

Abstract
Hyperglycemia is common in patients with acute stroke, even in those without preexisting diabetes, and denotes a bad outcome. However, the mechanisms underlying the detrimental effects of hyperglycemia are largely unclear. In a mouse model of ischemic stroke, we found that hyperglycemia increased the infarct volume and decreased the number of protective noninflammatory monocytes/macrophages in the ischemic brain. Ablation of peripheral monocytes blocked the detrimental effect of hyperglycemia, suggesting that monocytes are required. In hyperglycemic mice, α-dicarbonyl glucose metabolites, the precursors for advanced glycation end products, were significantly elevated in plasma and ischemic brain tissue. The receptor of advanced glycation end products, AGER (previously known as RAGE), interfered with polarization of macrophages to a noninflammatory phenotype. When Ager was deleted, hyperglycemia did not aggravate ischemic brain damage any longer. Independently of AGER, methylglyoxal reduced the release of endothelial CSF-1 (M-CSF), which stimulates polarization of macrophages to a noninflammatory phenotype in the microenvironment of the ischemic brain. In summary, our study identified α-dicarbonyls and AGER as mediators by which hyperglycemia lowers the number of protective noninflammatory macrophages and consequently increases ischemic brain damage. Modulating the metabolism of α-dicarbonyls or blocking AGER may improve the treatment of stroke patients with hyperglycemia.
SIGNIFICANCE STATEMENT Although glucose is the main energy substrate of the brain, hyperglycemia aggravates ischemic brain damage in acute stroke. So far, clinical trials have indicated that insulin treatment provides no solution to this common clinical problem. This study shows, in an experimental stroke model, that hyperglycemia interferes with the polarization of monocytes/macrophages to a protective cell type. Key players are α-dicarbonyls and the receptor for advanced glycation end products (AGER). Deletion of AGER normalized monocyte/macrophage polarization and reversed the detrimental effects of hyperglycemia, suggesting new avenues to treat stroke patients.
Keywords: cerebral ischemia, endothelial cells, inflammation, neuroprotection
Introduction
Hyperglycemia is a common clinical feature in patients with an acute ischemic stroke. Upon admission, up to 2/3 of stroke patients have plasma glucose concentrations >6.1 mmol/L (Scott et al., 1999). Often, the hyperglycemia is due to preexisting diabetes mellitus; however, there is now clinical evidence to suggest that stress induces hyperglycemia in nondiabetic stroke patients (Kruyt et al., 2010; Dziedzic et al., 2012). Epidemiological data have shown that high blood glucose concentrations on admission are a predictor for enhanced mortality rate and a bad functional outcome (Melamed, 1976; Kruyt et al., 2010). The predictive value of acute hyperglycemia is especially strong in nondiabetic patients, suggesting that the acute hyperglycemia is the causative factor rather than preexisting vascular damage (Capes et al., 2001; Kruyt et al., 2010). In support of this view, preclinical studies in rodents have confirmed that acute hyperglycemia can aggravate ischemic brain damage (Kruyt et al., 2010). Therefore, current guidelines from the American Heart Association and the European Stroke Organization recommend treating the hyperglycemia in patients with acute stroke. However, several large clinical trials have shown that treatment with insulin does not improve outcome in acute stroke and actually puts the patients at considerable risk for hypoglycemic episodes (Bellolio et al., 2014).
The mechanism(s) by which glucose metabolism aggravates ischemic brain injury is unknown. Previous studies have shown that the alternative energy substrate, β-hydroxybutyrate, modulates ischemic stroke by acting on noninflammatory monocytes and macrophages, also known as M2-type macrophages (Rahman et al., 2014). Monocytes/macrophages are plastic cells and present a wide range of phenotypes. Extremes of this phenotypic spectrum are, on one side, inflammatory cells, characterized by the markers Ly-6CHi, iNOS, IL-1β, and TNF, and, on the other side, noninflammatory cells with the markers Ly-6CLo, Arg1, RETNLA, and CHIL3 (Mosser and Edwards, 2008). The current concept is that inflammatory monocytes are recruited to the CNS during stroke and differentiate to noninflammatory macrophages, which play an important role in protecting the brain after ischemia (Gliem et al., 2012). Here, we report that acute hyperglycemia downregulates protective noninflammatory monocytes/macrophages in ischemic brain tissue. Furthermore, we provide evidence that α-dicarbonyl glucose metabolites and the receptor for advanced glycation end products (AGER) are potential mediators of the glucose effect on noninflammatory macrophages, suggesting new treatment strategies for hyperglycemic stroke.
Materials and Methods
Mice.
Mice were age and sex matched between experimental groups. CD11b-DTR, Ager−/−, and Cx3cr1GFP/+ mice have been described previously (Jung et al., 2000; Constien et al., 2001; Duffield et al., 2005). Ager−/− mice were backcrossed for >10 generations on a C57BL/6N background; therefore, we used C57BL/6N mice as wild-type controls. The mice were kept in individually ventilated cages in groups of four to five individuals with ad libitum access to food and water under a 12 h light/12 h dark cycle at 23°C. Studies were designed according to the ARRIVE guidelines. Therefore, investigators were blinded to treatment, genotype, or both in all experiments. All experiments were performed according to the German animal protection law and were approved by the local animal welfare authorities (Ministerium für Energiewende, Landwirtschaft, Umwelt und ländliche Räume, Kiel, Germany).
Stroke model.
To induce ischemic strokes, we permanently occluded the distal middle cerebral artery (MCA) in 10- to 12-week-old male mice (Lubjuhn et al., 2009). First, mice were anesthetized with pentobarbital (15 μl of 0.5% pentobarbital/g body weight, i.p.). A skin incision was made between the ear and the orbit on the left side. After removing the temporal muscle, a burr hole was drilled to expose the stem of the MCA. The MCA was then occluded by microbipolar electrocoagulation (Model ICC 50; Erbe). Finally, the skin incision was sutured and the mice were placed under a heating lamp until they fully recovered. The surgery was done under a microscope (Hund) and rectal temperature was maintained at 37°C with a heating pad. To determine the infarct volume, mice were deeply reanaesthetized with tribromoethanol (15 μl of 2.5% tribromoethanol/g body weight, i.p.) 48 h after MCA occlusion (MCAO) and perfused intracardially with 15–20 ml of Ringer's solution. Coronal cryosections (20 μm thick) were cut every 400 μm and stained using a silver technique (Lubjuhn et al., 2009). The infarct volume was determined using ImageJ and corrected for brain edema as described previously (Lubjuhn et al., 2009). Mice were only excluded from analysis if they died before perfusion. Mortality was <10% and did not differ between treatment groups. Mice were randomized to the treatment groups. To induce hyperglycemia, glucose (50 mg in 0.2 ml of normal saline) or vehicle were intraperitoneally injected immediately before MCAO.
Flow cytometry.
Blood samples (100 μl) were taken before or 48 h after MCAO. Mice were deeply anesthetized with tribromoethanol (15 μl of 2.5% tribromoethanol/g body weight, i.p.) 48 h after MCAO and perfused intracardially with Ringer's solution. After dissecting the brain and removing olfactory bulbs and cerebella, left hemispheres were digested in DMEM (Invitrogen) containing collagenase A (1 mg/ml; Roche) and DNase (0.1 mg/ml; Roche) for 30 min at 37°C (Rahman et al., 2014). Then, cells were filtered through a 40 μm nylon cell strainer (BD Biosciences) and the red blood cells were lysed on ice with standard erythrocyte lysis buffer. To separate cells from myelin and debris, a Percoll gradient (GE Healthcare; 78% and 30%) was used. The cells were carefully collected from the interface of the gradient and washed with 10 ml of PBS containing 0.5% BSA. After treatment with purified rat anti-mouse CD16/32 (Fc Block; BD PharMingen, 1:100) for 10 min on ice, cells were incubated with the antibodies or the respective isotype controls for 45 min on ice as follows: PE-labeled rat anti-mouse CD45 (BD PharMingen, 1:100), APC-labeled rat anti-mouse Ly-6C (BD PharMingen, 1:100), PerCP-labeled rat anti-mouse CD11b (BD PharMingen, 1:100), and FITC-labeled rat anti-mouse Ly-6G (BD PharMingen, 1:100). The cells were then analyzed on the Partec Cube8 with the laser lines 488 and 638 nm.
Immunofluorescence microscopy.
Arginase 1 (Arg1) staining was performed as reported previously with some modifications (Pomeshchik et al., 2015). Forty-eight hours after MCAO, wild-type, Ager−/−, or Cx3cr1GFP/+ > WT mice were deeply anesthetized with tribromoethanol (15 μl of 2.5% tribromoethanol/g body weight, i.p.) and perfused with Ringer's solution and 4% paraformaldehyde (PFA). Brains were postfixed in 4% PFA (24 h, 4°C), cryoprotected in 30% sucrose (24 h, 4°C), and snap frozen in isopentane. Coronal cryosections (20 μm thick) were air-dried for 1 h and rehydrated in PBS for 5 min. After antigen retrieval in citrate buffer, the slides were incubated overnight at room temperature with goat anti-mouse Arg1 (N-20; Santa Cruz Biotechnology, 1:200), together with rabbit anti-mouse Iba1 (Wako, 1:100; see Fig. 3) or with rabbit anti-mouse inducible NO synthase (iNOS; Calbiochem, 1:50) and chicken anti-GFP (Abcam, 1:2000; see Fig. 4). The next day, the slides were incubated with Cy3-labeled anti-goat (Jackson Immunoresearch), Alexa Fluor 488-labeled anti-rabbit (Invitrogen), Alexa Fluor 647-labeled anti-goat (Invitrogen), Alexa Fluor 594-labeled anti-rabbit (Invitrogen), or Cy2-labeled anti-chicken antibodies (Abcam). All secondary antibodies were used in 1:400 dilutions containing DAPI (1 μg/ml). Imaging was performed on a fluorescence (DMI 6000 B; Leica) or a confocal microscope (TCS SP5; Leica) using the same settings for the whole experiment. Immunostained cells were counted in the whole-brain section or in predefined fields (0.6 mm2) of the motor cortex, striatum, and insular cortex on 4 coronal sections per mouse (−0.22 mm relative to bregma) using ImageJ.
Figure 3.

Hyperglycemia reduces the number of Arg1 positive noninflammatory myeloid cells in the peri-infarct zone 48 h after MCAO. A–F, Representative confocal images of Iba1 (green, marker of microglia and monocytes/macrophages) and Arg1 (red) staining in normoglycemic and hyperglycemic mice. White arrows, Arg1+Iba+ cells; white arrowheads, Arg1−Iba+ cells. G, H, Quantification of Arg1+Iba+ and Arg1−Iba+ cells per square millimeter. Unpaired t test, **p < 0.003 (n = 6–8 mice/group) .Scale bar 100 μm. Data are shown as means ± SEM.
Figure 4.
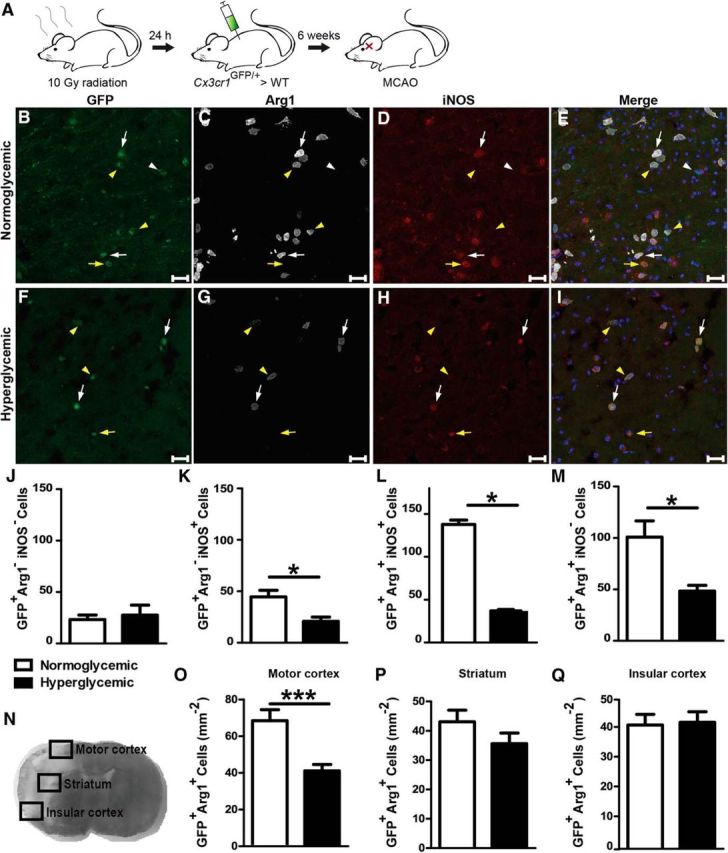
Acute hyperglycemia during MCAO modified the polarization of infiltrating monocytes/macrophages. A, Infiltrating monocytes/macrophages were labeled by transplantation of Cx3cr1GFP/+ bone marrow. B–I, Forty-eight hours after MCAO, Cx3cr1GFP/+ cells (green) were stained for Arg1 (white) and iNOS (red). Cx3cr1GFP/+ cells in the peri-infarct area of the motor cortex were Arg1−iNOS− (white arrowheads), Arg1−iNOS+ (yellow arrows, inflammatory macrophages), Arg1+iNOS+ (white arrows), or Arg1+iNOS− (yellow arrowheads, noninflammatory macrophages). Scale bar, 50 μm. J–M, Quantification of the four cell types in coronal sections of the whole brain revealed a lower density of inflammatory macrophages, but mainly the Arg1+iNOS+ and noninflammatory macrophages were reduced. N, To localize the hyperglycemic effect on noninflammatory polarization in the peri-infarct tissue, three distinct areas where analyzed separately. O–Q, Cx3cr1GFP/+Arg1+ cells in the targeted regions revealed a significant difference in the motor cortex, whereas there was no change in the striatum or insular cortex. Unpaired t test, *p < 0.05, ***p < 0.0005, data are shown as means ± SEM (n = 4/group for whole section and n = 10 mice/group for targeted regions).
Bone marrow transplantation.
Cx3cr1GFP/+ mice were anesthetized with isoflurane and decapitated. Femurs and tibias were dissected aseptically and the bone marrow was flushed out of the bones and dissolved in PBS using 23-gauge cannulae. The homogenous cell suspension was passed through a 40 μm cell strainer. After centrifugation (500 × g, 5 min, 4°C) cells were suspended in PBS and injected retro-orbitally in 8-week-old C57BL/6 mice (100 μl, 5 × 106 cells) that had been lethally irradiated (10 Gy) 1 d before. Six weeks after transplantation, the MCAO was performed under normoglycemic or hyperglycemic conditions as described previously.
Preparation and culture of primary cells.
Primary brain endothelial cells (PBECs) and bone-marrow-derived macrophages (BMDMs) were prepared from 8- to 10-week-old C57BL/6 (Charles River) or Ager−/− mice. The animals were anesthetized with isoflurane and killed by decapitation. The preparation of PBECs has been described previously (Ridder et al., 2011).
For BMDMs, bone marrow was prepared from femurs and tibias as described above. The cells were suspended in PBS, supplemented with 2% heat inactivated fetal calf serum (FCS; Biochrom), and passed through a 70 μm cell strainer. To remove red blood cells, 1 volume of NH4Cl (1.2% in PBS) was added and the suspension was incubated for 10 min on ice. After centrifugation (500 × g, 5 min, 4°C), cells were suspended in IMDM (25 mm glucose; Life Technologies) containing heat-inactivated FCS (10%), penicillin (100 U/ml), streptomycin (100 μg/ml), and colony stimulating factor 1 (CSF-1, 10 ng/ml; ProSpec) and seeded (2 × 106 cells/ml) on 24-well plates. On day 3, the medium was changed to DMEM (5 mm glucose; Life Technologies) containing the above supplements. On day 7, CSF-1 treatment was stopped and macrophages were polarized to an inflammatory cell type with lipopolysaccharides (LPS, E. coli 0111:B4, 100 ng/ml; Sigma-Aldrich) or a noninflammatory cell type with recombinant murine IL-4 (10 ng/ml; PeproTech) in DMEM or they were treated with the conditioned medium of methylglyoxal (MG)-challenged PBECs that were subjected to oxygen glucose deprivation (OGD) with or without goat anti-CSF-1 neutralizing antibodies (1.6 μg/ml; LSBio).
Measurement of α-dicarbonyls.
EDTA plasma was collected before (time point 0) and 30 and 50 min after MCAO. Ipsilateral and contralateral brain tissues were harvested and snap frozen in liquid nitrogen. The brain tissues were homogenized at 4°C in 100 μl of cold PBS using a high-throughput tissue homogenizer (Bullet Blender STORM BBY24M; Next Advance). After homogenization, protein concentrations were determined in the brain homogenates using Lowry's method. α-Dicarbonyls were then measured by LC/MS as described previously with some modifications (Thornalley and Rabbani, 2014). Proteins were precipitated by adding 10 μl of ice-cold trichloroacetic acid (20% weight/volume) to plasma samples (25 μl) or to brain homogenates (300 μg of protein) together with the isotopically labeled internal standard d4-MG, followed by centrifugation (20,000 × g, 4°C, 10 min). In the supernatant, α-dicarbonyls were derivatized to their quinoxaline derivatives by adding o-phenylenediamine (10 μl, 0.5 mm in 200 mm HCl/500 μm DETAPAC) for 4 h in the dark.
LC/MS analysis was performed on a TSQ Endura triple quadrupole mass spectrometer coupled to a Dionex Ultimate 3000 UHPLC system (Thermo Scientific). Derivatized α-dicarbonlys were separated on an Ascentis Express C18 column (100 mm × 2.1 mm × 5 μm; Sigma-Aldrich) using 0.1% formic acid in water as mobile phase A and acetonitrile as mobile phase B. The flow rate was set to 0.2 ml/min and the column temperature was kept at 30°C. The following gradient was used; 0–2 min isocratic gradient of 10% B, which increased to 50% between 2 and 10 min. Afterward, B increased to 100% between 10 and 11 min, which was followed by a washing step with 100% B between 11 and 15 min. Re-equilibration took place between 15 and 20 min back to 10% B. The retention times were as follows: derivatized 3-deoxyglucosone (3-DG), 3.16 min; derivatized glyoxal, 5.43 min; derivatized MG, 6.47 min; and derivatized d4-methylgyoxal, 6.41 min. A next-generation heated ion spray ionization source with the following parameters was used: spray voltage, 4600 V; vaporizer temperature, 100°C; and ion transfer tube temperature, 300°C. Multiple reaction monitoring was used to identify and quantify the derivatized α-dicarbonlys with collision-induced dissociation at 2.5 mTorr using argon and with the following scan parameters: 3-DG, m/z 235.1 (fragments m/z 145.1, 157.1, 171.1, and 199.2; CE = 24 V, 22 V, 22 and 18 V); glyoxal, m/z 131.1 (fragments m/z 77.2 and 104.1; CE = 30 and 21 V); MG, m/z 145.1 (fragments m/z 77.2 and 118.1; CE = 31 and 23 V), and d4-MG m/z 149.1 (fragments 77.2 and 121.1; CE = 32 and 24 V). Quantification of MG was performed using the stable isotope dilution method with a calibration curve using the ratio of the areas under the peaks of equal amounts MG (fragment m/z 118.1) to d4-MG (fragment m/z 121.1). Recovery was between 90% and 95% for plasma and brain homogenates and the imprecision (CV%) was between 2% and 7%. Quantification of other α-dicarbonyls was done semiquantitatively in relation to the MG calibration curve. Ten percent of the number of samples were run as duplicates and one quality control sample was run every 20–30 samples.
OGD and CSF-1 ELISA.
As an in vitro model of ischemia, we exposed PBECs to OGD. The medium was changed to glucose-free DMEM containing EC growth supplement and 2-deoxy-d-glucose (5 mm). Cells were treated with MG (500 μm) for 1 h before being transferred to an anaerobic chamber that was flushed with 5% CO2 and 95% N2 for 15 min and then sealed. After incubation for 4.5 h at 37°C, cells were switched back to EC-medium for additional 4 h before being lysed for mRNA preparation and the medium was collected for BMDM-treatment or CSF-1 measurement by ELISA (R&D Systems). ELISA was performed according to the manufacturer's instructions.
Real-time RT-PCR.
RNA was isolated from PBECs or BMDMs using the NucleoSpin 96 RNA purification kit (Macherey-Nagel) according to the manufacturer's instructions. Then, 400 ng of RNA was transcribed to cDNA with the Cloned AMV First-Strand cDNA Synthesis kit (Life Technologies) and oligo(dT)20 primers. The following primers were used for quantitative RT-PCR: Csf1 forward, 5′-GTG TCA GAA CAC TGT AGC CAC-3′, Csf1 reverse, 5′-TCA AAG GCA ATC TGG CAT GAA G-3′, PCR product 101 bp; Tnf forward, 5′-TGT AGC CCA CGT CGT AGC AAA-3′, Tnf reverse, 5′-GCT GGC ACC ACT AGT TGG TTG T-3′, PCR product 120 bp; Ager forward, 5′-ATT CAG CTG TTG GTT GAG CCT-3′, Ager reverse 5′-CCA TCC TTT ATC CAG TGG ACC T-3′, PCR product 113 bp; Chil3 forward, 5′-AGC CAG CAG AAG CTC TCC AGA AGC-3′, Chil3 reverse, 5′-TGC CAG ACC TGT GAC AAG AAT GAG C-3′, PCR product 75 bp; Retnla forward, 5′-TCC TGC CCT GCT GGG ATG ACT GCT A-3′, Retnla reverse, 5′-CAG CGG GCA GTG GTC CAG TCA A-3′, PCR product 125 bp; Arg1 forward 5′-TGG TGT GGT GGC AGA GGT CCA-3′, Arg1 reverse, 5′-ACT GCC AGA CTG TGG TCT CCA CC-3′, PCR product 72 bp; Il1b forward 5′-CTG TGA CTC ATG GGA TGA TGA TG-3′, Il1b reverse 5′-GGA GCC TGT AGT GCA GTT G-3′; PCR product 74 bp; Ppia forward, 5′-AGG TCC TGG CAT CTT GTC CAT-3′, Ppia reverse, PCR product 51 bp. Quantitative RT-PCR was performed with Platinum SYBR Green qPCR SuperMix (Invitrogen) at 40 cycles of the following protocol: 2 min at 50°C, 2 min at 95°C, 15 s at 95°C, and 1 min at 60°C with the ABI PRISM 7000 Sequence Detection System (Applied Bioscience). Quantified results were normalized to Ppia using the ΔΔCt method.
Statistical analysis.
Values are shown as means ± SEM. For comparison of two groups, t tests were used. For more than two groups, one-way ANOVA and Tukey's multiple-comparisons test or two-way ANOVA followed by Bonferroni post hoc test were used. On the basis of the SD of infarcts that we had determined in previous experiments (25% of means), we chose a sample size of n = 9–11 to detect a 40% reduction of infarct size with a power of 0.8 and an error of 0.05, depending on specific experimental conditions, when using two-way ANOVA with four groups. Data analysis was performed with GraphPad Prism 5 software.
Results
Establishment of a mouse model of hyperglycemic stroke
To analyze the effect of hyperglycemia on ischemic stroke, we subjected mice to permanent distal MCAO. We used pentobarbital for anesthesia because it does not elevate blood glucose levels, in contrast to tribromoethanol or other anesthetic agents (Fig. 1A; Brown et al., 2005; Muhammad et al., 2008). After injecting glucose (50 mg in 0.2 ml, intraperitoneally) at the time of vessel occlusion, blood glucose levels were higher than in the normoglycemic group treated with normal saline (Fig. 1A). Forty-eight hours after MCAO, infarct volumes were larger in hyperglycemic mice than in normoglycemic mice (Fig. 1B,C).
Figure 1.

Hyperglycemia increases the infarct volume. A, Blood glucose concentrations were significantly increased when mice were treated with 50 mg of glucose. Blood glucose concentrations were measured in anesthetized mice immediately before (0 min) and 30 min and 50 min after the glucose or normal saline injections. MCAO was performed after the first measurement. Two-way ANOVA, for glucose F(1, 36) = 12.52, p = 0.0023. ***p ≤ 0.001. **p ≤ 0.01 (Bonferroni posttests, n = 11 mice/group). B, Forty-eight hours after MCAO, the hyperglycemic group had larger infarcts than the normoglycemic group. Typical silver-stained coronal brain sections showing infarcts in light gray are depicted on top of the panel. Unpaired t test, **p < 0.001 (n = 13 mice/group). Scale bar, 2.5 mm. C, Distribution of infarcts on coronal sections in normoglycemic and hyperglycemic groups (F(1,240) = 13.9, p = 0.0013, repeated-measures ANOVA, n = 11 mice/group). Data are shown as means ± SEM.
Hyperglycemia downregulates noninflammatory monocytes/macrophages
β-hydroxybutyrate, an alternative energy substrate of the CNS, modulates ischemic brain damage by acting on monocytes and/or macrophages (Rahman et al., 2014). To determine whether this cell population would also mediate the effect of glucose, we analyzed myeloid cells 48 h after onset of MCAO by flow cytometry. The percentage of CD45LoCD11b+ cells corresponding to resident microglia in the ischemic hemisphere that were mostly Ly-6CLo were not significantly affected by hyperglycemia (Fig. 2A–D,G). CD45HiCD11b+ cells were detected only after MCAO in the ischemic brain and represent bone-marrow-derived cells, as described previously (Gliem et al., 2012). Among CD45HiCD11b+ cells, inflammatory monocytes/macrophages (Ly-6CHiLy-6G−) and neutrophils (Ly-6C−Ly-6G+) did not significantly change in number under hyperglycemic conditions (Fig. 2E,F,I,J; Mosser and Edwards, 2008). In contrast, hyperglycemia reduced noninflammatory monocytes/macrophages (Ly-6CLoLy-6G−) in the ischemic hemisphere (Fig. 2H), although the infarct volume increased under the same conditions (Fig. 1).
Figure 2.
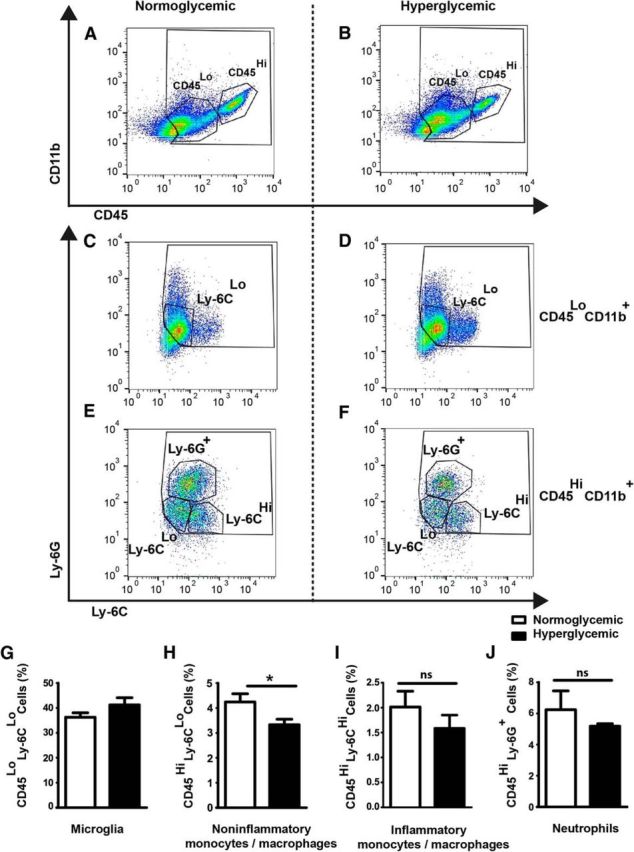
Flow cytometric characterization of immune cells in the ischemic brain under normoglycemic and hyperglycemic conditions. A–F, Forty-eight hours after MCAO, 40,000 events were analyzed. First, the cells were gated by CD45 and CD11b expression (A, B). Representative dot plots of gated CD45LoCD11b+ (C, D) and CD45HiCD11b+ cells (E, F) that were stained for Ly-6G and Ly-6C in the normoglycemic (A, C, E) and hyperglycemic groups (B, D, F) are shown. G–J, Quantification of CD45LoCD11b+Ly-6CLo microglia (p = 0.1684, t test), CD45HiCD11b+Ly-6CLo (*p < 0.03, unpaired t test), CD45HiCD11b+Ly-6CHi monocytes/macrophages (p = 0.3202, t test), and CD45HiCD11b+Ly-6G+ neutrophils are shown (p = 0.4012, t test). Data are presented as the mean percentage of all events counted during acquisition ± SEM (n = 11 mice/group).
Hyperglycemia reduces the number of infiltrating Arg1+ noninflammatory monocytes/macrophages
To verify the effect of hyperglycemia on the myeloid cell polarization, we stained brain sections for Iba1, a marker of monocytes/macrophages and microglia, and for Arg1, which is expressed by noninflammatory cells (Mosser and Edwards, 2008; Hickman et al., 2013). After stroke, hyperglycemia in mice did not affect the overall number of Iba1+ cells in the periphery of the ischemic lesion (normoglycemic group, 258 ± 16 Iba1+ cells/mm2; hyperglycemic group, 220 ± 16 Iba1+ cells/mm2, p = 0.13, t test), but did reduce the number of Iba1+ cells that expressed Arg1 (Fig. 3A–G), thereby confirming a downregulation of noninflammatory myeloid cells by hyperglycemia. In contrast, hyperglycemia did not alter the number of Arg1−Iba1+ cells (Fig. 3A–F,H).
Because flow cytometry had indicated that hyperglycemia affects polarization of monocytes/macrophages in the ischemic brain (Fig. 2), we labeled BMDMs by transplanting Cx3cr1GFP/+ bone marrow to C57BL/6 mice (Fig. 4A). Hyperglycemia had no effect on the number of Cx3cr1GFP/+ cells that were negative for Arg1 and the proinflammatory marker iNOS (Fig. 4B–J), but did reduce the number of Cx3cr1GFP/+Arg1−iNOS+ inflammatory cells (53 ± 25% reduction; Fig. 4B–I, K) and had a more pronounced effect on Cx3cr1GFP/+Arg1+iNOS+ cells, representing a population that presumably switches from a proinflammatory to a noninflammatory polarization (73 ± 4% reduction, Fig. 4B–I, L; Kigerl et al., 2009; Miron et al., 2013; Melero-Jerez et al., 2016). Furthermore, hyperglycemia significantly decreased the number of Cx3cr1GFP/+Arg1+iNOS− noninflammatory monocytes/macrophages (53 ± 19% reduction; Fig. 4B–I, M) and the Arg1 intensity per cell (data not shown). An analysis of the anatomical distribution of Cx3cr1GFP/+Arg1+ cells showed that the most prominent differences between normoglycemic and hyperglycemic mice were detected in the infarct border located in the motor cortex, whereas the effect lost its significance in the striatum and was undetectable in the insular cortex (Fig. 4N–Q). Notably, the motor cortex, where hyperglycemia had the most pronounced effects on monocyte/macrophage polarization, contains most salvageable tissue in the infarct model used here (Herrmann et al., 2005).
Identifying the role of monocytes/macrophages in hyperglycemic stroke
BMDMs infiltrate the ischemic brain as monocytes (Gliem et al., 2012). To elucidate the role of this cell population for hyperglycemic stroke, we ablated monocytes by treating CD11b-DTR mice with diphtheria toxin (DT). Consistent with previous data, DT had no effect on the blood count of neutrophils (Fig. 5A–G; Duffield et al., 2005). However, treatment with DT led to a >80% loss of Ly-6CLo and Ly-6CHi monocytes in CD11b-DTR mice, whereas treatment of the wild-type littermates had no effect (Fig. 5A–F). Ablation of monocytes slightly increased the infarct size under normoglycemic conditions, although this did not reach statistical significance (Fig. 5H). Hyperglycemia enlarged the infarct size. Post hoc testing showed that this effect was significant in wild-type animals, but not in CD11b-DTR mice in which monocytes were ablated (Fig. 5H), suggesting that monocytes are required for the detrimental effect of hyperglycemia. However, formal proof is missing because two-way ANOVA analysis showed that the interaction of genotype and glucose treatment was not statically significant, possibly because of the sample size.
Figure 5.
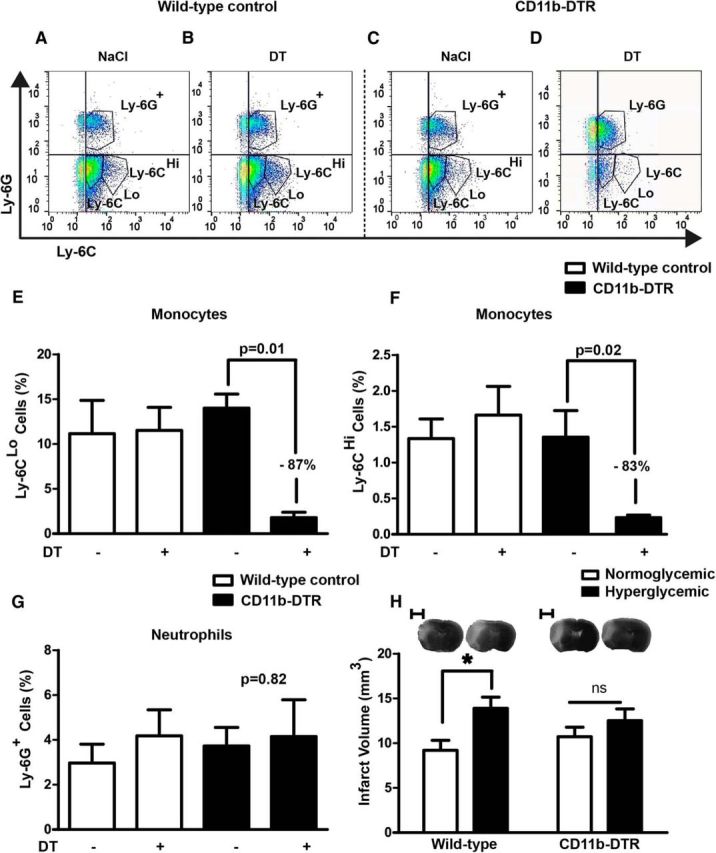
After ablation of monocytes hyperglycemia does not increase the infarct size. A–D, To ablate monocytes, we injected CD11b-DTR and wild-type littermates with DT. Flow cytometry on blood cells confirmed the ablation. Representative dot plots of the gated CD45+CD11b+ cell population stained for Ly-6C and Ly-6G are shown. E–G, Quantification of Ly-6CLo monocytes, Ly-6CHi monocytes, and Ly-6G+ neutrophils (n = 4 mice/group). Data are presented as a percentage of 50,000 events. H, The detrimental effect of hyperglycemia on the infarct size 48 h after MCAO was lost when CD11b+ cells had been ablated. Two-way ANOVA, for glucose treatment, F(1, 40) = 7.49, p = 0.0092; for genotype, F(1, 40) = 0, p = 0.9544; interaction, F(1, 40) = 1.5, p = 0.2289; *p < 0.05 (Bonferroni posttests, n = 11 mice/group). Scale bar, 2.5 mm. Data are shown as means ± SEM.
AGER mediates hyperglycemic brain damage during stroke
Glucose degradation leads to the formation of the α-dicarbonyls MG, glyoxal, and 3-DG (Rabbani and Thornalley, 2015). In humans, plasma concentrations of these three α-dicarbonyls have been reported previously to be elevated during acute hyperglycemia (Beisswenger et al., 2003; Maessen et al., 2015). In mice subjected to stroke, hyperglycemia elevated 3-DG concentrations in plasma (Fig. 6A–C), but had no statistically significant effect on glyoxal and MG levels, at least 30 and 50 min after stroke, when glucose levels were high. In extracts of the ischemic hemispheres, hyperglycemia increased glyoxal, but not MG levels; 3-DG could not be detected (Fig. 6D,E and data not shown). The fast metabolism of MG may explain why this compound was not elevated (Rabbani and Thornalley, 2015).
Figure 6.

AGER increases the infarct volume during hyperglycemic stroke and impairs macrophage polarization to a noninflammatory cell type. A, B, Concentrations of the α-dicarbonyls glyoxal, MG, and 3-DG were measured before glucose injection (0 min) and 30 min and 50 min after hyperglycemic stroke in plasma. Only 3-DG revealed a significant increase under hyperglycemic conditions (A–C; for glucose treatment, F(1, 56) = 107.07, p < 0.0001,***p < 0.001, Bonferroni posttests, n = 8 mice/group). In brain extracts, only glyoxal was elevated significantly by hyperglycemia (D, E; for glucose treatment, F(1, 27) = 14.47, p = 0.0007, **p < 0.01, Bonferroni posttests, n = 8 mice/group). F, Hyperglycemic Ager−/− mice had smaller infarcts than wild-type mice 48 h after MCAO. Typical silver-stained coronal brain sections showing infarcts in light gray are depicted on top of the panel. Two-way ANOVA, for glucose treatment F(1, 32) = 4.75, p = 0.0368. *p < 0.05, **p = < 0.01 (Bonferroni posttests, n = 9 mice/group). Scale bar, 2.5 mm. G, Hyperglycemia did not reduce noninflammatory Arg1+Iba1+ macrophages in the peri-infarct tissue of Ager−/− mice, in contrast to wild-type mice, 48 h after MCAO. The total number of Iba1+ cells did not differ between treatment groups. Two-way ANOVA, for interaction between glucose treatment and genotype F(1, 23) = 6.03, p = 0.022. *p < 0.05 (Bonferroni posttests, n = 8 mice/group). H–J, After polarization to a noninflammatory phenotype with IL-4, AGER-deficient macrophages had higher mRNA levels of the noninflammatory marker genes Arg1, Chil3, and Retlna than wild-type controls. K, L, Conversely, after polarization using LPS, AGER-deficient macrophages had lower mRNA levels of the inflammatory marker Il1b than wild-type controls, although Tnf levels did not differ. M, In vitro Ager mRNA expression was higher in inflammatory than in noninflammatory macrophages. *p < 0.05, **p < 0.01, ***p < 0.0003 (n = 12 with 4 wells in three independent experiments, unpaired t test). Data are shown as means ± SEM.
α-Dicarbonyl metabolites of glucose are important precursors of AGEs. To evaluate the role of AGEs in hyperglycemic stroke, we investigated mice deficient in AGER (also known as RAGE). Hyperglycemia enlarged the infarct volume in wild-type but not in Ager−/− animals (Fig. 6F), demonstrating that interaction of AGER is required for the effects of hyperglycemia on ischemic brain damage.
In Ager−/− animals, hyperglycemia did not significantly reduce the number of Arg1+Iba1+ noninflammatory cells, in contrast to its effect in wild-type mice (Fig. 6G), suggesting that AGER interferes with the noninflammatory polarization of monocytes/macrophages in the ischemic brain under hyperglycemic conditions (Juranek et al., 2013; Jin et al., 2015). This view was supported by in vitro experiments. The noninflammatory marker genes Arg1, Chil3 (Ym1), and Retnla (Fizz) were higher in Ager−/− than in wild-type BMDMs after polarizing cells with IL-4 to a noninflammatory phenotype (Fig. 6H–J). Conversely, the expression of Il-1β, a marker for inflammatory macrophages, was lower in Ager−/− than in wild-type cells after polarization to an inflammatory cell type by LPS treatment; however, mRNA expression of Tnf, another proinflammatory gene, did not differ (Fig. 6K,L). Interestingly, when we polarized wild-type macrophages to a noninflammatory cell type, they expressed less Ager mRNA than after polarization to an inflammatory cell type (Fig. 6M), indicating that the regulation of Ager expression may determine macrophage polarization.
The reactive glucose metabolite MG interferes with endothelium-mediated noninflammatory polarization of macrophages
When infiltrating the ischemic brain, monocytes have to pass the brain endothelium that expresses AGER. Therefore, we wondered whether PBECs could modulate the polarization of infiltrating monocytes/macrophages, potentially in a α-dicarbonyl- and AGER-dependent manner. Treating PBECs with nontoxic concentrations of MG (500 μm, data not shown), the most widely studied α-dicarbonyl (Rabbani and Thornalley, 2015), enhanced the expression of the proinflammatory cytokine Tnf and reduced the expression of Csf1 (Fig. 7A,B). CSF-1 has been shown to be neuroprotective in stroke (Lampron et al., 2013) and to shift the macrophage polarization toward a noninflammatory cell type (Mia et al., 2014), suggesting that lower endothelial Csf1 expression in response to MG might impair macrophage polarization to a noninflammatory cell type. Mimicking ischemic conditions with OGD stimulated secretion of CSF-1 from PBECs, but had no statistically significant effect on mRNA levels (Fig. 7B,C). Treatment with MG reduced CSF-1 secretion in both normal and OGD conditions, consistent with mRNA expression (Fig. 7B,C). To test the functional significance of paracrine factors released by endothelial cells, we treated BMDM with the medium conditioned by PBECs (Fig. 7D). Stimulation of BMDM with conditioned medium of PBECs, which had only been treated with OGD, increased expression of Retnla and Chil3 compared with treatment with unconditioned medium, indicating a shift to the noninflammatory phenotype (Fig. 7E,F). However, stimulation with the conditioned medium from PBECs cotreated with MG and OGD prevented this shift. Treatment with MG alone did not have an effect (Fig. 7E,F). To test whether the enhanced Retnla and Chil3 expression depends on CSF-1, we added CSF-1-neutralizing antibodies to the conditioned medium. This resulted in a similar reduction in Retnla and Chil3 expression as when the PBECs were cotreated with MG and OGD. Therefore, by reducing endothelial Csf1 expression, MG interferes with the noninflammatory polarization of macrophages. When performing these experiments in Ager−/− PBECs, the same results were obtained (data not shown), indicating that the effect of MG in PBECs is independent of AGER.
Figure 7.

By reducing expression and release of CSF-1 in PBECs, MG indirectly impairs noninflammatory polarization of macrophages. A, MG treatment increased Tnf expression in PBECs under control conditions and after OGD. Two-way ANOVA, F(1, 65) = 8.58, p = 0.0047. *p < 0.05, **p < 0.01, ***p < 0.001 (n = 18 with 6 wells in 3 independent experiments, Bonferroni posttests). B, C, In contrast, MG reduced mRNA levels of Csf1 (F(1, 68) = 23.78, p < 0.0001) and the release of CSF-1 from PBECs (F(1, 16) = 134.7, p < 0.0001, n = 18 with 6 wells in 3 independent experiments). D, To investigate the crosstalk of PBEC with BMDM by the secretion of CSF-1, BMDMs were treated with the conditioned medium (CM) of PBECs that were stimulated with MG and OGD as indicated. E, F, In BMDMs, the expression of Retnla and Chil3, typical marker genes of noninflammatory macrophages, was diminished by the CM of MG- and OGD-challenged PBECs. A comparable effect was obtained by adding CSF-1-neutralizing antibodies (α-CSF-1) to CM. Expression was normalized to controls that did not receive CM. One-way ANOVA, *p < 0.05, **p < 0.01, ***p < 0.005 (n = 15–28, Tukey's multiple-comparisons test). Data are shown as means ± SEM.
Discussion
Hyperglycemia is common in patients with acute stroke and denotes poor prognoses. So far, the mechanism(s) by which hyperglycemia aggravates ischemic brain damage was unclear, which poses an obstacle to the rational treatment for hyperglycemic stroke.
Our data suggest that hyperglycemia exerts its detrimental effect through monocytes. Inflammatory monocytes infiltrate the ischemic brain tissue and polarize under normoglycemic conditions to noninflammatory Arg1+ Ly-6CLo macrophages that have neuroprotective properties (Gliem et al., 2012; Rahman et al., 2014; Gliem et al., 2016). Hyperglycemia interferes with this process, as shown by lower numbers of noninflammatory Arg1+ or Ly-6CLo macrophages in the ischemic brain of hyperglycemic mice (Figs. 2F, 3G, 4M). In contrast, effects of hyperglycemia on inflammatory Ly-6CHi or iNOS+ cells were more variable (Figs. 2G, 4K). Interestingly, hyperglycemia also reduced the number of macrophages expressing both noninflammatory (Arg1+) and inflammatory (iNOS+) markers. These Arg1+iNOS+ cells might be caught in transition from an inflammatory to a noninflammatory phenotype (Kigerl et al., 2009; Miron et al., 2013). However, without cell fate studies, this interpretation is hypothetical. Alternatively, Arg1+iNOS+ cells might represent myeloid-derived suppressor cells, a heterogeneous cell population that has been reported in other CNS diseases (Melero-Jerez et al., 2016).
In addition to their neuroprotective effects, noninflammatory monocytes/macrophages promote tissue repair and regeneration. Ablation of these cells elevates the risk of hemorrhages after ischemic stroke (Gliem et al., 2012). Lower numbers of noninflammatory macrophages in hyperglycemic stroke are a likely cause of the higher rate of symptomatic intracerebral hemorrhages that has been found in hyperglycemic stroke patients treated with thrombolysis (Ahmed et al., 2010; Gliem et al., 2015).
By interfering with polarization toward a noninflammatory cell type, hyperglycemia may also obstruct endogenous protective mechanisms outside of the CNS. Noninflammatory macrophages are involved in tissue repair after myocardial infarction, kidney injury, or in limiting early stages of atherosclerosis (Nahrendorf et al., 2007; Ricardo et al., 2008; Shah et al., 2011; Brenner et al., 2015). On this background, it is even more important to know how hyperglycemia and glucose metabolism affect polarization of macrophages. Acute hyperglycemia increases plasma levels of the reactive α-dicarbonyl metabolites (Beisswenger et al., 2003; Maessen et al., 2015). In our mouse model, we observed elevated levels of 3-DG in plasma and glyoxal in ischemic brain tissue, but we might have missed an increase of MG by investigating only two time points. α-Dicarbonyls are the main precursors of AGEs that activate AGER. Accordingly, hyperglycemia did not increase ischemic brain damage in Ager−/− mice, providing evidence for the essential role of AGER in the damage induced by acute hyperglycemia (Fig. 6C). In support of this concept, AGER deficiency also reduced ischemic brain damage when hyperglycemia was induced as a side effect of tribromoethanol anesthesia during MCAO surgery (Brown et al., 2005; Muhammad et al., 2008). Under normoglycemic conditions, however, infarct volumes did not differ between Ager−/− and wild-type mice (Fig. 6C).
AGER is expressed by both BMDMs and PBECs that form the microenvironment in which monocytes polarize to protective noninflammatory macrophages (Vlassara et al., 1985; Deane et al., 2003). Consistent with previous studies, our data show that AGER inhibits noninflammatory polarization of BMDMs (Fig. 8; Juranek et al., 2013; Jin et al., 2015). Inhibition of noninflammatory polarization by AGER explains why AGER deficiency exclusively in bone marrow-derived cells reduced the infarct size under hyperglycemic conditions (Muhammad et al., 2008).
Figure 8.

In cerebral ischemia, inflammatory monocytes/macrophages (green) infiltrate the brain and polarize in the parenchyma to a protective phenotype (red). Hyperglycemia impairs noninflammatory polarization of macrophages. In monocytes/macrophages, AGER, the receptor for AGEs, inhibits polarization toward a protective phenotype. MG, an α-dicarbonyl derivative of glucose, reduces the expression and release of the cytokine CSF-1 from endothelial cells. The latter promotes the polarization of macrophages to a noninflammatory cell type.
In addition, our data reveal an AGER-independent mechanism by which the glucose metabolite MG can disturb the microenvironment that is responsible for noninflammatory polarization of macrophages in the brain. In cerebral ischemia, inflammatory monocytes infiltrate the ischemic brain and polarize to noninflammatory macrophages (Gliem et al., 2012). However, the mechanism(s) that triggers noninflammatory polarization in the brain was unclear. A key factor that promotes noninflammatory polarization is CSF-1 (Hamilton, 2014), which is expressed in PBECs and is upregulated by cerebral ischemia (Ryan et al., 2001; Schmidt-Kastner et al., 2002; Daneman et al., 2010). Our data show that ischemia induces the release of CSF-1 from PBECs (Fig. 7C). By releasing CSF-1, PBECs polarize macrophages toward a noninflammatory phenotype under ischemic conditions (Fig. 7D,E). MG can reduce the expression of Csf1, thereby preventing macrophage polarization (Fig. 7). In summary, we have identified two mechanisms by which glucose metabolites could interfere with macrophage polarization and ischemic brain injury. One comprises a cell-autonomous process in macrophages that depends on AGER and the other is AGER independent and relates to the crosstalk of PBECs and BMDMs (Fig. 8).
By increasing glucose degradation (Foufelle and Ferré, 2002), insulin may promote the intracellular production of α-dicarbonyls. Due to their detrimental effect, it may be questionable whether insulin is the correct choice in the treatment of patients with hyperglycemic stroke. Indeed, insulin was not effective in clinical trials of hyperglycemic stroke (Bellolio et al., 2014). Other treatment strategies that lower α-dicarbonyls are available (Beisswenger et al., 1999; Bierhaus et al., 2012) and should be tested to reverse the toxic effects of hyperglycemia in stroke.
Footnotes
The research leading to these results received funding from the Deutsche Forschungsgemeinschaft (Grant SCHW 413/7-1 to M.S. and Grant SFB 1118 to P.P.N. and T.F.) and the Spanish Ministerio de Economia y Competitividad (partially financed by FEDER; European Union “Una manera de hacer Europa”; Grant PI15/00963 to D.C.). We thank Prof. Dr Arnold von Eckardstein and Dr. Thorsten Hornemann for providing access to their mass spectrometry laboratory at the Institute of Clinical Chemistry, University of Zurich, during the early phases of the LC/MS method development for dicarbonyl measurement and Dr. Rodríguez-Alfaro and Dr. Mazarío for help with the confocal imaging.
The authors declare no competing financial interests.
References
- Ahmed N, Dávalos A, Eriksson N, Ford GA, Glahn J, Hennerici M, Mikulik R, Kaste M, Lees KR, Lindsberg PJ, Toni D. Association of admission blood glucose and outcome in patients treated with intravenous thrombolysis: results from the Safe Implementation of Treatments in Stroke International Stroke Thrombolysis Register (SITS-ISTR) Arch Neurol. 2010;67:1123–1130. doi: 10.1001/archneurol.2010.210. [DOI] [PubMed] [Google Scholar]
- Beisswenger PJ, Howell SK, Touchette AD, Lal S, Szwergold BS. Metformin reduces systemic methylglyoxal levels in type 2 diabetes. Diabetes. 1999;48:198–202. doi: 10.2337/diabetes.48.1.198. [DOI] [PubMed] [Google Scholar]
- Beisswenger PJ, Howell SK, Nelson RG, Mauer M, Szwergold BS. Alpha-oxoaldehyde metabolism and diabetic complications. Biochem Soc Trans. 2003;31:1358–1363. doi: 10.1042/bst0311358. [DOI] [PubMed] [Google Scholar]
- Bellolio MF, Gilmore RM, Ganti L. Insulin for glycaemic control in acute ischaemic stroke. Cochrane Database Syst Rev. 2014;1:CD005346. doi: 10.1002/14651858.CD005346.pub4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bierhaus A, Fleming T, Stoyanov S, Leffler A, Babes A, Neacsu C, Sauer SK, Eberhardt M, Schnölzer M, Lasitschka F, Neuhuber WL, Kichko TI, Konrade I, Elvert R, Mier W, Pirags V, Lukic IK, Morcos M, Dehmer T, Rabbani N, et al. Methylglyoxal modification of Nav1.8 facilitates nociceptive neuron firing and causes hyperalgesia in diabetic neuropathy. Nat Med. 2012;18:926–933. doi: 10.1038/nm.2750. [DOI] [PubMed] [Google Scholar]
- Brenner C, Franz WM, Kühlenthal S, Kuschnerus K, Remm F, Gross L, Theiss HD, Landmesser U, Kränkel N. DPP-4 inhibition ameliorates atherosclerosis by priming monocytes into M2 macrophages. Int J Cardiol. 2015;199:163–169. doi: 10.1016/j.ijcard.2015.07.044. [DOI] [PubMed] [Google Scholar]
- Brown ET, Umino Y, Loi T, Solessio E, Barlow R. Anesthesia can cause sustained hyperglycemia in C57/BL6J mice. Vis Neurosci. 2005;22:615–618. doi: 10.1017/S0952523805225105. [DOI] [PubMed] [Google Scholar]
- Capes SE, Hunt D, Malmberg K, Pathak P, Gerstein HC. Stress hyperglycemia and prognosis of stroke in nondiabetic and diabetic patients: a systematic overview. Stroke. 2001;32:2426–2432. doi: 10.1161/hs1001.096194. [DOI] [PubMed] [Google Scholar]
- Constien R, Forde A, Liliensiek B, Gröne HJ, Nawroth P, Hämmerling G, Arnold B. Characterization of a novel EGFP reporter mouse to monitor Cre recombination as demonstrated by a Tie2 Cre mouse line. Genesis. 2001;30:36–44. doi: 10.1002/gene.1030. [DOI] [PubMed] [Google Scholar]
- Daneman R, Zhou L, Agalliu D, Cahoy JD, Kaushal A, Barres BA. The mouse blood-brain barrier transcriptome: a new resource for understanding the development and function of brain endothelial cells. PLoS One. 2010;5:e13741. doi: 10.1371/journal.pone.0013741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deane R, Du Yan S, Submamaryan RK, LaRue B, Jovanovic S, Hogg E, Welch D, Manness L, Lin C, Yu J, Zhu H, Ghiso J, Frangione B, Stern A, Schmidt AM, Armstrong DL, Arnold B, Liliensiek B, Nawroth P, Hofman F, et al. RAGE mediates amyloid-beta peptide transport across the blood-brain barrier and accumulation in brain. Nat Med. 2003;9:907–913. doi: 10.1038/nm890. [DOI] [PubMed] [Google Scholar]
- Duffield JS, Forbes SJ, Constandinou CM, Clay S, Partolina M, Vuthoori S, Wu S, Lang R, Iredale JP. Selective depletion of macrophages reveals distinct, opposing roles during liver injury and repair. J Clin Invest. 2005;115:56–65. doi: 10.1172/JCI200522675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dziedzic T, Pera J, Zur-Wyrozumska K, Klimkowicz-Mrowiec A, Szczudlik A, Slowik A. Beta-blockers use and risk of hyperglycemia in acute stroke patients. Atherosclerosis. 2012;223:209–211. doi: 10.1016/j.atherosclerosis.2012.04.017. [DOI] [PubMed] [Google Scholar]
- Foufelle F, Ferré P. New perspectives in the regulation of hepatic glycolytic and lipogenic genes by insulin and glucose: a role for the transcription factor sterol regulatory element binding protein-1c. Biochem J. 2002;366:377–391. doi: 10.1042/bj20020430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gliem M, Mausberg AK, Lee JI, Simiantonakis I, van Rooijen N, Hartung HP, Jander S. Macrophages prevent hemorrhagic infarct transformation in murine stroke models. Ann Neurol. 2012;71:743–752. doi: 10.1002/ana.23529. [DOI] [PubMed] [Google Scholar]
- Gliem M, Klotz L, van Rooijen N, Hartung HP, Jander S. Hyperglycemia and PPARγ antagonistically influence macrophage polarization and infarct healing after ischemic stroke. Stroke. 2015;46:2935–2942. doi: 10.1161/STROKEAHA.115.010557. [DOI] [PubMed] [Google Scholar]
- Gliem M, Schwaninger M, Jander S. Protective features of peripheral monocytes/macrophages in stroke. Biochim Biophys Acta. 2016;1862:329–338. doi: 10.1016/j.bbadis.2015.11.004. [DOI] [PubMed] [Google Scholar]
- Hamilton TA. Myeloid colony stimulating factors as regulators of macrophage polarization. Front Immunol. 2014:5. doi: 10.3389/fimmu.2014.00554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrmann O, Baumann B, de Lorenzi R, Muhammad S, Zhang W, Kleesiek J, Malfertheiner M, Köhrmann M, Potrovita I, Maegele I, Beyer C, Burke JR, Hasan MT, Bujard H, Wirth T, Pasparakis M, Schwaninger M. IKK mediates ischemia-induced neuronal cell death. Nat Med. 2005;11:1322–1329. doi: 10.1038/nm1323. [DOI] [PubMed] [Google Scholar]
- Hickman SE, Kingery ND, Ohsumi TK, Borowsky ML, Wang LC, Means TK, El Khoury J. The microglial sensome revealed by direct RNA sequencing. Nat Neurosci. 2013;16:1896–1905. doi: 10.1038/nn.3554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin X, Yao T, Zhou Z, Zhu J, Zhang S, Hu W, Shen C. Advanced glycation end products enhance macrophages polarization into M1 phenotype through activating RAGE/NF-kappaB pathway. Biomed Res Int. 2015;2015:732450. doi: 10.1155/2015/732450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung S, Aliberti J, Graemmel P, Sunshine MJ, Kreutzberg GW, Sher A, Littman DR. Analysis of fractalkine receptor CX(3)CR1 function by targeted deletion and green fluorescent protein reporter gene insertion. Mol Cell Biol. 2000;20:4106–4114. doi: 10.1128/MCB.20.11.4106-4114.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juranek JK, Geddis MS, Song F, Zhang J, Garcia J, Rosario R, Yan SF, Brannagan TH, Schmidt AM. RAGE deficiency improves postinjury sciatic nerve regeneration in type 1 diabetic mice. Diabetes. 2013;62:931–943. doi: 10.2337/db12-0632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kigerl KA, Gensel JC, Ankeny DP, Alexander JK, Donnelly DJ, Popovich PG. Identification of two distinct macrophage subsets with divergent effects causing either neurotoxicity or regeneration in the injured mouse spinal cord. J Neurosci. 2009;29:13435–13444. doi: 10.1523/JNEUROSCI.3257-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruyt ND, Biessels GJ, Devries JH, Roos YB. Hyperglycemia in acute ischemic stroke: pathophysiology and clinical management. Nat Rev Neurol. 2010;6:145–155. doi: 10.1038/nrneurol.2009.231. [DOI] [PubMed] [Google Scholar]
- Lampron A, Pimentel-Coelho PM, Rivest S. Migration of bone marrow-derived cells into the central nervous system in models of neurodegeneration. J Comp Neurol. 2013;521:3863–3876. doi: 10.1002/cne.23363. [DOI] [PubMed] [Google Scholar]
- Lubjuhn J, Gastens A, von Wilpert G, Bargiotas P, Herrmann O, Murikinati S, Rabie T, Marti H, Amende I, Hampton TG, Schwaninger M. Functional testing in a mouse stroke model induced by occlusion of the distal middle cerebral artery. J Neurosci Methods. 2009;184:95–103. doi: 10.1016/j.jneumeth.2009.07.029. [DOI] [PubMed] [Google Scholar]
- Maessen DE, Hanssen NM, Scheijen JL, van der Kallen CJ, van Greevenbroek MM, Stehouwer CD, Schalkwijk CG. Post-glucose load plasma α-dicarbonyl concentrations are increased in individuals with impaired glucose metabolism and type 2 diabetes: the CODAM study. Diabetes Care. 2015;38:913–920. doi: 10.2337/dc14-2605. [DOI] [PubMed] [Google Scholar]
- Melamed E. Reactive hyperglycaemia in patients with acute stroke. J Neurol Sci. 1976;29:267–275. doi: 10.1016/0022-510X(76)90176-3. [DOI] [PubMed] [Google Scholar]
- Melero-Jerez C, Ortega MC, Moliné-Velazquez V, Clemente D. Myeloid derived suppressor cells in inflammatory conditions of the central nervous system. Biochim Biophys Acta. 2016;1862:368–380. doi: 10.1016/j.bbadis.2015.10.015. [DOI] [PubMed] [Google Scholar]
- Mia S, Warnecke A, Zhang XM, Malmström V, Harris RA. An optimized protocol for human M2 macrophages using M-CSF and IL-4/IL-10/TGF-β yields a dominant immunosuppressive phenotype. Scand J Immunol. 2014;79:305–314. doi: 10.1111/sji.12162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miron VE, Boyd A, Zhao JW, Yuen TJ, Ruckh JM, Shadrach JL, van Wijngaarden P, Wagers AJ, Williams A, Franklin RJ, ffrench-Constant C. M2 microglia and macrophages drive oligodendrocyte differentiation during CNS remyelination. Nat Neurosci. 2013;16:1211–1218. doi: 10.1038/nn.3469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. 2008;8:958–969. doi: 10.1038/nri2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muhammad S, Barakat W, Stoyanov S, Murikinati S, Yang H, Tracey KJ, Bendszus M, Rossetti G, Nawroth PP, Bierhaus A, Schwaninger M. The HMGB1 receptor RAGE mediates ischemic brain damage. J Neurosci. 2008;28:12023–12031. doi: 10.1523/JNEUROSCI.2435-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nahrendorf M, Swirski FK, Aikawa E, Stangenberg L, Wurdinger T, Figueiredo JL, Libby P, Weissleder R, Pittet MJ. The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J Exp Med. 2007;204:3037–3047. doi: 10.1084/jem.20070885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pomeshchik Y, Kidin I, Korhonen P, Savchenko E, Jaronen M, Lehtonen S, Wojciechowski S, Kanninen K, Koistinaho J, Malm T. Interleukin-33 treatment reduces secondary injury and improves functional recovery after contusion spinal cord injury. Brain Behav Immun. 2015;44:68–81. doi: 10.1016/j.bbi.2014.08.002. [DOI] [PubMed] [Google Scholar]
- Rabbani N, Thornalley PJ. Dicarbonyl stress in cell and tissue dysfunction contributing to ageing and disease. Biochem Biophys Res Commun. 2015;458:221–226. doi: 10.1016/j.bbrc.2015.01.140. [DOI] [PubMed] [Google Scholar]
- Rahman M, Muhammad S, Khan MA, Chen H, Ridder DA, Muller-Fielitz H, Pokorna B, Vollbrandt T, Stölting I, Nadrowitz R, Okun JG, Offermanns S, Schwaninger M. The beta-hydroxybutyrate receptor HCA2 activates a neuroprotective subset of macrophages. Nat Commun. 2014;5:3944. doi: 10.1038/ncomms4944. [DOI] [PubMed] [Google Scholar]
- Ricardo SD, van Goor H, Eddy AA. Macrophage diversity in renal injury and repair. J Clin Invest. 2008;118:3522–3530. doi: 10.1172/JCI36150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridder DA, Lang MF, Salinin S, Röderer JP, Struss M, Maser-Gluth C, Schwaninger M. TAK1 in brain endothelial cells mediates fever and lethargy. J Exp Med. 2011;208:2615–2623. doi: 10.1084/jem.20110398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan GR, Dai XM, Dominguez MG, Tong W, Chuan F, Chisholm O, Russell RG, Pollard JW, Stanley ER. Rescue of the colony-stimulating factor 1 (CSF-1)-nullizygous mouse (Csf1(op)/Csf1(op)) phenotype with a CSF-1 transgene and identification of sites of local CSF-1 synthesis. Blood. 2001;98:74–84. doi: 10.1182/blood.V98.1.74. [DOI] [PubMed] [Google Scholar]
- Schmidt-Kastner R, Zhang B, Belayev L, Khoutorova L, Amin R, Busto R, Ginsberg MD. DNA microarray analysis of cortical gene expression during early recirculation after focal brain ischemia in rat. Mol Brain Res. 2002;108:81–93. doi: 10.1016/S0169-328X(02)00516-8. [DOI] [PubMed] [Google Scholar]
- Scott JF, Robinson GM, French JM, O'Connell JE, Alberti KG, Gray CS. Prevalence of admission hyperglycaemia across clinical subtypes of acute stroke. Lancet. 1999;353:376–377. doi: 10.1016/S0140-6736(05)74948-5. [DOI] [PubMed] [Google Scholar]
- Shah Z, Kampfrath T, Deiuliis JA, Zhong J, Pineda C, Ying Z, Xu X, Lu B, Moffatt-Bruce S, Durairaj R, Sun Q, Mihai G, Maiseyeu A, Rajagopalan S. Long-term dipeptidyl-peptidase 4 inhibition reduces atherosclerosis and inflammation via effects on monocyte recruitment and chemotaxis. Circulation. 2011;124:2338–2349. doi: 10.1161/CIRCULATIONAHA.111.041418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornalley PJ, Rabbani N. Assay of methylglyoxal and glyoxal and control of peroxidase interference. Biochem Soc Trans. 2014;42:504–510. doi: 10.1042/BST20140009. [DOI] [PubMed] [Google Scholar]
- Vlassara H, Brownlee M, Cerami A. High-affinity-receptor-mediated uptake and degradation of glucose-modified proteins: a potential mechanism for the removal of senescent macromolecules. Proc Natl Acad Sci U S A. 1985;82:5588–5592. doi: 10.1073/pnas.82.17.5588. [DOI] [PMC free article] [PubMed] [Google Scholar]