 1996;66:286–295. doi: 10.1046/j.1471-4159.1996.66010286.x. [DOI] [PubMed] [Google Scholar]
- Shackelford DA, Yeh RY. Dephosphorylation of tau during transient forebrain ischemia in the rat. Mol Chem Neuropathol. 1998;34:103–120. doi: 10.1007/BF02815073. [DOI] [PubMed] [Google Scholar]
- Sit AJ. Intraocular pressure variations: causes and clinical significance. Can J Ophthalmol. 2014;49:484–488. doi: 10.1016/j.jcjo.2014.07.008. [DOI] [PubMed] [Google Scholar]
- Spires TL, Orne JD, SantaCruz K, Pitstick R, Carlson GA, Ashe KH, Hyman BT. Region-specific dissociation of neuronal loss and neurofibrillary pathology in a mouse model of tauopathy. Am J Pathol. 2006;168:1598–1607. doi: 10.2353/ajpath.2006.050840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tepper K, Biernat J, Kumar S, Wegmann S, Timm T, Hübschmann S, Redecke L, Mandelkow EM, Müller DJ, Mandelkow E. Oligomer formation of tau protein hyperphosphorylated in cells. J Biol Chem. 2014;289:34389–34407. doi: 10.1074/jbc.M114.611368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terry RD. Do neuronal inclusions kill the cell? J Neural Transm Suppl. 2000;59:91–93. doi: 10.1007/978-3-7091-6781-6_12. [DOI] [PubMed] [Google Scholar]
- Tham YC, Li X, Wong TY, Quigley HA, Aung T, Cheng CY. Global prevalence of glaucoma and projections of glaucoma burden through 2040: a systematic review and meta-analysis. Ophthalmology. 2014;121:2081–2090. doi: 10.1016/j.ophtha.2014.05.013. [DOI] [PubMed] [Google Scholar]
- van de Nes JA, Nafe R, Schlote W. Non-tau based neuronal degeneration in Alzheimer's disease—an immunocytochemical and quantitative study in the supragranular layers of the middle temporal neocortex. Brain Res. 2008;1213:152–165. doi: 10.1016/j.brainres.2008.03.043. [DOI] [PubMed] [Google Scholar]
- Vidal-Sanz M, Valiente-Soriano FJ, Ortín-Martínez A, Nadal-Nicolás FM, Jiménez-López M, Salinas-Navarro M, Alarcón-Martínez L, García-Ayuso D, Avilés-Trigueros M, Agudo-Barriuso M, Villegas-Pérez MP. Retinal neurodegeneration in experimental glaucoma. Prog Brain Res. 2015;220:1–35. doi: 10.1016/bs.pbr.2015.04.008. [DOI] [PubMed] [Google Scholar]
- Vogt BA, Vogt LJ, Vrana KE, Gioia L, Meadows RS, Challa VR, Hof PR, Van Hoesen GW. Multivariate analysis of laminar patterns of neurodegeneration in posterior cingulate cortex in Alzheimer's disease. Exp Neurol. 1998;153:8–22. doi: 10.1006/exnr.1998.6852. [DOI] [PubMed] [Google Scholar]
- Wang Y, Mandelkow E. Degradation of tau protein by autophagy and proteasomal pathways. Biochem Soc Trans. 2012;40:644–652. doi: 10.1042/BST20120071. [DOI] [PubMed] [Google Scholar]
- Wilson AM, Di Polo A. Gene therapy for retinal ganglion cell neuroprotection in glaucoma. Gene Ther. 2012;19:127–136. doi: 10.1038/gt.2011.142. [DOI] [PubMed] [Google Scholar]
- Wilson AM, Morquette B, Abdouh M, Unsain N, Barker PA, Feinstein E, Bernier G, Di Polo A. ASPP1/2 regulate p53-dependent death of retinal ganglion cells through PUMA and Fas/CD95 activation in vivo. J Neurosci. 2013;33:2205–2216. doi: 10.1523/JNEUROSCI.2635-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson AM, Chiodo VA, Boye SL, Brecha NC, Hauswirth WW, Di Polo A. Inhibitor of apoptosis-stimulating protein of p53 (iASPP) is required for neuronal survival after axonal injury. PLoS One. 2014;9:e94175. doi: 10.1371/journal.pone.0094175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittmann CW, Wszolek MF, Shulman JM, Salvaterra PM, Lewis J, Hutton M, Feany MB. Tauopathy in Drosophila: neurodegeneration without neurofibrillary tangles. Science. 2001;293:711–714. doi: 10.1126/science.1062382. [DOI] [PubMed] [Google Scholar]
- Yin Y, Cui Q, Li Y, Irwin N, Fischer D, Harvey AR, Benowitz LI. Macrophage-derived factors stimulate optic nerve regeneration. J Neurosci. 2003;23:2284–2293. doi: 10.1523/JNEUROSCI.23-06-02284.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoneda S, Hara H, Hirata A, Fukushima M, Inomata Y, Tanihara H. Vitreous fluid levels of β-amyloid (1–42) and tau in patients with retinal diseases. Jpn J Ophthalmol. 2005;49:106–108. doi: 10.1007/s10384-004-0156-x. [DOI] [PubMed] [Google Scholar]
- Yoshiyama Y, Higuchi M, Zhang B, Huang SM, Iwata N, Saido TC, Maeda J, Suhara T, Trojanowski JQ, Lee VM. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron. 2007;53:337–351. doi: 10.1016/j.neuron.2007.01.010. [DOI] [PubMed] [Google Scholar]
- Zempel H, Mandelkow E. Lost after translation: missorting of Tau protein and consequences for Alzheimer disease. Trends Neurosci. 2014;37:721–732. doi: 10.1016/j.tins.2014.08.004. [DOI] [PubMed] [Google Scholar]
- Zempel H, Luedtke J, Kumar Y, Biernat J, Dawson H, Mandelkow E, Mandelkow EM. Amyloid-β oligomers induce synaptic damage via Tau-dependent microtubule severing by TTLL6 and spastin. EMBO J. 2013;32:2920–2937. doi: 10.1038/emboj.2013.207. [DOI] [PMC free article] [PubMed] [Google Scholar]