

Abstract
The aggregation of amyloid-β peptide (Aβ) in brain is an early event and hallmark of Alzheimer's disease (AD). We combined the advantages of in vitro and in vivo approaches to study cerebral β-amyloidosis by establishing a long-term hippocampal slice culture (HSC) model. While no Aβ deposition was noted in untreated HSCs of postnatal Aβ precursor protein transgenic (APP tg) mice, Aβ deposition emerged in HSCs when cultures were treated once with brain extract from aged APP tg mice and the culture medium was continuously supplemented with synthetic Aβ. Seeded Aβ deposition was also observed under the same conditions in HSCs derived from wild-type or App-null mice but in no comparable way when HSCs were fixed before cultivation. Both the nature of the brain extract and the synthetic Aβ species determined the conformational characteristics of HSC Aβ deposition. HSC Aβ deposits induced a microglia response, spine loss, and neuritic dystrophy but no obvious neuron loss. Remarkably, in contrast to in vitro aggregated synthetic Aβ, homogenates of Aβ deposits containing HSCs induced cerebral β-amyloidosis upon intracerebral inoculation into young APP tg mice. Our results demonstrate that a living cellular environment promotes the seeded conversion of synthetic Aβ into a potent in vivo seeding-active form.
SIGNIFICANCE STATEMENT In this study, we report the seeded induction of Aβ aggregation and deposition in long-term hippocampal slice cultures. Remarkably, we find that the biological activities of the largely synthetic Aβ aggregates in the culture are very similar to those observed in vivo. This observation is the first to show that potent in vivo seeding-active Aβ aggregates can be obtained by seeded conversion of synthetic Aβ in a living (wild-type) cellular environment.
Keywords: alzheimer, amyloid, neurodegeneration, prion-like seeding, slice culture
Introduction
A hallmark and early trigger of Alzheimer's disease (AD) pathogenesis is the aggregation of amyloid-β peptides (Aβ) in brain (Hardy and Selkoe, 2002; Holtzman et al., 2011). Although Aβ aggregation studies in cell-free systems have provided important insights into the dynamics and kinetics of amyloid formation (Harper and Lansbury, 1997; Dasilva et al., 2010; Du et al., 2011), it was the development of Aβ precursor protein transgenic (APP tg) mouse models that has revolutionized the research of cerebral β-amyloidosis (Games et al., 1995; Hsiao et al., 1996; Sturchler-Pierrat et al., 1997; Duyckaerts et al., 2008; Jucker, 2010). The need of in vivo approaches to study cerebral β-amyloidosis was further bolstered by the observation that in vitro aggregated synthetic Aβ is a poor in vivo seed for cerebral β-amyloidosis (Meyer-Luehmann et al., 2006; Stöhr et al., 2012) likely due to conformational differences and/or cofactors compared with in vivo generated Aβ aggregates (Paravastu et al., 2009).
We and others (Harris-White et al., 1998; Johansson et al., 2006) have tried to model β-amyloidosis in hippocampal slice cultures (HSCs) from APP tg mice for more than a decade, however, with poor success. Now we report that seeding such cultures once with brain homogenates from aged APP tg mice while supplementing the medium continuously with synthetic Aβ induces robust Aβ aggregation and associated pathologies with biological activities remarkably similar to those observed in vivo in APP tg mice and AD brains. Our results demonstrate that in vitro formation of in vivo seeding-active Aβ aggregation can be achieved by seeded conversion of synthetic Aβ in a living cellular environment.
Materials and Methods
Mice for HSCs.
HSCs were prepared from pups of the following mice: male and female C57BL/6J mice, APP23 and APPPS1 tg mice, and crosses of APP23 × APPPS1 mice. APP23 tg mice overexpress human APP sevenfold and harbor the Swedish double mutation (Sturchler-Pierrat et al., 1997). APPPS1 tg mice overexpress human APP threefold and harbor Swedish double mutation and mutated human presenilin 1 (PS1) (Radde et al., 2006). We furthermore used App-null mice (Eisele et al., 2014) and mice expressing eGFP under the control of the neuron-specific Thy-1 promoter (Thy1-GFP) (Feng et al., 2000; Vlachos et al., 2012).
Preparation of HSCs.
HSCs were prepared from pups at postnatal day 2–3 according to previously published protocols (Brinks et al., 2004; Mayer et al., 2005). Postnatal pups were killed by decapitation, and brains were aseptically removed. Hippocampi were dissected and cut perpendicular to the longitudinal axis into 400 μm sections with a tissue chopper. Intact hippocampal sections were transferred into Petri dishes, containing cold buffer solution of minimum essential medium supplemented with 2 mm GlutaMAX (Invitrogen, Thermo Fisher Scientific) at pH 7.3. Four sections were placed onto one humidified porous membrane (Millicell Cell Culture Inserts CM30; Merck KGaA) per well in six well plates filled with 1.2 ml culture medium/well. The culture medium consisted of heat-inactivated horse serum (25%), HBSS (25%), and minimum essential medium (50%), complemented with GlutaMAX (2 mm) adjusted to pH 7.2. HSCs were kept at 37°C in humidified CO2-enriched atmosphere. The medium was changed three times per week. HSCs of Thy1-GFP mice also contained entorhinal cortex and were prepared according to a similar and previously described protocol (Del Turco and Deller, 2007).
Treatment of HSCs.
HSCs were kept for 10 d without any experimental treatment to obtain stable culturing conditions. At day 10 after the medium change, synthetic Aβ1–40 or Aβ1–42 (from 100 μm frozen stock solution; initially prepared by adding PBS to lyophilized Aβ, American Peptide; followed by 3 × 1 min vortexing on wet ice) was added to the medium to reach a final concentration of 1.5 μm. We cannot exclude that at least some Aβ oligomerization has taken place. Subsequently, 1 μl of brain extract of APP23 or APPPS1 tg mice or of non-tg wild-type mice (for a description of the extracts, see below) was pipetted once on the surface of each culture. Over the following 9 week cultivation period, all subsequent medium changes were done with culture medium containing 1.5 μm synthetic Aβ (no more extract was added). Some HSCs were prefixed after 1 week with 4% PFA in 0.1 m phosphate buffer (PB) at room temperature for 2 h, rinsed 3 times with PB for 10 min followed by the combined treatment as described above.
Histology and immunohistochemistry of HSCs.
HSCs were fixed with 4% PFA in 0.1 m PB, pH 7.4 for 2 h. Cultures were rinsed 3 times with 0.1 m PB for 10 min. The Millipore membrane with the fixed cultures was cut off, and cultures were mounted on a planar agar block and sliced into 50 μm sections on a vibratome (Leica VT 1000S Vibratome, Leica Biosystems). Typically 2–4 intact sections were obtained per culture and were collected in PBS. Immunohistochemistry was performed according to standard protocols on free-floating sections either using the Vectastain Elite ABC Kit (Vector Laboratories) or standard immunofluorescence with appropriate secondary antibodies (goat anti-rabbit or goat anti-mouse Cy3- or Alexa-488-, -546-, -568-, or -647-conjugated antibodies; Invitrogen, Thermo Fisher Scientific) (Mayer et al., 2005; Eisele et al., 2010). As primary antibodies were used: polyclonal rabbit CN3 antibody to human Aβ (Eisele et al., 2010); rabbit polyclonal antibody against ionized calcium binding adapter molecule 1 (Iba-1, Wako Chemicals); mouse monoclonal antibody against neurofilament light polypeptide (NF-L; MAB1615; Millipore); mouse monoclonal antibody AT8 (Thermo Scientific) directed against p-Tau position 191/194 (murine). Some of the sections were counterstained with Congo red and examined for birefringence under polarized light. Immunofluorescence-stained sections were routinely counterstained with DAPI nuclear stain (300 nm, Invitrogen, Thermo Fisher Scientific).
Extract preparation from brain and HSCs.
Brain extracts were prepared from aged (24-month-old) male or female APP23 tg mice, aged (21-month-old) APPPS1 tg mice, and age-matched non-tg control mice as described previously (Meyer-Luehmann et al., 2006). In brief, after removal of the cerebellum and lower brainstem, the forebrain was immediately fresh-frozen on dry ice and stored at −80°C until use. Tissue was homogenized (Ultra Turrax T8, IKA-Werke) at 10% (w/v) in sterile PBS (Lonza), vortexed, sonicated three times for 5 s each (LabSonic, B. Braun, Biotech International; 0.5 mm diameter sonotrode, cycle 1, amplitude 80%), and centrifuged at 3000 × g for 5 min. The extract is referred to the supernatant and typically contained 1–10 ng/μl total Aβ (Aβ40 and Aβ42 combined). Extracts were aliquoted and immediately frozen at −80°C until use. Slice culture homogenates were prepared from 15 identically treated HSCs. Cultures were removed from the membrane, pooled, and immediately frozen on dry ice and stored at −80°C until use. Frozen slice cultures were homogenized with EPPI pestles (Schuett Biotec) in 150 μl sterile PBS, vortexed, and sonicated three times for 5 s each. HSC homogenates were aliquoted and immediately frozen at −80°C until use.
SDS-PAGE and immunoblot analysis.
HSC homogenates/extracts were analyzed on NuPAGE Bis-Tris mini-gels using LDS sample buffer and MES running buffer (Invitrogen, Thermo Fisher Scientific). Proteins were transferred onto a nitrocellulose membrane, probed with monoclonal antibody m3.2 against murine Aβ (Morales-Corraliza et al., 2009), and visualized using SuperSignal West Dura chemiluminescent substrate (Invitrogen, Thermo Fisher Scientific).
Staining with luminescent conjugated oligothiophenes.
Luminescent conjugated oligothiophene (LCO) amyloid-binding dyes were used to distinguish between different Aβ morphotypes, by detecting fluorescence emission spectral differences. Specifically, the pentamer formyl acetic acid (pFTAA) LCO was used, as described previously (Fritschi et al., 2014). For visualizing Aβ deposition in HSCs from Thy1-GFP mice, an LCO with red-shifted fluorescence emission was chosen (the heptamer formyl acetic acid [hFTAA]) (Klingstedt et al., 2011), to avoid fluorescence cross talk between the eGFP signal and the LCO. Aβ deposition in microglia-depleted HSCs was visualized with the blue-shifted LCO qFTAA (quadro-formylthiophene acetic acid) (Klingstedt et al., 2011) to avoid bleed-through into the CN3 (secondary antibody conjugated to Alexa-546) and Iba-1 (secondary antibody conjugated to Alexa-647) channels. For the staining of either LCO, HSC sections were washed in PBS (3 × 10 min) and subsequently mounted on Superfrost slides. Staining with the LCOs (1.5 mm in deionized water, diluted 1:1000 in PBS) was performed similar to a previous description (Klingstedt et al., 2011). Sections were allowed to dry for 2 h at room temperature and then coverslipped with Dako Fluorescence Mounting Medium.
Quantification of total Aβ load.
The total Aβ load (%) of HSCs was determined on each section (50-μm-thick) from the resliced cultures by calculating the areal fraction occupied by CN3-positive (peroxidase-based) immunostaining in two-dimensional sectors over the whole section volume (20×/0.45 NA objective). The mean Aβ load of all the sections from each culture (typically 2–4 sections) was regarded as the total Aβ load of the corresponding culture.
The total Aβ load (%) induced by the injection of HSC extract into the dentate gyrus of young APP23 tg mice was determined on a set of every 12th systematically sampled, serial (25-μm-thick) sections throughout the entire dentate gyrus by calculating the areal fraction occupied by CN3- and Congo red-positive immunostaining in two-dimensional sectors at a single focal plane (20×/0.45 NA objective). All the stereological quantification was done with a microscope equipped with a motorized x-y-z stage coupled to a video microscopy system (Stereo Investigator; MicroBrightField) as previously described (Bondolfi et al., 2002). The investigator who performed analysis was blind toward the different groups.
Electron microscopy.
HSCs were immersed in a fixative solution (4% PFA, 0.05% glutaraldehyde, 15% saturated picric acid in 0.1 m PBS) for 2 h and then washed, osmicated, dehydrated, and flat-embedded in resin (Fluka Durkupan, EMS, Hatfield) on glass slides. Ultrathin sections were cut by an Ultracut (Leica EM UC7, Leica Biosystems) and collected on single-slot Formvar-coated nickel grids. For immunolabeling a postembedding protocol was used. After etching in 1% periodic acid for 10 min, an incubation in 1% sodium metaperiodate and glycine/sodium borohydrate solution followed (10 min each). Sections were blocked in 2% human serum albumin in TBST for 10 min and incubated with the polyclonal rabbit CN3 antibody to Aβ (Eisele et al., 2010) (1:100 overnight at 4°C). As secondary layer, a gold-conjugated (10 nm) anti-rabbit antibody (1:50, British Biocell Interaction) was used. Sections were exposed to lead citrate for 2 min. Digital images were taken by means of a transmission electron microscope (Leo 906 E, Carl Zeiss MicroImaging) equipped with a 2K Sharpeye CCD camera (Tröndle).
Spine counts at electron microscopic level.
For spine count analysis at electron microscopic level, 16-week-old untreated wild-type HSCs and HSCs treated with APPPS1 tg brain extract and synthetic Aβ1–42 were analyzed (n = 3 each). From each culture, 20 panoramic images (each composed of 3 × 3 attached and nonoverlapping photos) at the same magnification were taken randomly. The photographed area of each culture was ∼2450 μm2, and number of spines per area was manually counted. The investigator performing the analysis was blind toward the two groups. The Student's t test was used for statistical analysis.
Spine analysis at light microscopic level.
Spine analysis was done on HSCs derived from Thy1-GFP mice. Aβ plaques were visualized by hFTAA staining (see above for details). GFP and hFTAA were both excited using a 488 nm argon laser line. GFP fluorescence emission was collected using a narrow bandpass 505–530 nm filter, whereas the hFTAA emission was collected using a longpass 585 nm filter, ensuring no fluorescence cross talk between the two dyes. Dendritic spines were quantified in Fiji (public domain software, version 1.84, http://fiji.sc/Fiji) using 3D image stacks of side branches of principal apical dendrites (second- or higher-order dendritic segments) of CA1 pyramidal neurons within stratum radiatum. Image stacks were acquired using the Zeiss LSM 510 META (Axiovert 200M; Carl Zeiss MicroImaging) confocal microscope and a 63×/oil objective (1.4 NA, Zeiss). Spine counting was performed blind to experimental conditions on randomly chosen individual dendritic segments within each culture (4 cultures per condition, 3–8 dendritic segments per culture); the length of the dendritic segment was measured in 3D using the Simple Neurite Tracer plugin in Fiji. The number of spines on each segment was recorded using the Cell Counter plugin in Fiji. The average number of spines per μm on a dendritic segment was used as a measure of spine density. Spine density was calculated on all cultures per condition.
Spectral analysis of Aβ morphotypes.
Spectra of pFTAA-stained amyloid aggregates within HSC sections (for staining details, see above) were acquired on a Zeiss LSM 510 META (Axiovert 200M) confocal microscope (40× oil-immersion objective, 1.3 NA) equipped with a spectral detector. The dye was excited using the 458 nm argon laser line. Emission spectra were acquired from 470 to 695 nm at a step of 10.70 nm and normalized to their respective maxima. The ratio of the intensity of emitted light at the blue-shifted portion (502 nm) and red-shifted peak (599 nm) was used as a parameter for spectral distinction of different plaques. These peaks were selected to maximize the spectral differences of the pFTAA-stained Aβ plaques between HSC treated with brain APP23 or APPPS1 extract in combination with synthetic Aβ1–40 or Aβ1–42. The 599/502 ratio was computed for each plaque by determining the mean of the 599/502 ratio at 3 different ROIs within one Aβ plaque (core). All plaques analyzed were from the intermediate sections of the cultures (neither top nor bottom section) and border regions were excluded. In each culture, 4–15 Aβ plaques were analyzed and the mean was taken for statistical analysis (n = cultures; 11–13 cultures were analyzed per condition).
Microglia depletion.
To deplete microglia in culture (Kohl et al., 2003), the bisphosphonate clodronate (clodronate disodium salt, Merck KGaA) was dissolved in dH2O (1 mg/ml) and applied to the culture medium at 100 μg/ml. After 24 h, cultures were carefully rinsed with prewarmed PBS and replaced with fresh culture medium. Aβ deposition was induced on day 10 as described above. Seven weeks later, HSCs were fixed in 4% PFA. HSCs were sliced on a vibratome (as described above) and immune-stained for Iba-1 (with the secondary antibody conjugated to Alexa-647 fluorochrome) to assess the microglia depletion. Amyloid deposits were stained using polyclonal CN6 antibody (a follow-up version of CN3) with the secondary antibody conjugated to Alexa-546). Images of sectioned cultures were captured on a Zeiss Axioplan 2 microscope, using tile scanning (MosaiX on AxioVision 4.3) and a Zeiss Plan Neofluar ×10/0.3 objective. A semiautomated computer analysis was performed using a custom-made macro written for Fiji (public domain software, version 1.84, http://fiji.sc/Fiji) to quantify the percentage of Aβ immunostaining and microglia on each section. Gamma values were adjusted to reduce background tissue fluorescence before thresholding each channel. The ratio of the thresholded pixels to the total pixels yielded the microglia-stained area coverage, expressed as percentage microglia load and the Aβ stained area coverage, expressed as percentage Aβ load.
Electrochemiluminescence-linked immunoassay for Aβ.
Aβ (Aβx-40, Aβx-42) concentrations in brain extracts, culture extracts, and synthetic Aβ preparations were determined with an electrochemiluminescence-linked immunoassay as previously described (Langer et al., 2011) using the new V-PLEX Aβ peptide panel 1 (6E10) kit according to the manufacturer's instructions (Meso Scale Discovery). For total Aβ, Aβx-40 and Aβx-42 were combined.
Preparation of synthetic Aβ fibrils.
Aβ1–40 or Aβ1–42 (American Peptide) was dissolved in PBS at a concentration of 100 μm (i.e., 433 ng Aβ1–40/μl or 451 ng Aβ1–42/μl), vortexed 3 × 1 min on wet ice. Solutions were subsequently incubated at 37°C for 5 d on a rotator, as previously described (Meyer-Luehmann et al., 2006). Aliquots were stored at −80°C until use.
Assessing Aβ seeding activity of brain extracts and HSC homogenates.
Male and female 4-month-old APP23 tg mice on an App-null background (Eisele et al., 2014) served as host mice (gender was matched between the groups). The mice were anesthetized with a ketamine/xylazine mixture (ketamine 100 mg/kg, xylazine 10 mg/kg) in saline. Bilateral stereotactic injections of 2.5 μl brain extract or culture homogenate were made with a Hamilton syringe into the hippocampus (anteroposterior −2.5 mm, lateral ±2.0 mm, dorsoventral −1.8 mm). Injection speed was 1.25 μl/min, and the needle was kept in place for an additional 2 min before withdrawal. The surgical area was cleaned with sterile PBS, and the incision was sutured. The mice were kept under infrared light and monitored until recovery from anesthesia.
Four months later, animals were killed, and the brains were removed and immersion-fixed for 48 h in 4% PFA in PBS, followed by cryoprotection in 30% sucrose in PBS for an additional 48 h. After freezing, brains were cut into 25-μm-thick coronal sections using a freezing-sliding microtome (Leica SM 2000R, Leica Biosystems). Sections were collected in cryoprotectant (35% ethylene glycol, 25% glycerol in PBS) and stored at −20°C until use. Immunohistochemistry was performed according to standard protocols with the Vectastain Elite ABC Kit (Vector Laboratories) using the polyclonal rabbit CN3 antibody to Aβ and counterstained with Congo red as previously described (Eisele et al., 2010).
Study approval.
Experimental procedures were performed in accordance with German animal welfare legislation and approved by the local animal welfare officer and/or the local Animal Care and Use Committees.
Results
Aβ deposition in HSC is induced by seeded aggregation of synthetic Aβ
HSCs were prepared from postnatal APP23, APPPS1, and APP23 × APPPS1 double tg mice and cultivated for up to 10 weeks (Fig. 1). After this time, the cytoarchitecture was still largely intact. However, no Aβ deposition was detected in any of the untreated HSCs (Fig. 1C) despite robust Aβ deposition being present in 8-week-old APP23 × APPPS1 tg mice in vivo (F.L. and M.J., unpublished observation). To compensate for the release of the secreted Aβ from the slices into the medium the latter was supplemented continuously with 1.5 μm synthetic Aβ1–40. Again, no Aβ deposits emerged in the HSCs (Fig. 1D).
Figure 1.

Aβ deposition in HSCs is induced by seeded aggregation of synthetic Aβ. A, Experimental protocol. HSCs were kept 10 d to achieve stable culture conditions. Thereafter HSCs were treated once with brain extract, and the medium was supplemented with synthetic (syn) Aβ during the following 9 week culture period. B, Image of four HSCs placed on a membrane inset and illustration of one 150-μm-thick HSC. Slices were fixed and further sectioned into 50-μm-thick slices, enabling separate examination of top, intermediate, and bottom layers for each slice of the culture. C, Aβ-immunostained untreated HSCs of an APP23 × APPPS1 double tg mouse after 10 weeks of cultivation. No Aβ deposition could be detected. D, E, Similarly, no Aβ deposition was observed in HSCs of APP23 × APPPS1 mice when the medium was supplemented with 1.5 μm human syn Aβ1–40 (D) or when HSC was treated once with brain extract of an aged APP23 tg mouse (E). F, In contrast, robust Aβ deposition (Aβ immunostaining) was observed in HSCs of APP23 × APPPS1 mice treated once with APP23 brain extract and medium supplemented with 1.5 μm human syn Aβ1–40. G, H, Aβ deposition was also observed in HSCs of wild-type (G) and App-null mice (H) when treated on top once with brain extract and continuously with human syn Aβ1–40 in the medium. I, Two examples of top, intermediate, and bottom layers of wild-type HSCs treated with APP23 brain extract and 1.5 μm Aβ1–40 show dominant Aβ deposits at the rim of all sections but also smaller fibrillar deposits in the center of the culture. Boxed areas are shown at higher magnifications. J, K, Induced Aβ deposition in HSCs was time-dependent and dependent on the Aβ concentration in the medium. Quantification of the induced Aβ load (%) at 1, 4, and 9 weeks with 1.5 μm syn Aβ1–40 in the medium (n = 11 cultures/time point; ANOVA, F(2,30) = 13.45; p < 0.0001) (J). *p < 0.05 (Tukey's post hoc multiple-comparison test). ***p < 0.001 (Tukey's post hoc multiple-comparison test). Induced Aβ load (%) 9 weeks after treatment with various concentrations of human syn Aβ1–40 in the medium (n = 10–13 cultures/concentration; ANOVA, F(3,42) = 32.18; p < 0.0001) (K). ***p < 0.001 (Tukey's post hoc multiple-comparison test). L, Immunoblot analysis with an antibody specific to murine (m) Aβ revealed a faint Aβ band in the APP23 brain extract (lane 4) and, upon longer incubation, also in HSCs treated with APP23 extract and human syn Aβ in the medium (lane 3), but not in HSCs treated with APP23 brain extract alone (lane 2; lane 1 shows an untreated HSC). This indicates that at least some HSC-derived murine Aβ is codeposited with human Aβ in HSCs. GAPDH was used as loading control. Scale bars: C–H, I, 200 μm; I, boxes, 20 μm.
Previous work has shown that cerebral β-amyloidosis can be induced in APP tg mice by inoculation with Aβ-amyloid containing brain extracts (Meyer-Luehmann et al., 2006; Eisele et al., 2010). Inspired by these findings, a small volume (1 μl) of extract from an aged amyloid-laden APP23 tg mouse brain was applied once onto the top of the HSCs on day 10 after establishing the culture. Yet again, no Aβ deposits were induced even after 9 weeks in culture (Fig. 1E). However, when treatment with brain extract from aged APP23 tg mice (but not from age-matched wild-type control mice) was combined with the continuous application of synthetic human Aβ1–40 in the medium, robust Aβ deposition was induced in the slices (Fig. 1F). Remarkably, this treatment combination also induced Aβ deposition in HSCs derived from wild-type mice (Fig. 1G), suggesting that Aβ deposits develop also in the absence of transgenic HSC-generated human Aβ. This was also true for endogenous HSC-derived mouse Aβ because deposits were also found in HSCs from App-null mice using the same conditions (Fig. 1H). Thus, HSC-generated Aβ is not necessary for the induction of Aβ deposition; however, if present, HSC-derived murine Aβ appears to codeposit with the induced Aβ deposits (Fig. 1L). Strikingly, when tg brain extract and synthetic Aβ treatment were applied to formaldehyde-fixed (and thus inactivated) HSCs, Aβ deposition was limited to the rim of the culture (Fig. 2), whereas in living HSC Aβ deposition was much stronger and also present in the center of the sections (Figs. 1I, 2B). This argues that living hippocampal slices are mandatory for the development of Aβ deposits in HSCs. Furthermore, in living HSCs, Aβ deposition was dependent on time and on concentration of synthetic Aβ in the medium (Fig. 1J,K). With the highest Aβ concentration tested (1.5 μm synthetic Aβ1–40 in the medium), first Aβ deposits were detected 1 week after the combined treatment and increased further for up to 10 weeks in culture.
Figure 2.

HSCs fixed before treatment reveal only modest Aβ deposits and only at the rim of the culture. A, Wild-type HSCs were fixed in 4% PFA before treatment with APP23 brain homogenate and synthetic Aβ1–40. Aβ deposits (anti-Aβ immunostaining) were only observed at the edge of the HSC (inset), whereas no Aβ deposits were found in the center of the section (A). B, In contrast, numerous Aβ deposits were found with the same treatment in living wild-type HSC (see also Fig. 1). Scale bars: A, B, 50 μm; insets, 200 μm.
Brain extract and synthetic Aβ species determine the morphotype of HSC Aβ deposits
The requirement of adding both amyloid-laden APP tg brain extract on top of the culture and synthetic Aβ to the medium to induce HSC Aβ deposition suggests that Aβ aggregation in culture occurs via seeded conversion. To confirm such a mechanism, HSCs were treated with brain extracts from either aged APP23 or aged APPPS1 tg mice and were incubated with medium supplemented with either synthetic Aβ1–40 or Aβ1–42. Aβ deposits in APP23 tg mice differ in appearance and molecular architecture from deposits in APPPS1 tg mice (Heilbronner et al., 2013). While Aβ deposits in aged APP23 tg mice are fairly large and contain predominantly Aβ40 (i.e., Aβ ending with amino acid 40), Aβ plaques in aged APPPS1 tg mice are smaller, more numerous, highly compact, and contain predominantly Aβ42. Indeed, HSCs treated with APP23 brain extract and synthetic Aβ1–40 demonstrated larger plaques while deposits were smaller, more numerous, and highly compact when cultures were treated with APPPS1 extract and synthetic Aβ1–42 (Fig. 3). Based on morphological criteria, both the brain extract as well as the synthetic Aβ species in the medium determined the morphotypes of the induced Aβ deposits. Using the amyloid conformation-specific luminescent conjugated oligothiophene pFTAA (Klingstedt et al., 2011; Fritschi et al., 2014), this morphological observation was quantitatively confirmed (Fig. 3E).
Figure 3.

Aβ morphotypes in HSCs depend on the seed and synthetic Aβ species. A–D, Differences in structural appearance of induced Aβ deposits (anti-Aβ immunostaining) in 10-week-old wild-type HSCs inoculated with different brain extracts and incubated with 1.5 μm of either synthetic (syn) Aβ1–40 or Aβ1–42 in the culture medium. A, Large fibrillary deposits of Aβ (arrows) were observed with APP23 brain extract and syn Aβ1–40. B, Inoculation with APP23 brain extract and syn Aβ1–42 revealed a mixed pattern of small and large fibrillary deposits. C, APPPS1 brain extract combined with syn Aβ1–40 revealed large and compact Aβ deposits, whereas (D) numerous small Aβ deposits (arrows) were seen with APPPS1 brain extract and syn Aβ1–42. Scale bars: A–D, 20 μm. E, Spectral properties of the induced Aβ deposits using the conformation-dependent amyloid-specific dye pFTAA. For quantitative comparison, the ratio of light emitted at 599 and 502 nm was assessed for individual Aβ deposits and the mean ratio determined per HSC. Per condition, 11–13 HSCs were analyzed; data are mean ± SEM. ANOVA (extract × syn Aβ) revealed a significant effect for extract (F(1,43) = 6.41; p < 0.05) and syn Aβ (F(1,43) = 26.17; p < 0.001) but failed to reach significance for the interaction (F(1,43) = 1.61; p > 0.05).
HSC Aβ deposits recapitulate many aspects of cerebral β-amyloidosis
Electron microscopic analysis of HSC Aβ deposits revealed anti-Aβ-immunogold-labeled amyloid fibers arranged in typically packed parallel bundles (Fig. 4A) virtually identical as described for Aβ plaques in APP tg mice (Stalder et al., 1999, 2001) and AD patients (Terry et al., 1964). Larger deposits exhibited Congo red-positive staining with the characteristic birefringence under cross-polarized light (Fig. 4B,C).
Figure 4.
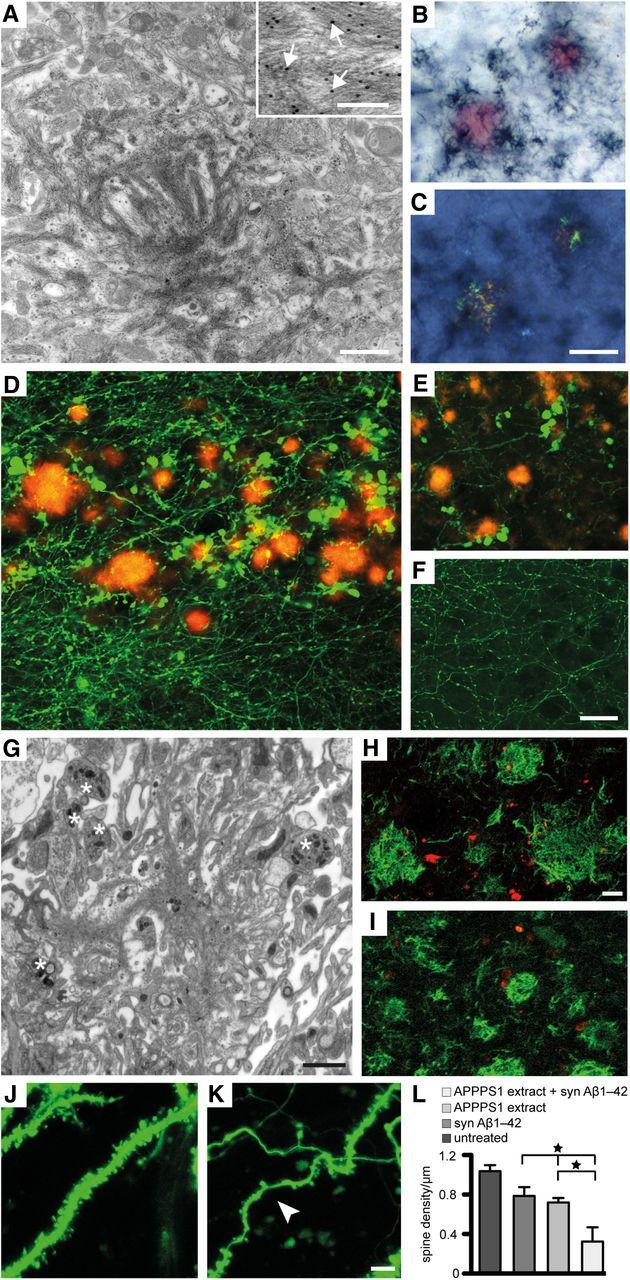
Pathology associated with seeded Aβ deposition in 10-week-old HSCs. A, Immunoelectron microscopy of induced Aβ deposits in wild-type HSCs treated with APP23 tg brain extract once on top and continuously supplemented with synthetic Aβ1–40 in the media revealed the typical bundles of amyloid fibrils interdigitated with cellular elements. Inset, Anti-Aβ-immunogold labeled amyloid fibers (arrows) at high magnification. B, C, Congo red-stained compact Aβ deposits in HSCs treated as in A combined with Aβ immunostaining (dark-blue) under normal light (B) and with the characteristic apple-green birefringence between crossed polarizers (C). D, E, Dystrophic GFP-positive structures in the vicinity of the induced Aβ deposits (Aβ immunostaining, red) in HSCs of Thy1-GFP mice treated with APPPS1 brain extract and syn Aβ1–42. Dystrophic neuritic elements were observed in stratum oriens (D), but they were also found in stratum radiatum and stratum lacunosum-moleculare of CA1 (E). Dystrophic elements were virtually absent in untreated cultures (F), cultures treated only with brain extract or only with syn Aβ1–42 (data not shown). Immunoelectron microscopy reveals an amyloid plaque in the stratum oriens (G) surrounded by several dystrophic boutons (asterisk). H, I, Dystrophic structures and boutons were (at least partly) also immunopositive for hyperphosphorylated tau (AT8 antibody; H, red) and neurofilament light chain (I, red). The Aβ deposits were visualized with anti-Aβ immunostaining (green). J, K, Dendritic spines in HSCs prepared from Thy1-GFP mice. Dendritic segments with numerous spines were observed in untreated HSCs (J). In contrast, curved and thinned (arrowhead) dendritic segments with only very few spines were present in HSCs treated with APPPS1 extract and syn Aβ1–42 (K). The number of dendritic spines/μm on side branches of apical dendrites (CA1 pyramidal neurons, stratum radiatum) was significantly reduced in HSCs with Aβ deposits (L) compared with control cultures treated only with syn Aβ1–42 or APPPS1 brain extract and compared with untreated cultures (n = 4 cultures per group; 3–8 segments analyzed in each culture; ANOVA, F(3,12) = 10.21; p < 0.001). *p < 0.05 (post hoc Tukey's multiple comparison). Scale bars: A,1 μm; inset, 0.1 μm; B, C, 20 μm; D–F, 20 μm; G, 1 μm; H, I, 5 μm; J, K, 5 μm.
To study amyloid-associated morphological alterations in more detail, HSCs were prepared from tg mice expressing eGFP in neurons (Thy1-GFP mice) and were treated once with brain extract from aged APPPS1 tg mice while the media was supplemented with synthetic Aβ1–42. Numerous GFP-positive dystrophic and neuritic structures were observed in vicinity of Aβ-plaques (Fig. 4D–F). Also, at the ultrastructural level, dystrophic boutons were observed in the vicinity of Aβ plaques (Fig. 4G). Some of these dystrophic structures were positive for phosphorylated Tau (AT8 antibody) and neurofilament light chain (Fig. 4H,I). Overall, the dystrophic elements appeared very similar to dystrophic neurites described in aged APP tg mice (Masliah et al., 1996; Phinney et al., 1999; Rupp et al., 2011) and AD brain (Probst et al., 1983). The same HSCs were then used for dendritic spine counts, which revealed an ∼50% decrease in the density of GFP-labeled spines (Fig. 4J–L). Spine loss appeared to be strongest in the vicinity of Aβ deposits, again similar to reports in aged APP tg mice (Spires et al., 2005; Bittner et al., 2010). A general decrease (39%) in the number of spines was also found at the ultrastructural level in wild-type HSCs treated with APPPS1 brain extract and synthetic Aβ1–42 compared with untreated wild-type HSCs (221.0 ± 31.79 vs 135.7 ± 13.22 spines per counting area, indicated is the mean ± SEM, for details, see Material and Methods; p < 0.05). Obvious neuron loss was not apparent in HSCs, similar to APP23 and APPPS1 mice, where neuron loss is also modest and develops only in aged mice (Calhoun et al., 1998; Rupp et al., 2011).
Aβ plaques in APP tg mice and human AD brain are surrounded by microglia (Itagaki et al., 1989; Mackenzie et al., 1995; Stalder et al., 1999; Grathwohl et al., 2009). Although microglia were evenly distributed in untreated HSCs, they were more numerous, hypertrophic, and intensely Iba1-positive in areas with Aβ deposition (Fig. 5A,B). Although individual microglia cells were attached to the amyloid deposits with apparent uptake of amyloid-positive material (Fig. 5C), the typical tight clustering of several microglia around a single Aβ deposit (plaque) as seen in APP tg mice and AD brain was not observed in HSCs. When microglia were depleted in HSCs, the total Aβ load was not significantly changed, but a significant increase of the more compact LCO-positive amyloid was noted (Fig. 5D).
Figure 5.

Microglia-amyloid interaction and microglia depletion in wild-type HSCs. A, B, Double immunolabeling of microglia (anti-Iba1, green) and Aβ (anti-Aβ, red) revealed the scattered distribution of microglial cells in untreated HSCs and thus in the absence of Aβ deposits (A). In contrast, in HSCs treated with APP23 brain extract and synthetic Aβ1–40, microglia were more numerous and appeared hypertrophic in areas rich in Aβ deposits (B). Scale bars: A, B, 20 μm. C, Confocal imaging and 3D surface reconstruction of microglia (Iba-1, green) and amyloid plaques (qFTAA, red) reveal a tight association of microglia processes with the amyloid suggestive of some uptake/interaction with the amyloid (yellow). Scale bar, 100 μm. D, Clodronate treatment of HSCs greatly depleted microglia. Shown are 8-week-old HSCs (n = 29 and n = 25 cultures/group for depleted and control, respectively. ***p < 0.001 (Mann–Whitney U test). A similar microglia depletion was already found after 1 week in culture consistent with previous reports (Kohl et al., 2003). Despite the microglia depletion, no significant effect on the induced total Aβ deposition was found (p = 0.640). However, when the amyloid-specific dye qFTAA was used, a significant increase was found (p < 0.001). Data are mean ± SEM.
HSCs with induced Aβ deposits efficiently seed β-amyloidosis in APP Tg animals
To assess whether such HSCs harbor β-amyloid inducing activity in vivo, 2.5 μl of homogenates from amyloid-laden cultures (treated with APP23 tg brain extract and synthetic Aβ1–40) or the same amount of homogenates from amyloid-free cultures (treated only with APP23 tg brain extract) was injected into the hippocampus of young, 4-month-old APP23 tg mice on an App-null background. For comparison, some hosts were intracerebrally injected with APP23 brain extract or with aggregated, synthetic Aβ1–40, which have previously been shown to have very potent and very poor in vivo β-amyloid-inducing activities, respectively (Meyer-Luehmann et al., 2006) (Fig. 6A,B). Analysis 4 months after inoculation revealed the expected induction of Aβ deposition in the hippocampus of mice receiving APP23 brain extract (Fig. 6C). Although less pronounced than for the APP23 extract (∼40% less), robust induction of Aβ deposition was observed with the amyloid-laden HSCs (Fig. 6D). In contrast, only minimal Aβ deposits were found with amyloid-free HSCs that were treated only with the APP23 extract (Fig. 6E). Fibrils of synthetic Aβ1–40, despite 40-fold higher concentration, induced only minimal Aβ deposition (Fig. 6F). These remarkable observations suggest that Aβ aggregates in HSCs reproduce seeding characteristics of brain-derived Aβ aggregates that are absent in synthetic in vitro aggregated Aβ.
Figure 6.

HSC homogenates with Aβ deposits induce β-amyloidosis upon inoculation in APP tg mice. A, Preparation of the different inoculates: (1) brain extract of an aged APP23 tg mouse was used as a control; (2) homogenates of HSCs with abundant Aβ deposits (induced with APP23 extract and syn Aβ1–40); (3) homogenates of HSCs that lack Aβ deposits (treated only with APP23 extract); and (4) preaggregated synthetic Aβ1–40. B, Aβ concentrations in the different preparations. C–F, Different preparations were injected into the hippocampus of young, 4-month-old APP23 tg mice on an App-null background (abbreviated here as APP23 tg mice). Brains were immunohistochemically analyzed for Aβ deposition together with Congo red staining 4 months later. The injection of the APP23 brain extract induced the expected Aβ deposition in hippocampus (dentate gyrus, C). Homogenates of amyloid-laden HSCs also induced overt, albeit less, Aβ deposition (D), whereas homogenates from HSCs only treated with APP23 extract revealed minor Aβ deposition (E). Preaggregated synthetic Aβ only induced sparse and very localized Aβ deposition (F). G, Stereological quantification of the induced Aβ load in the dentate gyrus (n = 6 or 7/group; ANOVA, F(3,21) = 19.45; p < 0.0001). *p < 0.05 (Tukey's post hoc multiple-comparison test). **p < 0.01 (Tukey's post hoc multiple-comparison test). Scale bars: C–F, 200 μm.
Discussion
In the present work, we demonstrate that HSCs promote the seeded aggregation of synthetic Aβ and the formation of amyloid plaques. Remarkably, the Aβ aggregates generated in the HSC model are active in vivo as seeds for inducing cerebral β-amyloidosis, which emphasizes their similarity to Aβ seeds formed in brain. Aβ aggregation has recently been described in APP-overexpressing 3D human neural stem cell cultures, but its in vivo activity has not been tested (Shi et al., 2012; Choi et al., 2014). Moreover, according to current insights, Aβ aggregation in the brain spreads along neuroanatomical pathways in a self-propagating non–cell-autonomous process (Jucker and Walker, 2013; Raj et al., 2015); therefore, an in vitro system more closely mimicking the in vivo brain environment (Frotscher and Heimrich, 1993; Bahr, 1995; Frotscher et al., 1995) is required. The development of amyloid plaque-associated pathologies in the HSCs underlines the similarities of this new in vitro model with cerebral β-amyloidosis in a transgenic mouse brain. Of note, the amyloid-associated changes in HSCs occur in wild-type brain tissue without APP overexpression and thus make seeded Aβ aggregation in HSCs a unique and valid model of cerebral β-amyloidosis, also for sporadic AD. Prefixation of the HSCs largely prevented Aβ aggregation and highlights the contribution of cellular processes to achieve biological active Aβ aggregates.
HSCs prepared from postnatal APP tg mice did not develop Aβ deposits, even after 10 weeks of cultivation and despite development of robust cerebral β-amyloidosis in the corresponding 8-week-old tg mice. This may be because Aβ generated in HSCs diffuses into the medium precluding the local buildup of sufficiently high Aβ levels for amyloid deposition (Waters, 2010). The recently reported Aβ deposition in human cell cultures (Shi et al., 2012; Choi et al., 2014) is probably related to the used Matrigel, a gelatinous mixture of extracellular matrix proteins that should trap Aβ. However, we achieved amyloid plaque formation in HSCs by repeatedly adding synthetic Aβ to the medium and brain-derived Aβ seeds once to the culture. This seeded Aβ aggregation can occur independent of endogenous Aβ generation and may be initiated at cellular membrane structures (Nagarathinam et al., 2013). Interestingly, however, seeded Aβ aggregation requires functional, living HSCs (membranes), as inactivation before cultivation largely eliminated amyloid deposition. Consistently, in previous work using a variety of cell lines and medium supplemented with Aβ (albeit in at least tenfold higher concentration than in the present work), it was found that Aβ aggregates occurred only in the presence of living cells (Friedrich et al., 2010), although again its in vivo activity was not tested.
Our data demonstrate that Aβ plaque formation occurs via templated conversion of synthetic Aβ exemplified by the achievement of different Aβ morphotypes depending on the nature of the synthetic Aβ species in the medium (Aβ1–40 vs Aβ1–42) and also on the applied brain extract (APP23 or APPPS1 mice). Polymorphic Aβ deposits and structural Aβ variants have also been described in APP tg mouse models (Heilbronner et al., 2013) and AD brain (Lu et al., 2013) and different Aβ conformers have different biological activities in vitro (Petkova et al., 2005; Eisenberg and Jucker, 2012).
Although in vitro aggregated synthetic Aβ can act as an in vivo seed in APP tg mice (Stöhr et al., 2012), its seeding activity is at least 100× less potent than brain-derived aggregated Aβ (Meyer-Luehmann et al., 2006; Stöhr et al., 2012). In contrast, HSC-derived aggregated Aβ shows seeding activity within the same order of magnitude as brain-derived aggregated Aβ. This is consistent with the idea that brain seeds impose their structure on the synthetic Aβ and render it seeding active. Moreover, the strong reduction of Aβ aggregation by fixation before treatment suggests a factor in living HSCs (and thus the brain), which contributes to the seeded Aβ aggregation.
Aβ plaque features (congophilic amyloid, star-like appearance, and parallel fibril bundles at ultrastructural level) and associated neural changes in HSCs (dystrophic and phosphorylated Tau-positive neurites, loss of dendritic spines) recapitulate largely the amyloid-associated neural changes in APP tg mouse models (Masliah et al., 1996; Phinney et al., 1999; Spires et al., 2005; Bittner et al., 2010; Rupp et al., 2011) and AD brain (Probst et al., 1983). However and most remarkably, these amyloid features and neural changes in HSCs occur in response to the induced aggregation of exogenously applied synthetic Aβ. Equally important and of note, all these changes occur in wild-type tissue and thus avoid the potential confounding overexpression of APP present in transgenic mice (Jucker, 2010).
Microglia in HSCs show a typical ramified morphology (Hailer et al., 1996; Skibo et al., 2000), which was retained throughout the 10 week HSC period. Although microglia were more numerous and hypertrophic in areas with Aβ plaques (typically around the rim of the culture), indicating that a local inflammatory and phagocytic response was taking place, the tight clustering of microglia observed around Aβ deposits in vivo was not observed in HSCs. This may be a result of the disruption of localized signals and microglial chemotaxis due to dilution of chemoattractants and/or the presence of synthetic Aβ in the culture medium. When microglia were depleted in HSCs, the total Aβ load was not changed, but a shift to more dense-cored plaques was found. Interestingly, this finding is reminiscent of our in vivo observations, where in microglia-depleted APP23 animals a small decrease in the diffuse Aβ load and a tendency toward an increase in the dense-cored amyloid load (albeit not significant) was observed after 4 weeks (Grathwohl et al., 2009). The lack of changes in total Aβ load after microglia-depletion in vitro and in vivo may at least partly be explained by overwhelming Aβ aggregation over (physiological) removal.
In conclusion, we have demonstrated that Aβ aggregation and many of the previously described neuronal changes in APP tg mice and AD brain can be achieved by the seeded conversion of synthetic Aβ in living wild-type brain tissue. HSCs combine advantages of in vitro tissue culture and in vivo analysis of mouse models. This easily accessible system is amenable to experimental manipulation and live imaging. Systems that model the complex brain environment are urgently needed to study the mechanisms governing Aβ aggregation in vivo, the downstream pathological processes of neurodegeneration, and to test therapeutic approaches. The finding of seeded conversion of synthetic Aβ into potent in vivo seeding-active Aβ aggregates should ease further studies aiming at deciphering the factors responsible for its conversion.
Footnotes
This work was supported by German Federal Ministry of Education and Research Grant BMBF-ALZKULT 031A198 to B.H. and M.J., EU Joint Programme on Neurodegenerative Diseases Grant JPND-NeuTARGETs, and German Research Foundation Grants DFG DE 551/11-2 and VL 72/1-2 to T.D. and A.V. We thank Ulrike Obermüller, Jay Rasmussen, Simone Zenker, Aileen Koch, Sigrun Nestel, Melanie Barth, Anke Biczysko, and the other members of our laboratories for help and discussions; and Paul Mathews (Nathan S. Kline Institute, Orangeburg, NJ) for antibody donation.
The authors declare no competing financial interests.
References
- Bahr BA. Long-term hippocampal slices: a model system for investigating synaptic mechanisms and pathologic processes. J Neurosci Res. 1995;42:294–305. doi: 10.1002/jnr.490420303. [DOI] [PubMed] [Google Scholar]
- Bittner T, Fuhrmann M, Burgold S, Ochs SM, Hoffmann N, Mitteregger G, Kretzschmar H, LaFerla FM, Herms J. Multiple events lead to dendritic spine loss in triple transgenic Alzheimer's disease mice. PLoS One. 2010;5:e15477. doi: 10.1371/journal.pone.0015477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondolfi L, Calhoun M, Ermini F, Kuhn HG, Wiederhold KH, Walker L, Staufenbiel M, Jucker M. Amyloid-associated neuron loss and gliogenesis in the neocortex of amyloid precursor protein transgenic mice. J Neurosci. 2002;22:515–522. doi: 10.1523/JNEUROSCI.22-02-00515.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinks H, Conrad S, Vogt J, Oldekamp J, Sierra A, Deitinghoff L, Bechmann I, Alvarez-Bolado G, Heimrich B, Monnier PP, Mueller BK, Skutella T. The repulsive guidance molecule RGMa is involved in the formation of afferent connections in the dentate gyrus. J Neurosci. 2004;24:3862–3869. doi: 10.1523/JNEUROSCI.5296-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calhoun ME, Wiederhold KH, Abramowski D, Phinney AL, Probst A, Sturchler-Pierrat C, Staufenbiel M, Sommer B, Jucker M. Neuron loss in APP transgenic mice. Nature. 1998;395:755–756. doi: 10.1038/27351. [DOI] [PubMed] [Google Scholar]
- Choi SH, Kim YH, Hebisch M, Sliwinski C, Lee S, D'Avanzo C, Chen H, Hooli B, Asselin C, Muffat J, Klee JB, Zhang C, Wainger BJ, Peitz M, Kovacs DM, Woolf CJ, Wagner SL, Tanzi RE, Kim DY. A three-dimensional human neural cell culture model of Alzheimer's disease. Nature. 2014;515:274–278. doi: 10.1038/nature13800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dasilva KA, Shaw JE, McLaurin J. Amyloid-beta fibrillogenesis: structural insight and therapeutic intervention. Exp Neurol. 2010;223:311–321. doi: 10.1016/j.expneurol.2009.08.032. [DOI] [PubMed] [Google Scholar]
- Del Turco D, Deller T. Organotypic entorhino-hippocampal slice cultures: a tool to study the molecular and cellular regulation of axonal regeneration and collateral sprouting in vitro. Methods Mol Biol. 2007;399:55–66. doi: 10.1007/978-1-59745-504-6_5. [DOI] [PubMed] [Google Scholar]
- Du D, Murray AN, Cohen E, Kim HE, Simkovsky R, Dillin A, Kelly JW. A kinetic aggregation assay allowing selective and sensitive amyloid-beta quantification in cells and tissues. Biochemistry. 2011;50:1607–1617. doi: 10.1021/bi1013744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duyckaerts C, Potier MC, Delatour B. Alzheimer disease models and human neuropathology: similarities and differences. Acta Neuropathol. 2008;115:5–38. doi: 10.1007/s00401-007-0312-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisele YS, Obermüller U, Heilbronner G, Baumann F, Kaeser SA, Wolburg H, Walker LC, Staufenbiel M, Heikenwalder M, Jucker M. Peripherally applied Abeta-containing inoculates induce cerebral beta-amyloidosis. Science. 2010;330:980–982. doi: 10.1126/science.1194516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisele YS, Fritschi SK, Hamaguchi T, Obermüller U, Füger P, Skodras A, Schäfer C, Odenthal J, Heikenwalder M, Staufenbiel M, Jucker M. Multiple factors contribute to the peripheral induction of cerebral beta-amyloidosis. J Neurosci. 2014;34:10264–10273. doi: 10.1523/JNEUROSCI.1608-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenberg D, Jucker M. The amyloid state of proteins in human diseases. Cell. 2012;148:1188–1203. doi: 10.1016/j.cell.2012.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng G, Mellor RH, Bernstein M, Keller-Peck C, Nguyen QT, Wallace M, Nerbonne JM, Lichtman JW, Sanes JR. Imaging neuronal subsets in transgenic mice expressing multiple spectral variants of GFP. Neuron. 2000;28:41–51. doi: 10.1016/S0896-6273(00)00084-2. [DOI] [PubMed] [Google Scholar]
- Friedrich RP, Tepper K, Rönicke R, Soom M, Westermann M, Reymann K, Kaether C, Fändrich M. Mechanism of amyloid plaque formation suggests an intracellular basis of Abeta pathogenicity. Proc Natl Acad Sci U S A. 2010;107:1942–1947. doi: 10.1073/pnas.0904532106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritschi SK, Cintron A, Ye L, Mahler J, Bühler A, Baumann F, Neumann M, Nilsson KP, Hammarström P, Walker LC, Jucker M. Abeta seeds resist inactivation by formaldehyde. Acta Neuropathol. 2014;128:477–484. doi: 10.1007/s00401-014-1339-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frotscher M, Heimrich B. Formation of layer-specific fiber projections to the hippocampus in vitro. Proc Natl Acad Sci U S A. 1993;90:10400–10403. doi: 10.1073/pnas.90.21.10400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frotscher M, Zafirov S, Heimrich B. Development of identified neuronal types and of specific synaptic connections in slice cultures of rat hippocampus. Prog Neurobiol. 1995;45:vii–xxviii. doi: 10.1016/0301-0082(95)90872-h. [DOI] [PubMed] [Google Scholar]
- Games D, Adams D, Alessandrini R, Barbour R, Berthelette P, Blackwell C, Carr T, Clemens J, Donaldson T, Gillespie F. Alzheimer-type neuropathology in transgenic mice overexpressing V717F beta-amyloid precursor protein. Nature. 1995;373:523–527. doi: 10.1038/373523a0. [DOI] [PubMed] [Google Scholar]
- Grathwohl SA, Kälin RE, Bolmont T, Prokop S, Winkelmann G, Kaeser SA, Odenthal J, Radde R, Eldh T, Gandy S, Aguzzi A, Staufenbiel M, Mathews PM, Wolburg H, Heppner FL, Jucker M. Formation and maintenance of Alzheimer's disease beta-amyloid plaques in the absence of microglia. Nat Neurosci. 2009;12:1361–1363. doi: 10.1038/nn.2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hailer NP, Jarhult JD, Nitsch R. Resting microglial cells in vitro: analysis of morphology and adhesion molecule expression in organotypic hippocampal slice cultures. Glia. 1996;18:319–331. doi: 10.1002/(SICI)1098-1136(199612)18:4<319::AID-GLIA6>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- Harper JD, Lansbury PT., Jr Models of amyloid seeding in Alzheimer's disease and scrapie: mechanistic truths and physiological consequences of the time-dependent solubility of amyloid proteins. Annu Rev Biochem. 1997;66:385–407. doi: 10.1146/annurev.biochem.66.1.385. [DOI] [PubMed] [Google Scholar]
- Harris-White ME, Chu T, Balverde Z, Sigel JJ, Flanders KC, Frautschy SA. Effects of transforming growth factor-beta (isoforms 1–3) on amyloid-beta deposition, inflammation, and cell targeting in organotypic hippocampal slice cultures. J Neurosci. 1998;18:10366–10374. doi: 10.1523/JNEUROSCI.18-24-10366.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heilbronner G, Eisele YS, Langer F, Kaeser SA, Novotny R, Nagarathinam A, Aslund A, Hammarström P, Nilsson KP, Jucker M. Seeded strain-like transmission of beta-amyloid morphotypes in APP transgenic mice. EMBO Rep. 2013;14:1017–1022. doi: 10.1038/embor.2013.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtzman DM, Morris JC, Goate AM. Alzheimer's disease: the challenge of the second century. Sci Transl Med. 2011;3:77sr71. doi: 10.1126/scitranslmed.3002369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G. Correlative memory deficits, abeta elevation, and amyloid plaques in transgenic mice. Science. 1996;274:99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- Itagaki S, McGeer PL, Akiyama H, Zhu S, Selkoe D. Relationship of microglia and astrocytes to amyloid deposits of Alzheimer disease. J Neuroimmunol. 1989;24:173–182. doi: 10.1016/0165-5728(89)90115-X. [DOI] [PubMed] [Google Scholar]
- Johansson S, Radesäter AC, Cowburn RF, Thyberg J, Luthman J. Modelling of amyloid beta-peptide induced lesions using roller-drum incubation of hippocampal slice cultures from neonatal rats. Exp Brain Res. 2006;168:11–24. doi: 10.1007/s00221-005-0069-z. [DOI] [PubMed] [Google Scholar]
- Jucker M. The benefits and limitations of animal models for translational research in neurodegenerative diseases. Nat Med. 2010;16:1210–1214. doi: 10.1038/nm.2224. [DOI] [PubMed] [Google Scholar]
- Jucker M, Walker LC. Self-propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature. 2013;501:45–51. doi: 10.1038/nature12481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klingstedt T, Aslund A, Simon RA, Johansson LB, Mason JJ, Nyström S, Hammarström P, Nilsson KP. Synthesis of a library of oligothiophenes and their utilization as fluorescent ligands for spectral assignment of protein aggregates. Org Biomol Chem. 2011;9:8356–8370. doi: 10.1039/c1ob05637a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohl A, Dehghani F, Korf HW, Hailer NP. The bisphosphonate clodronate depletes microglial cells in excitotoxically injured organotypic hippocampal slice cultures. Exp Neurol. 2003;181:1–11. doi: 10.1016/S0014-4886(02)00049-3. [DOI] [PubMed] [Google Scholar]
- Langer F, Eisele YS, Fritschi SK, Staufenbiel M, Walker LC, Jucker M. Soluble Abeta seeds are potent inducers of cerebral beta-amyloid deposition. J Neurosci. 2011;31:14488–14495. doi: 10.1523/JNEUROSCI.3088-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu JX, Qiang W, Yau WM, Schwieters CD, Meredith SC, Tycko R. Molecular structure of beta-amyloid fibrils in Alzheimer's disease brain tissue. Cell. 2013;154:1257–1268. doi: 10.1016/j.cell.2013.08.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie IR, Hao C, Munoz DG. Role of microglia in senile plaque formation. Neurobiol Aging. 1995;16:797–804. doi: 10.1016/0197-4580(95)00092-S. [DOI] [PubMed] [Google Scholar]
- Masliah E, Sisk A, Mallory M, Mucke L, Schenk D, Games D. Comparison of neurodegenerative pathology in transgenic mice overexpressing V717F beta-amyloid precursor protein and Alzheimer's disease. J Neurosci. 1996;16:5795–5811. doi: 10.1523/JNEUROSCI.16-18-05795.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer D, Fischer H, Schneider U, Heimrich B, Schwemmle M. Borna disease virus replication in organotypic hippocampal slice cultures from rats results in selective damage of dentate granule cells. J Virol. 2005;79:11716–11723. doi: 10.1128/JVI.79.18.11716-11723.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer-Luehmann M, Coomaraswamy J, Bolmont T, Kaeser S, Schaefer C, Kilger E, Neuenschwander A, Abramowski D, Frey P, Jaton AL, Vigouret JM, Paganetti P, Walsh DM, Mathews PM, Ghiso J, Staufenbiel M, Walker LC, Jucker M. Exogenous induction of cerebral beta-amyloidogenesis is governed by agent and host. Science. 2006;313:1781–1784. doi: 10.1126/science.1131864. [DOI] [PubMed] [Google Scholar]
- Morales-Corraliza J, Mazzella MJ, Berger JD, Diaz NS, Choi JH, Levy E, Matsuoka Y, Planel E, Mathews PM. In vivo turnover of tau and APP metabolites in the brains of wild-type and Tg2576 mice: greater stability of sAPP in the beta-amyloid depositing mice. PLoS One. 2009;4:e7134. doi: 10.1371/journal.pone.0007134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagarathinam A, Hoflinger P, Bühler A, Schäfer C, McGovern G, Jeffrey M, Staufenbiel M, Jucker M, Baumann F. Membrane-anchored Abeta accelerates amyloid formation and exacerbates amyloid-associated toxicity in mice. J Neurosci. 2013;33:19284–19294. doi: 10.1523/JNEUROSCI.2542-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paravastu AK, Qahwash I, Leapman RD, Meredith SC, Tycko R. Seeded growth of beta-amyloid fibrils from Alzheimer's brain-derived fibrils produces a distinct fibril structure. Proc Natl Acad Sci U S A. 2009;106:7443–7448. doi: 10.1073/pnas.0812033106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petkova AT, Leapman RD, Guo Z, Yau WM, Mattson MP, Tycko R. Self-propagating, molecular-level polymorphism in Alzheimer's beta-amyloid fibrils. Science. 2005;307:262–265. doi: 10.1126/science.1105850. [DOI] [PubMed] [Google Scholar]
- Phinney AL, Deller T, Stalder M, Calhoun ME, Frotscher M, Sommer B, Staufenbiel M, Jucker M. Cerebral amyloid induces aberrant axonal sprouting and ectopic terminal formation in amyloid precursor protein transgenic mice. J Neurosci. 1999;19:8552–8559. doi: 10.1523/JNEUROSCI.19-19-08552.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Probst A, Basler V, Bron B, Ulrich J. Neuritic plaques in senile dementia of Alzheimer type: a Golgi analysis in the hippocampal region. Brain Res. 1983;268:249–254. doi: 10.1016/0006-8993(83)90490-0. [DOI] [PubMed] [Google Scholar]
- Radde R, Bolmont T, Kaeser SA, Coomaraswamy J, Lindau D, Stoltze L, Calhoun ME, Jäggi F, Wolburg H, Gengler S, Haass C, Ghetti B, Czech C, Hölscher C, Mathews PM, Jucker M. Abeta42-driven cerebral amyloidosis in transgenic mice reveals early and robust pathology. EMBO Rep. 2006;7:940–946. doi: 10.1038/sj.embor.7400784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raj A, LoCastro E, Kuceyeski A, Tosun D, Relkin N, Weiner M. Network diffusion model of progression predicts longitudinal patterns of atrophy and metabolism in Alzheimer's disease. Cell Rep. 2015;10:359–369. doi: 10.1016/j.celrep.2014.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rupp NJ, Wegenast-Braun BM, Radde R, Calhoun ME, Jucker M. Early onset amyloid lesions lead to severe neuritic abnormalities and local, but not global neuron loss in APPPS1 transgenic mice. Neurobiol Aging. 2011;32:2324–2326. doi: 10.1016/j.neurobiolaging.2010.08.014. [DOI] [PubMed] [Google Scholar]
- Shi Y, Kirwan P, Smith J, MacLean G, Orkin SH, Livesey FJ. A human stem cell model of early Alzheimer's disease pathology in Down syndrome. Sci Transl Med. 2012;4:124ra129. doi: 10.1126/scitranslmed.3003771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skibo GG, Nikonenko IR, Savchenko VL, McKanna JA. Microglia in organotypic hippocampal slice culture and effects of hypoxia: ultrastructure and lipocortin-1 immunoreactivity. Neuroscience. 2000;96:427–438. doi: 10.1016/S0306-4522(99)00562-X. [DOI] [PubMed] [Google Scholar]
- Spires TL, Meyer-Luehmann M, Stern EA, McLean PJ, Skoch J, Nguyen PT, Bacskai BJ, Hyman BT. Dendritic spine abnormalities in amyloid precursor protein transgenic mice demonstrated by gene transfer and intravital multiphoton microscopy. J Neurosci. 2005;25:7278–7287. doi: 10.1523/JNEUROSCI.1879-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stalder M, Phinney A, Probst A, Sommer B, Staufenbiel M, Jucker M. Association of microglia with amyloid plaques in brains of APP23 transgenic mice. Am J Pathol. 1999;154:1673–1684. doi: 10.1016/S0002-9440(10)65423-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stalder M, Deller T, Staufenbiel M, Jucker M. 3D-Reconstruction of microglia and amyloid in APP23 transgenic mice: no evidence of intracellular amyloid. Neurobiol Aging. 2001;22:427–434. doi: 10.1016/S0197-4580(01)00209-3. [DOI] [PubMed] [Google Scholar]
- Stöhr J, Watts JC, Mensinger ZL, Oehler A, Grillo SK, DeArmond SJ, Prusiner SB, Giles K. Purified and synthetic Alzheimer's amyloid beta (Abeta) prions. Proc Natl Acad Sci U S A. 2012;109:11025–11030. doi: 10.1073/pnas.1206555109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturchler-Pierrat C, Abramowski D, Duke M, Wiederhold KH, Mistl C, Rothacher S, Ledermann B, Bürki K, Frey P, Paganetti PA, Waridel C, Calhoun ME, Jucker M, Probst A, Staufenbiel M, Sommer B. Two amyloid precursor protein transgenic mouse models with Alzheimer disease-like pathology. Proc Natl Acad Sci U S A. 1997;94:13287–13292. doi: 10.1073/pnas.94.24.13287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terry RD, Gonatas NK, Weiss M. Ultrastructural studies in Alzheimer's presenile dementia. Am J Pathol. 1964;44:269–297. [PMC free article] [PubMed] [Google Scholar]
- Vlachos A, Müller-Dahlhaus F, Rosskopp J, Lenz M, Ziemann U, Deller T. Repetitive magnetic stimulation induces functional and structural plasticity of excitatory postsynapses in mouse organotypic hippocampal slice cultures. J Neurosci. 2012;32:17514–17523. doi: 10.1523/JNEUROSCI.0409-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waters J. The concentration of soluble extracellular amyloid-beta protein in acute brain slices from CRND8 mice. PLoS One. 2010;5:e15709. doi: 10.1371/journal.pone.0015709. [DOI] [PMC free article] [PubMed] [Google Scholar]