

Abstract
All tissues are genetically programmed to acquire an optimal size that is defined by total cell number and individual cellular dimensions. The retina contains stereotyped proportions of one glial and six neuronal cell types that are generated in overlapping waves. How multipotent retinal progenitors know when to switch from making one cell type to the next so that appropriate numbers of each cell type are generated is poorly understood. Pten is a phosphatase that controls progenitor cell proliferation and differentiation in several lineages. Here, using a conditional loss-of-function strategy, we found that Pten regulates retinal cell division and is required to produce the full complement of rod photoreceptors and amacrine cells in mouse. We focused on amacrine cell number control, identifying three downstream Pten effector pathways. First, phosphoinositide 3-kinase/Akt signaling is hyperactivated in Pten conditional knock-out (cKO) retinas, and misexpression of constitutively active Akt (Akt-CA) in retinal explants phenocopies the reduction in amacrine cell production observed in Pten cKOs. Second, Akt-CA activates Tgfβ signaling in retinal explants, which is a negative feedback pathway for amacrine cell production. Accordingly, Tgfβ signaling is elevated in Pten cKO retinas, and epistatic analyses placed Pten downstream of TgfβRII in amacrine cell number control. Finally, Pten regulates Raf/Mek/Erk signaling levels to promote the differentiation of all amacrine cell subtypes, which are each reduced in number in Pten cKOs. Pten is thus a positive regulator of amacrine cell production, acting via multiple downstream pathways, highlighting its diverse actions as a mediator of cell number control.
SIGNIFICANCE STATEMENT Despite the importance of size for optimal organ function, how individual cell types are generated in correct proportions is poorly understood. There are several ways to control cell number, including readouts of organ function (e.g., secreted hormones reach functional levels when enough cells are made) or counting of cell divisions or cell number. The latter applies to the retina, where cell number is regulated by negative feedback signals, which arrest differentiation of particular cell types at threshold levels. Herein, we show that Pten is a critical regulator of amacrine cell number in the retina, acting via multiple downstream pathways. Our studies provide molecular insights into how PTEN loss in humans may lead to uncontrolled cell division in several pathological conditions.
Keywords: amacrine cells, Erk, negative feedback signaling, Pten, retina, Tgfβ
Introduction
Proper formation of functional neural networks requires that correct numbers of each cell type are generated during development. The neural retina contains one glial and six neuronal cell types that form three distinct cellular layers: the outer nuclear layer (ONL), inner nuclear layer (INL), and ganglion cell layer (GCL), each with stereotyped cellular compositions. In mouse, there are two differentiation peaks; the bulk of retinal ganglion cells (RGCs), cone photoreceptors, horizontal cells, and amacrine cells differentiate between embryonic day 11 (E11) and postnatal day 2 (P2), while differentiation of rod photoreceptors, bipolar cells and Müller glia peaks postnatally, ending at P11 (Young, 1985; Cepko et al., 1996). While some retinal cells arise from committed retinal precursors with fixed lineages (Pearson and Doe, 2004; Cayouette et al., 2006; He et al., 2012), others share a common lineage, arising from multipotent retinal progenitor cells (RPCs; Young, 1985; Alexiades and Cepko, 1997; Waid and McLoon, 1998). RPC fate decisions are stochastic, with the differentiation of more numerous retinal cell types occurring with higher probabilities (Close et al., 2005; Gomes et al., 2011). However, while the genes that specify individual retinal cell identities have been extensively studied, the molecules that “sense” cell number to bias RPC fate selection so that the same numbers of each cell type are made from individual to individual are just beginning to be explored.
An optimally sized organ requires correct numbers of each cell type to function. Cell number is regulated either by using readouts of organ function or by counting cell divisions or cell number (Wu et al., 2003). The latter applies to the retina, where cell number is regulated by secreted negative feedback signals, which stop the differentiation of a particular cell type upon reaching threshold levels (Reh and Tully, 1986; Belliveau and Cepko, 1999; Harmon et al., 2004). In the retina, a well-characterized feedback pathway controls amacrine cell production; the transcription factor Zac1 acts in amacrine cells to initiate transforming growth factor β 2 (TgfbII) expression, which negatively regulates RPC proliferation and amacrine cell differentiation (Tobin and Celeste, 2005; Ma et al., 2007). What remains unclear is how amacrine cell feedback signals are themselves regulated.
Several extracellular signals bias the decision by RPCs to proliferate or differentiate along a specific cell lineage (Marquardt, 2003; Hatakeyama and Kageyama, 2004; Ohsawa and Kageyama, 2008; Wallace, 2011). Included are ligands that signal through receptor tyrosine kinases (RTKs) and the two major downstream signal transduction cascades, Mek/Erk and phosphoinositide 3-kinase (PI3K) pathways (Stambolic et al., 1998; Downward, 2004; Comer and Parent, 2007). Here, we ask which RTK pathways influence the decision by RPCs to proliferate or differentiate in the retina and, in so doing, control cell number. It is difficult to genetically manipulate PI3K as it is a multisubunit enzyme encoded by several genes. We thus targeted Pten, which is a single gene-encoding lipid and protein phosphatase that is a major antagonist of PI3K signaling (Leslie et al., 2008). We previously found that Pten is required for the differentiation of the full complement of retinal amacrine cells and horizontal cells (Cantrup et al., 2012). In addition to defects in cell number, Pten mutations lead to neuronal hypertrophy and errors in migration and dendrite arborization in the retina, as well as cerebellum and cerebral cortex (Backman et al., 2001; Groszer et al., 2001; Marino et al., 2002; Yue et al., 2005; Kwon et al., 2006; Fraser et al., 2008; Lehtinen et al., 2011; Jo et al., 2012; Sakagami et al., 2012). Other studies have similarly found that retinal cell numbers are altered in Pten cKO retinas, but with conflicting findings, possibly because of the use of different Cre drivers (Sakagami et al., 2012) and a focus on the very peripheral retina (Jo et al., 2012). To further elucidate the role of Pten in retinal cell number control, we studied Pten cKO retinas, identifying three Pten effector pathways influencing amacrine cell differentiation.
Materials and Methods
Animals.
All animal procedures were approved by the University of Calgary Animal Care Committee in agreement with the guidelines of the Canadian Council of Animal Care. The Pax6::Cre driver (Marquardt et al., 2001) and Rosa26R-EGFP reporter (Gomer, 2001) lines were generated previously, and PCR genotyping was performed as described. The Ptenfl allele, in which exons 4 and 5 are flanked by loxP sites, was also generated previously, and PCR genotyping was performed as described (Backman et al., 2001).
Western blotting.
Retinas were collected from embryos and postnatal pups at the indicated stages, lysed in RIPA buffer with protease (1× protease inhibitor complete, 1 mm PMSF) and phosphatase (50 mm NaF, 1 mm NaOV) inhibitors, and 10 μg of lysate was run on SDS-PAGE gels for Western blot analysis as described previously (Ma et al., 2007). Primary antibodies included pAktSer473 (1:1000; Cell Signaling Technology, catalog #4060), total Akt (1:1000; Cell Signaling Technology, catalog #9272), pErk (1:1000; Cell Signaling Technology, catalog #9106), Erk (1:1000; Cell Signaling Technology, catalog #9102), Pten (1:1000; Cell Signaling Technology, catalog #9559), β-actin (1:10,000; Abcam, catalog #8227), pSmad2 (1:1000; Cell Signaling Technology, catalog #3101), Smad2/3 (1:1000; Cell Signaling Technology, catalog #5678), TgfβII (1:1000; Abcam, catalog #36495), and TgfβRII (1:2000; Abcam, catalog #186838). Each Western blot was performed a minimum of three times on three sets of independent samples, and densitometries were calculated using UN-SCAN-IT gel densitometry software (Silk Scientific). The average values of normalized expression levels were plotted.
Immunohistochemistry.
Whole eyes or retinal explants were fixed overnight in 4% paraformaldehyde diluted in PBS, washed three times for 10 min each in PBS, and then immersed in 20% sucrose overnight. Eyes were embedded in optimal cutting temperature compound and sectioned on a cryostat at 10 μm. Blocking solution (10% horse serum, 0.1% Triton X-100 in PBS, pH 7.5) was added for 1 h to limit nonspecific immunoreactivity before immunostaining. Primary antibodies diluted in blocking solution were incubated on slides overnight at 4°C. The following primary antibodies were used: pAktSer473 (1:500; Cell Signaling Technology, catalog #4060), Akt (1:500; Cell Signaling Technology, catalog #9272), AP2α (1:100; Developmental Studies Hybridoma Bank, catalog #3B5), Barhl2 (1:500; Sigma, catalog #AV31981), Beta3 (1:200; Santa Cruz Biotechnology, catalog #sc-6045), Brn3a (1:500; Millipore, catalog #AB5945), BrdU (1:50; Oxford Biotech, catalog #OBT0030), Chx10 (1:200; Santa Cruz Biotechnology, catalog #SC-21690), cone-arrestin (1:500; Millipore, catalog #AB15282), GAD65/67 (Abcam, catalog #ab11070), Glyt1 (1:200; Abcam, catalog #ab113823), phosphohistone H3 (pHH3; 1:1000; Millipore, catalog #06-570), Neurod6 (1:100; Abcam, catalog #ab85824), Pax6 (1:500; Covance, catalog #PRB-278P), Pten (1:500; Cell Signaling Technology, catalog #9559), rhodopsin (1:500; Millipore, catalog #MAB5356), and Sox9 (1:500; Millipore, catalog #AB5535). Slides were washed three times in PBS with 0.1% Triton X-100, and primary antibodies were detected using secondary antibodies conjugated with Alexa568 (1:500; Invitrogen, catalog #A11057) or Alexa488 (1:500; Invitrogen, A11055).
BrdU labeling.
To label S-phase progenitors, pregnant females were injected intraperitoneally with 100 μg/g body weight BrdU (Sigma) 30 min before being killed for proliferation assays, or were injected at E12.5, E14.5, or E18.5 for birthdating studies and killed at P7. Eyes were dissected and were processed for anti-BrdU staining as described above except for the addition of a pretreatment with 2N HCl for 30 min at 37°C.
Aggregation assay.
Aggregation assays were performed as described previously, with a slight modification (Assinder et al., 2009). E15.5 retinas were treated with 0.125% trypsin for 10 min at 37°C and triturated in Ca2+/Mg2+-free PBS/20% fetal bovine serum (FBS). Dissociated E15.5 retinal cells were then labeled with 10 μm BrdU for 1 h at 37°C. P2 Pten+/+ or Ptenfl/fl; Pax6::Cre retinas were dissociated with the same method. Following dissociation, cells were counted and then resuspended in culture media at 1 × 106 cells/ml (P2) or 5 × 105 cells/ml (E15.5). For cocultures, 100 μl (5 × 104 cells) of BrdU-labeled E15.5 progenitors were added to 1 ml (1 × 106 cells) of dissociated P2 Pten+/+ or Ptenfl/fl; Pax6::Cre retinal cells. A total of 1 × 106 cells of E15.5 cells were aggregated as a control. Aggregated cells were collected by low-speed centrifugation (100 rpm for 2 min), transferred as a pellet onto 0.25 μm nucleopore membranes, and cultured at 37°C, 5% CO2 for 8 d in vitro (DIV) in retinal explant medium (50% DMEM, 25% HBSS, 25% heat-inactivated horse serum, 200 μm l-glutamine, 0.6 mm HEPES, 1% Pen-Strep). Pellets were processed for anti-BrdU and anti-Pax6 staining as described above.
Ex utero electroporation for cell counts.
E18.5 CD1 retinas were dissected and retinal pigmented epithelia were removed in cold PBS. The tissues were then placed on a 2% agarose plug poured into the bottom of 24-well plates. All genes were cloned into pCIG2, a bicistronic expression vector containing a β-actin promoter/CMV enhancer and an internal ribosome entry site (IRES)-EGFP cassette (Hand et al., 2005). Genes cloned into pCIG2 included wild-type Pten [PTEN(wt); Spinelli et al., 2015], PTEN(C124S) (protein and lipid phosphatase dead; Spinelli et al., 2015), constitutively active Akt (Akt-CA; Eder et al., 1998), TgfβRII-CA (Wei et al., 2013), TgfβRII-DN (Wei et al., 2013), bRAF(V600E) (Addgene plasmid 15269; Boehm et al., 2007), hMAP2K1-CA (Yoshimura et al., 2006), and MEK-DN (dominant negative Mek; Yoshimura et al., 2006). Expression plasmids were purified using an endotoxin-free plasmid DNA purification kit (Qiagen, Maxi kit). Ten microliters of each expression plasmid (1 μg/μl) were applied directly on the retina, followed by the application of seven 50 ms pulses of 50 mV using a BTX electroporator. The retinas were then flat mounted on 0.25 μm nucleopore membranes and cultured at 37°C, 5% CO2 for 8 DIV in retinal explant medium. The explants were processed for anti-Pax6 staining as described above.
Transfections for Western blot analyses.
Eighty percent confluent HEK cells grown in six-well plates were transfected with 3 μg of plasmid DNA with Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. Twenty-four hours after transfection, cells were rinsed with PBS, transferred into 2.0 ml tubes, and lysed in RIPA buffer with protease (1× protease inhibitor complete, 1 mm PMSF) and phosphatase (50 mm NaF, 1 mm NaOV) inhibitors. Ten micrograms of lysate were run on SDS-PAGE gels for Western blot analysis as described previously (Ma et al., 2007).
In vitro electroporation for qPCR.
In vitro electroporation was performed on retinal explants as described previously (Matsuda and Cepko, 2004). Briefly, dissected retinas were placed in 2 mm gap cuvettes (VWR International) along with 30 μl of 0.5 μg/μl DNA and electroporated using five square 20 V pulses of 50 ms duration and 950 ms intervals using an ECM830 pulse generator (BTX Harvard Apparatus). The expression plasmids used are described above. Electroporated retinas were cultured on the 0.25 μm nucleopore membranes at 37°C, 5% CO2 for 3 DIV in retinal explant medium.
FACS.
Electroporated retinal explants were dissociated with 0.125% trypsin at 37°C for 10 min and triturated in 20% FBS diluted in Ca2+/Mg2+-free PBS. Dissociated cells were washed with cold 0.1% BSA dissolved in Ca2+/Mg2+-free PBS (hereafter, 0.1% BSA/PBS) twice, then resuspended in 1 ml of 0.1% BSA/PBS. One microliter of Viability Dye eFlour 780 (eBioscience) was added, and cells were kept on ice for 20 min. Cells were washed twice and resuspended in 500 μl of 0.1% BSA/PBS. Resuspended cells were filtered through a cell strainer (Falcon) to remove cell clumps. Cell suspensions were then sorted by FACS to sort out GFP+ cells. GFP+ cells were collected in TRIzol reagent (Thermo Fisher Scientific) for RNA extraction.
RNA extraction and qPCR.
Total RNA was extracted from sorted GFP+ cells using TRIzol reagent following the manufacturer's instructions. First-strand cDNA was synthesized from 25 ng of RNA using an RT2 First Strand kit (Qiagen, catalog #330401). Target gene mRNA levels were assessed by qPCR using RT2 SYBR Green Flour qPCR Mastermix (Qiagen, catalog #330500) and the CFX Connect Real-Time PCR Detection System (Bio-Rad). The relative change in Pax6 (RT2 qPCR primers; Qiagen, catalog #PPM04498B) expression was determined using CFX Manager (Bio-Rad), with values normalized to three housekeeping genes using the following RT2 qPCR primers (Qiagen): Gapdh (catalog #PPM02946E), B2m (catalog #PPM03562A), and Hrpt (catalog #PPM03559F). The ΔΔCt method was used to analyze qPCR data.
Measurements and statistical analysis.
Images were captured with a QImaging Retiga 2000R or QImaging Retiga EX digital camera and a Leica DMRXA2 optical microscope using OpenLab5 software (Improvision). All images for analysis were captured within 400 μm from the optic nerve, avoiding the centralmost retina, where Pten was not deleted, and the peripheralmost region, where the retina thins out and layering is altered. All analyses were performed on a minimum of three eyes per genotype or manipulation and a minimum of three photomicrographs per eye, which were used to count cell number per field. N values refer to the number of experimental repeats, whereas n values refer to the number of technical replicates within each experiment. Statistical significance for cell counts and Western blotting densitometry were calculated using two-way Student's t-tests (for two samples) or one-way ANOVA and post-hoc Tukey corrections (for multiple samples) using GraphPad Prism software, version 5.0. Error bars represent SEM.
Results
Generation of a conditional Pten loss-of-function allele in the retina
While previous reports suggested that Pten influenced final numbers of different retinal cell types at the end of the differentiation period, the results were conflicting. For example, while two studies reported a reduction in amacrine cell number in Pten cKO retinas (Cantrup et al., 2012; Jo et al., 2012), another reported an increase (Sakagami et al., 2012). As we did not quantitate all retinal cell types in our previous study (Cantrup et al., 2012), we set out to determine which cell types were most adversely affected by the loss of Pten in our model. Because Pten null mutations lead to early embryonic lethality (Di Cristofano et al., 1998), we conditionally deleted a floxed allele of Pten (Backman et al., 2001; Suzuki et al., 2001) using a Pax6::Cre retinal driver line (Marquardt et al., 2001) (Fig. 1A). To monitor Cre tissue specificity, we crossed Pax6::Cre driver mice with a Rosa26R-EGFP reporter line. In double heterozygous embryos harvested at P0, GFP expression was observed throughout the flat-mounted retina, except in a medial wedge that was wider on the dorsal side (Fig. 1B,B′), a spatial restriction that was consistent with previous reports (Marquardt et al., 2001). Accordingly, while Pten expression was detected throughout the mediolateral extent in sections of P0 wild-type retinas (Fig. 1C), in P0 Ptenfl/fl;Pax6::Cre (hereafter referred to as Pten cKO) sections, remaining Pten expression was confined to the central retina (Fig. 1D). We thus focused all further analyses on the ventral half of the retina but avoided the most medial domain, where Pten was not deleted. We also stayed away from the lateral edge, where the study by Jo et al. (2012) was focused, since the retina narrows and lamination differs in this domain.
Figure 1.
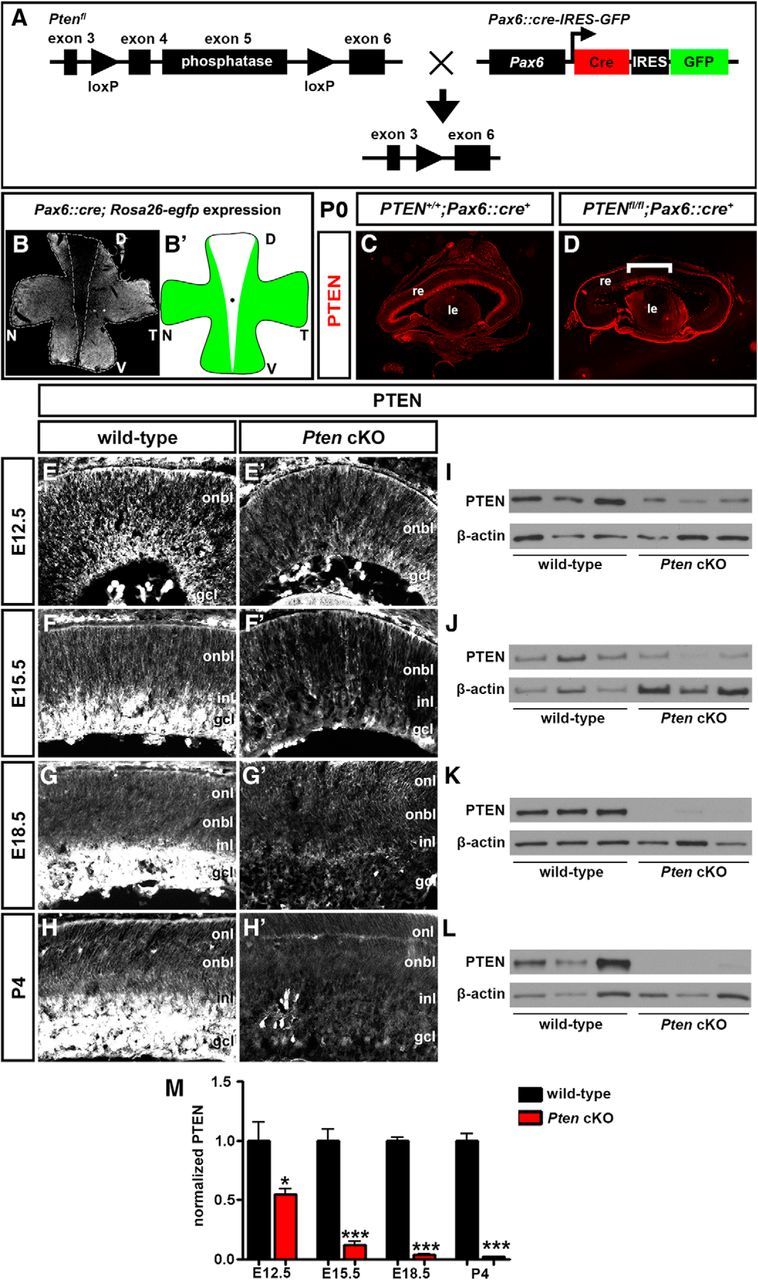
Generation of a retinal-specific Pten conditional mutation. A, Schematic illustration of crosses between transgenic animals carrying floxed Pten (Ptenfl) and Pax6α/P0::Cre-IRES-GFP transgenes. B, Flat-mount image of Pax6::Cre; Rosa26R-EGFP retina at P0. GFP is not expressed in the central retina. The dotted line outlines the domain where Cre was not active. B′, Schematic illustration of Cre activity domain. C, D, Expression of Pten in P7 wild-type (C) and Pten cKO (D) retinal transverse sections. The bracket in D shows central region where Pten is not deleted and expression is maintained. E–M, Western blot analysis and densitometry of Pten expression levels in wild-type and Pten cKO retinal lysates at E12.5 (E, E′, I), E15.5 (F, F′, J), E18.5 (G, G′, K), and P4 (H, H′, L). Pten levels were reduced at all stages analyzed (M). *p < 0.05; **p < 0.01; ***p < 0.001. D, Dorsal; le, lens; N; nasal, re, retina; T, temporal; V, ventral.
To determine in which retinal layers Pten was expressed, and when in retinal development expression was lost in Pten cKOs, we conducted a series of immunostaining experiments from E12.5 to P4. In wild-type retinal tissue at E12.5, Pten expression was detected at low levels in the outer neuroblast layer (ONBL), where proliferating RPCs reside, as well as at higher levels in the developing GCL (Fig. 1E). By E15.5 and until P4, Pten expression was detected at low levels in the ONBL and at higher levels in the developing GCL, INL, and the intervening inner plexiform layer (IPL; Fig. 1F–H). In Pten cKO retinas, a clear reduction in Pten expression levels was observed as early as E12.5 (Fig. 1E′), with an almost complete ablation at later developmental stages (E15.5–P4; Fig. 1F′,G′,H′). The reduction in Pten expression was confirmed by Western blots, with approximately half the normal Pten levels observed in Pten cKO retinas at E12.5 (1.83-fold decrease; N = 3, n = 9; p = 0.0488), and a progressive, almost complete loss of expression between E15.5 and P4 (8.07-fold decrease at E15.5, p = 0.0009; 26.1-fold decrease at E18.5, p < 0.0001; 46.3-fold decrease at P4, p < 0.0001; N = 3, n = 9; Fig. 1I–M).
Pten is thus expressed in both RPCs and differentiating cells in the GCL/INL in the embryonic/early postnatal retina, and this expression is rapidly lost in Pten cKOs starting as early as E12.5, except in the medialmost retina.
Biphasic effects on RPC proliferation and cellular differentiation in Pten cKO retinas
Pten is a tumor suppressor, while Akt1 is an oncogene, suggesting that Pten prevents, while Akt1 promotes, cellular proliferation (Sinor and Lillien, 2004; Cully et al., 2006). Indeed, the deletion of Pten and consequent activation of PI3K/Akt signaling in tumor cells accelerates the progression through G1 and into S-phase of the cell cycle (Ramaswamy et al., 1999). To test whether the deletion of Pten in the retina would similarly promote RPC proliferation, we quantitated the number of S-phase progenitors labeled with a 30 min BrdU pulse and the number of G2/M-phase progenitors expressing pHH3. Proliferation was assessed at E12.5, E15.5, E18.5, and P4, a protracted window of time when RPCs are still actively proliferating in the retina (Dyer and Cepko, 2001).
At E12.5 (1.45-fold increase; n = 3; p = 0.0004; Fig. 2C) and E15.5 (1.18-fold increase; n = 3; p = 0.0217; Fig. 2A,A′,C), total numbers of BrdU+ S-phase progenitors were elevated in Pten cKO retinas compared to wild-type retinas. In contrast, by E18.5, BrdU+ cells were detected at wild-type levels in Pten cKO retinas (n = 3; p = 0.1974; Fig. 2C), and by P4, the total number of S-phase cells began to decline (3.04-fold decrease; n = 3; p = 0.0006; Fig. 2B,C). A similar biphasic effect of Pten loss was observed when examining the mitotic marker pHH3, with more mitotic cells observed in Pten cKO retinas at E12.5 (1.56-fold increase; n = 3; p = 0.0106; Fig. 2F) and E15.5 (1.45-fold increase; n = 3; p = 0.0107; Fig. 2D,D′,F), followed by a normalization at E18.5 (n = 3; p = 0.507; Fig. 2F) and a decline by P4 (1.55-fold decrease; n = 3; p = 0.0280; Fig. 2E,F). Notably, the biphasic effect of Pten loss on cell proliferation was consistent with a previous report (Jo et al., 2012). Pten-deficient RPCs thus hyperproliferate during early retinal development, but the proliferative pool is depleted by the early postnatal period.
Figure 2.

Loss of Pten alters proliferation and retinal cell number. A–C, Immunolabeling of wild-type and Pten cKO retinas at E15.5 (A, A′) and P4 (B, B′) for BrdU (red). Blue is a DAPI counterstain. The graph (C) shows the number of BrdU+ cells in wild-type and Pten cKO retinas at E12.5, E15.5, E18.5, and P4. D–F, Immunolabeling of wild-type and Pten cKO retinas at E15.5 (D, D′) and P4 (E, E′) for pHH3 (red). The graph (F) shows the number of pHH3+ cells in wild-type and Pten cKOs at E12.5, E15.5, E18.5, and P4. G–L, DAPI staining of wild-type and Pten cKO retinas at E15.5 (G, G′), E18.5 (I, I′), and P7 (K, K′). The total number of cells in wild-type and Pten cKO retinas at three different stages are shown in the graphs (H, J, L). M–R, Immunolabeling of P7 wild-type and Pten cKO retinas for BrdU after administering at E12.5 (M, M′), E14.5 (O, O′), and E18.5 (Q, Q′). The graphs show the percentages of BrdU+ cells in each retinal layer (left) and the total number of BrdU+ cells (right; N, P, R). *p < 0.05; **p < 0.01; ***p < 0.001.
We next asked whether hyperproliferation of Pten cKO RPCs at early embryonic stages translated into an overall increase in retinal cell number. To test this possibility, we quantitated total DAPI-labeled nuclei of the retina at three different developmental stages (E15.5, E18.5, and P4), when the proliferative pool of RPCs is still expanding. Quantitation of DAPI+ nuclei at E15.5 showed no significance difference in Pten cKO retinas versus wild-type controls (n = 5; p = 0.6974; Fig. 2G,H). However, by E18.5, the number of total DAPI+ nuclei increased by 12% in Pten cKOs compared to wild-type controls (n = 5; p = 0.0162; Fig. 2I,J), probably because of elevated proliferation at earlier stages (E12.5 to E15.5; Fig. 2C). Finally, at P7, 39% fewer DAPI+ nuclei were present in Pten cKO retinas (n = 5; p = 0.0006; Fig. 2K,L). Notably, we do not attribute these differences in cell number to cell death, as the proportion of cleaved caspase 3+ apoptotic cells in wild-type and Pten cKO retinas was similar at E15.5, P0, and P7 (Cantrup et al., 2012).
Shifts in retinal cell birthdating in each cellular layer in the Pten cKO retina
We next asked whether the temporal sequence of cellular differentiation was shifted by the loss of Pten in RPCs. We conducted BrdU birthdating experiments at distinct days of gestation to mark newly differentiated retinal cells undergoing their final cell division. P7 retinas that were BrdU pulse labeled at E12.5 (Fig. 2M,M′), E14.5 (O,O′), or E18.5 (Q,Q′) in wild-type (M,O,Q) and Pten cKO (M′,O′,Q′) retinas were immunostained with BrdU and counterstained with DAPI. When BrdU was injected at E12.5 and labeled cells were quantitated at P7, there was an eightfold increase (n = 6; p < 0.0001; Fig. 2N) in BrdU+ cells in the ONL in the Pten cKO relative to wild-type retinas. Conversely, the INL contained 17% fewer (n = 6; p = 0.0008; Fig. 2N) BrdU+ cells in Pten cKO retinas relative to wild-type, while the GCL showed no significant difference in BrdU+ cells (n = 6; p = 0.2303; Fig. 2N). When BrdU was injected at E14.5 and immunostaining quantitated at P7, we observed an 8.6-fold increase (n = 6; p = 0.0673; Fig. 2P) in BrdU+ cells in the ONL in the Pten cKO retina relative to wild-type. Conversely, the INL contained 18% fewer (n = 6; p = 0.0002) BrdU+ cells in the Pten cKO retina relative to wild-type, while the GCL showed a 1.32-fold increase (n = 6; p = 0.0001; Fig. 2P). Finally, when BrdU was injected at E18.5 and immunostaining quantitated at P7, we found a 1.64-fold increase (n = 6; p = 0.0151; Fig. 2R) in BrdU+ cells in the ONL in the Pten cKO retina relative to wild-type. Conversely, the INL contained 11% fewer (n = 6; p = 0.0100; Fig. 2R) BrdU+ cells in Pten cKO retinas relative to wild-type, while the GCL showed no significant differences (n = 6; p = 0.9614; Fig. 2R). Notably, at each of the three developmental stages tested, no change in the total number of BrdU+ cells was detected, suggesting that it is the proportion of cells differentiating in each layer that is altered (n = 6; p = 0.3044 at E12.5, p = 0.5655 at E14.5, p = 0.0869 at E18.5; Fig. 2N,P,R, right). Collectively, these data show that the lack of Pten in RPCs alters the birthdates of cells destined for distinct retinal cell layers.
Pten is required to produce the full complement of retinal amacrine cells and rod photoreceptors
We speculated that the differences in RPC proliferation and birthdates in Pten cKO retinas might influence the final numbers of individual retinal cells that differentiated. To test this possibility, we quantified the number of cells expressing cell type-specific markers, focusing on P7 retinas, when differentiation is mostly complete except in peripheralmost domains, which we avoided. We first examined retinal cells whose differentiation windows peaked in the embryonic period. Brn3a+ RGCs (Pan et al., 2005; Nadal-Nicolas et al., 2009), which are the first retinal cells to differentiate, were produced in similar numbers in P7 wild-type and Pten cKO retinas (n = 3; p = 0.1188; Fig. 3A,B,M). The numbers of cone arrestin+ pedicles was also similar in P7 wild-type and Pten cKO retinas (n = 3; p = 0.4723; Fig. 3E,F,M). In contrast, 20% fewer Pax6-labeled amacrine cells (de Melo et al., 2003) were detected in the Pten cKO INL at P7 (n = 3; p = 0.0167; Fig. 3C,D,M). These data were consistent with our previous demonstration that there are fewer amacrine cells in Pten cKO retinas at P21, which we attributed to a decrease in the birthrate of these cells at all embryonic stages examined, including E12.5, E15.5, and E18.5 (Cantrup et al., 2012). Notably, we also demonstrated previously that horizontal cells were reduced in number in Pten cKO retinas (Cantrup et al., 2012), but because these cells are so sparse in number, cell counts could not be performed in sections (as in this study), but rather were performed on retinal flat mounts (Cantrup et al., 2012). Thus, of the retinal cells whose production peaks in the embryonic period, only horizontal cells and amacrine cells were reduced in number in Pten cKO retinas.
Figure 3.

Loss of Pten alters the generation of rod photoreceptors and amacrine cells. A–L, Immunolabeling of wild-type and Pten cKO retinas at P7 for Brn3a (A, B), Pax6 (C, D), cone arrestin (E, F), Chx10 (G, H), rhodopsin (I, J), and Sox9 (K, L). Blue is a DAPI counterstain. M, The graph shows the number of cell-type marker positive cells in wild-type and Pten cKO retinas. *p < 0.05; **p < 0.01. ACs, Amacrine cells; BCs, bipolar cells; PR, photoreceptor.
We next analyzed the differentiation of later-born retinal cell types. The numbers of Chx10+ bipolar cells (n = 3; p = 0.6608; Fig. 3G,H,M) and Sox9+ Müller glial cells (n = 3; p = 0.1461; Fig. 3K–M) were similar in P7 wild-type and Pten cKO retinas, although the positioning of Sox9+ cells was clearly disrupted. In contrast, DAPI+ nuclei were present in reduced numbers in the ONL of P7 Pten cKO retinas (1.21-fold decrease; n = 3; p = 0.0031; Fig. 3I,J,M). Notably, 97% of ONL nuclei were rod (as opposed to cone) photoreceptors (Carter-Dawson and LaVail, 1979; Volland et al., 2015), allowing us to conclude that changes in DAPI+ nuclei in the ONL reflect changes in rod number, in particular given that cone numbers were unchanged in Pten cKO retinas (Fig. 3E,F,M). Indeed, the rhodopsin+ rod photoreceptor layer was noticeably thinner in P7 Pten cKO retinas (Fig. 3I,J).
Collectively, we conclude that Pten regulates the production of early-born horizontal and amacrine cells, as well as later-born rod photoreceptors, but is dispensable for the production of early-born RGCs and cone photoreceptors and late-born bipolar cells and Müller glia. Pten thus plays a selective role in regulating cellular production during retinal neurogenesis, and its function is not restricted to a specific temporal window. To begin to understand mechanisms of action, we set out to determine how Pten specifically influences amacrine cell number.
Akt is activated in Pten cKO retinas and is sufficient to inhibit amacrine cell differentiation
PI3K phosphorylates and activates membrane inositol phospholipids by converting PIP2 (phosphatidylinositol-4,5-bisphosphate) into PIP3 (phosphatidylinositol-3,4,5-triphosphate), which serves as a lipid second messenger, activating downstream Akt kinases (Whitman et al., 1988; Engelman et al., 2006; Kriplani et al., 2015). Conversely, Pten removes the 3′-phosphate in PIP3 to block downstream signaling, including PI3K/Akt activation. To determine when in development Pten was functionally deleted, we assessed pAktSer473 levels, which serves as a readout of PI3K activation. At E12.5, pAktSer473 was detected at very low levels in the wild-type retina, but by E15.5, pAktSer473, expression was detected in the developing GCL, INL, and IPL, with only weak immunoreactivity in the ONBL, a pattern that was maintained at E18.5 and P4 (Fig. 4A–D). In Pten cKO retinas, a similar expression profile was observed, except that from E15.5 onward, pAktSer473 levels were strikingly upregulated in the GCL, INL, and IPL, while weak ONBL expression remained (Fig. 4B′,C′,D′). To confirm the increases in pAktSer473 levels in Pten cKOs, especially after E15.5, Western blotting was performed. While there was only a 1.34-fold increase in pAktSer473 levels in Pten cKOs at E12.5, levels were increased 3.17-fold, 9.46-fold, and 14.2-fold, respectively, at E15.5, E18.5, and P4 (N = 3, n = 9; p = 0.0054 at E12.5; p = 0.0004 at E15.5; p = 0.0001 at E18.5; p = 0.0051 at P4; Fig. 4E–I). Together, these data indicate that conditional deletion of Pten in the retina elevated pAktSer473 levels as early as E12.5.
Figure 4.
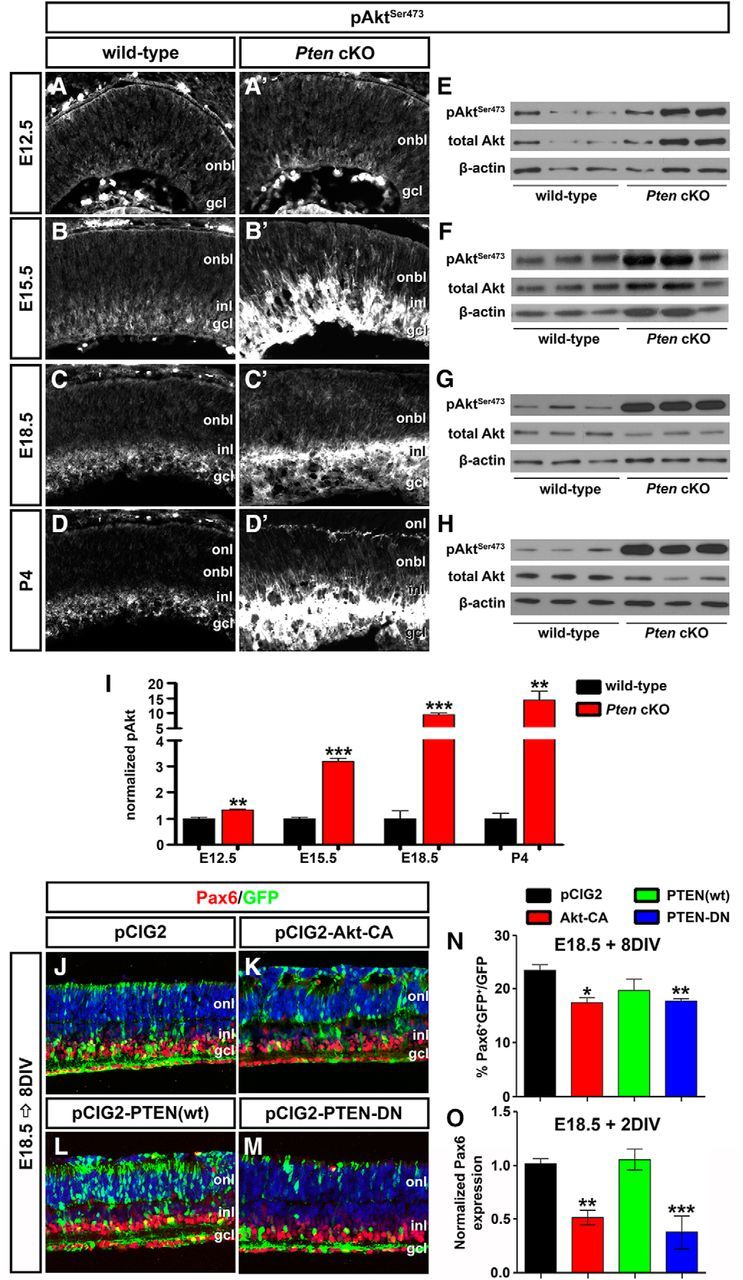
Hyperactivated Akt in Pten cKO retinas contributes to the decline in amacrine cell differentiation. A–D′, Immunolabeling of wild-type and Pten cKO retinas at E12.5 (A, A′), E15.5 (B, B′), E18.5 (C, C′), and P4 (D, D′) for pAktSer473. E–I, Western blot analysis and densitometry of pAktSer473 in wild-type and Pten cKO retinal lysates at E12.5 (E), E15.5 (F), E18.5 (G), and P4 (H). Levels of pAktSer473 are elevated at all stages analyzed when Pten is deleted (I). J–M, E18.5 retinas electroporated with pCIG2 control (J), Akt-CA (K), PTEN(wt) (L), or PTEN-DN (M) and cultured for 8 DIV. GFP+ (green) electroporated amacrine cells were identified by Pax6 immunolabeling (red). Blue is a DAPI counterstain. N, Percentages of GFP+ amacrine cells (GFP+Pax6+) after electroporation of pCIG2, Akt-CA, PTEN(wt), or PTEN-DN. O, qPCR to assess Pax6 transcript levels in GFP+ cells sorted by FACS after electroporation of pCIG2, Akt-CA, PTEN(wt), or PTEN-DN. *p < 0.05; **p < 0.01; ***p < 0.001.
We next asked whether activation of Akt in Pten cKO retinas contributed to the inhibition of amacrine cell production. To address this question, we used ex utero electroporation to overexpress Akt-CA in E18.5 retinal explants, comparing the effects on amacrine cell production to those observed when PTEN(wt) or dominant negative PTEN (PTEN-DN) was overexpressed. To visualize transfected cells, each gene was cloned into pCIG2. After culturing electroporated retinas for 8 DIV, the number of GFP+ cells that acquired an amacrine cell fate (Pax6+) was enumerated. Notably, while Pax6 is expressed in both amacrine cells and RGCs, RGCs rapidly die upon optical nerve transection, so the only remaining Pax6+ cells in retinal explants are amacrine cells. As expected, when PTEN-DN was overexpressed in E18.5 retinal explants, fewer amacrine cells were produced compared to pCIG2 controls (1.22-fold decrease; N = 3, n = 9; p = 0.0010; Fig. 4J,M,N), phenocopying the Pten cKO phenotype, whereas PTEN(wt) had no effect on amacrine cell production (N = 3, n = 9; p = 0.3892; Fig. 4J,L,N). Strikingly, Akt-CA had a similar ability to reduce amacrine cell production in E18.5 retinal explants (1.47-fold decrease; N = 3, n = 9; p = 0.0170; Fig. 4J,K,N). To confirm alterations in number of Pax6+ amacrine cells, electroporated cells were sorted by FACS, and qPCR was performed after 2 DIV to quantify Pax6 transcript levels. Both the overexpression of Akt-CA and PTEN-DN reduced Pax6 transcript levels compared to pCIG2 controls (Akt-CA, 1.96-fold decrease, N = 3, n = 9, p = 0.0060; PTEN-DN, 2.75-fold decrease, N = 3, n = 9, p = 0.0004; Fig. 4O). In contrast, consistent with the cell count data, PTEN(wt) did not alter Pax6 transcript levels (N = 3, n = 9; p = 0.7399; Fig. 4O). Notably, while Pax6 is also expressed in retinal progenitors at P1 (the equivalent of E18.5 explants cultured 2 DIV), it is expressed at significantly lower levels in progenitors versus amacrine cells in the early postnatal retina (Dixit et al., 2014). Together with the enumeration of Pax6+ amacrine cells, we thus conclude that Pten controls amacrine cell number at least in part by regulating Akt activation, which must be kept at low levels to allow amacrine cell differentiation to ensue.
Tgfβ signaling is activated by Akt and acts cell autonomously to block amacrine cell differentiation
While our data suggested that activated Akt could influence amacrine cell production, the underlying mechanisms were not known. It was shown previously that amacrine cell number is controlled by TgfβII-mediated negative feedback signaling (Ma et al., 2007), suggesting that Akt could influence this pathway. To assess interactions between TgfβII and Akt signaling, we first asked whether the manipulation of TgfβII signaling via ex utero electroporation influenced amacrine cell production. TgfβII binds to two related dual specificity transmembrane kinase receptors (TgfβRI and TgfβRII; Akhurst and Padgett, 2015). To manipulate this pathway, TgfβRII-CA and TgfβRII-DN expression vectors were electroporated into E18.5 retinas, at a time when amacrine cell genesis begins to decline as feedback signaling becomes operational. As expected, misexpression of activated TgfβRII-CA reduced the production of Pax6+ amacrine cells after 8 DIV (1.83-fold decrease; N = 3, n = 9; p = 0.0084; Fig. 5A,B,D), whereas TgfβRII-DN enhanced amacrine cell differentiation (1.29-fold increase; N = 3, n = 9; p = 0.0171; Fig. 5A,C,D). These data were consistent with the increased production of amacrine cells observed in TgfβRII-cKOs (Ma et al., 2007). Further validating these findings, FACS of GFP+ electroporated cells 2 d after electroporation followed by qPCR analysis confirmed that Pax6 mRNA levels were indeed reduced by TgfβRII-CA (2.63-fold decrease; n = 3; p = 0.0082) and increased by TgfβRII-DN (1.43-fold increase; n = 3; p = 0.0467) overexpression (Fig. 5E).
Figure 5.

Tgfβ signaling is promoted by Akt and acts cell autonomously to block amacrine cell differentiation. A–C, E18.5 retinas electroporated with pCIG2 control (A), TgfβRII-CA (B) or TgfβRII-DN (C) and cultured for 8 DIV. GFP+ (green) electroporated amacrine cells were identified by Pax6 immunolabeling (red). Blue is a DAPI counterstain. D, Percentages of GFP+ amacrine cells (GFP+Pax6+) after electroporation of pCIG2, TgfβRII-CA, or TgfβRII-DN. E, qPCR to assess Pax6 transcript levels in GFP+ cells sorted by FACS after electroporation of pCIG2, TgfβRII-CA, or TgfβRII-DN. F, GFP+ HEK cells 24 h after transfection. G–I, Western blot analysis and densitometry of pSmad2 after transfection of TgfβRII-CA (G), TgfβRII-DN (H), and Akt-CA (I). J, Schematic model of how amacrine cell differentiation is regulated by both Tgfβ and Pten signaling pathways. Note that pAkt could regulate pSmad2 via directly influencing phosphorylation of Smad2 or the upstream receptor TgfβRII. *p < 0.05; **p < 0.01; ***p < 0.001.
To confirm that TgfβII signaling was altered in this assay, we examined the phosphorylation status of Smad2/3, which is phosphorylated upon TgfβII ligand binding to TgfβRI/RII (Akhurst and Padgett, 2015). For that we transfected HEK cells with TgfβRII-CA or TgfβRII-DN (Fig. 5F), followed by Western blot. As expected, 24 h after overexpression of TgfβRII-CA in HEK cells, levels of pSmad2Ser465/467 were elevated (2.43-fold increase; N = 3, n = 9; p = 0.0004; Fig. 5G), whereas TgfβRII-DN reduced Smad2 phosphorylation (2.7-fold decrease; N = 3, n = 9; p = 0.0006; Fig. 5H). Finally, we asked whether Akt signaling could influence TgfβII signaling. Strikingly, overexpression of Akt-CA in HEK cells led to a significant increase in pSmad2Ser465/467 levels after 24 h (4.95-fold increase; N = 3, n = 9; p = 0.0004 Fig. 5I).
Together, these data suggest that Akt may influence amacrine cell production by enhancing the TgfβII negative feedback response (Fig. 5J), a working model that we further validate below.
Pten acts downstream of Tgfβ signaling to control amacrine cell number
To further examine the potential link between activated Akt and Tgfβ/Smad signaling, we examined Pten cKO retinas, in which Akt signaling is hyperactivated. Specifically, we asked whether Tgfβ signaling was enhanced in Pten cKO retinas by performing Western blot analyses of Tgfβ signaling pathway components, including the ligand (TgfβII), receptor (TgfβRII), and downstream effector (pSmad2Ser465/467). In E18.5 retinas, at a stage when feedback signaling is active, TgfβII ligand levels were unchanged in Pten cKO retinas (N = 3, n = 10; p = 0.6148; Fig. 6A,B). In contrast, there were significant increases in TgfβRII (2.25-fold increase; N = 3, n = 10; p = 0.0061; Fig. 6A,C), pSmad2 (1.75-fold increase; N = 3, n = 10; p = 0.0017; Fig. 6A,D) and Smad2 (1.16-fold increase; N = 3, n = 10; p = 0.0017; Fig. 6A,E) protein levels in Pten cKO retinas, suggesting that in the absence of Pten, Tgfβ signaling is elevated.
Figure 6.

Pten acts downstream of Tgfβ signaling to control amacrine cell number. A–E, Western blot analysis and densitometry of TgfβII (B), TgfβRII (C), pSmad2 (D), and Smad2 (E) in wild-type and Pten cKO retinal lysates at E18.5. TgfβRII, pSmad2, and Smad2 were significantly increased in Pten cKO retinas. K–N, E18.5 retinas electroporated with pCIG2 control (K), TgfβRII-DN (L), PTEN-DN (M), or in combination (TgfβRII-DN + PTEN-DN; N) and cultured for 8 DIV. GFP+ (green) electroporated amacrine cells were identified by Pax6 immunolabeling (red). Blue is a DAPI counterstain. O, Percentages of GFP+ amacrine cells (GFP+Pax6+) after electroporation of pCIG2, TgfβRII-DN, PTEN-DN or a combination. P, Schematic pathway of how the Tgfβ signaling pathway and Pten/Akt signaling pathway simultaneously regulate amacrine cell differentiation. *p < 0.05; **p < 0.01; ***p < 0.001.
Next, to test TgfβII-Pten epistasis, we used ex utero electroporation to inhibit both TgfβRII and Pten functions at the same time. In double knock-downs, the downstream phenotype should prevail, allowing us to determine upstream/downstream relationships (i.e., fewer amacrine cells if Pten is downstream of TgfβRII, or more if vice versa). Consistent with previous data (Figs. 4N, 5D), misexpression of TgfβRII-DN increased (1.55-fold increase; N = 3, n = 9; p = 0.0002; Fig. 6K,L,O) while PTEN-DN decreased amacrine cell numbers (1.29-fold decrease; N = 3, n = 9; p = 0.0246; Fig. 6K,M,O) 8 d after electroporation at E18.5. However, when the two constructs were coelectroporated in E18.5 retinas, the ability of TgfβRII-DN to enhance amacrine cell production was suppressed by Pten (N = 3, n = 9; p = 0.4649; Fig. 6K,N,O).
Together with the enhanced Smad2/3 phosphorylation observed in Pten cKO retinas, these data strongly suggest that Pten is required downstream of TgfβII to limit responsiveness of RPCs to this negative feedback signal (Fig. 6P).
Pten acts in RPCs to control responsiveness to amacrine cell negative feedback signals
Our data suggested that the Tgfβ negative feedback signaling pathway that normally limits amacrine cell production later in development was overactive in Pten cKO retinas. To test whether Pten was an essential component of this negative feedback loop, we performed heterochronic aggregation assays, mixing early stage (E15.5) BrdU-labeled RPCs (peak amacrine cell production) with a 20-fold excess of late-stage (P2) retinal cells (amacrine cell feedback signal source; Fig. 7A). This assay measures the ability of factors secreted from mature amacrine cells to inhibit amacrine cell fate choice in younger RPCs and tests the site of Pten function by combining wild-type and mutant cells (Ma et al., 2007). The latter feature is important since Pten is expressed in both RPCs and amacrine cells, and it may function in either cell type to control amacrine cell number.
Figure 7.

Pten acts in RPCs to control responsiveness to amacrine cell negative feedback signals. A, Schematic illustration of aggregation assay protocol. RPCs are shown in green and amacrine cells in red. B–G, The combinations of aggregate conditions are shown in B, D, and F. Immunolabeling of aggregated cell pellets with Pax6 (green) and BrdU (red) is shown with DAPI counterstain (blue). E15.5 RPCs were cultured alone (B, C) or with P2 wild-type (D, E) or Pten cKO (F, G) retinal cells. Arrowheads mark Pax6/BrdU double+ nuclei (C, E, G). H, Percentage of BrdU+ E15.5 cells differentiated into Pax6+ amacrine cells when cultured alone (black bar) or with P2 wild-type (white bar) or Pten cKO (red bar) retinal cells. I–P, Immunolabeling of aggregated cell pellets with Pax6 (green) and BrdU (red) with DAPI counterstain (blue). E15.5 RPCs were cultured alone (I, J) or with P2 wild-type (K, L), and E15.5 Pten cKO RPCs were cultured alone (M, N) or with P2 wild-type (O, P) retinal cells. Arrowheads mark Pax6/BrdU double+ nuclei (J, L, N, P). Q, Percentage of BrdU+ E15.5 cells differentiated into Pax6+ amacrine cells when cultured alone (wild-type, black bar; Pten cKO, white bar) or with P2 wild-type (blue bar, light blue bar) retinal cells. *p < 0.05; **p < 0.01; ***p < 0.001.
To first test Pten function in amacrine cells, we generated aggregates of E15.5 wild-type BrdU-labeled RPCs alone (control) or E15.5 wild-type BrdU-labeled RPCs mixed with a 20-fold excess of P2 wild-type or Pten cKO RPCs. After 8 DIV, the number of Pax6+ amacrine cells that arose from BrdU-labeled RPCs was quantified. In E15.5 RPC pellet cultures, 39.0% of BrdU+ RPCs became Pax6+BrdU+ amacrine cells (N = 3, n = 9; Fig. 7B,C,H), consistent with previous results (Ma et al., 2007). In contrast, amacrine cell differentiation from the E15.5-labeled cohort was reduced 1.2-fold in cocultures with wild-type P2 retinal cells (N = 3, n = 9; p = 0.0218; Fig. 7D,E,H), and a similar 1.25-fold reduction was observed in P2 Pten cKO cocultures (n = 3; p = 0.0059; Fig. 7F–H). Thus, P2 retinal cells emit negative feedback signals that curtail further amacrine cell production, and these feedback signals are released independently of Pten.
To next test Pten function in RPCs, we performed similar heterochronic aggregation assays, but this time cultured E15.5 wild-type or Pten cKO BrdU-labeled RPCs with a 20-fold excess of wild-type P2 retinal cells. When wild-type E15.5 BrdU-labeled RPCs were cultured alone, 40.1% of BrdU+ RPCs became BrdU+Pax6+ amacrine cells (N = 3, n = 9; Fig. 7I,J,Q), consistent with the previous experiment (Fig. 7B,C,H). Similarly, 37.2% of Pten cKO BrdU-labeled RPCs differentiated into BrdU+Pax6+ amacrine cells (N = 3, n = 9; Fig. 7M,N,Q). While these data suggest that the loss of Pten does not affect the intrinsic ability of RPCs to differentiate into amacrine cells, it does not assess feedback responsiveness. Indeed, when comparing the two coculturing conditions (i.e., E15.5 wild-type RPCs plus P2 wild-type retinal cells vs E15.5 Pten cKO RPCs plus P2 wild-type retinal cells; Fig. 7K,L,O,P,Q), there was a further 1.2-fold reduction in amacrine cell production when Pten cKO RPCs were exposed to a source of amacrine cell feedback signals (N = 3, n = 9; p = 0.0062; Fig. 7Q), suggesting that mutant RPCs are hypersensitive to feedback signaling.
Pten is thus required in RPCs to curtail responsiveness to feedback signals. Together, our data support a model in which Pten acts in RPCs to limit responsiveness to TgfβII-mediated amacrine cell negative feedback signaling.
Erk signaling declines in Pten cKO retinas and is required to promote amacrine cell differentiation
PI3K/Akt signaling can be activated by a variety of extracellular stimuli that also target the Raf/Mek/Erk signaling pathway (Myers et al., 1994; Foncea et al., 1997). Importantly, these two signaling pathways can also cross talk, with active Akt (pAktThr308/Ser473) inhibiting Raf by phosphorylating Ser-259 (Zimmermann and Moelling, 1999; Moelling et al., 2002; Steelman et al., 2011). Thus, we posited that Pten might also control the Raf/Mek/Erk signaling pathway to regulate amacrine cell genesis. To assess whether Raf/Mek/Erk signaling was altered in E18.5 Pten cKO retinas, we performed Western blotting with anti-pErk (phospho-p44/42 MAPKThr202/Tyr204; hereafter, pErk), revealing a 3.75-fold reduction in pErk levels in mutant retinas (N = 3, n = 9; p = 0.0001; Fig. 8A,B). To next test whether PI3K/Akt signaling influenced activity of the Raf/Mek/Erk pathway, Akt-CA was transfected into HEK cells, followed 24 h later by pErk Western blotting. Misexpression of activated Akt-CA led to a 1.93-fold reduction in pErk levels (N = 3, n = 9; p = 0.0094; Fig. 8C,D). Upregulation of PI3K/Akt signaling thus reduces Raf/Mek/Erk activity levels, consistent with the observations made in Pten cKOs.
Figure 8.
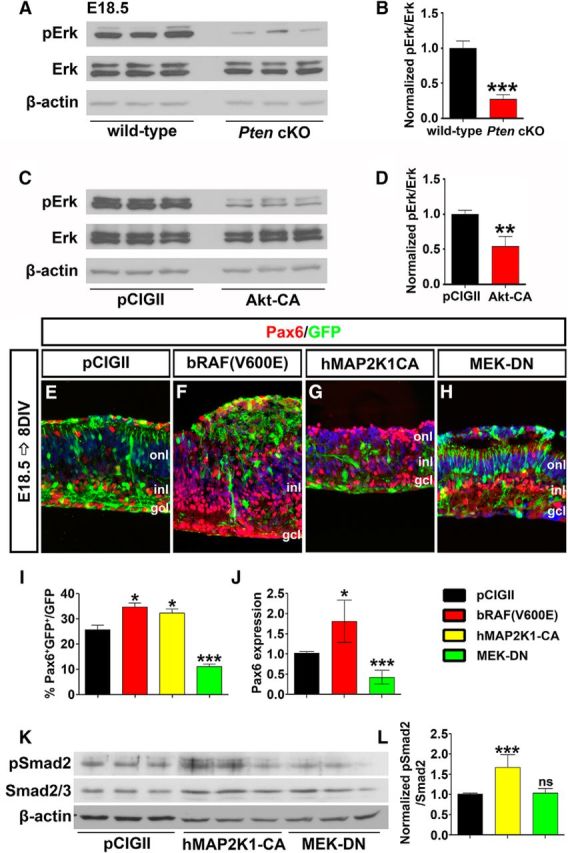
Pten regulates amacrine cell differentiation via Raf/Mek/Erk signaling. A, B, Western blot analysis (A) and densitometry (B) of pErk in E18.5 wild-type and Pten cKO retinal lysates. C, D, Western blot analysis (C) and densitometry (D) of pErk in Akt-CA transfected HEK cells. Loss of Pten or constitutive activation of Akt reduces pErk levels. E–H, E18.5 retinas electroporated with pCIG2 control (E), bRAF(V600E) (F), hMAP2K1-CA (G), or MEK-DN (H) and cultured for 8 DIV. GFP+ (green) electroporated amacrine cells were identified by Pax6 immunolabeling (red). Blue is a DAPI counterstain. I, Percentages of GFP+ amacrine cells (GFP+Pax6+) after electroporation. J, qPCR to assess Pax6 transcript levels in GFP+ cells sorted by FACS 2 d after electroporation of pCIG2, bRAF(V600E), or MEK-DN. K, L, Western blot analysis (K) and densitometry (L) for pSmad2 24 h after transfection of pCIG2, hMAP2K1-CA, or MEK-DN. *p < 0.05; **p < 0.01; ***p < 0.001.
We next tested whether activation of Raf/Mek/Erk signaling could influence amacrine cell production by electoporating constitutively active Raf [bRAF(V600E)] and Mek (hMAP2K1-CA) or MEK-DN into E18.5 retinal explants (Fig. 8E–I). After 8 DIV, bRAF(V600E) and hMAP2K1-CA induced 1.35-fold (N = 3, n = 9; p = 0.0159; Fig. 8E,F,I) and 1.26-fold (N = 3, n = 9; p = 0.0344; Fig. 8E,G,I) increases in Pax6+GFP+ amacrine cells, respectively, whereas the numbers of amacrine cells produced were decreased 2.32-fold by MEK-DN (N = 3, n = 9; p = 0.0009; Fig. 8E,H,I). The changes in Pax6 expression were also confirmed by qPCR of E18.5 electroporated retinal explants after 2 DIV, with a 1.75-fold decrease in Pax6 transcript levels following activation of the pathway with bRAF(V600E) (n = 3; p = 0.0173; Fig. 8J), and a 2.61-fold increase in Pax6 transcript levels following the electroporation of MEK-DN (n = 3; p = 0.0002; Fig. 8J). Activation of Erk signaling thus promotes amacrine cell production in the retina.
Finally, we wanted to determine whether Erk signaling influenced amacrine cell production via modulating TgfβII signaling. Transfection of MEK-CA into HEK cells led to a 1.98-fold increase in pSmad2 levels (N = 6, n = 18; p = 0.0006; Fig. 8K,L). In contrast, MEK-DN did not alter phosphorylation of Smad2 (N = 6, n = 18; p = 0.7781; Fig. 8K,L). The Raf/Mek/Erk signaling is thus sufficient but not necessary to influence TgfβII negative feedback signaling.
Pten controls the differentiation of all amacrine cell subtypes via the Erk signaling pathway
Amacrine cells can be classified based on their neurotransmitter phenotypes, with the major groups including glycinergic (∼35%), GABAergic (∼40%), and nonglycinergic/non-GABAergic (nGnG; ∼25%; Kay et al., 2011; Zhang and McCall, 2012). To determine whether the loss of Pten influenced the generation of a particular class of amacrine cells, we immunolabeled P7 wild-type and Pten cKO retinas with different amacrine cell subtype markers. Similar to our observations with Pax6, the numbers of cells expressing the pan-amacrine cell markers AP2α (Jin et al., 2015) and Barhl2 (Mo et al., 2004) were reduced in Pten cKO retinas (AP2α+, 1.24-fold decrease, n = 3, p = 0.0071; Barhl2+, 1.35-fold decrease, n = 3, p = 0.0008; Fig. 9A–D). Of note, Barhl2 was also ectopically expressed in the upper INL in Pten cKO retinas (Fig. 9C′), where bipolar cells and Müller glia primarily reside, so colabeling was performed with Pax6 to identify amacrine cells. This reduction in total amacrine cell number influenced all amacrine cell subclasses, as there were reductions in the numbers of GABAergic amacrine cells expressing GAD65+ (Feng et al., 2006; 1.15-fold decrease; n = 3; p = 0.0104; Fig. 9E,E′,F), glycinergic amacrine cells expressing GlyT1+ (Pow and Hendrickson, 1999; 1.46-fold decrease; n = 4; p = 0.0386; Fig. 9G,G′,H), and nGnG amacrine cells expressing Neurod6+ (Kay et al., 2011; 1.40-fold decrease; n = 3; p = 0.0033; Fig. 9I,I′,J) in Pten cKO retinas.
Figure 9.
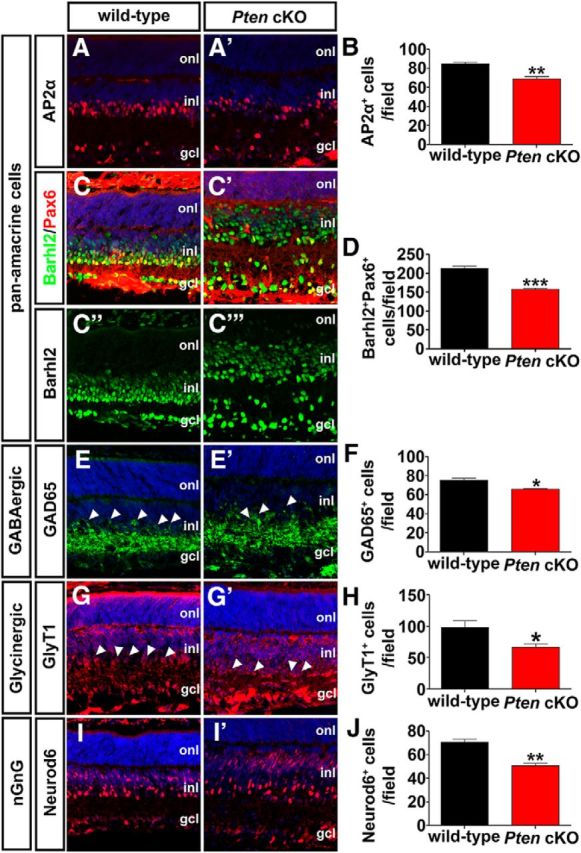
A–J, Pten regulates differentiation of all amacrine cell subtypes. Immunolabeling of P7 wild-type and Pten cKO retinas with antibodies against AP2α (A, A′), Pax6 (C–C‴, green), Barhl2 (C–C‴, red), GAD65 (E, E′), GlyT1 (G, G′), and Neurod6 (I, I′). Barhl2 and Pax6 are colabeled, as ectopic Barhl2 expression is seen in Pten cKO retinas (C′). Blue is a DAPI counterstain. Arrowheads in E,E′,G,G′ mark amacrine cell bodies in the INL. The graphs show the number of AP2α+ (B), Barhl2+Pax6+ (D), GAD65+ (F), GlyT1+ (H), and Neurod6+ (J) cells per field in P7 wild-type and Pten cKO retinas. *p < 0.05; **p < 0.01; ***p < 0.001.
Finally, we used our electroporation assay to determine whether the Erk signaling pathway influenced the production of a particular amacrine cell subtype. Analysis of the pan-amacrine cell marker Barhl2 revealed that activation of the Erk pathway by electroporation of bRAF(V600E) into E18.5 retinal explants increased the number of Barhl2+ amacrine cells after 8 DIV (3.07-fold increase; p = 0.0054; N = 3, n = 9; Fig. 10A–B″,M), whereas MEK-DN reduced amacrine cell production (1.33-fold decrease; p = 0.0246; N = 3, n = 9; Fig. 10A–A″,C–C″,M). These results are similar to those of our analyses of expression of Pax6, which is also a pan-amacrine cell marker (Fig. 8I). Similarly, electroporation of bRAF(V600E) into E18.5 retinal explants increased the production of GABAergic (GAD65+; 3.53-fold increase; N = 3, n = 9; p = 0.0072; Fig. 10D–E″,N), glycinergic (GlyT1+; 1.59-fold increase; N = 3, n = 9; p = 0.0017; Fig. 10G–H″,O), and nGnG (Neurod6+; 1.75-fold increase; N = 3, n = 9; p = 0.0020; Fig. 10J–K″,P) amacrine cells after 8 DIV. Conversely, electroporation of MEK-DN decreased the production of GAD65+ (1.90-fold decrease; N = 3, n = 9; p = 0.0474; Fig. 10D–D″,F–F″,N), GlyT1+ (1.83-fold decrease; N = 3, n = 9; p = 0.0058; Fig. 10G–G″,I–I″,O), and Neurod6+ (2.21-fold increase; N = 3, n = 9; p = 0.0034; Fig. 10J–J″,L–L″,P) amacrine cells.
Figure 10.

Pten regulates amacrine cell differentiation via Erk. A–L″, E18.5 retinas electroporated with pCIG2 control (A–A″, D–D″, G–G″, J–J″), bRAF(V600E) (B–B″, E–E″, H–H″, K–K″), or MEK-DN (C–C″, F–F″, I–I″, L–L″) and cultured for 8 DIV. GFP+ (green) electroporated cells were immunolabled for Barhl2 (A–C″), GAD65 (D–F″), GlyT1 (G–I″), and Neurod6 (J–L″). Arrowheads mark GFP+marker+ double positive cells. M–P, The number of amacrine cells increases when Raf is constitutively active, whereas it decreases when Mek activity is downregulated. *p < 0.05; **p < 0.01.
Pten and the downstream Erk pathway are thus both necessary and sufficient to influence the differentiation of all amacrine cell subtypes.
Discussion
Amacrine cell differentiation is regulated by intrinsic factors that also specify subtype identities, conferring morphological and neurotransmitter phenotypes onto postmitotic cells. However, the molecular mechanisms that ensure that appropriate numbers of the correct types of amacrine cells are generated are poorly understood. Here, we reveal that Pten, an intracellular phosphatase that functions downstream of several extracellular signals, is a critical regulator of amacrine cell number control, impacting differentiation through several downstream pathways (Fig. 11). First, we show that in the retina, as in other tissues, Pten functions as a negative regulator of PI3K signaling, and we demonstrate that elevated activity of the downstream effector Akt in the absence of Pten negatively impacts amacrine cell production by enhancing RPC responsiveness to TgfβII negative feedback signals. Specifically, Pten normally limits Smad2 phosphorylation, such that in the absence of Pten, Smad2 is hyperphosphorylated and amacrine cell production is inhibited. In addition, Pten is a positive regulator of Raf/Mek/Erk signaling, which promotes amacrine cell differentiation. Thus, in Pten cKO retinas, pErk levels are reduced, and glycinergic, GABAergic, and nGnG amacrine cells all decline in number. We have thus provided new molecular insights into how appropriate numbers of amacrine cells arise from multipotent RPCs.
Figure 11.

Pten controls amacrine cell production via multiple downstream pathways. Upon TgfβII binding to the receptor on the RPC plasma membrane, Smad2 is activated through phosphorylation. Elevation in pSmad2 levels within RPCs inhibits the acquisition of an amacrine cell fate, and thus amacrine cell production is inhibited. Pten/Akt signaling also leads to elevated Smad2 phosphorylation. Finally, Pten signaling also controls amacrine cell differentiation via Raf/Mek/Erk signaling. Upon activation of Erk, RPCs favor an amacrine cell fate, which will differentiate into all subtypes. Akt signaling blocks this event by inhibition of Erk.
Pten differentially regulates RPC proliferation in embryonic and early postnatal development
Pten is a tumor suppressor gene in neural and nonneural lineages, such that a reduction in activity or loss of Pten expression leads to uncontrolled cell division (Cully et al., 2006; Knobbe et al., 2008; Alimonti, 2010). We found that Pten is a critical regulator of RPC proliferation as more RPCs cycle in Pten cKO retinas in early embryogenesis (E12.5 and E15.5), culminating in an increase in retinal cell number by P0. This trend reverses by E18.5, when proliferation normalizes in Pten cKOs, followed by a sharp decline in RPC division by P4. As a consequence, fewer retinal cells are detected in Pten cKOs by P7. Such a biphasic effect on proliferation in Pten cKO retinas was also observed by Jo et al. (2012); however, their examination of peripheral retina suggested that there was an overall increase in cell number at P8. Jo et al. (2012) suggested that early on, RPCs hyperproliferate in Pten cKOs, followed by premature differentiation, which leads to a hypercellular retina and a subsequent depletion of the progenitor pool. Furthermore, they likened their observations to a similar requirement for Pten in hematopoietic stem cell maintenance, with overproduction of hematopoietic cells observed in Pten cKOs (Yilmaz et al., 2006). While we did not observe a similar increase in retinal cells in Pten cKOs at P7, cell number was increased at P0. Moreover, our birthdating studies suggested that there are defects in temporal identity transitions in Pten cKO retinas. Indeed, we have evidence for precocious neurogenesis in Pten cKO retinas, as more ONL cells (i.e., 97% rod photoreceptors) differentiated at E12.5, E14.5, and E18.5, even though rod photoreceptors are normally one of the latest born cell types in the retina. However, at the same time, fewer retinal cells were generated in the INL at these three stages, a reduction that we attributed to the underproduction of amacrine cells (Cantrup et al., 2012). The normal timing of retinal cell differentiation is thus perturbed by Pten loss.
Pten and retinal cell number control
In addition to our work (Cantrup et al., 2012; current study), two additional groups assessed the role of Pten in controlling the differentiation of individual retinal cell types, but in several instances the results were conflicting. With regard to amacrine cells, we found that fewer Pax6+ amacrine cells were produced in Pten cKO retinas (Cantrup et al., 2012; current study), as did Jo et al. (2012), albeit to a lesser extent. Importantly, while Jo et al. (2012) used the same Cre driver (Pax6-α-Cre) and amacrine cell marker (Pax6), they focused on lateralmost domains, where retina width tapers off and cell numbers are generally lower. In contrast, using a different Cre driver (Chx10-Cre), Sakagami et al. (2012) found a small 4.2% increase in amacrine cell number at P10. One difference is that the Chx10-Cre driver may be active earlier than the Pax6-α-Cre transgene, as GFP expression initiates at E11 in Chx10-Cre-GFP transgenic animals (Sakagami et al., 2012), whereas Pax6-α-Cre only starts to be active at ∼E12.5, when we see an ∼50% reduction in Pten levels in Pten cKO retinas (current study). Consistent with earlier Chx10-Cre activity, there was a 45.5% decrease in RGCs, which are the earliest-born cells in the retina, in Pten cKO animals generated with this Cre driver (Sakagami et al., 2012). In contrast, we (current) and Jo et al. (2012) did not observe a significant decline in RGCs at early postnatal stages (P7 and P8), possibly because Pax6-Cre activity is initiated after RGC differentiation has already begun. However, Jo et al. (2012) did observe more RGCs in E12.5 retinas, suggesting that differentiation is accelerated in the absence of Pten, which we also have evidence for (see Figure 2M–R). It is more difficult to reconcile our data with those from the study by Sakagami et al. (2012) based on marker analysis, since we found reduced numbers of AP2α+ amacrine cells in Pten cKO retinas. AP2α is expressed in a large number but not all amacrine cells (Bassett et al., 2007), suggesting that Cre-driver activity most likely accounts for differences in our studies.
The two major amacrine cell subtypes are glycinergic and GABAergic (Balasubramanian and Gan, 2014). Recent studies have revealed that these amacrine cell subtypes are born in a defined order, with GABAergic neurons born first, followed by glycinergic amacrine cells (Voinescu et al., 2009). Our data indicate that Pten is required for the genesis of both amacrine cell subtypes, even though negative feedback signaling is only active during the genesis of late-born amacrine cells (Ma et al., 2007). Accordingly, in our prior birthdating studies, amacrine cell production was reduced at E12.5, E14.5, and E18.5 (Cantrup et al., 2012). These data suggest that Pten may function through pathways in addition to TgfβII to regulate amacrine cell production. Indeed, we show here that activated Erk, which is reduced in Pten cKO retinas, is required and sufficient to influence the production of glycinergic, GABAergic, and nGnG amacrine cells, providing another mechanism by which Pten specifies an amacrine cell fate.
While we did not perform an in-depth analysis of other retinal cell types, we did observe a 1.2-fold decrease in rod photoreceptor number. This result more closely resembles the report by Sakagami et al. (2012), in which there was a 36.4% decrease in photoreceptor number, whereas Jo et al. (2012) reported a 27% increase. Again, a notable difference between our study and the study by Jo et al. (2012) is that they focused on the lateralmost retina, where tapering of the cell layers suggests that cell number control is differentially regulated in this domain. Notably, while amacrine cell differentiation peaks in the embryonic period, rod photoreceptor differentiation is at its highest in the early postnatal period, suggesting that Pten function is not restricted to a specific developmental window. Further studies will be required to assess how Pten controls rod development.
Pten and Tgfβ negative feedback signaling
We found that one of the ways in which Pten controls retinal amacrine cell number is by limiting RPC responsiveness to TgfβII negative feedback signaling. Specifically, we found that activated Akt, which is elevated in Pten cKO retinas, increases Smad2 phosphorylation, which is a readout of active TgfβII signaling. Our aggregation assays provided further support for a role for Pten in limiting the negative feedback response by RPCs. What remains unclear is how amacrine cell feedback signals interact with fate determinants. For example, in skeletal muscle, myostatin (Gdf8) is a Tgfβ family member that controls skeletal muscle mass through negative feedback signaling. Upon activation, myostatin induces Smad2/3 phosphorylation, which directly inhibits MyoD and Myogenin activity, which are bHLH transcription factors that specify a skeletal muscle identity (Langley et al., 2002). This raises the possibility that within the amacrine cell lineage, transcription factors might be directly regulated by TgfβII and/or PI3K/Pten signaling. There are several candidate amacrine cell fate determinants that may be directly impacted by these signaling pathways, including Foxn4 (Li et al., 2004), Rorβ1 (Liu et al., 2013), and Ptf1a (Nakhai et al., 2007), the loss of all of which leads to a sharp decline in amacrine cell production. We showed previously that another tumor suppressor gene, Zac1, which encodes a zinc finger transcription factor, also selectively regulates final numbers of amacrine cells, but rather than functioning in RPCs, Zac1 controls TgfβII secretion by postmitotic amacrine cells (Ma et al., 2007). Further dissection of the amacrine cell regulatory pathways are thus likely to lead to the identification of regulators of amacrine cell number that function both within RPCs and amacrine cells.
In summary, our study builds on the prior identification of key regulators of amacrine cell development, filling in gaps in our understanding of how the retina acquires a predefined size and cell number.
Footnotes
This work was supported by the March of Dimes (C.S.); Canadian Institutes of Health Research (CIHR) Grant 89994 (C.S.); the Lion's Sight Centre (C.S.); a CIHR/Alberta Children's Hospital Research Institute Training Grant in Genetics, Child Health and Development (N.T., R.C.); a Foundation Fighting Blindness studentship (R.C.); and a CIHR Canada Hope Fellowship (R.D.). We thank Natasha Klenin, Yoon Su Mee, and Eko Raharjo for technical assistance and Pamela Hoodless, Britta Eickholt, William Hahn, Franck Polleux, Jonathan Ashwell, and Christopher Marshall for reagents.
The authors declare no competing financial interests.
References
- Akhurst RJ, Padgett RW. Matters of context guide future research in TGFbeta superfamily signaling. Sci Signal. 2015;8:re10. doi: 10.1126/scisignal.aad0416. [DOI] [PubMed] [Google Scholar]
- Alexiades MR, Cepko CL. Subsets of retinal progenitors display temporally regulated and distinct biases in the fates of their progeny. Development. 1997;124:1119–1131. doi: 10.1242/dev.124.6.1119. [DOI] [PubMed] [Google Scholar]
- Alimonti A. PTEN breast cancer susceptibility: a matter of dose. Ecancermedicalscience. 2010;4:192. doi: 10.3332/ecancer.2010.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Assinder SJ, Dong Q, Kovacevic Z, Richardson DR. The TGF-beta, PI3K/Akt and PTEN pathways: established and proposed biochemical integration in prostate cancer. Biochem J. 2009;417:411–421. doi: 10.1042/BJ20081610. [DOI] [PubMed] [Google Scholar]
- Backman SA, Stambolic V, Suzuki A, Haight J, Elia A, Pretorius J, Tsao MS, Shannon P, Bolon B, Ivy GO, Mak TW. Deletion of Pten in mouse brain causes seizures, ataxia and defects in soma size resembling Lhermitte-Duclos disease. Nat Genet. 2001;29:396–403. doi: 10.1038/ng782. [DOI] [PubMed] [Google Scholar]
- Balasubramanian R, Gan L. Development of retinal amacrine cells and their dendritic stratification. Curr Ophthalmol Rep. 2014;2:100–106. doi: 10.1007/s40135-014-0048-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassett EA, Pontoriero GF, Feng W, Marquardt T, Fini ME, Williams T, West-Mays JA. Conditional deletion of activating protein 2alpha (AP-2alpha) in the developing retina demonstrates non-cell-autonomous roles for AP-2alpha in optic cup development. Mol Cell Biol. 2007;27:7497–7510. doi: 10.1128/MCB.00687-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belliveau MJ, Cepko CL. Extrinsic and intrinsic factors control the genesis of amacrine and cone cells in the rat retina. Development. 1999;126:555–566. doi: 10.1242/dev.126.3.555. [DOI] [PubMed] [Google Scholar]
- Boehm JS, Zhao JJ, Yao J, Kim SY, Firestein R, Dunn IF, Sjostrom SK, Garraway LA, Weremowicz S, Richardson AL, Greulich H, Stewart CJ, Mulvey LA, Shen RR, Ambrogio L, Hirozane-Kishikawa T, Hill DE, Vidal M, Meyerson M, et al. Integrative genomic approaches identify IKBKE as a breast cancer oncogene. Cell. 2007;129:1065–1079. doi: 10.1016/j.cell.2007.03.052. [DOI] [PubMed] [Google Scholar]
- Cantrup R, Dixit R, Palmesino E, Bonfield S, Shaker T, Tachibana N, Zinyk D, Dalesman S, Yamakawa K, Stell WK, Wong RO, Reese BE, Kania A, Sauvé Y, Schuurmans C. Cell-type specific roles for PTEN in establishing a functional retinal architecture. PLoS One. 2012;7:e32795. doi: 10.1371/journal.pone.0032795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter-Dawson LD, LaVail MM. Rods and cones in the mouse retina. I. Structural analysis using light and electron microscopy. J Comp Neurol. 1979;188:245–262. doi: 10.1002/cne.901880204. [DOI] [PubMed] [Google Scholar]
- Cayouette M, Poggi L, Harris WA. Lineage in the vertebrate retina. Trends Neurosci. 2006;29:563–570. doi: 10.1016/j.tins.2006.08.003. [DOI] [PubMed] [Google Scholar]
- Cepko CL, Austin CP, Yang X, Alexiades M, Ezzeddine D. Cell fate determination in the vertebrate retina. Proc Natl Acad Sci U S A. 1996;93:589–595. doi: 10.1073/pnas.93.2.589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Close JL, Gumuscu B, Reh TA. Retinal neurons regulate proliferation of postnatal progenitors and Muller glia in the rat retina via TGF beta signaling. Development. 2005;132:3015–3026. doi: 10.1242/dev.01882. [DOI] [PubMed] [Google Scholar]
- Comer FI, Parent CA. Phosphoinositides specify polarity during epithelial organ development. Cell. 2007;128:239–240. doi: 10.1016/j.cell.2007.01.010. [DOI] [PubMed] [Google Scholar]
- Cully M, You H, Levine AJ, Mak TW. Beyond PTEN mutations: the PI3K pathway as an integrator of multiple inputs during tumorigenesis. Nat Rev Cancer. 2006;6:184–192. doi: 10.1038/nrc1819. [DOI] [PubMed] [Google Scholar]
- de Melo J, Qiu X, Du G, Cristante L, Eisenstat DD. Dlx1, Dlx2, Pax6, Brn3b, and Chx10 homeobox gene expression defines the retinal ganglion and inner nuclear layers of the developing and adult mouse retina. J Comp Neurol. 2003;461:187–204. doi: 10.1002/cne.10674. [DOI] [PubMed] [Google Scholar]
- Di Cristofano A, Pesce B, Cordon-Cardo C, Pandolfi PP. Pten is essential for embryonic development and tumour suppression. Nat Genet. 1998;19:348–355. doi: 10.1038/1235. [DOI] [PubMed] [Google Scholar]
- Dixit R, Tachibana N, Touahri Y, Zinyk D, Logan C, Schuurmans C. Gene expression is dynamically regulated in retinal progenitor cells prior to and during overt cellular differentiation. Gene Exp Patterns. 2014;14:42–54. doi: 10.1016/j.gep.2013.10.003. [DOI] [PubMed] [Google Scholar]
- Downward J. PI 3-kinase, Akt and cell survival. Semin Cell Dev Biol. 2004;15:177–182. doi: 10.1016/j.semcdb.2004.01.002. [DOI] [PubMed] [Google Scholar]
- Dyer MA, Cepko CL. p27Kip1 and p57Kip2 regulate proliferation in distinct retinal progenitor cell populations. J Neurosci. 2001;21:4259–4271. doi: 10.1523/JNEUROSCI.21-12-04259.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eder AM, Dominguez L, Franke TF, Ashwell JD. Phosphoinositide 3-kinase regulation of T cell receptor-mediated interleukin-2 gene expression in normal T cells. J Biol Chem. 1998;273:28025–28031. doi: 10.1074/jbc.273.43.28025. [DOI] [PubMed] [Google Scholar]
- Engelman JA, Luo J, Cantley LC. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat Rev Genet. 2006;7:606–619. doi: 10.1038/nrg1879. [DOI] [PubMed] [Google Scholar]
- Feng L, Xie X, Joshi PS, Yang Z, Shibasaki K, Chow RL, Gan L. Requirement for Bhlhb5 in the specification of amacrine and cone bipolar subtypes in mouse retina. Development. 2006;133:4815–4825. doi: 10.1242/dev.02664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foncea R, Andersson M, Ketterman A, Blakesley V, Sapag-Hagar M, Sugden PH, LeRoith D, Lavandero S. Insulin-like growth factor-I rapidly activates multiple signal transduction pathways in cultured rat cardiac myocytes. J Biol Chem. 1997;272:19115–19124. doi: 10.1074/jbc.272.31.19115. [DOI] [PubMed] [Google Scholar]
- Fraser MM, Bayazitov IT, Zakharenko SS, Baker SJ. Phosphatase and tensin homolog, deleted on chromosome 10 deficiency in brain causes defects in synaptic structure, transmission and plasticity, and myelination abnormalities. Neuroscience. 2008;151:476–488. doi: 10.1016/j.neuroscience.2007.10.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomer RH. Not being the wrong size. Nat Rev Mol Cell Biol. 2001;2:48–54. doi: 10.1038/35048058. [DOI] [PubMed] [Google Scholar]
- Gomes FL, Zhang G, Carbonell F, Correa JA, Harris WA, Simons BD, Cayouette M. Reconstruction of rat retinal progenitor cell lineages in vitro reveals a surprising degree of stochasticity in cell fate decisions. Development. 2011;138:227–235. doi: 10.1242/dev.059683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groszer M, Erickson R, Scripture-Adams DD, Lesche R, Trumpp A, Zack JA, Kornblum HI, Liu X, Wu H. Negative regulation of neural stem/progenitor cell proliferation by the Pten tumor suppressor gene in vivo. Science. 2001;294:2186–2189. doi: 10.1126/science.1065518. [DOI] [PubMed] [Google Scholar]
- Hand R, Bortone D, Mattar P, Nguyen L, Heng JI, Guerrier S, Boutt E, Peters E, Barnes AP, Parras C, Schuurmans C, Guillemot F, Polleux F. Phosphorylation of Neurogenin2 specifies the migration properties and the dendritic morphology of pyramidal neurons in the neocortex. Neuron. 2005;48:45–62. doi: 10.1016/j.neuron.2005.08.032. [DOI] [PubMed] [Google Scholar]
- Harmon EB, Apelqvist AA, Smart NG, Gu X, Osborne DH, Kim SK. GDF11 modulates NGN3+ islet progenitor cell number and promotes beta-cell differentiation in pancreas development. Development. 2004;131:6163–6174. doi: 10.1242/dev.01535. [DOI] [PubMed] [Google Scholar]
- Hatakeyama J, Kageyama R. Retinal cell fate determination and bHLH factors. Semin Cell Dev Biol. 2004;15:83–89. doi: 10.1016/j.semcdb.2003.09.005. [DOI] [PubMed] [Google Scholar]
- He J, Zhang G, Almeida AD, Cayouette M, Simons BD, Harris WA. How variable clones build an invariant retina. Neuron. 2012;75:786–798. doi: 10.1016/j.neuron.2012.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin K, Jiang H, Xiao D, Zou M, Zhu J, Xiang M. Tfap2a and 2b act downstream of Ptf1a to promote amacrine cell differentiation during retinogenesis. Mol Brain. 2015;8:28. doi: 10.1186/s13041-015-0118-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo HS, Kang KH, Joe CO, Kim JW. Pten coordinates retinal neurogenesis by regulating Notch signalling. EMBO J. 2012;31:817–828. doi: 10.1038/emboj.2011.443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kay JN, Voinescu PE, Chu MW, Sanes JR. Neurod6 expression defines new retinal amacrine cell subtypes and regulates their fate. Nat Neurosci. 2011;14:965–972. doi: 10.1038/nn.2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knobbe CB, Lapin V, Suzuki A, Mak TW. The roles of PTEN in development, physiology and tumorigenesis in mouse models: a tissue-by-tissue survey. Oncogene. 2008;27:5398–5415. doi: 10.1038/onc.2008.238. [DOI] [PubMed] [Google Scholar]
- Kriplani N, Hermida MA, Brown ER, Leslie NR. Class I PI 3-kinases: function and evolution. Adv Biol Regul. 2015;59:53–64. doi: 10.1016/j.jbior.2015.05.002. [DOI] [PubMed] [Google Scholar]
- Kwon CH, Luikart BW, Powell CM, Zhou J, Matheny SA, Zhang W, Li Y, Baker SJ, Parada LF. Pten regulates neuronal arborization and social interaction in mice. Neuron. 2006;50:377–388. doi: 10.1016/j.neuron.2006.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langley B, Thomas M, Bishop A, Sharma M, Gilmour S, Kambadur R. Myostatin inhibits myoblast differentiation by down-regulating MyoD expression. J Biol Chem. 2002;277:49831–49840. doi: 10.1074/jbc.M204291200. [DOI] [PubMed] [Google Scholar]
- Lehtinen MK, Zappaterra MW, Chen X, Yang YJ, Hill AD, Lun M, Maynard T, Gonzalez D, Kim S, Ye P, D'Ercole AJ, Wong ET, LaMantia AS, Walsh CA. The cerebrospinal fluid provides a proliferative niche for neural progenitor cells. Neuron. 2011;69:893–905. doi: 10.1016/j.neuron.2011.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leslie NR, Batty IH, Maccario H, Davidson L, Downes CP. Understanding PTEN regulation: PIP2, polarity and protein stability. Oncogene. 2008;27:5464–5476. doi: 10.1038/onc.2008.243. [DOI] [PubMed] [Google Scholar]
- Li S, Mo Z, Yang X, Price SM, Shen MM, Xiang M. Foxn4 controls the genesis of amacrine and horizontal cells by retinal progenitors. Neuron. 2004;43:795–807. doi: 10.1016/j.neuron.2004.08.041. [DOI] [PubMed] [Google Scholar]
- Liu H, Kim SY, Fu Y, Wu X, Ng L, Swaroop A, Forrest D. An isoform of retinoid-related orphan receptor beta directs differentiation of retinal amacrine and horizontal interneurons. Nat Commun. 2013;4:1813. doi: 10.1038/ncomms2793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma L, Cantrup R, Varrault A, Colak D, Klenin N, Götz M, McFarlane S, Journot L, Schuurmans C. Zac1 functions through TGFbetaII to negatively regulate cell number in the developing retina. Neural Dev. 2007;2:11. doi: 10.1186/1749-8104-2-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marino S, Krimpenfort P, Leung C, van der Korput HA, Trapman J, Camenisch I, Berns A, Brandner S. PTEN is essential for cell migration but not for fate determination and tumourigenesis in the cerebellum. Development. 2002;129:3513–3522. doi: 10.1242/dev.129.14.3513. [DOI] [PubMed] [Google Scholar]
- Marquardt T. Transcriptional control of neuronal diversification in the retina. Prog Retinal Eye Res. 2003;22:567–577. doi: 10.1016/S1350-9462(03)00036-3. [DOI] [PubMed] [Google Scholar]
- Marquardt T, Ashery-Padan R, Andrejewski N, Scardigli R, Guillemot F, Gruss P. Pax6 is required for the multipotent state of retinal progenitor cells. Cell. 2001;105:43–55. doi: 10.1016/S0092-8674(01)00295-1. [DOI] [PubMed] [Google Scholar]
- Matsuda T, Cepko CL. Electroporation and RNA interference in the rodent retina in vivo and in vitro. Proc Natl Acad Sci U S A. 2004;101:16–22. doi: 10.1073/pnas.2235688100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mo Z, Li S, Yang X, Xiang M. Role of the Barhl2 homeobox gene in the specification of glycinergic amacrine cells. Development. 2004;131:1607–1618. doi: 10.1242/dev.01071. [DOI] [PubMed] [Google Scholar]
- Moelling K, Schad K, Bosse M, Zimmermann S, Schweneker M. Regulation of Raf-Akt cross-talk. J Biol Chem. 2002;277:31099–31106. doi: 10.1074/jbc.M111974200. [DOI] [PubMed] [Google Scholar]
- Myers MG, Jr, Grammer TC, Wang LM, Sun XJ, Pierce JH, Blenis J, White MF. Insulin receptor substrate-1 mediates phosphatidylinositol 3′-kinase and p70S6k signaling during insulin, insulin-like growth factor-1, and interleukin-4 stimulation. J Biol Chem. 1994;269:28783–28789. [PubMed] [Google Scholar]
- Nadal-Nicolas FM, Jiménez-López M, Sobrado-Calvo P, Nieto-López L, Cánovas-Martínez I, Salinas-Navarro M, Vidal-Sanz M, Agudo M. Brn3a as a marker of retinal ganglion cells: qualitative and quantitative time course studies in naive and optic nerve-injured retinas. Invest Ophthalmol Vis Sci. 2009;50:3860–3868. doi: 10.1167/iovs.08-3267. [DOI] [PubMed] [Google Scholar]
- Nakhai H, Sel S, Favor J, Mendoza-Torres L, Paulsen F, Duncker GI, Schmid RM. Ptf1a is essential for the differentiation of GABAergic and glycinergic amacrine cells and horizontal cells in the mouse retina. Development. 2007;134:1151–1160. doi: 10.1242/dev.02781. [DOI] [PubMed] [Google Scholar]
- Ohsawa R, Kageyama R. Regulation of retinal cell fate specification by multiple transcription factors. Brain Res. 2008;1192:90–98. doi: 10.1016/j.brainres.2007.04.014. [DOI] [PubMed] [Google Scholar]
- Pan L, Yang Z, Feng L, Gan L. Functional equivalence of Brn3 POU-domain transcription factors in mouse retinal neurogenesis. Development. 2005;132:703–712. doi: 10.1242/dev.01646. [DOI] [PubMed] [Google Scholar]
- Pearson BJ, Doe CQ. Specification of temporal identity in the developing nervous system. Annu Rev Cell Dev Biol. 2004;20:619–647. doi: 10.1146/annurev.cellbio.19.111301.115142. [DOI] [PubMed] [Google Scholar]
- Pow DV, Hendrickson AE. Distribution of the glycine transporter glyt-1 in mammalian and nonmammalian retinae. Vis Neurosci. 1999;16:231–239. doi: 10.1017/s0952523899162047. [DOI] [PubMed] [Google Scholar]
- Ramaswamy S, Nakamura N, Vazquez F, Batt DB, Perera S, Roberts TM, Sellers WR. Regulation of G1 progression by the PTEN tumor suppressor protein is linked to inhibition of the phosphatidylinositol 3-kinase/Akt pathway. Proc Natl Acad Sci U S A. 1999;96:2110–2115. doi: 10.1073/pnas.96.5.2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reh TA, Tully T. Regulation of tyrosine hydroxylase-containing amacrine cell number in larval frog retina. Dev Biol. 1986;114:463–469. doi: 10.1016/0012-1606(86)90210-1. [DOI] [PubMed] [Google Scholar]
- Sakagami K, Chen B, Nusinowitz S, Wu H, Yang XJ. PTEN regulates retinal interneuron morphogenesis and synaptic layer formation. Mol Cell Neurosci. 2012;49:171–183. doi: 10.1016/j.mcn.2011.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinor AD, Lillien L. Akt-1 expression level regulates CNS precursors. J Neurosci. 2004;24:8531–8541. doi: 10.1523/JNEUROSCI.1470-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spinelli L, Black FM, Berg JN, Eickholt BJ, Leslie NR. Functionally distinct groups of inherited PTEN mutations in autism and tumour syndromes. J Med Genet. 2015;52:128–134. doi: 10.1136/jmedgenet-2014-102803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stambolic V, Suzuki A, de la Pompa JL, Brothers GM, Mirtsos C, Sasaki T, Ruland J, Penninger JM, Siderovski DP, Mak TW. Negative regulation of PKB/Akt-dependent cell survival by the tumor suppressor PTEN. Cell. 1998;95:29–39. doi: 10.1016/S0092-8674(00)81780-8. [DOI] [PubMed] [Google Scholar]
- Steelman LS, Chappell WH, Abrams SL, Kempf RC, Long J, Laidler P, Mijatovic S, Maksimovic-Ivanic D, Stivala F, Mazzarino MC, Donia M, Fagone P, Malaponte G, Nicoletti F, Libra M, Milella M, Tafuri A, Bonati A, Bäsecke J, Cocco L, et al. Roles of the Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR pathways in controlling growth and sensitivity to therapy-implications for cancer and aging. Aging. 2011;3:192–222. doi: 10.18632/aging.100296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki A, Yamaguchi MT, Ohteki T, Sasaki T, Kaisho T, Kimura Y, Yoshida R, Wakeham A, Higuchi T, Fukumoto M, Tsubata T, Ohashi PS, Koyasu S, Penninger JM, Nakano T, Mak TW. T cell-specific loss of Pten leads to defects in central and peripheral tolerance. Immunity. 2001;14:523–534. doi: 10.1016/S1074-7613(01)00134-0. [DOI] [PubMed] [Google Scholar]
- Tobin JF, Celeste AJ. Myostatin, a negative regulator of muscle mass: implications for muscle degenerative diseases. Curr Opin Pharmacol. 2005;5:328–332. doi: 10.1016/j.coph.2005.01.011. [DOI] [PubMed] [Google Scholar]
- Voinescu PE, Kay JN, Sanes JR. Birthdays of retinal amacrine cell subtypes are systematically related to their molecular identity and soma position. J Comp Neurol. 2009;517:737–750. doi: 10.1002/cne.22200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volland S, Esteve-Rudd J, Hoo J, Yee C, Williams DS. A comparison of some organizational characteristics of the mouse central retina and the human macula. PLoS One. 2015;10:e0125631. doi: 10.1371/journal.pone.0125631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waid DK, McLoon SC. Ganglion cells influence the fate of dividing retinal cells in culture. Development. 1998;125:1059–1066. doi: 10.1242/dev.125.6.1059. [DOI] [PubMed] [Google Scholar]
- Wallace VA. Concise review: making a retina–from the building blocks to clinical applications. Stem Cells. 2011;29:412–417. doi: 10.1002/stem.602. [DOI] [PubMed] [Google Scholar]
- Wei W, Hou J, Alder O, Ye X, Lee S, Cullum R, Chu A, Zhao Y, Warner SM, Knight DA, Yang D, Jones SJ, Marra MA, Hoodless PA. Genome-wide microRNA and messenger RNA profiling in rodent liver development implicates mir302b and mir20a in repressing transforming growth factor-beta signaling. Hepatology. 2013;57:2491–2501. doi: 10.1002/hep.26252. [DOI] [PubMed] [Google Scholar]
- Whitman M, Downes CP, Keeler M, Keller T, Cantley L. Type I phosphatidylinositol kinase makes a novel inositol phospholipid, phosphatidylinositol-3-phosphate. Nature. 1988;332:644–646. doi: 10.1038/332644a0. [DOI] [PubMed] [Google Scholar]
- Wu HH, Ivkovic S, Murray RC, Jaramillo S, Lyons KM, Johnson JE, Calof AL. Autoregulation of neurogenesis by GDF11. Neuron. 2003;37:197–207. doi: 10.1016/S0896-6273(02)01172-8. [DOI] [PubMed] [Google Scholar]
- Yilmaz OH, Valdez R, Theisen BK, Guo W, Ferguson DO, Wu H, Morrison SJ. Pten dependence distinguishes haematopoietic stem cells from leukaemia-initiating cells. Nature. 2006;441:475–482. doi: 10.1038/nature04703. [DOI] [PubMed] [Google Scholar]
- Yoshimura T, Arimura N, Kawano Y, Kawabata S, Wang S, Kaibuchi K. Ras regulates neuronal polarity via the PI3-kinase/Akt/GSK-3beta/CRMP-2 pathway. Biochem Biophys Res Commun. 2006;340:62–68. doi: 10.1016/j.bbrc.2005.11.147. [DOI] [PubMed] [Google Scholar]
- Young RW. Cell differentiation in the retina of the mouse. Anat Rec. 1985;212:199–205. doi: 10.1002/ar.1092120215. [DOI] [PubMed] [Google Scholar]
- Yue Q, Groszer M, Gil JS, Berk AJ, Messing A, Wu H, Liu X. PTEN deletion in Bergmann glia leads to premature differentiation and affects laminar organization. Development. 2005;132:3281–3291. doi: 10.1242/dev.01891. [DOI] [PubMed] [Google Scholar]
- Zhang C, McCall MA. Receptor targets of amacrine cells. Vis Neurosci. 2012;29:11–29. doi: 10.1017/S0952523812000028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann S, Moelling K. Phosphorylation and regulation of Raf by Akt (protein kinase B) Science. 1999;286:1741–1744. doi: 10.1126/science.286.5445.1741. [DOI] [PubMed] [Google Scholar]