

Abstract
Type 1 metabotropic glutamate receptor (mGluR1)-dependent signaling at parallel fiber to Purkinje neuron synapses is critical for cerebellar function. In a mouse model of human spino-cerebellar ataxia type 1 (early SCA1, 12 weeks) we find prolonged parallel fiber mGluR1-dependent synaptic currents and calcium signaling. Acute treatment with a low dose of the potent and specific activity-dependent mGluR1-negative allosteric modulator JNJ16259685 shortened the prolonged mGluR1 currents and rescued the moderate ataxia. Our results provide exciting new momentum for developing mGluR1-based pharmacology to treat ataxia.
SIGNIFICANCE STATEMENT Ataxia is a progressive and devastating degenerative movement disorder commonly associated with loss of cerebellar function and with no known cure. In the early stages of a mouse model of human spinocerebellar ataxia type 1, SCA1, where mice exhibit only moderate motor impairment, we detect excess “gain of function” of metabotropic glutamate receptor signaling at an important cerebellar synapse. Because careful control of this type of signaling is critical for cerebellar function in mice and humans, we sought to remove the excess signaling with a powerful, readily available pharmacological modulator. Remarkably, this pharmacological treatment acutely restored normal motor function in the ataxic mice. Our results pave the way for exploring a new avenue for early treatment of human ataxias.
Keywords: ataxia, calcium, cerebellum, mGluR1, parallel fibers
Introduction
Normal function of cerebellar Purkinje neurons (PNs) depends on type 1 metabotropic glutamate receptors (mGluR1) that drive phosphoinositide-mediated signaling events at parallel fiber (PF) synapses (Batchelor and Garthwaite, 1997). Dysfunction of this receptor and its downstream signaling effectors such as Ca2+ mobilization, protein kinase C and the transient receptor potential current, TRPC3, are all implicated in the pathophysiology of cerebellar ataxias (Becker et al., 2009; Kasumu et al., 2012; Kato et al., 2012). A central role for PN mGluR1 is best illustrated by the ataxic phenotype of knock-out mice that can be reversed by PN-specific re-expression of mGluR1 (Ohtani et al., 2014). Human ataxic patients also express mGluR1 autoantibodies (Sillevis Smitt et al., 2000), and mutations to mGluR1 and TRPC3 occur in two rare, early onset autosomal-recessive ataxias (Guergueltcheva et al., 2012; Fogel et al., 2015).
mGluR1s may therefore provide a promising target for pharmacological treatment of cerebellar ataxias, especially as advances in medicinal chemistry now provide powerful and specific activity-dependent positive allosteric modulators (PAMs) and negative allosteric modulators (NAMs) for mGluR1 (Wu et al., 2014). In several mouse models of autosomal-dominant human spinocerebellar ataxias (SCAs) CAG trinucleotide (Q) expansion disrupts nuclear transcription programs. In SCA1, loss of ROR-α-mediated (Serra et al., 2006) signaling drives reduced expression of mGluR1, TRPC3, and the PN-specific excitatory amino acid transporter EAAT4 (Serra et al., 2004). mGluR1 expression and function are also reduced in mouse models of SCA5 and SCA3 (Armbrust et al., 2014; Konno et al., 2014) and the mGluR1 PAM improves motor function in severe SCA1 (Notartomaso et al., 2013). Conversely, in SCA28 mice, mGluR1 activity might instead be pathologically elevated, because reducing mGluR1 expression reduces Ca2+ rises and alleviates the ataxia (Maltecca et al., 2015). Further, TRPC3 currents are enhanced in SCA14 and ataxic moonwalker and hotfoot-4J mice, consistent with excessive mGluR1 activity (Hartmann et al., 2008; Becker et al., 2009; Shuvaev et al., 2011; Kato et al., 2012). Therefore, mGluR1 PAMs and NAMs may both have potential for treating a variety of cerebellar ataxias.
Here, we show that moderate SCA1 ataxia involves prolonged mGluR1 synaptic signaling at cerebellar PF synapses, and is reversed by an mGluR1 NAM. These findings provide exciting new momentum for mGluR1-based treatment of ataxias.
Materials and Methods
Mice.
The University of Otago Animal Ethics Committee approved all procedures. We used 12-week-old male and female SCA1 82Q Tre/Tre;tTA/tTA, called 82Q mice, and wild-type FVB/pcp2 tTA/tTA, called WT mice kindly provided by H. T. Orr (University of Minnesota, USA) (Zu et al., 2004).
Motor behavior.
We tested groups of male and female mice separately, at the same time each day in a purpose-built room with sodium lighting. Chronic motor performance was assessed with an accelerating rotarod (latency to fall; Rotamex, Columbus Instruments 4–40 rpm over 5 min, 10 min between trials, 4 trials/d for 4 d; Clark et al., 1997). Acute motor performance was assessed with a fixed speed rotarod (latency to fall; 8 rpm for 2 min, minimum 2 min between trials). We measured hindpaw base-of-support (Catwalk, Noldus) from 3 to 5 runway crossings per mouse. Thirty minutes before acute testing, mice received subcutaneous injections of JNJ16259685 (0.03 mg/kg, <0.3 ml; Tocris Bioscience) in sterile saline with 10% hydroxypropyl-betacyclodextrin (ThermoFisher) as vehicle, or vehicle alone. Mice were not pretrained, poor performers (latency <10 s) were immediately re-trialed and persistent poor performers excluded.
Electrophysiology and calcium imaging.
Mice were rapidly euthanized with CO2 and cerebellar slices (300 μm) prepared using the following (in mm): 75 sucrose, 87 NaCl, 2.5 KCl, 1.25 NaH2PO4, 6 MgCl2, 0.5 CaCl2, 25 NaHCO3, and 25 glucose (Sigma-Aldrich) and maintained in artificial CSF (aCSF) containing the following (in mm): 126 NaCl, 3 KCl, 1 NaH2PO4, 26 NaHCO3, 2.4 CaCl2, MgCl2, and 10 glucose before perfusion (2.5 ml/min) with aCSF containing 50 μm picrotoxin, 25 μm CNQX (1,2,3,4-tetrahydro-6-nitro-2,3-dioxo-benzo[f]quinoxaline-7-sulfonamide) followed by 10 min 50 μm TBOA (DL-threo-β-benzyloxyaspartic acid) or 200 μm (S)MCPG ((S)-α-methyl-4-carboxyphenylglycine) or 10 min 5 nm, then 20 nm JNJ16259685 (Tocris Bioscience).
Whole-cell voltage-clamp recordings from PNs from folia III to V (−70 mV) used electrodes containing the following (in mm): 4.5 KCl, 20 KOH, 3.48 MgCl2, 4 NaCl, 120 K gluconate, 10 HEPES, 8 sucrose, 10 EGTA, 4 Na2ATP, and 0.4 Na2GTP, 8 biocytin (Sigma-Aldrich); osmolarity 295–305 mOsm, pH 7.3, resistance 3–5 MΩ. For Ca2+ imaging, HEPES and 100 μm Oregon Green BAPTA-1 (OGB-1, Life Technologies) replaced EGTA. Cell-attached recordings in picrotoxin used electrodes containing aCSF. Recordings used an Axopatch 200B (whole-cell) or Multiclamp (cell-attached) (Molecular Devices) digitized at 10 kHz (Digidata 1440A, Molecular Devices; or 1401plus CED) and analyzed with pClamp 10 (Molecular Devices). We excluded recordings with holding current <−200 pA or series resistance >35 MΩ with >10% change throughout the experiment. PF stimulation (DS2A Digitimer, 200 μs duration, 10–30 V, 0.03 Hz interval) evoked a 400–600 pA peak EPSC at 10× stimulation (200 Hz; 3–5 sweep average). During Ca2+ imaging, we depolarized voltage-clamped PNs to 0 mV for 400 ms to measure synapse-independent Ca2+ responses and to fill Ca2+ stores.
Wide-field fluorescence-based Ca2+ imaging (100 Hz, 4× binned; Hamamatsu C9100) commenced 20 min after establishing whole-cell configuration. PF stimulation evoked a local rise in fluorescence corrected for bleaching (no stimulation) normalized to baseline fluorescence, F(ΔF/F) average of 5, using Simple PCI and Excel.
Immunohistochemistry.
Slices containing biocytin-filled PNs were fixed in cold 4% paraformaldehyde in PBS (75 mm Na2HPO4 and 25 mm NaH2PO4), 2.7 mm KCl, 137 mm NaCl, pH 7.4 (all Sigma-Aldrich). After PBS washes and permeabilization (PBS + 0.3% Triton X-100) slices were incubated with Streptavidin AlexaFluor 647 (Life Technologies; 2 μg/ml, 4 h, room temperature) before reconstruction (Nikon A1R confocal;, 0.21 μm/pixel x, y, 1 μm z-step; 638 nm laser excitation, Coherent Scientific; 630 ± 50 nm emission). Immunohistochemistry used anti-calbindin, to measure molecular layer (ML) height, and anti-vGlut2 to measure climbing fiber (CF) extension (guinea-pig, 214005; rabbit, 135405 respectively, both Synaptic Systems; 1:1000 overnight, room temperature) with secondary detection (AlexaFluor 647 and 555-conjugated antibodies, respectively; 1:500, 4 h, room temperature; Life Technologies) before confocal imaging (0.4 μm/pixel x, y, 543 and 638 nm laser excitation 525 ± 50, 630 ± 50 nm emission, respectively). We did not detect labeling in the absence of primary antibody.
Analysis and statistics.
We measured mGluR1 current duration from 3 to 5 sweeps and spike firing frequency from a 3 min recording. ΔF/F recovery kinetics used a single exponential fit and integrated the area under the curve to quantify PF-evoked long-lasting Ca2+ signals (GraphPad, Prism). We obtained average ML height and CF extension from five regions in folia III-V across two to three slices per mouse using ImageJ. Sholl analysis (http://fiji.sc/Sholl_Analysis; Ferreira et al., 2014) used a separation radius of 10 μm.
Statistical analysis used two-way repeated-measures ANOVA with Bonferroni's multiple comparisons and Students unpaired t tests where appropriate (GraphPad Prism).
Results
We confirmed behavioral and neuronal deficits in 12-week-old mice with PN-specific 82Q repeats in the ataxin-1 gene (82Q mice) as a model of moderate SCA1. 82Q mice exhibited reduced motor performance (Fig. 1A,B; Clark et al., 1997; Zu et al., 2004) and reduced complexity of PN outer dendrites, a reduced PN height with CF retraction (Fig. 1C–G; Barnes et al., 2011) and reduced PN firing (Fig. 1H,I; Hourez et al., 2011).
Figure 1.
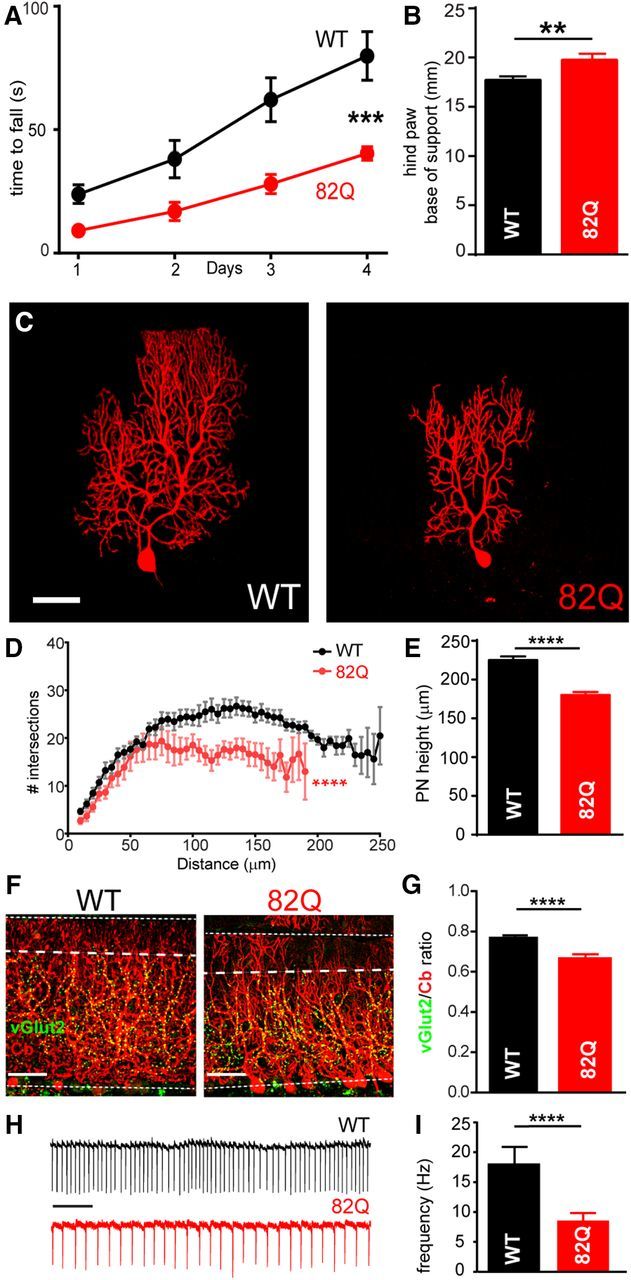
Moderate ataxia and cellular changes in 12-week-old 82Q mice. A, Reduced performance on the accelerating rotarod (***F(1,19) = 15.41, p = 0.005, n = 10/group, two-way ANOVA) and (B) increased hindpaw base-of-support (with paw prints). **p = 0.008, t test in 82Q compared with WT mice. Data are mean ± SEM. C, Representative reconstructed PNs, with Sholl plots in D showing a reduced number of intersections in 82Q PN dendrites (****F(1,21) = 62.4, p < 0.0001, n = 8/group, two-way ANOVA) and E shows their reduced height. ****p < 0.0001 t test. F, G, Reduced vGlut2-positive CF extension in the thinner calbindin-positive (Cb) ML of 82Q mice. ****p < 0.0001(n = 11) t test. H, I, Reduced simple spike firing in 82Q PNs. Scale bar, 500 ms. ****p < 0.0001, (n = 8) t test. Scale bars: A, C, 50 μm. Data are mean ± SEM.
We next sought to assess the physiological contribution of mGluR1 to PF–PN synaptic signaling in moderately ataxic 82Q PNs. We recorded PF-evoked mGluR1-dependent slow EPSCs in PNs, with and without the excitatory amino acid transporter (EAAT) blocker TBOA. Slow EPSCs were significantly prolonged in 82Q compared with WT PNs but of similar amplitude (Fig. 2A, dark traces, B,C dark bars). These slow currents were largely abolished by 200 μm (S)MCPG and 20 nm JNJ16259685 confirming their mGluR1origin (both n = 3) and previous work shows that these currents are mediated by TRPC3 activation (Hartmann et al., 2008). The longer currents in 82Q PNs occurred at all stimulation intensities so were not a consequence of altered PF recruitment (Fig. 2D). To test whether reduced expression of EAAT4 was responsible for the prolonged mGluR1 currents we eliminated EAATs with TBOA (Fig. 2A, light traces). TBOA increased the amplitude and duration of PF-evoked mGluR1 currents in WT but not 82Q PNs (Fig. 2A–C) consistent with a loss of EAAT4 in 82Q PNs.
Figure 2.
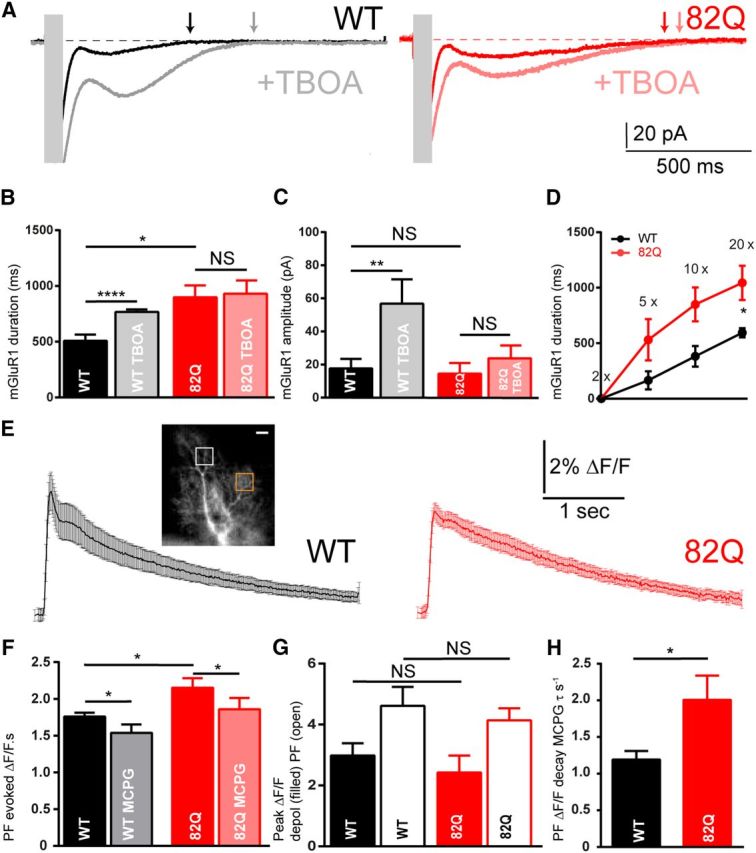
Prolonged mGluR1 synaptic signaling in 82Q mice. A, Prolonged PF-evoked mGluR1-mediated synaptic currents (10× stimulation, 200 Hz, stimulus artifacts obscured with gray boxes) in PNs from WT and 82Q mice in the presence and absence of TBOA to block glutamate transporters. B, Longer duration mGluR1 current in 82Q PNs versus WT: *F(1,9) = 8.03, p = 0.02, n = 5 each, two-way ANOVA and increased by TBOA in WT PNs but not 82Q PNs; ****p < 0.0001 two-way ANOVA, multiple comparison. C, mGluR1 current peak amplitude is similar between WT and 82Q PNs: F(1,9) = 2.24, p = 0.17, two-way ANOVA and increased by TBOA in WT but less so in 82Q; **p < 0.01 two-way ANOVA. D, Longer mGluR1 currents with increasing stimulation (2×–20×) in 82Q versus WT PNs: *F(1,44) = 21.6, p < 0.0001, n = 9, two-way ANOVA; NS, not significant. E, Mean (and SEM) of Ca2+ signals in PN outer dendrites from WT and 82Q mice following 10× PF stimulation (200 Hz). Inset, An OGB-1 filled PN dendrite (scale bar, 20 μm) with locally responsive dendrite (white box) used to extract fluorescence changes and remote unresponsive area (orange box). F, The increased mean integrated slow PF-mediated Ca2+ signal in 82Q PNs: *F(1,10) = 8.13, p = 0.017, n = 6 each, two-way ANOVA; (S)MCPG reduced the Ca2+ signal in both WT and 82Q PN dendrites: F(1,10) = 6.3, p = 0.013, to similar values; p = 0.12, t test. F, Peak Ca2+ rises were similar across all PNs; p = 0.4, t test (n = 6); NS, nonsignificant. G, In the presence of (S)MCPG, the mean PF-evoked Ca2+ decay time constant, τ, is longer in 82Q PN dendrites; *p = 0.018, t test (n = 6). Error bars are mean ± SEM.
PF-evoked mGluR1 signaling elevates Ca2+ in PN dendrites (Canepari and Ogden, 2006), so we tested whether the longer PF-evoked mGluR1 current also prolonged PF-evoked dendritic Ca2+ responses. 82Q PNs exhibited modestly prolonged Ca2+ signals in their outer dendrites compared with WT (Fig. 2E,F, filled bars). Although subtle, these enhanced long-lasting Ca2+ signals were synapse-specific, because depolarization-evoked Ca2+ responses were unchanged between WT and 82Q (Fig. 2G, filled bars). Similarly, the fast component of the PF-evoked Ca2+ signals were similar in WT and 82Q PN dendrites (Fig. 2G, open bars) confirming a specific increase of long-lasting Ca2+signals in 82Q PNs. In all cases the long-lasting Ca2+ signals were reduced by the mGluR1 antagonist (S)MCPG (Fig. 2F, lighter bars). Removal of the mGluR1 component revealed PF-evoked dendritic Ca2+ signals that took longer to recover in 82Q compared with WT PNs (Fig. 2H).
To address the relevance of prolonged cerebellar PF-evoked mGluR1 signaling for the ataxic phenotype, we treated mice with JNJ16259685 (JNJ), a potent and specific mGluR1 NAM. JNJ binds deep in the receptor complex only after glutamate binding (Wu et al., 2014) and behaves as an activity-dependent NAM. We administered 0.03 mg/kg JNJ to reduce cerebellar mGluR1 occupancy by ∼20–30% in vivo (Lavreysen et al., 2004) predicting a greater effect on motor behavior in 82Q mice where mGluR1 signaling is increased. As seen in Figure 3, A and B, JNJ restored motor performance in 82Q mice without influencing WT mice. To help explain these findings we simulated the in vivo treatment by applying 5 nm JNJ to cerebellar slices that reduced mGluR1 current duration in 82Q but not WT PNs, and to WT levels (Fig. 3C–E). Importantly, 20 nm JNJ reduced mGluR1 currents (by ∼90%) in both WT and 82Q cells (n = 3) consistent with its actions in normal cerebellar slices (Fukunaga et al., 2007). Together our findings provide a compelling mechanistic link between removal of overactive mGluR1-mediated synaptic signaling by JNJ in 82Q PNs and temporary relief from moderate ataxia in 82Q mice.
Figure 3.

The mGluR1 NAM, JNJ16259685 improves acute motor performance of 82Q mice. JNJ16259685 (0.03 mg/kg) restores (A) rotarod performance: *F(3,25) = 3.3, p = 0.03, two-way ANOVA; and (B) hindpaw base-of-support: *F(3,25) = 3.8, p = 0.02, two-way ANOVA, in 82Q but not WT mice, n = 7–8/group. C, JNJ16259685 (5 nm) reduces the amplitude (D) and duration (E) of 82Q but not WT mGluR1 currents: **F(1,9) = 19.9, p < 0.005; ***F(1,9) = 27, p < 0.001, n = 5 each group, two-way ANOVA. Symbols and error bars represent mean ± SEM.
Discussion
We found prolonged mGluR1 activity at cerebellar PF–PN synapses from moderately ataxic SCA1 mice. Removing this excessive activity with an mGluR1 NAM improved motor performance in these mice and may provide a promising approach to treat cerebellar ataxia.
82Q model of SCA1
The PN-specific 82Q mouse model of SCA1 used here recapitulates many aspects of human SCA1. The mice performed poorly on the accelerating rotarod and exhibited an increased hindpaw stance consistent with moderate ataxia (Fig. 1), as in the B05 SCA1 mouse (Clark et al., 1997). 82Q expression resulted in shrunken PNs with decreased complexity in outer dendrites and CF retraction (Barnes et al., 2011). 82Q PN simple spike firing was also slower (Fig. 1), as seen earlier (Hourez et al., 2011) and later (Dell'Orco et al., 2015) in SCA1.
Prolonged mGluR1-mediated PF synaptic signaling in SCA1
We observed functionally prolonged PF-evoked mGluR1-mediated inward currents in PNs from ataxic mice (Fig. 2A–C), that were consistent with mGluR1-dependent TRPC3 activation. These currents are also enhanced in spontaneously ataxic moonwalker and hotfoot-4J mice and in SCA14 mice (Becker et al., 2009; Shuvaev et al., 2011; Kato et al., 2012), suggesting that enhanced mGluR1synaptic signaling may be a common feature of several ataxias. Interestingly, mGluR1/TRPC3-positive PNs exhibit particularly high-frequency simple spikes (Zhou et al., 2014) so altering their signaling could be especially disruptive.
The prolonged mGluR1 current in 82Q PNs seen here contrasts with reduced mGluR1 (and TRPC3) expression in mid- and late-stage SCA1 (Zu et al., 2004; Notartomaso et al., 2013). Instead, we show that the functionally prolonged mGluR1 current is explained by a loss of glutamate transporter activity (Fig. 2B,C), most likely the high affinity, low capacity PN-specific EAAT4 that declines early in SCA1 (Serra et al., 2004). EAAT4 is also disrupted in the β-III spectrin knock-out SCA5 mouse and in human SCA5 (Ikeda et al., 2006; Perkins et al., 2010). Perhaps, as the influence of EAAT4 declines in SCA1, reduced mGluR1 expression is necessary to control excessive mGluR1 signaling.
The longer mGluR1 currents in SCA1 may also arise in response to 82Q toxicity when PKCγ is removed from the PN dendrite into somatic vacuoles (Skinner et al., 2001). PKCγ normally inactivates TRPC3 (Kwan et al., 2006) so its removal could allow slow mGluR1 currents to persist, particularly in the dendrites. Interestingly, mutations to PKCγ in SCA14 also enhance TRPC3 currents and disrupt cerebellar synaptic plasticity (Shuvaev et al., 2011).
We also observed prolonged mGluR1-dependent slow dendritic Ca2+ responses in SCA1 PN dendrites (Fig. 2E,F) that may arise from increased Ca2+ entry during the longer mGluR1-mediated current. However, these currents normally make a minor contribution to mGluR1-dependent dendritic Ca2+ signals (Knöpfel et al., 2000; Canepari and Ogden, 2006) so other mechanisms must also contribute. Possibilities include increased mGluR1-dependent release of Ca2+ from InsP3 stores, as in SCA2 (Kasumu et al., 2012), slower uptake of Ca2+ (Fig. 2H) caused by reduced SERCA expression in this SCA1 model (Serra et al., 2004) or increased mGluR1-mediated inactivation of Kv4-type K+ channels leading to hyperexcitable 82Q PN outer dendrites (Otsu et al., 2014). But, like others (Inoue et al., 2001), we detected healthy depolarization-induced PN Ca2+ responses and fast synaptic Ca2+ responses indicating remarkably effective adaptive remodeling of Ca2+ handling in 82Q PN dendrites.
Together our results support enhanced mGluR1 signaling at PFs in moderate SCA1. We predict that with early loss of PN-specific EAAT4 (Serra et al., 2004), later loss of EAAT1 (Cvetanovic, 2015), altered K+ currents (Hourez et al., 2011; Dell'Orco et al., 2015) and slowed Ca2+ uptake (Serra et al., 2004) the enhanced mGluR1 signaling in more compact PN dendrites ultimately promotes PN Ca2+ overload, excitotoxicity, death, and end-stage ataxia.
mGluR1 NAM treatment restores motor function in SCA1
Nevertheless, earlier in the SCA1model the prolonged mGluR1 signaling provides an exciting therapeutic opportunity. Significantly, 82Q mice treated with a very low dose of the mGluR1 NAM, JNJ16259685 improved their motor performance (Fig. 3A,B). A similarly low concentration of JNJ16259685 in vitro (5 nm) also reduced the prolonged 82Q mGluR1 currents to WT levels (Fig. 3C–E), thus providing a compelling link between prolonged PF–PN mGluR1 synaptic signaling and moderate SCA1 ataxia. It seems surprising that acute JNJ16259685 treatment improved motor performance in SCA1 mice given their permanently shrunken PNs (Fig. 1). However, in moderate SCA1, shrinkage is adaptive rather than degenerative and allows 82Q PNs to sustain their firing (Dell'Orco et al., 2015) and may also allow them to respond to acute mGluR1 manipulation. JNJ could also influence other, unknown sites of elevated mGluR1 activity in 82Q mice, but 82Q overexpression is restricted to PNs in these SCA1 mice suggesting that PN dysfunction is at the core of their ataxia and the probable target for our pharmacological intervention.
Given its critical role in cerebellar function, the role of mGluR1 in ataxia is likely to be far-reaching. Although successful therapy in late-stage SCA1 ataxia recently used a PAM to positively boost remaining mGluR1s (Notartomaso et al., 2013), reducing mGluR1 expression in SCA28 mice lowered Ca2+ responses and relieved ataxia (Maltecca et al., 2015). Our findings here suggest that removing excessive mGluR1 function successfully restores acute motor function in moderate SCA1 and provides exciting new momentum for mGluR1-based pharmacological approaches to treat ataxia.
Footnotes
This work was supported by a University of Otago PhD scholarship to E.M.P., and an OSMS Dean's Bequest Grant and Otago Medical Research Foundation summer scholarship to A.M.
The authors declare no competing financial interests.
References
- Armbrust KR, Wang X, Hathorn TJ, Cramer SW, Chen G, Zu T, Kangas T, Zink AN, Öz G, Ebner TJ, Ranum LP. Mutant beta-III spectrin causes mGluR1alpha mislocalization and functional deficits in a mouse model of spinocerebellar ataxia type 5. J Neurosci. 2014;34:9891–9904. doi: 10.1523/JNEUROSCI.0876-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes JA, Ebner BA, Duvick LA, Gao W, Chen G, Orr HT, Ebner TJ. Abnormalities in the climbing fiber-Purkinje cell circuitry contribute to neuronal dysfunction in ATXN1[82Q] mice. J Neurosci. 2011;31:12778–12789. doi: 10.1523/JNEUROSCI.2579-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batchelor AM, Garthwaite J. Frequency detection and temporally dispersed synaptic signal association through a metabotropic glutamate receptor pathway. Nature. 1997;385:74–77. doi: 10.1038/385074a0. [DOI] [PubMed] [Google Scholar]
- Becker EB, Oliver PL, Glitsch MD, Banks GT, Achilli F, Hardy A, Nolan PM, Fisher EM, Davies KE. A point mutation in TRPC3 causes abnormal Purkinje cell development and cerebellar ataxia in moonwalker mice. Proc Natl Acad Sci U S A. 2009;106:6706–6711. doi: 10.1073/pnas.0810599106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canepari M, Ogden D. Kinetic, pharmacological and activity-dependent separation of two Ca2+ signalling pathways mediated by type 1 metabotropic glutamate receptors in rat Purkinje neurones. J Physiol. 2006;573:65–82. doi: 10.1113/jphysiol.2005.103770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark HB, Burright EN, Yunis WS, Larson S, Wilcox C, Hartman B, Matilla A, Zoghbi HY, Orr HT. Purkinje cell expression of a mutant allele of SCA1 in transgenic mice leads to disparate effects on motor behaviors, followed by a progressive cerebellar dysfunction and histological alterations. J Neurosci. 1997;17:7385–7395. doi: 10.1523/JNEUROSCI.17-19-07385.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cvetanovic M. Decreased expression of glutamate transporter GLAST in bergmann glia is associated with the loss of Purkinje neurons in the spinocerebellar ataxia type 1. Cerebellum. 2015;14:8–11. doi: 10.1007/s12311-014-0605-0. [DOI] [PubMed] [Google Scholar]
- Dell'Orco JM, Wasserman AH, Chopra R, Ingram MA, Hu YS, Singh V, Wulff H, Opal P, Orr HT, Shakkottai VG. Neuronal atrophy early in degenerative ataxia is a compensatory mechanism to regulate membrane excitability. J Neurosci. 2015;35:11292–11307. doi: 10.1523/JNEUROSCI.1357-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira TA, Blackman AV, Oyrer J, Jayabal S, Chung AJ, Watt AJ, Sjostrom PJ, van Meyel DJ. Neuronal morphometry directly from bitmap images. Nat Meth. 2014;11:982–984. doi: 10.1038/nmeth.3125. http://www.nature.com/nmeth/journal/v11/n10/full/nmeth.3125.html. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fogel BL, Hanson SM, Becker EB. Do mutations in the murine ataxia gene TRPC3 cause cerebellar ataxia in humans? Mov Disord. 2015;30:284–286. doi: 10.1002/mds.26096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukunaga I, Yeo CH, Batchelor AM. Potent and specific action of the mGlu1 antagonists YM-298198 and JNJ16259685 on synaptic transmission in rat cerebellar slices. Br J Pharmacol. 2007;151:870–876. doi: 10.1038/sj.bjp.0707286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guergueltcheva V, Azmanov DN, Angelicheva D, Smith KR, Chamova T, Florez L, Bynevelt M, Nguyen T, Cherninkova S, Bojinova V, Kaprelyan A, Angelova L, Morar B, Chandler D, Kaneva R, Bahlo M, Tournev I, Kalaydjieva L. Autosomal-recessive congenital cerebellar ataxia is caused by mutations in metabotropic glutamate receptor 1. Am J Hum Genet. 2012;91:553–564. doi: 10.1016/j.ajhg.2012.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmann J, Dragicevic E, Adelsberger H, Henning HA, Sumser M, Abramowitz J, Blum R, Dietrich A, Freichel M, Flockerzi V, Birnbaumer L, Konnerth A. TRPC3 channels are required for synaptic transmission and motor coordination. Neuron. 2008;59:392–398. doi: 10.1016/j.neuron.2008.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hourez R, Servais L, Orduz D, Gall D, Millard I, de Kerchove d'Exaerde A, Cheron G, Orr HT, Pandolfo M, Schiffmann SN. Aminopyridines correct early dysfunction and delay neurodegeneration in a mouse model of spinocerebellar ataxia type 1. J Neurosci. 2011;31:11795–11807. doi: 10.1523/JNEUROSCI.0905-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda Y, Dick KA, Weatherspoon MR, Gincel D, Armbrust KR, Dalton JC, Stevanin G, Dürr A, Zühlke C, Bürk K, Clark HB, Brice A, Rothstein JD, Schut LJ, Day JW, Ranum LP. Spectrin mutations cause spinocerebellar ataxia type 5. Nat Genet. 2006;38:184–190. doi: 10.1038/ng1728. [DOI] [PubMed] [Google Scholar]
- Inoue T, Lin X, Kohlmeier KA, Orr HT, Zoghbi HY, Ross WN. Calcium dynamics and electrophysiological properties of cerebellar Purkinje cells in SCA1 transgenic mice. J Neurophysiol. 2001;85:1750–1760. doi: 10.1152/jn.2001.85.4.1750. [DOI] [PubMed] [Google Scholar]
- Kasumu AW, Liang X, Egorova P, Vorontsova D, Bezprozvanny I. Chronic suppression of inositol 1,4,5-triphosphate receptor-mediated calcium signaling in cerebellar purkinje cells alleviates pathological phenotype in spinocerebellar ataxia 2 mice. J Neurosci. 2012;32:12786–12796. doi: 10.1523/JNEUROSCI.1643-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato AS, Knierman MD, Siuda ER, Isaac JT, Nisenbaum ES, Bredt DS. Glutamate receptor δ2 associates with metabotropic glutamate receptor 1 (mGluR1), protein kinase Cγ, and canonical transient receptor potential 3 and regulates mGluR1-mediated synaptic transmission in cerebellar Purkinje neurons. J Neurosci. 2012;32:15296–15308. doi: 10.1523/JNEUROSCI.0705-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knöpfel T, Anchisi D, Alojado ME, Tempia F, Strata P. Elevation of intradendritic sodium concentration mediated by synaptic activation of metabotropic glutamate receptors in cerebellar Purkinje cells. Eur J Neurosci. 2000;12:2199–2204. doi: 10.1046/j.1460-9568.2000.00122.x. [DOI] [PubMed] [Google Scholar]
- Konno A, Shuvaev AN, Miyake N, Miyake K, Iizuka A, Matsuura S, Huda F, Nakamura K, Yanagi S, Shimada T, Hirai H. Mutant ataxin-3 with an abnormally expanded polyglutamine chain disrupts dendritic development and metabotropic glutamate receptor signaling in mouse cerebellar Purkinje cells. Cerebellum. 2014;13:29–41. doi: 10.1007/s12311-013-0516-5. [DOI] [PubMed] [Google Scholar]
- Kwan HY, Huang Y, Yao X. Protein kinase C can inhibit TRPC3 channels indirectly via stimulating protein kinase G. J Cell Physiol. 2006;207:315–321. doi: 10.1002/jcp.20567. [DOI] [PubMed] [Google Scholar]
- Lavreysen H, Wouters R, Bischoff F, Nóbrega Pereira S, Langlois X, Blokland S, Somers M, Dillen L, Lesage AS. JNJ16259685, a highly potent, selective and systemically active mGlu1 receptor antagonist. Neuropharmacology. 2004;47:961–972. doi: 10.1016/j.neuropharm.2004.08.007. [DOI] [PubMed] [Google Scholar]
- Maltecca F, Baseggio E, Consolato F, Mazza D, Podini P, Young SM, Jr, Drago I, Bahr BA, Puliti A, Codazzi F, Quattrini A, Casari G. Purkinje neuron Ca2+ influx reduction rescues ataxia in SCA28 model. J Clin Invest. 2015;125:263–274. doi: 10.1172/JCI74770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Notartomaso S, Zappulla C, Biagioni F, Cannella M, Bucci D, Mascio G, Scarselli P, Fazio F, Weisz F, Lionetto L, Simmaco M, Gradini R, Battaglia G, Signore M, Puliti A, Nicoletti F. Pharmacological enhancement of mGlu1 metabotropic glutamate receptors causes a prolonged symptomatic benefit in a mouse model of spinocerebellar ataxia type 1. Mol Brain. 2013;6:48. doi: 10.1186/1756-6606-6-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohtani Y, Miyata M, Hashimoto K, Tabata T, Kishimoto Y, Fukaya M, Kase D, Kassai H, Nakao K, Hirata T, Watanabe M, Kano M, Aiba A. The synaptic targeting of mGluR1 by its carboxyl-terminal domain is crucial for cerebellar function. J Neurosci. 2014;34:2702–2712. doi: 10.1523/JNEUROSCI.3542-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otsu Y, Marcaggi P, Feltz A, Isope P, Kollo M, Nusser Z, Mathieu B, Kano M, Tsujita M, Sakimura K, Dieudonné S. Activity-dependent gating of calcium spikes by A-type K+ channels controls climbing fiber signaling in Purkinje cell dendrites. Neuron. 2014;84:137–151. doi: 10.1016/j.neuron.2014.08.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkins EM, Clarkson YL, Sabatier N, Longhurst DM, Millward CP, Jack J, Toraiwa J, Watanabe M, Rothstein JD, Lyndon AR, Wyllie DJ, Dutia MB, Jackson M. Loss of beta-III spectrin leads to Purkinje cell dysfunction recapitulating the behavior and neuropathology of spinocerebellar ataxia type 5 in humans. J Neurosci. 2010;30:4857–4867. doi: 10.1523/JNEUROSCI.6065-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serra HG, Byam CE, Lande JD, Tousey SK, Zoghbi HY, Orr HT. Gene profiling links SCA1 pathophysiology to glutamate signaling in Purkinje cells of transgenic mice. Hum Mol Genet. 2004;13:2535–2543. doi: 10.1093/hmg/ddh268. [DOI] [PubMed] [Google Scholar]
- Serra HG, Duvick L, Zu T, Carlson K, Stevens S, Jorgensen N, Lysholm A, Burright E, Zoghbi HY, Clark HB, Andresen JM, Orr HT. RORalpha-mediated Purkinje cell development determines disease severity in adult SCA1 mice. Cell. 2006;127:697–708. doi: 10.1016/j.cell.2006.09.036. [DOI] [PubMed] [Google Scholar]
- Shuvaev AN, Horiuchi H, Seki T, Goenawan H, Irie T, Iizuka A, Sakai N, Hirai H. Mutant PKCγ in spinocerebellar ataxia type 14 disrupts synapse elimination and long-term depression in Purkinje cells in vivo. J Neurosci. 2011;31:14324–14334. doi: 10.1523/JNEUROSCI.5530-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sillevis Smitt P, Kinoshita A, De Leeuw B, Moll W, Coesmans M, Jaarsma D, Henzen-Logmans S, Vecht C, De Zeeuw C, Sekiyama N, Nakanishi S, Shigemoto R. Paraneoplastic cerebellar ataxia due to autoantibodies against a glutamate receptor. N Engl J Med. 2000;342:21–27. doi: 10.1056/NEJM200001063420104. [DOI] [PubMed] [Google Scholar]
- Skinner PJ, Vierra-Green CA, Clark HB, Zoghbi HY, Orr HT. Altered trafficking of membrane proteins in purkinje cells of SCA1 transgenic mice. Am J Pathol. 2001;159:905–913. doi: 10.1016/S0002-9440(10)61766-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, Wang C, Gregory KJ, Han GW, Cho HP, Xia Y, Niswender CM, Katritch V, Meiler J, Cherezov V, Conn PJ, Stevens RC. Structure of a class C GPCR metabotropic glutamate receptor 1 bound to an allosteric modulator. Science. 2014;344:58–64. doi: 10.1126/science.1249489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H, Lin Z, Voges K, Ju C, Gao Z, Bosman LW, Ruigrok TJ, Hoebeek FE, De Zeeuw CI, Schonewille M. Cerebellar modules operate at different frequencies. Elife. 2014;3:e02536. doi: 10.7554/eLife.02536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zu T, Duvick LA, Kaytor MD, Berlinger MS, Zoghbi HY, Clark HB, Orr HT. Recovery from polyglutamine-induced neurodegeneration in conditional SCA1 transgenic mice. J Neurosci. 2004;24:8853–8861. doi: 10.1523/JNEUROSCI.2978-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]