

Abstract
Potentiation of excitatory inputs onto dopamine neurons of the ventral tegmental area (VTA) induced by cocaine exposure allows remodeling of the mesocorticolimbic circuitry, which ultimately drives drug-adaptive behavior. This potentiation is mediated by changes in NMDAR and AMPAR subunit composition. It remains unknown how this synaptic plasticity affects the activity of dopamine neurons. Here, using rodents, we demonstrate that a single cocaine injection increases the firing rate and bursting activity of VTA dopamine neurons, and that these increases persist for 7 d. This enhanced activity depends on the insertion of low-conductance, Ca2+-impermeable NMDARs that contain GluN3A. Since such receptors are not capable of activating small-conductance potassium channels, the intrinsic excitability of VTA dopamine neurons increases. Activation of group I mGluRs rescues synaptic plasticity and restores small-conductance calcium-dependent potassium channel function, normalizing the firing activity of dopamine neurons. Our study characterizes a mechanism linking drug-evoked synaptic plasticity to neural activity, revealing novel targets for therapeutic interventions.
SIGNIFICANCE STATEMENT We show that cocaine-evoked synaptic changes onto ventral tegmental area (VTA) dopamine (DA) neurons leads to long-lasting increases in their burst firing. This increase is due to impaired function of Ca2+-activated small-conductance calcium-dependent potassium (SK) channels; SK channels regulate firing of VTA DA neurons, but this regulation was absent after cocaine. Cocaine exposure drives the insertion of GluN3A-containing NMDARs onto VTA DA neurons. These receptors are Ca2+-impermeable, and thus SK channels are not efficiently activated by synaptic activity. In GluN3A knock-out mice, cocaine did not alter SK channel function or VTA DA neuron firing. This study directly links synaptic changes to increased intrinsic excitability of VTA DA neurons after cocaine, and explains how acute cocaine induces long-lasting remodeling of the mesolimbic DA system.
Keywords: dopamine, firing rate, GluN3A, NMDA
Introduction
All addictive drugs induce synaptic plasticity at glutamatergic synapses onto dopamine (DA) neurons in the ventral tegmental area (VTA; Ungless et al., 2001; Bellone and Lüscher, 2006; Pascoli et al., 2012; Yuan et al., 2013). Within hours of a single injection of cocaine, synaptic strength of excitatory inputs onto VTA DA neurons is potentiated (Ungless et al., 2001; Bellone and Lüscher, 2006). The expression of this long-lasting plasticity is expressed by changes in AMPAR and NMDAR subunit composition (Bellone and Lüscher, 2006; Yuan et al., 2013) and promotes staged adaptations in target regions that eventually cause addictive behavior (Mameli et al., 2011; Pascoli et al., 2012). This drug-evoked synaptic plasticity at excitatory inputs onto VTA DA neurons contributes to drug-seeking behavior (Chen et al., 2008) and acts as an incentive signal to promote reward seeking (Berridge and Robinson, 1998; Wise, 2004).
NMDARs are heteromeric receptors; GluN2A and GluN2B are the most abundant subunits in the CNS and they assemble with GluN1 to form canonical heterodimeric or heterotrimeric receptors (Paoletti et al., 2013). These receptors are characterized by a strong voltage-dependent Mg2+ block at negative membrane potentials and they readily flux Ca2+. In contrast, the GluN3A subunit forms noncanonical NMDARs, which are quasi-Ca2+-impermeable, exhibit reduced Mg2+ sensitivity, and have lower conductance (Henson et al., 2010). After a single cocaine injection, GluN3A-containing NMDARs are inserted at excitatory inputs onto VTA DA neurons, where the NMDARs allow the insertion of GluA2-lacking, Ca2+-permeable AMPARs (Yuan et al., 2013).
In light of the redistribution of both AMPAR and NMDAR subunit composition, changes in firing of VTA DA neurons are difficult to predict, but may contribute to plasticity in target regions and to the expression of maladaptive behaviors. Midbrain DA neurons release DA most efficiently when the cells fire in short bursts of 5–10 action potentials (APs) at frequencies of >20 Hz (Overton and Clark, 1997). Glutamatergic transmission may drive the switch from pacemaker to burst activity of DA neurons in the VTA (Gonon, 1988; Zweifel et al., 2009). In particular, activation of NMDARs triggers burst firing of DA neurons, promotes release of DA in the nucleus accumbens, and regulates reward-predictive, cue-induced phasic DA release (Suaud-Chagny et al., 1992; Sombers et al., 2009).
The activity of VTA DA neurons is also modulated by small-conductance calcium-dependent potassium (SK) channels, which mediate the afterhyperpolarization (AHP) following each AP (Wolfart and Roeper, 2002). When SK channels are blocked, the firing frequency of VTA DA neurons in the pacemaker mode increases and more burst firing is observed (Shepard and Bunney, 1988). SK channels can be activated by synaptically evoked Ca2+ transients. This activation engages a negative-feedback loop that inactivates NMDARs due to voltage-dependent Mg2+ block, thus reducing the Ca2+ influx through NMDARs and consequently the amplitude of EPSPs (Ngo-Anh et al., 2005).
Extensive work has characterized changes in synaptic transmission and intrinsic activity of VTA DA neurons following cocaine exposure. However, it is not known whether these changes are causally related and, if so, how changes in synaptic transmission contribute to altered activity of VTA DA neurons. Here, we establish a causal link between cocaine-evoked insertion of GluN3A-containing receptors, SK channel function, and increased intrinsic activity of VTA DA neurons. These alterations may contribute to enhanced DA release, synaptic plasticity in target regions, and ultimately to drug-adaptive behavior.
Materials and Methods
Animals
Sprague Dawley rats (300–400 g; Janvier Labs) were used for in vivo recordings. Pixt3-GFP and GluN3A−/− mice (males and females crossed to C57B6 background for ≥10 generations, aged 25–28 or 84–98 d) were used for slice physiology experiments (https://www.jax.org/strain/013636, https://www.jax.org/strain/021962). Both rats and mice were housed three or four per cage under controlled conditions (22–23°C; 12 h light/dark cycle; lights on 7:00 A.M.) with food and water available ad libitum. All procedures were conducted in accordance with European Directive 2010-63-EU and with approval from the Bordeaux University Animal Care and Use Committee (N°50120205-A) and from the Veterinary Office Canton de Genève (Switzerland).
Drugs
Rats and mice received intraperitoneal injections of cocaine (15 mg/kg) or vehicle (0.9% saline) and were returned to their home cage until recordings were performed. Where indicated, rats were injected intraperitoneally with MK-801 (1 mg/kg) or vehicle (0.9% saline) 15 min before cocaine or saline injections, and VTA recordings were done after 24 h. Local VTA injections of apamin (100 μm) were done using double-barrel micropipettes (Georges and Aston-Jones, 2002). For ex vivo experiments, apamin and 3,5-dihydroxyphenylglycine (DHPG) were used at 100 nm and 20 μm respectively. Cocaine-hydrochloride was obtained from Sigma-Aldrich Biosciences, all other drugs used in ex vivo experiments were obtained from Tocris Bioscience.
Surgery
Rats were anesthetized with 4% isoflurane 2 l/min air and O2 for induction and placed in the stereotaxic frame. During surgical procedures and recordings, 1.5% isoflurane 2 l/min air and O2 were delivered through a facial mask via spontaneous respiration. Body temperature was maintained at 36–38°C with a thermistor-controlled electric heating pad during the procedure. For recordings, the skull was exposed and holes were drilled above the VTA (5.6 mm caudal to bregma, 0.6 mm lateral to bregma, and −7.2 mm ventral from dura) or above the tail of the VTA (tVTA; 6.8 mm caudal to bregma, 0.4 mm lateral to bregma, and 7.4 mm ventral from dura).
In vivo single-unit recordings
VTA DA neuron recordings.
A glass micropipette (1–2 μm, 10–12 mΩ) filled with 2.0% pontamine sky blue in 0.5 m sodium acetate was used for recording. The extracellular potential was recorded with an Axoclamp-2B amplifier and filter (300/0.5 Hz; (Georges and Aston-Jones, 2002). Single-neuron spikes were collected on-line (CED 1401, SPIKE 2, Cambridge Electronic Design). For VTA recordings, electrophysiological criteria used to identify putative DA neurons were similar to those of previous studies (Grace and Bunney, 1984; Ungless et al., 2004; Ungless and Grace, 2012). These included (1) AP with biphasic or triphasic waveform ≥2.5 ms in duration, (2) ≥1.1 ms from spike onset to negative trough, and (3) slow spontaneous firing rate of <10 spikes/s. Local injections of apamin (60–120 nl) during VTA dopaminergic recording were done as described above.
tVTA/right middle temporal gyrus and VTA GABA neuron recordings.
A glass micropipette (tip diameter, 1–2 μm; 10–12 MΩ) was lowered into the tVTA/right rostromedial tegmental nucleus (RMTg) or the VTA. Recordings were similar to those described above for VTA DA. As in previous reports (Jalabert et al., 2011; Lecca et al., 2012), we considered tVTA units to be putative GABA neurons if the early spike duration [peak neurons using as criteria: (1) an AP <1.1 ms in duration (peak from spike onset to initial negative trough) was ≤1.1 ms. tVTA GABA cells are reported to have highly variable basal firing rates [ranging from 1 to 60 Hz in the study by Jalabert et al., (2011)]. Therefore, we did not use this parameter to identify them. At the end of each recording experiment, the recording electrode placement was marked with an iontophoretic deposit of pontamine sky blue dye (−10 μA, pulsed current for 30 min). After the experimental procedures, the rats were deeply anesthetized with isoflurane (5%) and decapitated. Brains were removed and snap-frozen in a solution of isopentane at −80°C. Coronal, 40-μm-thick sections were cut on a cryostat and counterstained with neutral red. Histological localization of recording sites enabled us to verify the localization of the electrode tips.
Patch-clamp electrophysiology
Horizontal brain slices (180 μm) were prepared in cooled artificial CSF containing the following (in mm): 119 NaCl, 2.5 KCl, 1.3 MgCl, 2.5 CaCl2, 1.0 Na2HPO4, 26.2 NaHCO3, and 11 glucose, bubbled with 95% O2 and 5% CO2. Slices were kept at 30–34°C in a recording chamber superfused with 2.5 ml/min artificial CSF. Visualized whole-cell recording techniques were used to measure holding and synaptic responses of DA neurons in the VTA.
Spontaneous firing rate.
Recordings were performed in a cell-attached configuration with saline in the patch pipette. Recordings were included if spontaneous activity was maintained for 10 min with <20% variability; firing rate was analyzed during the last 5 min of recording. For apamin experiments, the drug was applied after 10 min of baseline and the firing rate was analyzed for a further 5 min.
AHP current.
Holding potential was maintained at −60 mV, and access resistance was monitored by a depolarizing step of −14 mV each sweep, every 20 s. The liquid junction potential was small (−3 mV); therefore traces were not corrected. Experiments were discarded if the access resistance varied by >20%. Currents were amplified, filtered at 5 kHz, and digitized at 20 kHz. AHP current (IAHP) was measured following a hyperpolarizing step of 60 mV applied for 100 ms. The SK-mediated component of the IAHP was determined by subtraction after the bath application of apamin (100 nm). The internal solution contained (in mm) 130 CsCl, 4 NaCl, 5 creatine phosphate, 2 MgCl2, 2 Na2ATP, 0.6 Na3 GTP, 1.1 EGTA, and 5 HEPES.
NMDA EPSP.
Recordings were made in current-clamp configuration, in Mg2+-free aCSF in the presence of NBQX (10 μm) and picrotoxin (50 μm) to isolate the NMDA component. Potentials were evoked with a bipolar stimulating electrode placed anterior to the VTA.
Data analysis and statistical analysis
To compare in vivo basal firing rates of neurons between groups, each cell was recorded for ≥120 s. In addition, for VTA DA neurons, burst analysis was performed. The onset of a burst was defined as the occurrence of two spikes with an interspike interval of <80 ms (Grace and Bunney, 1984). We evaluated the amount of bursting activity by calculating the bursting rate (number of burst events per second) and the mean spike per burst. To analyze the effect of apamin on VTA dopaminergic activity, we compared the firing rate and the bursting rate of recorded VTA DA neurons 60 s before injection to 60 s after injection. A maximum of one infusion of apamin was performed in each hemisphere. For ex vivo firing rate comparisons, cells were recorded for ≥10 min to determine a stable firing rate. For ex vivo LTP experiments, the final 5 min of the baseline measurement was compared with the final 5 min of the recording. Two group comparisons were performed with Student's t tests or Wilcoxon matched-pairs test. For multiple comparisons, values were subjected to a one-way ANOVA (with repeated measures where appropriate) followed by a Newman–Keuls post hoc test.
Results
Acute cocaine exposure induces long-term increases in firing rate and burst firing of VTA DA neurons
To directly examine the in vivo activity of VTA DA neurons 24 h after a single cocaine exposure, we recorded the extracellular activity from single VTA DA neurons identified according to their electrophysiological properties (Grace and Bunney, 1984; Jalabert et al., 2011; Ungless and Grace, 2012). We found a shift in activity distribution of VTA DA neurons toward higher frequencies following cocaine exposure (Fig. 1A–C; saline, 24 h, n = 45 cells/9 rats; cocaine, 24 h, n = 46 cells/8 rats; Kolmogorov–Smirnov comparison: D(91) = 0.2271; p < 0.05), and a significant increase in both firing rate and bursting activity of VTA DA neurons in cocaine-treated compared with saline-treated rats (Fig. 1D; saline, 24 h, n = 45 cells/9 rats; cocaine, 24 h, n = 46 cells/9 rats; firing rate: saline, 24 h, 3.79 ± 0.3 Hz; cocaine, 24 h, 5.98 ± 0.35 Hz, t(89) = 4.8, p < 0.0001; bursting rate: saline, 24 h, 0.64 ± 0.08 Hz; cocaine, 24 h, 0.98 ± 0.1 Hz, t(89) = 2.7, p = 0.0074; mean spike/burst: saline, 24 h, 2.47 ± 0.11; cocaine, 24 h, 5.98 ± 0.23, t(89) = 3.7, p = 0.0003). The increase in activity was apparent 3 h after a single injection and persisted up to 5 d (Fig. 1E; saline, n = 85 cells/15 rats, 3.89 ± 0.2 Hz; cocaine, 3 h, n = 33 cells/5 rats, 5.59 ± 0.4 Hz; cocaine, 6 h, n = 38 cells/5 rats, 5.35 ± 0.4 Hz; cocaine, 24 h, n = 46 cells/9 rats, 5.98 ± 0.35 Hz; cocaine, 5 d, n = 35 cells/5 rats, 5.18 ± 0.42 Hz; F(4,232) = 8.3 p < 0.001), mimicking the time course of cocaine-induced plasticity previously reported (Ungless et al., 2001; Mameli et al., 2007). Pretreatment with the NMDAR antagonist MK-801 blocked cocaine-evoked increases in firing rate and bursting activity (Fig. 1F,G; saline/vehicle, n = 45 cells/9 rats; vehicle/cocaine, n = 46 cells/8 rats; MK-801/saline, n = 37 cells/5 rats; MK-801/cocaine, n = 42 cells/4 rats; firing rate: saline/vehicle, 3.79 ± 0.2 Hz; vehicle/cocaine, 5.98 ± 0.35 Hz; MK-801/saline, 4.16 ± 0.44 Hz; MK-801/cocaine, 4.76 ± 0. 42 Hz, F(3,166) = 7.0, p = 0.0002; bursting rate: saline/vehicle, 0.63 ± 0.08 Hz; vehicle/cocaine, 0.98 ± 0.1 Hz; MK-801/saline, 0.42 ± 0.78 Hz; MK-801/cocaine, 0.38 ± 0.07 Hz, F(3,166) = 11.1, p < 0.0001; mean spike/burst: saline/vehicle, 2.47 ± 0.11; vehicle/cocaine, 3.46 ± 24; MK-801/saline, 2.47 ± 0.37; MK-801/cocaine, 2.49 ± 0.23 Hz, F(3,166) = 4.5, p = 0.0044), suggesting that NMDAR activation is necessary for the expression of cocaine-evoked activity changes. Although earlier reports suggested a correlation between sleep-like brain states and the activity pattern of DA neurons (Brown et al., 2009), the use of isoflurane in the experiment presented here promotes a predominant state of cortical activation and prevents cyclic brain state alternations (MacIver and Bland 2014). It is then unlikely that the changes in DA neuron firing patterns we observed several hours after acute cocaine could be due to cocaine-evoked changes in brain state. Critically, we did not find any significant difference in firing rate of putative GABA neurons recorded in the VTA (Fig. 1 H–J; saline, 24 h, n = 15 cells/8 rats, 17.8 ± 4.6 Hz; cocaine, 24 h, n = 23 cells/5 rats, 17.4 ± 2.7 Hz, t(36) = 0.075, p = 0.94) or tVTA/RMTg (Fig. 1K–M; saline, 24 h, n = 26 cells/7 rats, 19.2 ± 3.6 Hz; cocaine, 24 h, n = 26 cells/8 rats, 16.9 ± 2.8 Hz, t(50) = 0.49, p = 0.61), indicating that increased activity of VTA DA neurons observed 24 h after acute cocaine injection is not the consequence of inhibition of the mesencephalic GABAergic transmission.
Figure 1.
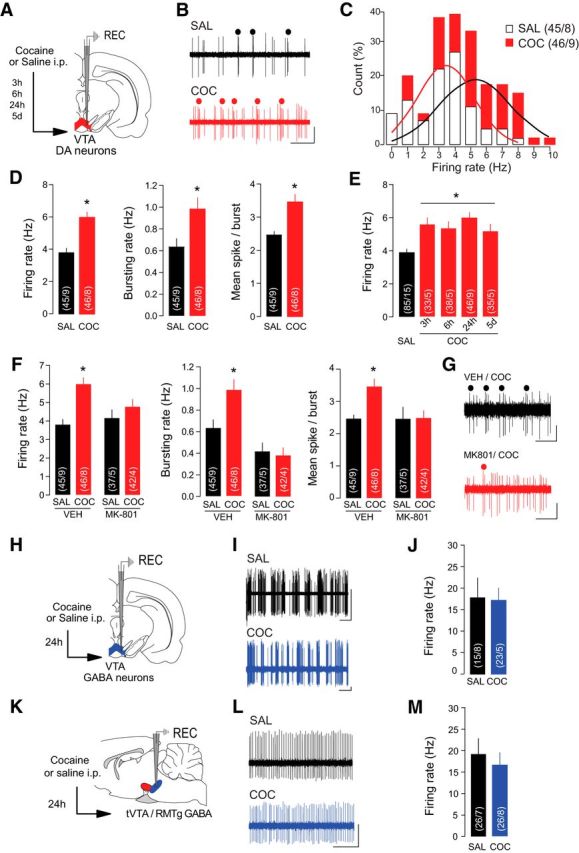
Cocaine increases firing rate and burst firing of VTA DA neurons in vivo. A, Schematic of the experimental design. Rats received an injection of cocaine (COC) or saline (SAL) 24 h before recordings of VTA DA neurons except where indicated (E). B, Representative traces from VTA DA neurons with burst events indicated. Scale bars: horizontal, 1 s; vertical, 1 mV. C, Distribution of VTA DA neurons by their firing rate. Solid line represents the best-fit normal distribution curve for the histogram data. D, Firing rate (left), bursting rate (middle), and mean spikes per burst (right) of VTA DA neurons were increased in COC-treated rats. E, The increase in VTA DA firing rate was significant when recordings were performed 3 h, 6 h, 24 h, and 5 d after COC exposure. F, Pretreatment with NMDAR antagonist MK-801 occluded COC-induced increases in firing rate (left), bursting rate (middle), and mean spikes per burst (right) of VTA DA neurons. G, Representative traces of VTA DA neurons from COC-treated rats with or without MK-801 pretreatment. Scale bars: horizontal, 1 s; vertical, 0.5 mV. H, Schematic of the experimental design. Rats were treated with SAL or COC 24 h before recording putative VTA GABA neurons. I, Sample traces from putative VTA GABA neurons. Scale bars: horizontal, 2 s; vertical, 0.5 mV. J, Firing rate of VTA GABA neurons was not altered by COC. K, Schematic of the experimental design. Rats were treated with SAL or COC 24 h before recording GABA neurons in the tVTA/RMTg. L, Sample traces from GABA tVTA/RMTg neurons. Scale bars: horizontal, 0.5 s; vertical, 1 mV. M, Firing rate of tVTA/RMTg GABA neurons was not altered by COC. *p < 0.05.
SK channels contribute to the cocaine-induced increases in neuronal activity
Burst activity of VTA DA neurons is modulated by both synaptic activity and intrinsic mechanisms. SK channels regulate the firing pattern of DA neurons (Grace and Bunney, 1983), as the SK antagonist apamin increases spontaneous bursting in vivo (Shepard and Bunney, 1991). In vivo recordings revealed that intra-VTA infusion of apamin (Fig. 2A) significantly increased the firing rate and mean number of spikes/burst in DA neurons recorded from saline-treated rats (Fig. 2B; saline, 24 h, n = 10 cells/8 rats; variation of firing rate: 99.88 ± 2.93% before apamin vs 135.3 ± 14.01% after apamin, Wilcoxon match-paired test w(10) = 39, p = 0.0244; variation of bursting rate: 105.2 ± 6.93% before apamin vs 164.7% after apamin, w(10) = 25 p = 0.116; variation of mean spike in burst: 99.91 ± 3.59% before apamin vs 146.6 ± 18.53%, w(10) = 21, p = 0.0156) but did not have any effect on VTA DA neurons recorded in cocaine-treated rats (Fig. 2C; cocaine, n = 7 cells/6 rats; variation of firing rate: 102.4 ± 1.94% before apamin vs 90.53 ± 27.02%, w(7) = −14.0, p = 0.148; variation of bursting rate: 101.6 ± 1.2% before apamin vs 104.5% ± 8 0.85 after apamin, w(7) = 7.0 p = 0.281; variation of mean spike in burst: 101.3 ± 1.3% before apamin vs 101.1% after apamin, w(7) = 0.0, p = 0.5). Our data suggest that cocaine exposure induces long-lasting changes in intrinsic activity of VTA DA neurons by affecting the function of the SK channel. To better characterize the cellular mechanisms underlying these cocaine-induced changes in firing activity neurons, we performed ex vivo electrophysiology recordings of VTA DA neurons (Fig. 2D). In agreement with in vivo findings, cell-attached recordings revealed a 2.26-fold increase in DA activity after apamin perfusion in slices from saline-treated mice (Fig. 2E; n = 8 cells/5 mice, t(7) = 5.02, p = 0.002). Given the potential age-dependent effects of cocaine, we compared the effects of cocaine on the firing rate of VTA DA neurons in young (P28—P35) and adult (P84–P98) mice. Following cocaine treatment, the absolute firing rate was increased relative to saline-treated controls in both age groups (young: saline, n = 18 cells/7 mice, 1.02 ± 0.072 Hz; cocaine, n = 11 cells/7 mice, 2.51 ± 0.180 Hz; t(27) = 9.11, p < 0.001; adult: saline, n = 15 cells/5 mice, 0.966 ± 0.213 Hz; cocaine, n = 7 cells/4 mice, 3.27 ± 0.819 Hz; t(20) = 3.73, p = 0.0013) and apamin application did not significantly change the activity of VTA DA neurons (Fig. 2F,G; young: n = 8 cells/6 mice, p = 0.6575; adult: n = 11 cells/4 mice, t(17) = 0.25, p = 0.809).
Figure 2.
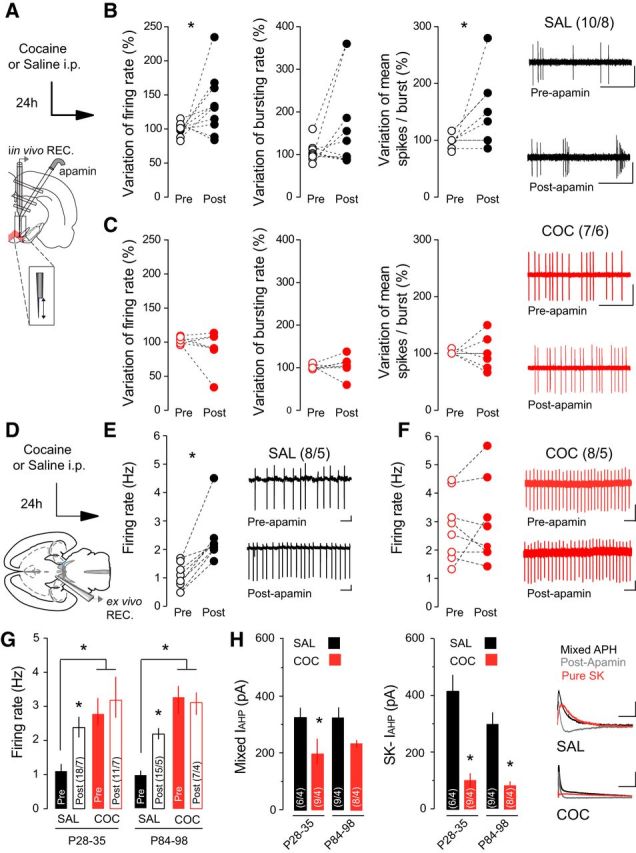
SK channels contribute to increased burst firing of VTA DA neurons following cocaine (COC). A, Schematic of the in vivo experimental design using double-barrel pipette for microinjection. B, In saline (SAL)-treated rats, application of apamin increased the firing rate (left) and spikes per burst (right) of VTA DA neurons. No effect is observed on the bursting rate (middle). Representative traces from SAL-treated rats. Scale bars: horizontal, 1 s; vertical, 1 mV. C, Application of apamin had no effect on firing rate, burst firing, or spikes per burst in COC-treated rats. Representative traces from COC-treated rats. Scale bars: horizontal, 1 s; vertical, 1 mV. D, Schematic of ex vivo recording experiments. E, F, In cell-attached recordings, apamin application increased the firing rate of VTA DA neurons in SAL-treated mice, but had no effect in mice receiving COC. Inset, Representative traces. Scale bars: horizontal, 1 s; vertical, 20 mV. G, In both adolescent (P28–P35) and adult (P84–P98) mice, basal firing rate was increased in COC-treated relative to SAL-treated mice, and apamin application increased the firing rate in SAL-treated mice only. H, The mixed and SK-mediated components of the IAHP were reduced following COC treatment in both adolescent and adult mice. Right, Representative traces. Scale bars: horizontal, 50 ms; vertical, 50 pA. *p < 0.05.
Activation of SK channels contributes to the IAHP potential that follows an AP (Waroux et al., 2005). To investigate whether cocaine exposure alters SK-dependent contribution to IAHP, we first measured the IAHP evoked by a 100 ms depolarizing voltage pulse. We then applied apamin to obtain the apamin-sensitive, SK-mediated IAHP. Cocaine treatment significantly decreased both the mixed IAHP and the apamin-sensitive IAHP component (Fig. 2H; young: saline/cocaine, n = 6/9 cells from 4 mice/condition; mixed IAHP: 419.77 ± 51.89 pA/202.88 ± 43.42 pA, t(13) = 2.87, p = 0.009, SK-IAHP: 330.32 ± 23.16 pA/106.29 ± 23.23 pA, t(13) = 4.42, p < 0.001; adult: saline/cocaine, n = 9/8 cells from 4 mice/condition; mixed IAHP: 333.21 ± 67.46 pA/218.5 ± 27.398 pA, t(15) = 1.93, p = 0.153, SK-IAHP: 298.1 ± 81.88 pA/85.06 ± 13.01 pA, t(15) = 2.52, p = 0.024) suggesting that impaired SK channel function contributes to the increased firing activity of VTA DA neurons.
GluN3A-containing NMDARs are necessary for cocaine-evoked changes in SK channel function and increased activity of VTA DA neurons.
Apamin-sensitive SK channels are activated by synaptically evoked Ca2+ transient through NMDARs and influence the EPSPs by providing a local shunting current to promote rapid membrane hyperpolarization and consequent Mg2+ block of the NMDARs (Ngo-Anh et al., 2005). To investigate whether this Ca2+-mediated feedback loop was affected in VTA DA neurons after cocaine treatment, we performed whole-cell current-clamp recordings of VTA DA neurons, pharmacologically isolating NMDA EPSPs. Apamin increased the amplitude of pharmacologically isolated NMDA EPSPs in the control condition (n = 5 cells/3 mice; +196.22 ± 16.24%), but did not have any effect in slices from mice treated with cocaine (Fig. 3A; n = 6 cells/3 mice, +115.22 ± 17.05%; repeated-measures ANOVA, F(1,5) = 21.4, p < 0.001). We have previously shown by the insertion of noncanonical GluN3A-containing NMDARs that cocaine-evoked synaptic plasticity is expressed at excitatory synapses onto DA neurons of the VTA (Yuan et al., 2013). GluN3A-containing NMDARs are Ca2+-impermeable, and their insertion following cocaine treatment would lead to reduced Ca2+ influx, which could then alter SK channel function. Consistent with this hypothesis, in GluN3A−/− mice, apamin increased the amplitude of NMDA EPSPs to a similar extent in cocaine-treated and saline-treated mice (Fig. 3B; saline: n = 10 cells/4 mice, 219.71 ± 36.10%; cocaine: n = 12 cells/4 mice, 201.13 ± 37.46%; repeated-measures ANOVA, F(1,20) = 0.2, p = 0.638). The mixed IAHP (saline: n = 11 cells/5 mice, 422.78 ± 207.52 pA; cocaine: n = 16 cells/5 mice, +474.19 ± 229.63 pA), apamin-sensitive IAHP (Fig. 3C; saline: 300.305 ± 177.84 pA; cocaine: 297.23 ± 142.84 pA), and firing rate (Fig. 3D; saline: n = 9 cells/4 mice, 1.12 ± 0.29 Hz; cocaine: n = 13 cells/4 mice, 1.67 ± 0.34 Hz) recorded in VTA DA neurons of GluN3A−/− mice were not significantly different between saline and cocaine treatment. Together, our experiments indicate that the cocaine-induced changes in intrinsic properties of VTA DA neurons depend on the presence of GluN3A-containing, Ca2+-impermeable NMDARs, which fail to efficiently activate SK channels.
Figure 3.

SK channel function in VTA DA neurons in GluN3A−/− mice is preserved after cocaine (COC). A, In VTA DA neurons, bath application of apamin potentiated NMDA-mediated EPSP amplitude, which was occluded 24 h after COC treatment. Inset, Representative traces showing averaged EPSP 5 min immediately before apamin application and 20 min after application. Scale bars: horizontal, 50 ms; vertical, 1 mV. B, COC treatment did not occlude apamin-induced LTP of NMDA-mediated EPSPs in GluN3A−/− mice. Inset, Representative traces showing averaged EPSP 5 min immediately before apamin application and 20 min after application. Scale bars: horizontal, 50 ms; vertical, 1 mV. C, The mixed and SK-mediated components of the IAHP were not reduced by COC treatment in GluN3A−/− mice. Inset, Representative traces. Scale bars: horizontal, 100 ms; vertical, 50 pA. D, COC-induced increase in firing rate of VTA DA neurons was not significant in GRIN3A knock-out mice. Scale bars: horizontal, 1 s; vertical, 20 mV. *p < 0.05. SAL, Saline.
Activation of Group I mGluRs rescues the cocaine-induced changes in intrinsic activity of VTA DA neurons
To demonstrate a direct link between cocaine-evoked synaptic changes and increased firing activity of VTA DA neurons, we attempted to rescue SK channel function and the firing properties with pharmacological Group I mGluR activation. We have previously shown that Group I mGluR activation rescues cocaine-evoked synaptic plasticity both in vivo and in vitro (Bellone and Lüscher, 2006; Yuan et al., 2013), by inducing the removal of GluN3A-containing NMDARs, which are substituted with canonical GluN2A-containing NMDARs. In slices that had been incubated with the Group I mGluR-agonist DHPG, the apamin-induced increase of NMDA-EPSP amplitude in cocaine-treated mice was restored and was not significantly different from saline-treated controls (Fig. 4A; saline, n = 6 cells/5 mice, 171.17 ± 27.47%; cocaine, n = 7 cells/5 mice, 204.96 ± 43.62%). To preclude the possibility that DHPG incubation induced an increase in NMDA EPSP and thus occluded the effects of apamin application, we show that while acute application of DHPG alone did in fact increase the amplitude of the NMDA EPSP in cocaine-treated mice (n = 11 cells/7 mice; 175.17 ± 43.42%), the application of apamin further enhanced this increase (Fig. 4B; n = 8 cells/6 mice; 263.75 ± 52.64%), although this result did not achieve statistical significance (t test DHPG vs DHPG + apamin t(17) = 1.3, p = 0.209). We have previously shown that in vitro and in vivo activation of mGluR1 specifically reversed cocaine-evoked synaptic plasticity (Bellone and Lüscher, 2006; Yuan et al., 2013). Here, we extended these findings and we showed that mGluR1-specific positive allosteric modulator Ro 67-7476 changed intrinsic activity of VTA DA neurons. Incubation with either DHPG or Ro 67-7476 rescued the mixed and apamin-sensitive IAHP (Fig. 4C; saline: n = 8 cells/4 mice, mixed IAHP = 384.86 ± 75.18 pA, SK-IAHP = 212.42 ± 63.40 pA; cocaine: n = 9 cells/4 mice, mixed IAHP = 193.92 ± 28.95 pA, SK-IAHP = 57.13 ± 16.77 pA, cocaine plus DHPG: n = 12 cells/4 mice, mixed IAHP = 299.65 ± 84.86 pA, SK-IAHP = 185.93 ± 66.37 pA, COC + Ro 67–7476: n = 13 cells/4 mice, mixed IAHP = 338.56 ± 69.44 pA, SK-IAHP = 187.42 ± 26.41 pA; t test, saline vs cocaine SK-IAHP: t(15) = 2.5, p = 0.025, t test saline vs cocaine + DHPG; SK-IAHP, t(18) = 2.1, p = 0.050, t test cocaine vs cocaine + Ro 67-7476 SK-IAHP, t(20) = 3.8, p = 0.0011) and the firing rate of DA neurons ex vivo (Fig. 4D; saline, n = 11 cells/4 mice, 1.08 ± 0.43 Hz; cocaine, n = 13 cells/4 mice, 2.93 ± 0.60 Hz; cocaine + DHPG, n = 11 cells/4 mice, 1.26 ± 0.42 Hz, t test, cocaine + Ro 67-7476, n = 13 cells/4 mice, 1.09 ± 0.17 Hz; saline vs cocaine, t(20) = 2.4, p = 0.025, t test cocaine vs cocaine + DHPG, t(20) = 2.2, p = 0.038, t test cocaine vs cocaine + Ro 67-7476, t(22) = 3.6, p = 0.0011).
Figure 4.

mGluR1 activation with DHPG rescues SK channel function after cocaine (COC). A, Incubation with DHPG rescued apamin-induced potentiation of the NMDA-mediated EPSP. B, Acute application of DHPG also rescued apamin-induced potentiation of the NMDA-mediated EPSP. C, Summary plots and representative traces showing that incubation with DHPG or Ro 67-7476 rescued the SK-mediated component of the IAHP in COC-treated mice. Scale bars: horizontal, 100 ms; vertical, 50 pA. D, Summary plots and representative traces showing that incubation with DHPG or Ro 67-7476 rescued COC-induced increase in firing rate measured with cell-attached recordings. Scale bars: horizontal, 1 s; vertical, 20 mV. *p < 0.05. SAL, Saline.
Discussion
Here, we observe that a single exposure to cocaine increases the firing rate and burst firing activity of VTA DA neurons. This increase is observed between 3 h and 5 d after the injection, and parallels synaptic potentiation onto these neurons (Ungless et al., 2001; Mameli et al., 2007). Cocaine-evoked plasticity in the VTA is driven first by the insertion of noncanonical GluN3A-containing NMDAs, followed by a switch in AMPAR composition (Bellone and Lüscher, 2006; Yuan et al., 2013). We identify the absence of SK channel activation as the link between potentiation of excitatory transmission onto VTA DA neurons and increased intrinsic activity.
Contrary to expectations, the increase in DA firing rate and burst firing activity does not result from disinhibition due to reduced mesencephalic GABAergic input. In line with this interpretation, our in vivo recordings of putative GABA neurons in the VTA and tVTA/RMTg revealed no difference in firing rate 24 h following cocaine exposure. Instead, “noncanonical” GluN3A-containing, low-conductance, Ca2+-impermeable NMDARs seem necessary for the increase in firing rate and bursting activity of VTA DA neurons. This is based on the observation that the NMDA antagonist MK-801, as well the genetic deletion of the GluN3 subunit, precluded increased firing.
How can the insertion of GluN3A-containing NMDARs enhance the activity of VTA DA neurons? Under physiological conditions, Ca2+ entering the cell via NMDARs activates SK channels that influence the EPSPs by providing a local shunting current to promote rapid membrane hyperpolarization and consequent Mg2+ block of the NMDARs (Ngo-Anh et al., 2005). Following cocaine exposure, the insertion of GluN3A-containing NMDARs—which have very low Ca2+ permeability—reduces the activation of SK channels (Fig. 5).
Figure 5.
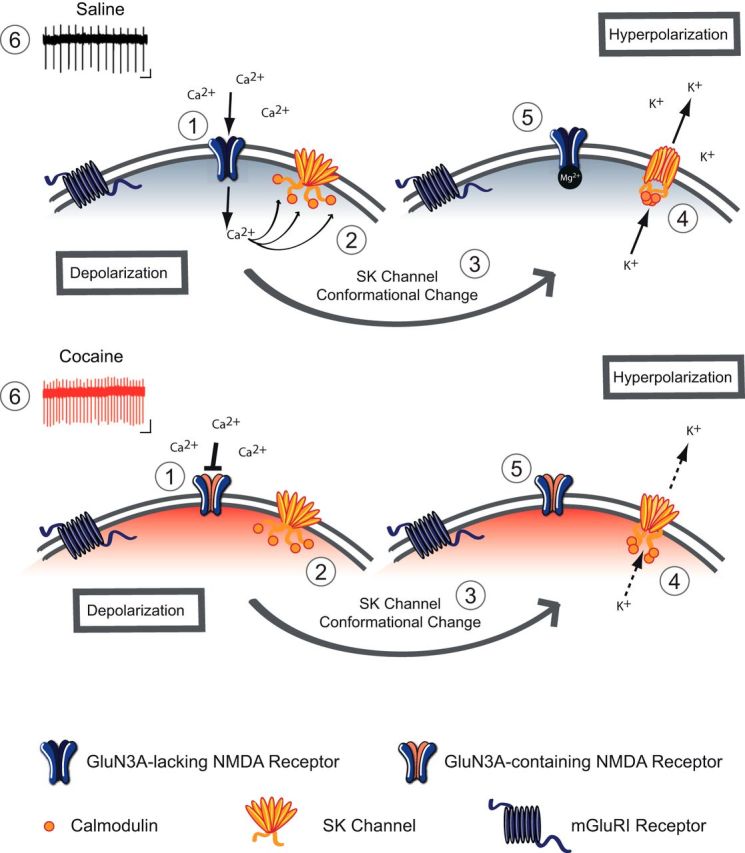
Model of altered Ca2+-SK channel feedback following cocaine. Schematic of a VTA DA neuron 24 h after saline treatment (top) and cocaine treatment (bottom). 1. Under baseline conditions, excitatory transmission is mediated by GluN3A-lacking NMDA receptors, which are Ca2+-permeable and sensitive to Mg2+ block at negative resting membrane potential. After cocaine, the switch in NMDA subunit composition to GluN3A-containing NMDA receptors, which are quasi-Ca2+-impermeable and are insensitive to Mg2+ block. 2. Under basal conditions Ca2+ influx through NMDARs binds to calmodulin through calmodulin-binding domains on SK channels. 3. Ca2+ binding induces a conformational change in the SK channel. 4. Upon activation, K+ efflux through the SK channel contributes to IAHP potential of the membrane. 5. At hyperpolarized potentials, NMDARs are subject to Mg2+ block and no longer flux Ca2+. 6. The net result is an increase in firing rate after cocaine, due to insertion of GluN3A-containing NMDARs.
We have previously shown that pharmacological activation of group I mGluRs removes GluN3A-containing NMDARs and restores basal synaptic transmission onto VTA DA neurons (Yuan et al., 2013). In this study, we did not examine the involvement of SK subtypes, which are differentially expressed in the VTA and the substantia nigra (Wolfart et al., 2001). The difference in SK channel expression within different populations of midbrain DA neurons could reconcile our results with previous reports that mGluR5 signaling is necessary for cocaine-induced reduction of mGluR1-dependent SK channel function (Kramer and Williams 2015). Here, we extend previous work specifically implicating mGluR1 in VTA DA neurons in cocaine-evoked synaptic plasticity (Bellone and Lüscher, 2006). Using a positive allosteric modulator of mGluR1, we now establish a causal link between cocaine-induced synaptic alterations and increases in neuronal activity. Indeed activation of mGluR1 restores not only basal synaptic transmission, but also SK channel function and normalizes the firing rate of VTA DA neurons after cocaine exposure. Although we cannot exclude a direct effect of mGluR signaling on SK channel function, our data indicate that the rescue of basal NMDAR properties contributes to the normalization of SK channel function and consequently decreases neuronal activity.
The demonstration of a causal link between synaptic plasticity and activity of VTA DA neurons may help us understand the effects of cocaine beyond the VTA. In fact, the synaptic plasticity observed in VTA DA neurons after cocaine suggests the possibility of altered synaptic plasticity in downstream projection regions, such as the NAc (Mameli et al., 2009), which mediates distinct components of drug-adaptive behavior (Pascoli et al., 2014). However, while plasticity in the VTA occurs within hours following acute cocaine exposure, downstream plasticity in the NAc is only observed after a withdrawal period of several days (Pascoli et al., 2014; Creed et al., 2015). By increasing the activity of VTA DA neurons through alterations in SK channel function, synaptic plasticity in the VTA may contribute to enhanced DA tone in VTA projection areas. Impaired function of SK channels may therefore represent a crucial step in the progression of drug-evoked synaptic plasticity throughout the mesolimbic DA system. Enhancing SK channel activity, for example through positive allosteric modulation, may thus emerge as a therapeutic strategy to halt the progression from cocaine exposure to addiction.
Footnotes
This work was supported by the Swiss National Science Foundation (C.B.), the Pierre Mercier Foundation (C.B.), the National Competence Center for Research Synapsy (C.B.), the Synapsis Foundation (C.B.), the Centre National de la Recherche Scientifique (F.G.), the University of Bordeaux (F.G.), Agence Nationale de la Recherche (ANR-12-BSV4-0022; F.G.), Labex Brain ANR-10-LABX-43 (F.G.), and Region Aquitaine (F.G.). We thank members of the Bellone Laboratory for helpful discussions and suggestions regarding this manuscript. GluN3A knock-out mice were kindly provided by Nobuki Nakanishi and Stuart Lipton.
The authors declare no competing financial interests.
References
- Bellone C, Lüscher C. Cocaine triggered AMPA receptor redistribution is reversed in vivo by mGluR-dependent long-term depression. Nat Neurosci. 2006;9:636–641. doi: 10.1038/nn1682. [DOI] [PubMed] [Google Scholar]
- Berridge KC, Robinson TE. What is the role of dopamine in reward: hedonic impact, reward learning, or incentive salience? Brain Res Brain Res Rev. 1998;28:309–369. doi: 10.1016/S0165-0173(98)00019-8. [DOI] [PubMed] [Google Scholar]
- Brown MT, Henny P, Bolam JP, Magill PJ. Activity of neurochemically heterogeneous dopaminergic neurons in the substantia nigra during spontaneous and driven changes in brain state. J Neurosci. 2009;29:2915–2925. doi: 10.1523/JNEUROSCI.4423-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen BT, Bowers MS, Martin M, Hopf FW, Guillory AM, Carelli RM, Chou JK, Bonci A. Cocaine but not natural reward self-administration nor passive cocaine infusion produces persistent LTP in the VTA. Neuron. 2008;59:288–297. doi: 10.1016/j.neuron.2008.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creed M, Pascoli VJ, Lüscher C. Addiction therapy. Refining deep brain stimulation to emulate optogenetic treatment of synaptic pathology. Science. 2015;347:659–664. doi: 10.1126/science.1260776. [DOI] [PubMed] [Google Scholar]
- Georges F, Aston-Jones G. Activation of ventral tegmental area cells by the bed nucleus of the stria terminalis: a novel excitatory amino acid input to midbrain dopamine neurons. J Neurosci. 2002;22:5173–5187. doi: 10.1523/JNEUROSCI.22-12-05173.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonon FG. Nonlinear relationship between impulse flow and dopamine released by rat midbrain dopaminergic neurons as studied by in vivo electrochemistry. Neuroscience. 1988;24:19–28. doi: 10.1016/0306-4522(88)90307-7. [DOI] [PubMed] [Google Scholar]
- Grace AA, Bunney BS. Intracellular and extracellular electrophysiology of nigral dopaminergic neurons—1. Identification and characterization. Neuroscience. 1983;10:301–315. doi: 10.1016/0306-4522(83)90135-5. [DOI] [PubMed] [Google Scholar]
- Grace AA, Bunney BS. The control of firing pattern in nigral dopamine neurons: single spike firing. J Neurosci. 1984;4:2866–2876. doi: 10.1523/JNEUROSCI.04-11-02866.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henson MA, Roberts AC, Pérez-Otaño I, Philpot BD. Influence of the NR3A subunit on NMDA receptor functions. Prog Neurobiol. 2010;91:23–37. doi: 10.1016/j.pneurobio.2010.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jalabert M, Bourdy R, Courtin J, Veinante P, Manzoni OJ, Barrot M, Georges F. Neuronal circuits underlying acute morphine action on dopamine neurons. Proc Natl Acad Sci U S A. 2011;108:16446–16450. doi: 10.1073/pnas.1105418108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer PF, Williams JT. Cocaine decreases metabotropic glutamate receptor mGluR1 currents in dopamine neurons by activating mGluR5. Neuropsychopharmacology. 2015;40:2418–2424. doi: 10.1038/npp.2015.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lecca S, Melis M, Luchicchi A, Muntoni AL, Pistis M. Inhibitory inputs from rostromedial tegmental neurons regulate spontaneous activity of midbrain dopamine cells and their responses to drugs of abuse. Neuropsychopharmacology. 2012;37:1164–1176. doi: 10.1038/npp.2011.302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacIver MB, Bland BH. Chaos analysis of EEG during isoflurane-induced loss of righting in rats. Front Syst Neurosci. 2014;8:203. doi: 10.3389/fnsys.2014.00203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mameli M, Balland B, Luján R, Lüscher C. Rapid synthesis and synaptic insertion of GluR2 for mGluR-LTD in the ventral tegmental area. Science. 2007;317:530–533. doi: 10.1126/science.1142365. [DOI] [PubMed] [Google Scholar]
- Mameli M, Halbout B, Creton C, Engblom D, Parkitna JR, Spanagel R, Lüscher C. Cocaine-evoked synaptic plasticity: persistence in the VTA triggers adaptations in the NAc. Nat Neurosci. 2009;12:1036–1041. doi: 10.1038/nn.2367. [DOI] [PubMed] [Google Scholar]
- Mameli M, Bellone C, Brown MT, Lüscher C. Cocaine inverts rules for synaptic plasticity of glutamate transmission in the ventral tegmental area. Nat Neurosci. 2011;14:414–416. doi: 10.1038/nn.2763. [DOI] [PubMed] [Google Scholar]
- Ngo-Anh TJ, Bloodgood BL, Lin M, Sabatini BL, Maylie J, Adelman JP. SK channels and NMDA receptors form a Ca2+-mediated feedback loop in dendritic spines. Nat Neurosci. 2005;8:642–649. doi: 10.1038/nn1449. [DOI] [PubMed] [Google Scholar]
- Overton PG, Clark D. Burst firing in midbrain dopaminergic neurons. Brain Res Brain Res Rev. 1997;25:312–334. doi: 10.1016/S0165-0173(97)00039-8. [DOI] [PubMed] [Google Scholar]
- Paoletti P, Bellone C, Zhou Q. NMDA receptor subunit diversity: impact on receptor properties, synaptic plasticity and disease. Nat Rev Neurosci. 2013;14:383–400. doi: 10.1038/nrn3504. [DOI] [PubMed] [Google Scholar]
- Pascoli V, Turiault M, Lüscher C. Reversal of cocaine-evoked synaptic potentiation resets drug-induced adaptive behaviour. Nature. 2012;481:71–75. doi: 10.1038/nature10709. [DOI] [PubMed] [Google Scholar]
- Pascoli V, Terrier J, Espallergues J, Valjent E, O'Connor EC, Lüscher C. Contrasting forms of cocaine-evoked plasticity control components of relapse. Nature. 2014;509:459–464. doi: 10.1038/nature13257. [DOI] [PubMed] [Google Scholar]
- Shepard PD, Bunney BS. Effects of apamin on the discharge properties of putative dopamine-containing neurons in vitro. Brain Res. 1988;463:380–384. doi: 10.1016/0006-8993(88)90414-3. [DOI] [PubMed] [Google Scholar]
- Shepard PD, Bunney BS. Repetitive firing properties of putative dopamine-containing neurons in vitro: regulation by an apamin-sensitive Ca(2+)-activated K+ conductance. Exp Brain Res. 1991;86:141–150. doi: 10.1007/BF00231048. [DOI] [PubMed] [Google Scholar]
- Sombers LA, Beyene M, Carelli RM, Wightman RM. Synaptic overflow of dopamine in the nucleus accumbens arises from neuronal activity in the ventral tegmental area. J Neurosci. 2009;29:1735–1742. doi: 10.1523/JNEUROSCI.5562-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suaud-Chagny MF, Chergui K, Chouvet G, Gonon F. Relationship between dopamine release in the rat nucleus accumbens and the discharge activity of dopaminergic neurons during local in vivo application of amino acids in the ventral tegmental area. Neuroscience. 1992;49:63–72. doi: 10.1016/0306-4522(92)90076-E. [DOI] [PubMed] [Google Scholar]
- Ungless MA, Grace AA. Are you or aren't you? Challenges associated with physiologically identifying dopamine neurons. Trends Neurosci. 2012;35:422–430. doi: 10.1016/j.tins.2012.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ungless MA, Whistler JL, Malenka RC, Bonci A. Single cocaine exposure in vivo induces long-term potentiation in dopamine neurons. Nature. 2001;411:583–587. doi: 10.1038/35079077. [DOI] [PubMed] [Google Scholar]
- Ungless MA, Magill PJ, Bolam JP. Uniform inhibition of dopamine neurons in the ventral tegmental area by aversive stimuli. Science. 2004;303:2040–2042. doi: 10.1126/science.1093360. [DOI] [PubMed] [Google Scholar]
- Waroux O, Massotte L, Alleva L, Graulich A, Thomas E, Liégeois JF, Scuvée-Moreau J, Seutin V. SK channels control the firing pattern of midbrain dopaminergic neurons in vivo. Eur J Neurosci. 2005;22:3111–3121. doi: 10.1111/j.1460-9568.2005.04484.x. [DOI] [PubMed] [Google Scholar]
- Wise RA. Dopamine, learning and motivation. Nat Rev Neurosci. 2004;5:483–494. doi: 10.1038/nrn1406. [DOI] [PubMed] [Google Scholar]
- Wolfart J, Roeper J. Selective coupling of T-type calcium channels to SK potassium channels prevents intrinsic bursting in dopaminergic midbrain neurons. J Neurosci. 2002;22:3404–3413. doi: 10.1523/JNEUROSCI.22-09-03404.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfart J, Neuhoff H, Franz O, Roeper J. Differential expression of the small-conductance, calcium-activated potassium channel SK3 is critical for pacemaker control in dopaminergic midbrain neurons. J Neurosci. 2001;21:3443–3456. doi: 10.1523/JNEUROSCI.21-10-03443.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan T, Mameli M, O'Connor EC, Dey PN, Verpelli C, Sala C, Perez-Otano I, Lüscher C, Bellone C. Expression of cocaine-evoked synaptic plasticity by GluN3A-containing NMDA receptors. Neuron. 2013;80:1025–1038. doi: 10.1016/j.neuron.2013.07.050. [DOI] [PubMed] [Google Scholar]
- Zweifel LS, Parker JG, Lobb CJ, Rainwater A, Wall VZ, Fadok JP, Darvas M, Kim MJ, Mizumori SJ, Paladini CA, Phillips PE, Palmiter RD. Disruption of NMDAR-dependent burst firing by dopamine neurons provides selective assessment of phasic dopamine-dependent behavior. Proc Natl Acad Sci U S A. 2009;106:7281–7288. doi: 10.1073/pnas.0813415106. [DOI] [PMC free article] [PubMed] [Google Scholar]