

Abstract
Huntington's disease (HD) is a heritable neurodegenerative disorder caused by expansion of CAG (glutamine) repeats in the HTT gene. A prodromal stage characterized by psychiatric disturbances normally precedes primary motor symptoms and suppressed motivation represents one of the earliest and most common psychiatric symptoms. Although dopamine in the nucleus accumbens (NAc) critically regulates motivation and altered dopamine signaling is implicated in HD, the nature of dopaminergic deficits and contribution to symptoms in HD is poorly understood. We therefore tested whether altered NAc dopamine release accompanies motivational deficits in the Q175 knock-in HD mouse model. Q175 mice express a CAG expansion of the human mutant huntingtin allele in the native mouse genome and gradually manifest symptoms late in life, closely mimicking the genotypic context and disease progression in human HD. Sub-second extracellular dopamine release dynamics were monitored using fast-scan cyclic voltammetry, whereas motivation was assessed using a progressive ratio reinforcement schedule. As the response ratio (lever presses per reward) escalated, Q175 mice exerted less effort to earn fewer rewards versus wild-type (WT). Moreover, dopamine released at reward delivery dynamically encoded increasing reward cost in WT but not Q175 mice. Deficits were specific to situations of high effortful demand as no difference was observed in locomotion, free feeding, hedonic processing, or reward seeking when the response requirement was low. This compromised dopaminergic encoding of reward delivery coincident with suppressed motivation to work for reward in Q175 mice provides novel, neurobiological insight into an established and clinically relevant endophenotype of prodromal HD.
SIGNIFICANCE STATEMENT Psychiatric impairments in Huntington's disease (HD) typically manifest early in disease progression, before motor deficits. However, the neurobiological factors contributing to psychiatric symptoms are poorly understood. We used a mouse HD model and assessed whether impaired dopamine release in the nucleus accumbens (NAc), a brain region critical to goal-directed behaviors, accompanies motivational deficits, one of the most common early HD symptoms. HD mice exhibited blunted motivation to work for food reward coincident with diminished dopamine release to reward receipt. Motivational and NAc dopaminergic deficits were not associated with gross motor deficits or impaired food seeking when effortful demands were low. This work identifies a specific prodromal HD phenotype associated with a prominent and previously unidentified neurobiological impairment.
Keywords: accumbens, dopamine, Huntington's, motivation, progressive ratio, voltammetry
Introduction
Psychiatric deficits in Huntington's disease (HD) appear early during a prodromal stage that precedes motor deficits (eg, chorea) by a decade or more (Paulsen et al., 2008; Epping et al., 2016). These nonmotor symptoms severely impact daily functioning and quality of life for HD patients and caregivers (Hamilton et al., 2003; Papoutsi et al., 2014), but are poorly understood and insufficiently treated (Frank, 2014). Whereas striatal medium spiny neuron (MSN) degeneration represents the canonical, histopathological hallmark of HD (Reiner et al., 1988; Vonsattel and DiFiglia, 1998), altered dopamine input onto striatal MSNs is also implicated (Klawans et al., 1970; Spokes, 1980) and may promote early psychiatric impairments (Bäckman et al., 1997). However, the functional nature of dopaminergic deficits and their behavioral relevance in HD remains controversial as estimates of extracellular dopamine levels in HD patients and animal models have been inconsistent (Cepeda et al., 2014; Schwab et al., 2015). Addressing this question is particularly important because the only FDA-approved pharmaceutical treatment option for HD targets chorea and is confined to agents that inhibit the vesicular monoamine transporter-2 (VMAT-2) to disrupt extracellular dopamine release (Venuto et al., 2012).
Apathy, characterized by diminished motivation to engage reinforcers, is one of the most common psychiatric symptoms in HD (Paulsen et al., 2001; Naarding et al., 2009; Tabrizi et al., 2013; van Duijn et al., 2014). Motivational deficits appear early in the prodromal stage and worsen over disease progression. The mesolimbic dopamine system projects onto MSNs in the nucleus accumbens (NAc) and fundamentally regulates motivated behavior (Salamone and Correa, 2002; Salamone et al., 2015), such that elevating or suppressing NAc dopamine signaling, respectively, increases or decreases the propensity to overcome high opportunity costs to acquire rewards. Yet, these same manipulations do not affect motor or hedonic aspects of reward consumption, highlighting a specific role for NAc dopamine release in incentive motivational processing of effortful costs. It remains unclear, however, whether altered dopamine input to NAc MSNs accompanies motivational deficits in HD.
In the current study, we simultaneously monitored NAc dopamine release and motivation in a mouse model of HD that possesses high face and construct validity. Specifically, experiments were performed in the knock-in Q175 HD mouse model, which expresses the human Htt allele containing expanded CAG repeats (∼179) in a heterozygous manner within the native mouse HTT gene (Menalled et al., 2012). Sub-second extracellular dopamine concentration dynamics were monitored in the NAc using fast-scan cyclic voltammetry (FSCV), whereas motivation was quantified using a progressive ratio (PR) reinforcement schedule. During the PR task, the number of lever presses required to earn sucrose pellet reinforcement increased on each successive trial, whereas reward magnitude remained fixed. Q175 mice exerted less effort (lever presses) to acquire fewer rewards and exhibited compromised dopaminergic encoding of reward delivery specifically when response cost was greatest. No difference, however, was observed in hedonic processing of taste, food seeking under low cost conditions, or locomotion, indicating a selective disruption in the motivation to overcome effortful costs to gain reward. Collectively, this work identifies a prodromal HD phenotype associated with a specific deficit in sub-second dopamine release in the NAc.
Materials and Methods
Animals.
All mice (20–30 g) were male and received from Jackson Laboratories at 30 weeks of age. Dopamine measures during the PR task were obtained from heterozygous Q175 mice (n = 12) and wild-type (WT; n = 8) littermates. A separate cohort of Q175 (n = 19) and WT (n = 14) mice were used to compare feeding behavior and locomotion. Mice were housed in a temperature-controlled room maintained on a reverse 12 h light/dark cycle (07:00–19:00 h). Following surgery, mice were housed individually and allowed ad libitum access to water and food. Mice were restricted to 85–90% bodyweight for the duration of behavioral measures. All experiments were conducted in the animal's light cycle. Animal care and experimental procedures conformed to the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Use and Care Committee at the University of Maryland, Baltimore.
Carbon-fiber microelectrodes.
Chronically implantable electrodes were constructed as previously described (Clark et al., 2010). Individual carbon fibers (r = 3.5 μm, Hexcel) were aspirated into a 5-mm-length segment of fused silica. The seal between the silica and carbon fiber was created by applying a two-part epoxy (Super Glue; TQs12 Epoxy). The exposed carbon fiber was then cut to ∼150 μm and a silver connector was attached on the opposite end for conductivity.
Surgery.
Mice were anesthetized with isoflurane in O2 (4% induction and 1% maintenance) and implanted with a chronic voltammetry electrode targeting the NAc core (+1.2 AP, +1.1 ML, −3.7 DV relative to bregma) and a Ag/AgCl reference electrode in the contralateral superficial cortex. All components were permanently affixed with dental cement.
Recording sessions.
FSCV was used to monitor dopamine by applying a triangular waveform (−0.4 V to +1.3 V at 400 V/s) at 10 Hz to implanted carbon fiber microelectrodes. Principal component regression (PCR) was used to statistically extract the dopamine component from the voltammetric recording of current (Heien et al., 2005; Keithley et al., 2009). The training set for PCR consisted of five background-subtracted dopamine, and five basic pH shift voltammograms. Measured current was converted to concentration based on a dataset developed in vitro using a flow cell apparatus to quantify dopamine oxidation current versus nonfaradaic background current (Roberts et al., 2013). Electrochemical measures were normalized or “background-subtracted” relative to 0.5 s preceding reward delivery and compared across the PR session and animals. Measured current following reward presentation was plotted across the applied potential to generate “cyclic voltammograms” (CVs), which display the electrochemical properties of the recorded analyte. Individual CVs pertaining to the reward-evoked dopamine signal were obtained from the current at the maximal dopamine oxidation potential (∼0.65 V) during the 3 s period following reward presentation. Sequential CVs were plotted across time in color plots. Individual CVs and color plots were binned into thirds of each animal's PR session and averaged across all animals in each group, and therefore reflect all trials from all animals.
Apparatus.
Mice were tested in operant chambers (21.6 × 17.6 × 14 cm; Med Associates) housed within sound-attenuating enclosures. Each chamber was equipped with two retractable levers (located 2 cm above the floor) and one LED stimulus light located above each lever (4.6 cm above the lever). An external food magazine was connected to a dispenser, centrally located between the two levers that delivered sucrose pellets (14 mg; Bio-Serv). A houselight and a white-noise speaker (80 dB, masking noise background) were located on the opposite wall.
Behavioral testing.
Mice were initially trained under a fixed-ratio (FR) 1 schedule with a 10 s timeout and switched to PR after stable responding was established (<15% variation in response rate across 3 consecutive sessions). The PR schedule of positive reinforcement was used to quantify the effort animals were willing to expend for a reward (Hodos, 1961). The number of lever presses required to earn each reward (ie, response ratio) increased exponentially on successive trials [response ratio = (5 × e(0.2 × reward number) − 5)], yielding ratios of 1, 2, 4, 6, 9, 12, 15, 20, 25, 32, 40, 50, 62, 77, 95, 118, etc, after rounding to the closest integer. The final ratio attained (ie, breakpoint) is a metric of inherent motivation for the reward. FSCV recordings were performed after individual animals displayed stable responding on the PR task (varying by no more than ±1 response ratio over 3 consecutive sessions). Session onset was signaled by both levers extending and illumination of the house light and cue light above the active lever. Responses on the inactive lever were recorded but had no programmed consequences. Upon reaching the response requirement on each trial, a single pellet was delivered while both levers retracted and lights dimmed for a 10 s period before the next trial began. Sessions were terminated after 20 min passed without reward delivery.
To assess feeding under free-access conditions, mice were restricted to 85–90% bodyweight and given 1 h ad libitum access to sucrose pellets once/d for 3 d. To measure hedonic processing, a saccharin preference test was performed under ad libitum feeding conditions, (Harkin et al., 2002; Sclafani et al., 2010). WT and Q175 mice were given a series of two-bottle choice tests between saccharin and water. To obtain a concentration–response function, seven saccharin concentrations (% saccharin; 0%, 0.01%, 0.03%, 0.1%, 0.3%, 1%, and 3% w/v) were presented concurrently with water. Each concentration was presented for 2 d each in increasing order of concentration, and average 24 h intake of saccharin was compared with intake of water to obtain percentage saccharin preference [(24 h saccharin intake)/(24 h saccharin + water intake) × 100] at each concentration. Placement of the water and saccharin container was counterbalanced across animals and alternated on the second day. To assess overall motor function, an open-field locomotor test was performed in a 1 × 1 m arena for 1 h. Behavior was recorded with a digital video camera, positioned overhead. Data were analyzed using TopScan systems (Cleversys).
Statistics.
Individual voltammetric recordings of dopamine concentration and behavioral measures were compared across the PR session (time factor) and between genotypes (group factor) using a two-way repeated-measures ANOVA. FR1 behavior was also compared between genotypes (group factor) and the first four FR1 sessions (time factor) using a two-way repeated-measures ANOVA. Saccharin preference (%) was also compared between genotypes and across saccharin concentrations using a two-way repeated-measures ANOVA. Holm–Sidak post hoc tests were used to make comparisons between groups when main effects or the interaction were significant. Breakpoint, maximal reward-evoked dopamine concentrations, locomotion, sucrose consumption, and bodyweight were compared using Student's t tests. Deming regression was used to compare active presses or time to complete each trial versus reward-evoked dopamine concentrations across the final eight trials of each animal's PR session. This analysis is used when error is present in both x and y dimensions and tests whether the slope of this relationship differs from zero. Statistical tests were performed with SigmaPlot (v12.5) and significance was set at p < 0.05.
Results
Suppressed motivation and NAc dopamine release in Q175 mice
Q175 mice displayed compromised motivation for a sucrose pellet reinforcer. Figure 1A depicts a cumulative record of active lever presses in Q175 and WT mice during representative PR sessions. Responding diverges as the session progresses and Q175 mice exert less effort, as demonstrated by the significantly lower breakpoint (Fig. 1B), to earn fewer rewards (WT: 20.5 ± 0.5 pellets, Q175: 16.7 ± 1.1 pellets; p < 0.05). To assess whether differences in dopamine function accompany motivational deficits, we recorded sub-second dopamine release in the NAc with FSCV during the PR task. The average dopamine concentration following reward receipt was significantly greater in WT versus Q175 mice (Fig. 1C). Moreover, the single, largest dopamine release event elicited by reward receipt during the entire PR session was significantly greater, on average, in WT versus Q175 mice (Fig. 1D), suggesting compromised dopaminergic function in this HD model.
Figure 1.

Q175 mice display compromised motivation and reward-evoked dopamine release in the NAc. A, Representative cumulative response record during a PR session in the two genotypes. Vertical ticks demarcate reward receipt. B, Mean (+SEM) breakpoints in WT (n = 8) and Q175 (n = 12) mice. C, Mean dopamine concentration ([DA]; +SEM) across all trials. D, Mean of the single, largest dopamine concentration increase (+SEM) in each animal. **p < 0.01, ***p < 0.001.
To compare changes in dopamine release as the PR session progresses, data were binned into thirds of the session according to the total number of trials for each animal. This allows the same proportion of the PR session to be compared across animals, despite a greater overall number of trials in WT mice due to a higher breakpoint. In WT mice, average dopamine release evoked by reward receipt increased as the PR session progressed (Fig. 2). Average CVs (Fig. 2, inset) and pseudo-color plots (Fig. 2, bottom) verify that the recorded analyte was dopamine The concentration of dopamine released during the “Early” and “Middle” trials was similar, whereas a large increase occurred during “Late” trials when the response cost was greatest. Figure 3 displays dopamine released to reward receipt in Q175 mice. In contrast to WT mice (Fig. 2), the amount of dopamine released did not change as the PR session progressed (Fig. 3). Statistical analysis confirmed that Q175 mice exhibit compromised dopaminergic encoding of reward delivery (Fig. 4). Reward-evoked dopamine release, divided into thirds of the PR session, was significantly different between genotypes and across the session (Fig. 4A: genotype × epoch: F(2,36) = 7.55, p < 0.01). Genotypic differences were due to increased reward-evoked dopamine release during the final third of the session (Late trials) in WT mice versus earlier time points and versus Q175 mice at the final time point. In contrast, no difference in dopamine release was observed in Q175 mice across the PR session (all p values >0.20).
Figure 2.

NAc dopamine release dynamically encodes reward delivery across PR session in WT mice (n = 8). Mean (+SEM) dopamine concentration across all WT mice aligned to reward delivery (vertical dashed line at 5 s) and divided into thirds of the entire session for each animal. Inset, Mean (+SEM) cyclic voltammograms across all WT mice during the peak dopamine concentration following reward delivery. Bottom, Pseudo-color plots of cyclic voltammograms displaying changes in current across the applied potential (E) plotted over time and averaged across all WT mice.
Figure 3.

Suppressed dopaminergic encoding of reward delivery in Q175 mice (n = 12). Mean (+SEM) dopamine concentration across all Q175 mice aligned to reward delivery (vertical dashed line at 5 s) and divided into thirds of the entire session for each animal. Inset, Mean (+SEM) cyclic voltammograms across all Q175 mice during the peak dopamine concentration following reward delivery. Bottom, Pseudo-color plots of cyclic voltammograms plotted over time and averaged across all Q175 mice.
Figure 4.
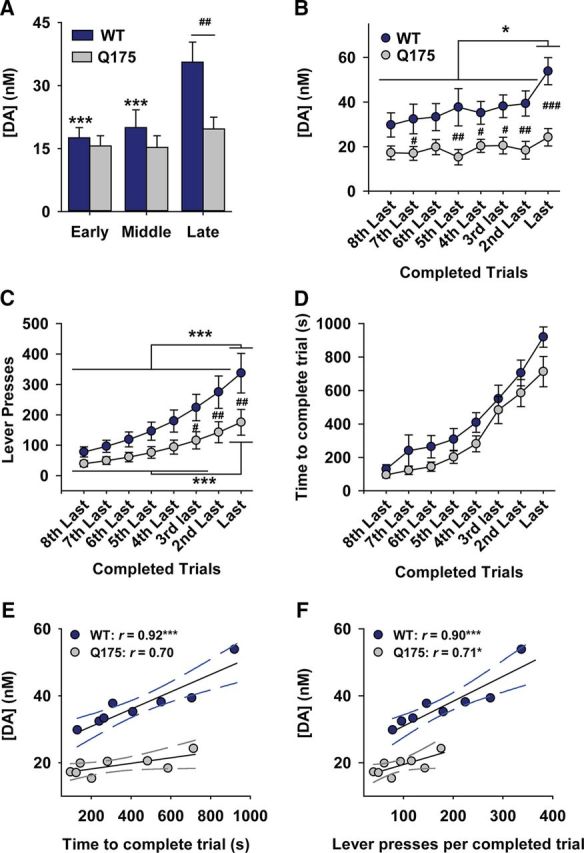
Dopaminergic encoding of effortful costs is compromised in Q175 mice. A, Mean dopamine concentration (+SEM) following reward delivery divided into thirds of the PR session. B, Mean dopamine concentration (+SEM) aligned to the last trial increased with escalating costs as WT mice (n = 8), but not Q175 mice (n = 12), reached their respective breakpoints. C, WT mice exerted more effort but (D) incurred similar temporal costs versus Q175 mice. E, Temporal costs correlated with the amount of dopamine released by reward in WT, but not Q175 mice. F, Effortful costs correlated with the amount of dopamine released by reward in WT and Q175 mice. ***p < 0.001, *p < 0.05 versus WT (within-group comparison). ###p < 0.001, ##p < 0.01, #p < 0.05 versus Q175 (between-group comparison).
Dopaminergic deficits emerge as effortful demands increase
The final trials of the PR session encompass the highest effortful demands and the breakpoint reflects each animal's inherent willingness to no longer meet these demands. Moreover, the largest difference in dopamine release between genotypes occurred in response to reward receipt late in the PR session (Fig. 4A). We therefore further assessed how dopamine encodes behavior during the final eight completed trials of each animal's PR session (Wanat et al., 2010). As mice reached their individual breakpoint, reward-evoked dopamine release differed between Q175 and WT mice and across trials (Fig. 4B; genotype: F(1,126) = 11.13, p < 0.01; trial: F(7,126) = 4.36, p < 0.001). Dopamine release was greater in WT mice during the last trial versus all prior time points and versus Q175 mice during the final five trials. No change was observed at any time point in Q175 mice (all p values >0.58).
In naturalistic settings, effortful requirements and delay to reinforcement both function as costs during reward seeking (Stevens et al., 2005). This is reflected in the PR schedule, as each successive trial incorporates increasing effortful (ie, lever press response ratio) and temporal (ie, time to complete response ratio) costs that are necessarily linked (Wanat et al., 2010). Indeed, the number of lever presses and time to complete each trial increased exponentially during the final eight trials of each animal's PR session and were highly correlated (r = 0.995, r = 0.989 in WT and Q175, respectively). To assess whether dopamine release or behavior differentially respond to terminal costs in Q175 versus WT mice, we compared the number of lever presses (ie, response ratio; Fig. 4C) and time to complete each trial (Fig. 4D) as mice approached their individual breakpoint. Lever press count significantly differed across trials and between genotypes (Fig. 4C; genotype × trial: F(7,126) = 5.02, p < 0.001) and was significantly greater in WT versus Q175 mice during the final 3 trials, in agreement with a greater overall breakpoint (Fig. 1). Although the time to complete each trial increased (Fig. 4C; time: F(7,126) = 54.93, p < 0.001), surprisingly, this did not differ between genotypes (genotype: F(1,126) = 3.02, p = 0.10; time × genotype: F(7,126) = 0.54, p = 0.80), despite Q175 mice completing fewer lever presses, reflecting a lower response rate. Thus, Q175 mice exert less effort but incur similar temporal costs per reinforcer.
To assess how dopamine encodes effortful and temporal costs, reward-evoked dopamine concentrations during the final eight trials were compared with number of lever presses (Fig. 4E) and time (Fig. 4F) per completed trial. Dopamine was highly and positively (ie, slope > 0) correlated with both time and lever presses in WT mice. In contrast, although dopamine was correlated with time in Q175 mice the slope of this relationship versus zero did not reach statistical significance (p = 0.054) due to the signal not changing over time. Alternatively, a significant positive relationship was observed relative to lever presses (p = 0.036), suggesting a slightly stronger association between dopamine and effortful versus temporal costs in Q175 mice. Nevertheless, the small dynamic range of the dopamine signal for both comparisons, particularly when compared with WT, suggests a profound disruption in dopaminergic encoding of reward-associated costs in Q175 mice.
Although the lower dopamine response to reward observed in the Q175 mice may indicate compromised dopaminergic function, it may also reflect the distinct experience of the two groups. Indeed, prior work demonstrates that reward-evoked dopamine release in the NAc increases as a function of the delay to receive reward (Wanat et al., 2010). Thus, differences might not arise from an altered dopamine system, per se, but may reflect the larger temporal delays and response ratios endured by WT mice. In support of this hypothesis, as the PR session progressed, the amount of dopamine released to reward receipt (Fig. 5A) and the proportion of mice reaching their individual breakpoints (Fig. 5A, inset) sharply diverged. This rapid decline in the number of Q175 mice may confound interpretations of differences in dopamine function during these later trials. To assess whether dopamine function is indeed suppressed in Q175 mice, reward-evoked dopamine release was assessed in a subset of mice (7 of 8 WT; 7 of 12 Q175) that completed at least 16 trials (Fig. 5B), permitting direct comparisons under identical response requirements. Similar to what was observed when data were normalized to each animal's breakpoint (Fig. 4B), dopamine released to the reward diverged in the two groups as the PR session progressed (genotype: F(1,180) = 4.86, p = 0.048; trial: F(15,180) = 3.27, p < 0.001; genotype × trial: F(15,180) = 1.72, p = 0.05). The average reward-evoked dopamine release and the maximal dopamine release to a given reward across these matched trials was significantly greater in WT versus Q175 mice (Fig. 5B, inset), in agreement with the comparison across all animals and trials (Fig. 1). Moreover, although the time to complete each response ratio increased on successive trials, this did not differ across groups (genotype: F(1,180) = 0.25, p = 0.63; trial: F(15,180) = 20.22, p < 0.001; genotype × trial: F(15,180) = 1.09, p = 0.37, data not shown), similar to the prior analysis (Fig. 4D). Thus, dopamine released to reward receipt increases as a function of effortful demands in WT, but not Q175 mice, and this is associated with an increased propensity to overcome these demands.
Figure 5.

Divergent dopaminergic encoding of the same motivational cost in Q175 mice. A, Mean dopamine concentration (+SEM) following reward delivery in all animals across all trials of the PR session. The number of animals remaining on each trial is indicated. Inset, Proportion of each group reaching their breakpoint as trials progressed. B, Mean dopamine concentration (+SEM) following reward delivery in a subset of animals matched according to the same response ratio. Inset, Mean dopamine concentration (+SEM) across all trials and mean of the single largest dopamine concentration increase (+SEM) in each animal.
Motivational deficit is specific to situations of high effortful demand
A number of biological factors contribute to motivated feeding, including hedonic processing, appetite, and motor function. To better understand the amotivational state in Q175 mice, we assessed individual dimensions of food taking (Fig. 5). WT and Q175 mice displayed a similar preference for a sweet substance as assessed by the saccharin preference test (Fig. 5A; % sach: F(6,186) = 16.96, p < 0.001), but this did not differ between genotypes (genotype: F(1,186) = 0.005, p = 0.94; % sach × genotype: F(6,186) = 0.31, p = 0.93), indicating similar levels of hedonic processing. Both groups also consumed similar quantities of sucrose under free-feeding conditions (Fig. 5B; p = 0.65). Average bodyweight (Fig. 5C; p = 0.14) and open-field locomotion (Fig. 5D; p = 0.69) were also comparable, suggesting similar energetic stores and motor capacity, respectively, in both groups. Finally, the number of lever presses during the first 4 d of training on an FR1 schedule of reinforcement increased across sessions (Fig. 5E; day: F(3,54) = 72.29, p < 0.001), but this did not differ between genotypes (genotype: F(1,54) = 3.67, p = 0.08; day × genotype: F(3,54) = 2.07, p = 0.12). Thus, Q175 mice exhibit no deficit in the acquisition or expression of appetitive behavior when response costs are low.
Discussion
Altered dopaminergic function has long been implicated in HD, but the precise nature of dopamine deficits and the behavioral relevance to HD symptoms has been difficult to determine. By simultaneously monitoring sub-second dopamine release dynamics and motivated behavior in an HD mouse model, this work identifies a dopaminergic deficit associated with a specific and major behavioral symptom of prodromal HD. We found that compromised dopaminergic encoding of reward delivery by phasic dopamine release in the NAc was associated with a pronounced motivational deficit in Q175 mice. Differences versus WT cannot be explained by an alteration in the rewarding or hedonic value of the food reinforcer, locomotion, or temporal cost. Rather, the dopaminergic deficit in Q175 mice is specifically associated with reduced willingness to overcome effortful costs to gain reinforcement.
Dopamine dysfunction in HD
Dopamine dysfunction is a recognized pathogenic feature of HD but is poorly characterized. Historically, a hyperdopaminergic state was hypothesized to underlie the dyskinetic movements that characterize Huntington's chorea (Klawans et al., 1970; Bird, 1980; Spokes, 1980). However, more recent measures demonstrate reduced dopaminergic markers in postmortem tissue from HD patients (Yohrling et al., 2003; Bédard et al., 2011) and positron emission tomography measures show reduced striatal VMAT-2 (Bohnen et al., 2000), dopamine receptor (D1 and D2-type), and dopamine transporter (DAT) function in asymptomatic HD mutation carriers (Richfield et al., 1991; Weeks et al., 1996; van Oostrom et al., 2009). Moreover, decreased striatal dopaminergic indices predict early cognitive decline in presymptomatic and early stage HD patients (Bäckman et al., 1997) and progressively deteriorate as symptoms worsen over the duration of illness (Ginovart et al., 1997). Although general consensus has been that the NAc region of the striatum is relatively spared at the expense of the caudate nucleus and putamen, decreased NAc dopaminergic markers are also observed in HD (Bédard et al., 2011).
Preclinical studies using HD animal models also support suppressed dopamine function (but see, Jahanshahi et al., 2010). In particular, microdialysis measures show decreased extracellular dopamine concentration in the striatum of R6/1 or R6/2 mice (Hickey et al., 2002; Petersén et al., 2002; Callahan and Abercrombie, 2011), both of which represent more aggressive HD models compared with Q175. Moreover, temporally resolved FSCV measures of electrically evoked dopamine release in striatal brain slices (Johnson et al., 2006; Ortiz et al., 2010, 2012) or individual vesicular release events in cell isolates (Johnson et al., 2007) from R6/2 mice demonstrate compromised exocytotic release mechanisms, but intact DAT function (but see, Ortiz et al., 2011), suggesting intact terminal expression of DAT, indicative of intact dopaminergic innervation. Compromised exocytosis may result from reduced dopamine content per vesicle (Johnson et al., 2007), fewer vesicles, or disrupted mobilization of reserve vesicular pools (Ortiz et al., 2010). One possibility is that aggregates of the Htt protein, which can impair vesicular protein function (Morton et al., 2001; Li et al., 2003), disrupt vesicular mobilization, and dopamine release.
Compromised vesicular dopamine release may underlie the suppressed dopaminergic encoding of reward observed in the current study. Although dopamine release in WT mice increased as a function of response cost, dopamine release was fixed across the session in Q175 mice. Static dopamine release despite increasing response costs in Q175 mice suggests that a ceiling effect narrows the dynamic range of the dopamine signal. Suppressed vesicular release mechanisms are implicated because the peak concentration of these phasic dopamine signals is dependent on the integrity of releasable dopamine stores (Covey et al., 2013). The nature of the PR task and FSCV monitoring of dopamine release were necessary for revealing this pronounced deficit. Because reward-evoked dopamine release during Early and Middle sessions was similar, compromised dopamine signaling may not have been detected using a low effort instrumental task, which Q175 and WT mice performed at a similar level (Fig. 5E). Moreover, the sub-second dopamine measures afforded by FSCV revealed a precise circumstance when dopaminergic deficits emerge, in response to reward receipt under conditions of high motivational demand. Alternative neurochemical monitoring approaches with low temporal resolution, such as microdialysis or PET, are unlikely to detect this specific difference.
Compromised NAc dopamine release selectively accompanies disrupted motivational aspects of food seeking
Extensive work on the role of NAc dopamine in instrumental behavior suggests that relatively greater dopaminergic encoding of response costs in WT mice supported an increased propensity to invest effort to obtain rewards. The NAc has traditionally been conceptualized as a “limbic-motor interface” (Mogenson et al., 1980) by allowing information related to motivation and emotion to influence basal ganglia motor circuits. Dopamine release in the NAc is critical to this process and acts to facilitate the persistence of responding when response costs are high (Salamone et al., 2015), such as during the final trials of the PR task. Alternatively, disrupting NAc dopamine function does not alter food seeking when response costs are low (Aberman et al., 1998; Aberman and Salamone, 1999; Salamone et al., 2001; Ishiwari et al., 2004) or the motivation to overcome temporal costs for reinforcement (Mingote et al., 2005), indicating a selective role in motivating effortful decisions. The degree to which dopamine released to the reward on successive PR trials reflected the prior circumstance (ie, response or delay cost) or acts to influence future responding remains unclear; although these functions are not mutually exclusive (Hamid et al., 2016). Nevertheless, compromised dopamine release in Q175 mice despite comparable effortful and temporal costs incurred during PR (Figs. 4, 5), normal responding under “low-cost” conditions during the FR1 schedule, and intact motor, ingestive, and hedonic aspects of food seeking (Fig. 6) suggests a critical and highly specific role for compromised reward-evoked NAc dopamine release in the amotivational phenotype of Q175 mice. Moreover, prior work indicates that Q175 mice, at the age assessed in the current study, do not exhibit deficits in muscle strength (Menalled et al., 2012), but progressive motor impairments including reduced locomotion and coordination have been reported in this mouse strain at later ages that may be further exacerbated during the dark cycle (Menalled et al., 2012). However, the intact motor function and normal feeding behavior observed in the current study suggests that motivation, specifically to overcome effortful costs, and dopaminergic deficits are linked and may arise early in a prodromal HD state before full phenoconversion.
Figure 6.

Q175 display intact reward taking and locomotion. WT and Q175 mice displayed similar (A) preference for saccharin solution, (B) sucrose pellet consumption during free-feeding conditions, (C) bodyweight on the day of the FSCV PR session, (D) locomotion during a 60 min period, and (E) acquisition of lever pressing for sucrose pellet reinforcement on an FR1 schedule. A–D, WT, n = 14; Q175, n = 19. E, WT, n = 8; Q175, n = 12.
Amotivational syndrome in HD
Apathy, a disorder of motivation characterized by diminished goal-directed behavior is one of the most common psychiatric symptoms in HD (Hamilton et al., 2003; Naarding et al., 2009). Although frequently associated with depression, motivational deficits represent a distinct disorder (Naarding et al., 2009) driven by unique neural mechanisms. Indeed, Q175 mice exhibited suppressed motivation but normal “liking” of a reward, as demonstrated by the saccharin preference test, a common test for depressive symptoms (Harkin et al., 2002). Based on the concomitant decrease in NAc dopamine release, we hypothesize that compromised phasic dopamine function in situations of high motivational demand is more specifically associated with the amovitational phenotype seen in HD. Paradoxically, the only FDA-approved treatment for HD is the dopamine-depleting agent, tetrabenazine (TBZ; Huntington Study Group, 2006; Paleacu, 2007; Frank, 2010). Although effective for suppressing chorea, TBZ is associated with several adverse side effects (Frank, 2014; Killoran and Biglan, 2014). Moreover, in WT rats, TBZ suppresses the propensity to overcome high response costs to gain rewards (Nunes et al., 2013; Randall et al., 2014), an effect also observed following NAc dopamine depletions (Aberman et al., 1998; Aberman and Salamone, 1999; Salamone et al., 2001; Ishiwari et al., 2004). Thus, findings from the current study, which suggest that disrupted dopamine function in HD promotes an amotivational phenotype, indicate that blocking dopamine function with TBZ to treat chorea may exacerbate psychiatric symptoms, such as apathy.
Conclusions
Overall, this work demonstrates that compromised dynamic encoding of reward delivery by NAc dopamine release accompanies motivational deficits in Q175 HD mice, identifying a specific neurobiological factor associated with a prominent psychiatric impairment in HD. These findings broaden our limited understanding of non-motor aspects of HD and inform future treatment targets for reducing the impact of this devastating disorder.
Footnotes
This work was supported by a research grant from CHDI to J.F.C.
The authors declare no competing financial interests.
References
- Aberman JE, Salamone JD. Nucleus accumbens dopamine depletions make rats more sensitive to high ratio requirements but do not impair primary food reinforcement. Neuroscience. 1999;92:545–552. doi: 10.1016/S0306-4522(99)00004-4. [DOI] [PubMed] [Google Scholar]
- Aberman JE, Ward SJ, Salamone JD. Effects of dopamine antagonists and accumbens dopamine depletions on time-constrained progressive-ratio performance. Pharmacol Biochem Behav. 1998;61:341–348. doi: 10.1016/S0091-3057(98)00112-9. [DOI] [PubMed] [Google Scholar]
- Bäckman L, Robins-Wahlin TB, Lundin A, Ginovart N, Farde L. Cognitive deficits in Huntington's disease are predicted by dopaminergic PET markers and brain volumes. Brain. 1997;120:2207–2217. doi: 10.1093/brain/120.12.2207. [DOI] [PubMed] [Google Scholar]
- Bédard C, Wallman MJ, Pourcher E, Gould PV, Parent A, Parent M. Serotonin and dopamine striatal innervation in Parkinson's disease and Huntington's chorea. Parkinsonism Relat Disord. 2011;17:593–598. doi: 10.1016/j.parkreldis.2011.05.012. [DOI] [PubMed] [Google Scholar]
- Bird ED. Chemical pathology of Huntington's disease. Annu Rev Pharmacol Toxicol. 1980;20:533–551. doi: 10.1146/annurev.pa.20.040180.002533. [DOI] [PubMed] [Google Scholar]
- Bohnen NI, Koeppe RA, Meyer P, Ficaro E, Wernette K, Kilbourn MR, Kuhl DE, Frey KA, Albin RL. Decreased striatal monoaminergic terminals in Huntington disease. Neurology. 2000;54:1753–1759. doi: 10.1212/WNL.54.9.1753. [DOI] [PubMed] [Google Scholar]
- Callahan JW, Abercrombie ED. In vivo dopamine efflux is decreased in striatum of both fragmented (R6/2) and full-length (YAC128) transgenic mouse models of Huntington's disease. Front Syst Neurosci. 2011;5:61. doi: 10.3389/fnsys.2011.00061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cepeda C, Murphy KP, Parent M, Levine MS. The role of dopamine in Huntington's disease. Prog Brain Res. 2014;211:235–254. doi: 10.1016/B978-0-444-63425-2.00010-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark JJ, Sandberg SG, Wanat MJ, Gan JO, Horne EA, Hart AS, Akers CA, Parker JG, Willuhn I, Martinez V, Evans SB, Stella N, Phillips PE. Chronic microsensors for longitudinal, subsecond dopamine detection in behaving animals. Nat Methods. 2010;7:126–129. doi: 10.1038/nmeth.1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covey DP, Juliano SA, Garris PA. Amphetamine elicits opposing actions on readily releasable and reserve pools for dopamine. PLoS One. 2013;8:e60763. doi: 10.1371/journal.pone.0060763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epping EA, Kim JI, Craufurd D, Brashers-Krug TM, Anderson KE, McCusker E, Luther J, Long JD, Paulsen JS, PREDICT-HD Investigators and Coordinators of the Huntington Study Group Longitudinal psychiatric symptoms in prodromal Huntington's disease: a decade of data. Am J Psychiatry. 2016;173:184–192. doi: 10.1176/appi.ajp.2015.14121551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank S. Tetrabenazine: the first approved drug for the treatment of chorea in US patients with Huntington disease. Neuropsychiatr Dis Treat. 2010;6:657–665. doi: 10.2147/NDT.S6430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank S. Treatment of Huntington's disease. Neurotherapeutics. 2014;11:153–160. doi: 10.1007/s13311-013-0244-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginovart N, Lundin A, Farde L, Halldin C, Bäckman L, Swahn CG, Pauli S, Sedvall G. PET study of the pre- and post-synaptic dopaminergic markers for the neurodegenerative process in Huntington's disease. Brain. 1997;120:503–514. doi: 10.1093/brain/120.3.503. [DOI] [PubMed] [Google Scholar]
- Hamid AA, Pettibone JR, Mabrouk OS, Hetrick VL, Schmidt R, Vander Weele CM, Kennedy RT, Aragona BJ, Berke JD. Mesolimbic dopamine signals the value of work. Nat Neurosci. 2016;19:117–126. doi: 10.1038/nn.4173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton JM, Salmon DP, Corey-Bloom J, Gamst A, Paulsen JS, Jerkins S, Jacobson MW, Peavy G. Behavioural abnormalities contribute to functional decline in Huntington's disease. J Neurol Neurosurg Psychiatry. 2003;74:120–122. doi: 10.1136/jnnp.74.1.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harkin A, Houlihan DD, Kelly JP. Reduction in preference for saccharin by repeated unpredictable stress in mice and its prevention by imipramine. J Psychopharmacol. 2002;16:115–123. doi: 10.1177/026988110201600201. [DOI] [PubMed] [Google Scholar]
- Heien ML, Khan AS, Ariansen JL, Cheer JF, Phillips PE, Wassum KM, Wightman RM. Real-time measurement of dopamine fluctuations after cocaine in the brain of behaving rats. Proc Natl Acad Sci U S A. 2005;102:10023–10028. doi: 10.1073/pnas.0504657102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickey MA, Reynolds GP, Morton AJ. The role of dopamine in motor symptoms in the R6/2 transgenic mouse model of Huntington's disease. J Neurochem. 2002;81:46–59. doi: 10.1046/j.1471-4159.2002.00804.x. [DOI] [PubMed] [Google Scholar]
- Hodos W. Progressive ratio as a measure of reward strength. Science. 1961;134:943–944. doi: 10.1126/science.134.3483.943. [DOI] [PubMed] [Google Scholar]
- Huntington Study Group. Tetrabenazine as antichorea therapy in Huntington disease: a randomized controlled trial. Neurology. 2006;66:366–372. doi: 10.1212/01.wnl.0000198586.85250.13. [DOI] [PubMed] [Google Scholar]
- Ishiwari K, Weber SM, Mingote S, Correa M, Salamone JD. Accumbens dopamine and the regulation of effort in food-seeking behavior: modulation of work output by different ratio or force requirements. Behav Brain Res. 2004;151:83–91. doi: 10.1016/j.bbr.2003.08.007. [DOI] [PubMed] [Google Scholar]
- Jahanshahi A, Vlamings R, Kaya AH, Lim LW, Janssen ML, Tan S, Visser-Vandewalle V, Steinbusch HW, Temel Y. Hyperdopaminergic status in experimental Huntington disease. J Neuropathol Exp Neurol. 2010;69:910–917. doi: 10.1097/NEN.0b013e3181ee005d. [DOI] [PubMed] [Google Scholar]
- Johnson MA, Rajan V, Miller CE, Wightman RM. Dopamine release is severely compromised in the R6/2 mouse model of Huntington's disease. J Neurochem. 2006;97:737–746. doi: 10.1111/j.1471-4159.2006.03762.x. [DOI] [PubMed] [Google Scholar]
- Johnson MA, Villanueva M, Haynes CL, Seipel AT, Buhler LA, Wightman RM. Catecholamine exocytosis is diminished in R6/2 Huntington's disease model mice. J Neurochem. 2007;103:2102–2110. doi: 10.1111/j.1471-4159.2007.04908.x. [DOI] [PubMed] [Google Scholar]
- Keithley RB, Heien ML, Wightman RM. Multivariate concentration determination using principal component regression with residual analysis. Trends Analyt Chem. 2009;28:1127–1136. doi: 10.1016/j.trac.2009.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Killoran A, Biglan KM. Current therapeutic options for Huntington's disease: good clinical practice versus evidence-based approaches? Mov Disord. 2014;29:1404–1413. doi: 10.1002/mds.26014. [DOI] [PubMed] [Google Scholar]
- Klawans HC, Paulson GW, Barbeau A. Predictive test for Huntington's chorea. Lancet. 1970;2:1185–1186. doi: 10.1016/s0140-6736(70)90367-3. [DOI] [PubMed] [Google Scholar]
- Li H, Wyman T, Yu ZX, Li SH, Li XJ. Abnormal association of mutant huntingtin with synaptic vesicles inhibits glutamate release. Hum Mol Genet. 2003;12:2021–2030. doi: 10.1093/hmg/ddg218. [DOI] [PubMed] [Google Scholar]
- Menalled LB, Kudwa AE, Miller S, Fitzpatrick J, Watson-Johnson J, Keating N, Ruiz M, Mushlin R, Alosio W, McConnell K, Connor D, Murphy C, Oakeshott S, Kwan M, Beltran J, Ghavami A, Brunner D, Park LC, Ramboz S, Howland D. Comprehensive behavioral and molecular characterization of a new knock-in mouse model of Huntington's disease: zQ175. PLoS One. 2012;7:e49838. doi: 10.1371/journal.pone.0049838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mingote S, Weber SM, Ishiwari K, Correa M, Salamone JD. Ratio and time requirements on operant schedules: effort-related effects of nucleus accumbens dopamine depletions. Eur J Neurosci. 2005;21:1749–1757. doi: 10.1111/j.1460-9568.2005.03972.x. [DOI] [PubMed] [Google Scholar]
- Mogenson GJ, Jones DL, Yim CY. From motivation to action: functional interface between the limbic system and the motor system. Prog Neurobiol. 1980;14:69–97. doi: 10.1016/0301-0082(80)90018-0. [DOI] [PubMed] [Google Scholar]
- Morton AJ, Faull RL, Edwardson JM. Abnormalities in the synaptic vesicle fusion machinery in Huntington's disease. Brain Res Bull. 2001;56:111–117. doi: 10.1016/S0361-9230(01)00611-6. [DOI] [PubMed] [Google Scholar]
- Naarding P, Janzing JG, Eling P, van der Werf S, Kremer B. Apathy is not depression in Huntington's disease. J Neuropsychiatry Clin Neurosci. 2009;21:266–270. doi: 10.1176/jnp.2009.21.3.266. [DOI] [PubMed] [Google Scholar]
- Nunes EJ, Randall PA, Hart EE, Freeland C, Yohn SE, Baqi Y, Müller CE, López-Cruz L, Correa M, Salamone JD. Effort-related motivational effects of the VMAT-2 inhibitor tetrabenazine: implications for animal models of the motivational symptoms of depression. J Neurosci. 2013;33:19120–19130. doi: 10.1523/JNEUROSCI.2730-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortiz AN, Kurth BJ, Osterhaus GL, Johnson MA. Dysregulation of intracellular dopamine stores revealed in the R6/2 mouse striatum. J Neurochem. 2010;112:755–761. doi: 10.1111/j.1471-4159.2009.06501.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortiz AN, Kurth BJ, Osterhaus GL, Johnson MA. Impaired dopamine release and uptake in R6/1 Huntington's disease model mice. Neurosci Lett. 2011;492:11–14. doi: 10.1016/j.neulet.2011.01.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortiz AN, Osterhaus GL, Lauderdale K, Mahoney L, Fowler SC, von Hörsten S, Riess O, Johnson MA. Motor function and dopamine release measurements in transgenic Huntington's disease model rats. Brain Res. 2012;1450:148–156. doi: 10.1016/j.brainres.2012.02.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paleacu D. Tetrabenazine in the treatment of Huntington's disease. Neuropsychiatr Dis Treat. 2007;3:545–551. [PMC free article] [PubMed] [Google Scholar]
- Papoutsi M, Labuschagne I, Tabrizi SJ, Stout JC. The cognitive burden in Huntington's disease: pathology, phenotype, and mechanisms of compensation. Mov Disord. 2014;29:673–683. doi: 10.1002/mds.25864. [DOI] [PubMed] [Google Scholar]
- Paulsen JS, Ready RE, Hamilton JM, Mega MS, Cummings JL. Neuropsychiatric aspects of Huntington's disease. J Neurol Neurosurg Psychiatry. 2001;71:310–314. doi: 10.1136/jnnp.71.3.310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulsen JS, Langbehn DR, Stout JC, Aylward E, Ross CA, Nance M, Guttman M, Johnson S, MacDonald M, Beglinger LJ, Duff K, Kayson E, Biglan K, Shoulson I, Oakes D, Hayden M. Detection of Huntington's disease decades before diagnosis: the predict-HD study. J Neurol Neurosurg Psychiatry. 2008;79:874–880. doi: 10.1136/jnnp.2007.128728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersén A, Puschban Z, Lotharius J, NicNiocaill B, Wiekop P, O'Connor WT, Brundin P. Evidence for dysfunction of the nigrostriatal pathway in the R6/1 line of transgenic Huntington's disease mice. Neurobiol Dis. 2002;11:134–146. doi: 10.1006/nbdi.2002.0534. [DOI] [PubMed] [Google Scholar]
- Randall PA, Lee CA, Nunes EJ, Yohn SE, Nowak V, Khan B, Shah P, Pandit S, Vemuri VK, Makriyannis A, Baqi Y, Müller CE, Correa M, Salamone JD. The VMAT-2 inhibitor tetrabenazine affects effort-related decision making in a progressive ratio/chow feeding choice task: reversal with antidepressant drugs. PLoS One. 2014;9:e99320. doi: 10.1371/journal.pone.0099320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiner A, Albin RL, Anderson KD, D'Amato CJ, Penney JB, Young AB. Differential loss of striatal projection neurons in Huntington disease. Proc Natl Acad Sci U S A. 1988;85:5733–5737. doi: 10.1073/pnas.85.15.5733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richfield EK, O'Brien CF, Eskin T, Shoulson I. Heterogeneous dopamine receptor changes in early and late Huntington's disease. Neurosci Lett. 1991;132:121–126. doi: 10.1016/0304-3940(91)90448-3. [DOI] [PubMed] [Google Scholar]
- Roberts JG, Toups JV, Eyualem E, McCarty GS, Sombers LA. In situ electrode calibration strategy for voltammetric measurements in vivo. Anal Chem. 2013;85:11568–11575. doi: 10.1021/ac402884n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salamone JD, Correa M. Motivational views of reinforcement: implications for understanding the behavioral functions of nucleus accumbens dopamine. Behav Brain Res. 2002;137:3–25. doi: 10.1016/S0166-4328(02)00282-6. [DOI] [PubMed] [Google Scholar]
- Salamone JD, Wisniecki A, Carlson BB, Correa M. Nucleus accumbens dopamine depletions make animals highly sensitive to high fixed ratio requirements but do not impair primary food reinforcement. Neuroscience. 2001;105:863–870. doi: 10.1016/S0306-4522(01)00249-4. [DOI] [PubMed] [Google Scholar]
- Salamone JD, Pardo M, Yohn SE, López-Cruz L, SanMiguel N, Correa M. Mesolimbic dopamine and the regulation of motivated behavior. Curr Top Behav Neurosci. 2015 doi: 10.1007/7854_2015_383. Advance online publication. Retrieved March 24, 2016. [DOI] [PubMed] [Google Scholar]
- Schwab LC, Garas SN, Drouin-Ouellet J, Mason SL, Stott SR, Barker RA. Dopamine and Huntington's disease. Expert Rev Neurother. 2015;15:445–458. doi: 10.1586/14737175.2015.1025383. [DOI] [PubMed] [Google Scholar]
- Sclafani A, Bahrani M, Zukerman S, Ackroff K. Stevia and saccharin preferences in rats and mice. Chem Senses. 2010;35:433–443. doi: 10.1093/chemse/bjq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spokes EG. Neurochemical alterations in Huntington's chorea: a study of post-mortem brain tissue. Brain. 1980;103:179–210. doi: 10.1093/brain/103.1.179. [DOI] [PubMed] [Google Scholar]
- Stevens JR, Rosati AG, Ross KR, Hauser MD. Will travel for food: spatial discounting in two new world monkeys. Curr Biol. 2005;15:1855–1860. doi: 10.1016/j.cub.2005.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabrizi SJ, Scahill RI, Owen G, Durr A, Leavitt BR, Roos RA, Borowsky B, Landwehrmeyer B, Frost C, Johnson H, Craufurd D, Reilmann R, Stout JC, Langbehn DR, TRACK-HD Investigators Predictors of phenotypic progression and disease onset in premanifest and early stage Huntington's disease in the TRACK-HD study: analysis of 36-month observational data. Lancet Neurol. 2013;12:637–649. doi: 10.1016/S1474-4422(13)70088-7. [DOI] [PubMed] [Google Scholar]
- van Duijn E, Craufurd D, Hubers AA, Giltay EJ, Bonelli R, Rickards H, Anderson KE, van Walsem MR, van der Mast RC, Orth M, Landwehrmeyer GB, European Huntington's Disease Network Behavioural Phenotype Working Group Neuropsychiatric symptoms in a european Huntington's disease cohort (REGISTRY) J Neurol Neurosurg Psychiatry. 2014;85:1411–1418. doi: 10.1136/jnnp-2013-307343. [DOI] [PubMed] [Google Scholar]
- van Oostrom JC, Dekker M, Willemsen AT, de Jong BM, Roos RA, Leenders KL. Changes in striatal dopamine D2 receptor binding in pre-clinical Huntington's disease. Eur J Neurol. 2009;16:226–231. doi: 10.1111/j.1468-1331.2008.02390.x. [DOI] [PubMed] [Google Scholar]
- Venuto CS, McGarry A, MA Q, Kieburtz K. Pharmacologic approaches to the treatment of Huntington's disease. Mov Disord. 2012;27:31–41. doi: 10.1002/mds.23953. [DOI] [PubMed] [Google Scholar]
- Vonsattel JP, DiFiglia M. Huntington disease. J Neuropathol Exp Neurol. 1998;57:369–384. doi: 10.1097/00005072-199805000-00001. [DOI] [PubMed] [Google Scholar]
- Wanat MJ, Kuhnen CM, Phillips PE. Delays conferred by escalating costs modulate dopamine release to rewards but not their predictors. J Neurosci. 2010;30:12020–12027. doi: 10.1523/JNEUROSCI.2691-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weeks RA, Piccini P, Harding AE, Brooks DJ. Striatal D1 and D2 dopamine receptor loss in asymptomatic mutation carriers of Huntington's disease. Ann Neurol. 1996;40:49–54. doi: 10.1002/ana.410400110. [DOI] [PubMed] [Google Scholar]
- Yohrling GJ, 4th, Jiang GC, DeJohn MM, Miller DW, Young AB, Vrana KE, Cha JH. Analysis of cellular, transgenic and human models of Huntington's disease reveals tyrosine hydroxylase alterations and substantia nigra neuropathology. Brain Res Mol Brain Res. 2003;119:28–36. doi: 10.1016/j.molbrainres.2003.08.009. [DOI] [PubMed] [Google Scholar]