

Abstract
How the integrity of laminar structures in the postnatal brain is maintained impacts neuronal functions. Ndel1, the mammalian homolog of NuDE from the filamentous fungus Aspergillus nidulans, is an atypical microtubule (MT)-associated protein that was initially investigated in the contexts of neurogenesis and neuronal migration. Constitutive knock-out mice for Ndel1 are embryonic lethal, thereby necessitating the creation a conditional knock-out to probe the roles of Ndel1 in postnatal brains. Here we report that CA1 pyramidal neurons from mice postnatally lacking Ndel1 (Ndel1 conditional knock-out) exhibit fragmented MTs, dendritic/synaptic pathologies, are intrinsically hyperexcitable and undergo dispersion independently of neuronal migration defect. Secondary to the pyramidal cell changes is the decreased inhibitory drive onto pyramidal cells from interneurons. Levels of the glycoprotein Reelin that regulates MTs, neuronal plasticity, and cell compaction are significantly reduced in hippocampus of mutant mice. Strikingly, a single injection of Reelin into the hippocampus of Ndel1 conditional knock-out mice ameliorates ultrastructural, cellular, morphological, and anatomical CA1 defects. Thus, Ndel1 and Reelin contribute to maintain postnatal CA1 integrity.
SIGNIFICANCE STATEMENT The significance of this study rests in the elucidation of a role for Nde1l and Reelin in postnatal CA1 integrity using a new conditional knock-out mouse model for the cytoskeletal protein Ndel1, one that circumvents the defects associated with neuronal migration and embryonic lethality. Our study serves as a basis for understanding the mechanisms underlying postnatal hippocampal maintenance and function, and the significance of decreased levels of Ndel1 and Reelin observed in patients with neurological disorders.
Keywords: cytoskeleton, dendrites, hippocampus, knock-out, microtubules, synapses
Introduction
Ndel1 (pronounced “noodle”), the mammalian homolog of the aspergillus nidulans NuDE, is an atypical microtubule (MT)-associated protein (MAP) that was initially investigated in the context of cytoskeletal organization during nucleokinesis and neuronal migration (for review, see Chansard et al., 2011). Ndel1 is now recognized as an integrator of the cytoskeleton, impacting MTs, microfilaments, intermediate filaments, as well as their associated proteins. In addition to its cytoskeletal functions, Ndel1 also serves as a modulating platform for signaling proteins, such as kinases and GTPases. Most of the functions assigned to Ndel1 were discovered in cultured cells, ex vivo tissues, or using in utero electroporation on a limited number of embryonic progenitor cells. The early embryonic lethality of Ndel1 knock-out (KO) mice precludes a clear definition of the roles of the protein in postnatal brain neurons. Unequivocal proof for an in vivo role for Ndel1 in MT organization has not yet been forthcoming.
Ndel1 has been associated with several neuropsychiatric disorders exhibiting cognitive impairment (Lipska et al., 2006; Burdick et al., 2008; Nicodemus et al., 2010). Transcribed in an activity-dependent manner by the cAMP-responsive element binding CREB protein (Impey et al., 2004), Ndel1 is a robust interacting partner of Disrupted-in-schizophrenia 1 (DISC-1), another protein genetically associated with schizophrenia, major depression and bipolar disorders in some populations (Porteous et al., 2006; Brandon, 2007; Mackie et al., 2007; Bradshaw et al., 2008). Through interactions with the disease-gene product and atypical MAP Lis1, Ndel1 is also linked to lissencephaly, a neuronal migration disorder with concomitant memory deficits (Kerjan and Gleeson, 2007; Wynshaw-Boris et al., 2010). Finally, Ndel1 expression is decreased in the hippocampus of patients with schizophrenia (Lipska et al., 2006).
Like Ndel1, the large extracellular matrix glycoprotein Reelin (D'Arcangelo et al., 1995) regulates neurodevelopment and has been implicated in neurological diseases (Förster et al., 2006; Stranahan et al., 2013). Binding of Cajal-Retzius cell-secreted Reelin lipoprotein receptors triggers a cascade of events resulting in the activation of kinases that modulate MTs during neuronal migration and hippocampal dendrite development (Förster et al., 2006; Herz and Chen, 2006; Herz et al., 2009; Lakatosova and Ostatnikova, 2012; Stranahan et al., 2013). Mutations in Reelin underlie human cases with thickened cortex, pachygyria (indicative of impaired neuronal migration), significant cerebellar hypoplasia, and seizures (Förster et al., 2006), whereas Reelin mutant mice (i.e., Reeler) display defects in neuronal migration. In the mature brain, Reelin is expressed by GABAergic interneurons where it may regulate neuronal plasticity and alignment (Herz and Chen, 2006; Lakatosova and Ostatnikova, 2012; Stranahan et al., 2013). Exogenous application of Reelin enhances LTP in hippocampal slice cultures (Weeber et al., 2002) and inhibition of Reelin signaling by injection of CR-50 Reelin blocking antibodies (D'Arcangelo et al., 1997; Miyata et al., 1997; Nakajima et al., 1997; Heinrich et al., 2006) in the adult dentate gyrus or cerebellum causes slight neuronal dispersion (D'Arcangelo et al., 1997; Miyata et al., 1997; Nakajima et al., 1997; Heinrich et al., 2006).
In this study, through generation of Ndel1 conditional-null mice that bypass embryonic lethality and show reduced levels of Reelin, we uncovered key roles for both proteins in postnatal CA1 integrity.
Materials and Methods
Generation of CaMKIIα-Cre Ndel1 conditional KO (CKO) or Ndel1 CKO (−/−)
Forebrain CA1 excitatory neuron-specific KO mice for Ndel1 were created by breeding CaMKIIα-Cre transgenic mice line 29–1 (The Jackson Laboratory) (Tsien et al., 1996) with Ndel1-LoxP mice generated previously (Sasaki et al., 2005). Our breeding strategy (of Ndel1 KO deleted/wild-type [WT]; CaMKII-Cre heterozygote mice with Ndel1 KO deleted/WT; CaMKII-Cre heterozygote mice) predicts that 3 of 16 animals have both Ndel1 alleles deleted (data not shown). These animals have one or two CaMKIIα-Cre alleles and are designated Ndel1 CKO (−/−) at ∼1 month of age. They consistently show the same phenotypes with high penetrance. Only CKO animals were used for this study. All mice were genotyped by PCR before experimentation. Genotypes, as well as knock-out of Ndel1, were confirmed by PCR and Western blots, respectively. For all experiments, WT (+/+) and heterozygous KO littermates with one Ndel1-LoxP deleted (+/−) were used as controls; +/− mice were indistinguishable from WT littermates. Phenotypes of littermates with heterozygous CaMKII-Cre insertion are indistinguishable from littermates with homozygous CaMKII-Cre insertion. All mice were housed and handled according to the Canadian Council on Animal Care guidelines and experimentation approved by the Health Sciences Animal Care Committee at the University of Calgary.
Genotyping of CaMKIIα-Cre Ndel1 CKO or Ndel1 CKO (−/−)
Genotyping by PCRs was performed with three different set of primers to detect the presence of the LoxP-Ndel1 alleles, Cre expression, and the excision of the LoxP-alleles (corresponding to the presence of gene KO) using DNA isolated from mouse ears and brain tissues (cortex and hippocampus). The same approach was used in a time course experiment to detect the earliest time point of Ndel1 gene excision at P15. Our result is consistent with the expression of the cre recombinase between the second and third week reported in the original paper by Tsien et al. (1996) for the CaMKIIα-Cre mouse line 29–1. Briefly, pups were collected blindly from multiple females and analyzed at different time points (P4, P5, P10, P15, P17, P19, P21, P23, P32, and P70) on different days. Only 3 pups of 16 may actually become CKO. Because of the random distribution of the unknown genotypes (CKO, WT, etc.) on PCR gels, the potential absence of litters with the wanted genotypes, and the analyses performed on different days/weeks, the results were collected in no specific order. PCR experiments were run in parallel with Western blots experiments (see Fig. 1) to determine when mice with Ndel1-LoxP alleles deleted are actually CKO (knockdown of Ndel1 protein levels).
Figure 1.
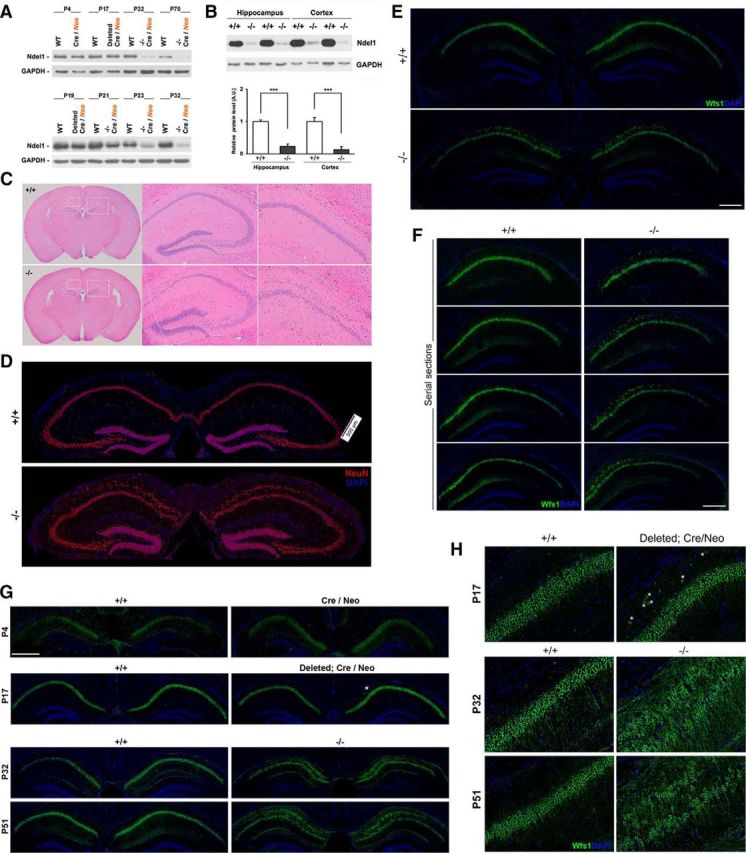
Hippocampal Ndel1 expression levels and postnatal dispersion of CA1 pyramidal neurons in Ndel1 CKO mice. A, Levels of Ndel1 protein in forebrain excitatory neurons of Ndel1 CKO mice start to decrease around P19 and the decline is accentuated at P21 and P23. Minimal levels of Ndel1 that persist throughout lifespan are detected at P32. Residual levels of Ndel1 are expressed by glial cells and other neuronal cell types not targeted by the KO strategy. Because of the turnover of the Ndel1 polypeptide, the protein levels are not totally downregulated until 1 month of age despite the excision of the Ndel1 LoxP alleles at P15. B, Western blots showing levels of Ndel1 in the hippocampus and cortex of WT (+/+) and Ndel1 CKO (−/−) mice and summary bar graph data. Arbitrary levels of Ndel1 in the hippocampus: WT (+/+): 1.00 ± 0.06 versus Ndel1 CKO (−/−): 0.27 ± 0.11; mean ± SD (n = 3 animals). ***p < 0.001 (Student's t test). Arbitrary levels of Ndel1 in the cortex: WT (+/+): 1.00 ± 0.17 versus Ndel1 CKO (−/−): 0.27 ± 0.12; mean ± SD (n = 3 animals). ***p < 0.005 (Student's t test). C, CA1 pyramidal bilayer in dorsal hippocampus and enlarged ventricles in 7-week-old Ndel1 CKO brain (sections stained with H&E). D, E, Bilayer of CA1 pyramidal neurons in the dorsal hippocampus of 10-week-old Ndel1 CKO brain as detected by staining with (D) the neuronal marker NeuN and (E) the CA1 pyramidal neuron marker Wfs1, respectively. F, Serial sections of 7-week-old Ndel1 CKO brains stained with Wfs1 marker confirm the bilayer phenotype (from bregma ∼−1.7 mm to bregma ∼−2.7 mm). G, H, Time course analysis of CA1 anatomy reveals postnatal dispersion of principal cells. Dispersion of CA1 principal cells does not occur at P4 (LoxP-Ndel1 alleles not deleted). A few cells disperse at P17 in animals with the LoxP-Ndel1 alleles deleted (white asterisks). Dispersion is clearly evident at P32 and 51 when Ndel1 protein is significantly knocked down. Expression of Wfs1 that is associated with the endoplasmic reticulum and regulates calcium homeostasis is lower in P4 samples. Scale bars: D–F, 500 μm; G, 800 μm.
Histology and immunofluorescence labeling
Histology.
Brains were removed from perfused mice and fixed in 10% buffered formalin, then embedded in paraffin; 5-μm-thick coronal sections were cut and stained with H&E.
Immunofluorescence labeling.
Mice were perfused in 4% PFA, brains were then removed and postfixed in 4% PFA for 48 h at 4°C, individually cryoprotected in 30% sucrose for 48 h, then embedded in OCT, and 20 μm coronal sections were cut on a cryostat. Sections were washed with PBS, blocked in 1% normal goat serum, 0.1% Triton X-100 in PBS for 1 h, then incubated in anti-NeuN antibody (1:100, Millipore, catalog #MAB377) for 1 h. After washing in PBS three times, the sections were incubated with anti-mouse IgG conjugated to Cy3 (1:300) for 1 h. Double-labeling of Reelin and GAD67 was performed sequentially as both primary antibodies were raised in mouse. Sections were blocked in 5% normal goat serum, 1% Triton X-100 in PBS for 1 h followed by incubating with anti-Reelin antibody (CR-50, 1:100, MBL, catalog #D223-3) for 1 h. After washing, the sections were incubated with FITC-anti-mouse IgG (1:100) for 1 h before blocking again overnight at 4°C to avoid cross-reactivity. Next, incubation with antibodies against GAD-67 (1:200, Millipore, catalog #MAB5406), Reelin (1:250 for cells; 1:100 for tissues), NeuN (1:1000 for cells; 1:200 for tissues), MAP2 (1:1000), or Wfs1 (1:250), was again performed for 1 h or overnight followed by 1 h incubation with Cy3 or Alexa-488 anti-mouse or anti-rabbit IgG (1:200) and DAPI. The sections were washed with PBS and mounted with AQUA-MOUNT (Thermo Scientific).
Western blot
Total protein extracts of mouse brains were obtained by homogenization in SDS-urea (0.5% SDS, 8 m urea in 7.4 phosphate buffer) or Triton X-100 (10 mm Tris-HCl, pH 7.5, 150 mm NaCl, 1 mm EDTA, pH 8.0, and 1% Triton X-100) buffer. The protein concentration was estimated by the Bradford or DC assay (Bio-Rad Laboratories). Proteins were fractionated by SDS-PAGE and blotted on a nitrocellulose or PVDF membrane for Western blot analysis. Membranes were incubated with specific antibodies. The Western blots were examined using a chemiluminescence kit from NEN Life Science. Quantitations were corrected with levels of housekeeping proteins, such as actin and α-tubulin, and performed with the Labscan program (Image Master, 2D software version 3.10, GE Healthcare Pharmacia Biotech).
Golgi staining
Golgi staining procedures were performed following the manufacturer's instructions (FD NeuroTechnologies, FD Rapid GolgiStain Kit, catalog #PK401A). Coronal sections (100 μm thick) were cut using Cryostat and sections were mounted on slides. Images (20×) were captured using Olympus microscope (BX61VS) running VS- ASWFLL software (version 2.6).
Image reconstruction and dendrite/synapse quantifications
Raw images of neurons were obtained from Golgi-stained hippocampal slices. Neurons were selected based on their minimal dendrite overlapping with other neurons and the absence of artifact. The Golgi-stained pictures were then edited in Photo-Paint (Corel). The entire dendritic tree was quantitatively reconstructed using Imaris imaging software (Bitplane). CA1 pyramidal neurons from control (+/+, n = 4 or 5) and Ndel1 CKO animals (−/−, n = 4 or 5) mice were used to determine total dendritic length, number of dendrites, and spine density. Measurements were made by two independent blinded researchers.
Electron microscopy, synapse, and microtubule length quantification
Brains from control (+/+, n = 3) and Ndel1 CKO animals (−/−, n = 3) were perfused and fixed with 2.5% glutaraldehyde and 2% PFA solution. These brains were then sectioned into 350-μm-thick slices. After postfixation and labeling, the samples were sectioned into ultrathin slices (70 nm) and mounted on grids. Samples were visualized with transmission electron microscopy at 2500× magnification. Sampling areas were defined as 5-μm-long segments of the dendrite of interest. All cytoskeletal microtubule elements were measured against a standard length of 1 μm and separated into two categories: >1 μm and ≤1 μm. The counts were performed manually using images cropped to represent a 5 μm length of the dendrite using Adobe Photoshop Elements 9, and relative measurements were made by comparison with a 1 μm movable scale-bar. MT filaments that were cut off by the boundary of the sampling area were negated, and only those filaments contained within the sampling area were included in the measurement. To reduce the impact of the varying thickness of selected dendrites, the relative lengths were presented as percentages rather than absolute numbers. Statistical significance was determined using the Student's t test. Quantifications were made by at least two independent blinded researchers.
For synapse quantification, brain ultrathin slices (70 nm) were made, and electron microscopic images were made at 8000× magnification. Synapses were counted in 3 different areas of CA1. Seven fields per area were photographed for a total of 21 fields per mouse. Synapses were classified as asymmetric or symmetric and counted accordingly. Synaptic vesicles were counted according to the method of Applegate and Landfield (1988). Electron microscopic images were prepared at 15,000× magnification. The symmetrical vesicles within 150 nm of the active zone (up to 150 nm from the presynaptic membrane) were counted. Vesicle number in the active zone area per μm2 was measured and the two groups were compared by t test. Both counts were quantified by two independent blinded researchers.
Electrophysiology and immunofluorescent labeling of brain slices
Anesthetized mice (n = 6; ketamine/xylazine/acepromazine mixture; i.p.) were perfused through the heart using an ice-cold Ringer's solution as previously described (Gavrilovici et al., 2012). The brain was removed and temporal lobe was coronally sliced (350 μm) using a Leica VT 1000S vibratome. The slices were incubated at 37°C for 30 min and subsequently moved to a room temperature (22°C) bath for at least 45 min. All solutions were maintained at pH 7.4 and bubbled with 5% CO2/95% O2 (carbogen). Patch electrodes (3–5 mΩ) were pulled from borosilicate glass capillaries and filled with K+-gluconate based solution, as described by Gavrilovici et al. (2012). Whole-cell patch-clamp recordings from neurons in CA1 stratum pyramidale were obtained using a Multiclamp 700A (Molecular Devices) and digitized with a Digidata 1440A and pClamp software (Molecular Devices). All experiments were performed at 32°C. Cell responses were obtained after forming whole-cell configuration by injecting hyperpolarizing and depolarizing current steps (500 ms, 20 pA) using current-clamp protocols (Clampex, Molecular Devices). Input resistance (Ri) was calculated by linear regression of the current-voltage relationship in response to hyperpolarizing steps (see Gavrilovici et al., 2012) using Origin software (Microcal). Firing pattern analysis was performed at the current level that produced reliable repetitive firing (i.e., twice the firing threshold) in the presence of NMDA, non-NMDA blockers (50 μm APV; 10 μm DNQX; Research Biochemicals) and a GABAA receptor blocker (50 μm picrotoxin; Sigma). Spontaneously occurring miniature IPSCs were recorded using a KCl electrode solution in the presence of TTX, APV, and DNQX and then selected and analyzed with Mini Analysis software (Synaptosoft), as previously described (Gavrilovici et al., 2006). Comparisons among different electrophysiological parameters (control vs Ndel1 CKO groups) were made using Student's t tests (p < 0.05 was considered to be significant). To reconstruct neuronal morphology, patch electrodes included 0.2% Biocytin. Slice immunohistochemistry using streptavidin-conjugated AlexaFluor-594 dye (5 μg/ml; Invitrogen), as well as confocal microscopy imaging and morphological reconstruction of patched cells (Imaris software; Bitplane) were all used following protocols described previously (Gavrilovici et al., 2012).
Purification and injection of Reelin
Reelin purification.
Supernatants containing recombinant full-length Reelin proteins were produced from a stable HEK293 cell line (a gift from Dr. Joachim Herz laboratory, University of Texas Southwestern Medical Center, and Dr. Tom Curran, University of Pennsylvania). Conditioned medium from the Reelin-secreting HEK293 cell line or an HEK293 cell line (control for injection) was collected after 48 h of incubation in serum-free Opti-MEM media. The conditioned media were concentrated ∼30-fold using Amicon Ultra 100,000 molecular weight cutoff filters (Millipore) and dialyzed in PBS according to previously published methods (Förster et al., 2002; Weeber et al., 2002). The Reelin content was confirmed by Western blotting using the anti-Reelin antibody G10 (Abcam, catalog #ab78544).
Reelin injection.
Mice were anesthetized with 2% isoflurane in 100% O2, then positioned between the ear bars of a stereotaxic frame. To maintain anesthesia, isoflurane was continuously delivered through a small facemask custom-fitted to the stereotaxic frame. A midline incision was made in the skin overlying the skull. Two holes (anteroposterior −2.3; mediolateral ±2.0; dorsoventral 2.0) were drilled through the skull and solution containing Reelin proteins (210 nm) or control was injected using a stereotaxic injector and Hamilton syringe at a rate of 1.0 μl/min and left in place for 2–5 min. Immediately after removal of the needle, the skin was closed with sutures.
Cell dispersion counting.
Images of Wfs1-stained brain sections (n = 4) from 3 different animals per genotype and condition (WT, CKO, CKO + control, CKO + Reelin) were calibrated to a 200 μm scale using CellSens. Wfs1-positive cells from full-length CA1 or randomly selected in 800 μm × 800 μm boxes were counted by hand using Adobe Photoshop CS6 or automatically using ImageJ. To quantify the percentage of cells dispersed, the stratum pyramidale was outlined using the line tool to separate the dispersed neurons from the pyramidal cell layer. Up to 700 cells were counted per section. No significant difference was found between results obtained with Adobe Photoshop CS6 and ImageJ. The results presented are from hand counting.
Results
Postnatal knockdown of Ndel1 in forebrain excitatory neurons
Mice with a targeted disruption of Ndel1 are embryonic lethal, whereas heterozygous KO animals reveal little about Ndel1's postnatal functions when analyzed in the Lis1 hemizygote mutant background (Sasaki et al., 2005). Furthermore, our understanding of the cellular functions of Ndel1 in the adult brain is mostly derived from cell culture experiments (for review, see Chansard et al., 2011). To understand the in vivo roles of Ndel1 in differentiated neurons, we generated CKO mice lacking Ndel1 specifically in forebrain CA1 excitatory neurons after birth using the CaMKIIα-Cre/LoxP system (CaMKIIα-Cre Ndel1 CKO, Ndel1 CKO or −/−; see Material and Methods). The LoxP-Ndel1 alleles are deleted at postnatal day 15 (P15; see Materials and Methods), consistent with the time of activation of CaMKIIα promoter between the second and third week after birth in this Cre mouse line 29–1 (Tsien et al., 1996). Ndel11 protein levels are slightly reduced at P19 and further decline at P21, P23 to reach a minimum at P32 (Fig. 1A). The significant decrease then persists throughout the lifespan of the animals (Fig. 1A). The slight delay between Ndel1 gene excision at P15 and protein knockdown at later stage is likely due to the turnover of the protein. In both hippocampus and cortex, residual levels of Ndel1 (∼10%) are likely expressed by glial cells and other neuronal types that are not targeted by our KO strategy (Fig. 1B). This is the first animal model hypomorph for Ndel1 in the postnatal brain at a developmental stage equivalent to a ∼2.5-year-old human and that circumvents the known defects in neurogenesis and neuronal migration caused by embryonic deletion of Ndel1. The average lifespan of these animals is ∼11 weeks, and only a few such mice (∼5%) surpass 20 weeks of age (see Discussion). Between 4 and 11 weeks of age, these mice display several phenotypes that highlight the key roles of Ndel1 in postnatal neurons.
Dispersion of CA1 pyramidal cells in Ndel1 CKO mice
We first analyzed the gross brain anatomy of Ndel1 CKO with H&E staining. At ∼7 weeks of age, these mice exhibit enlarged ventricles (Fig. 1C). Remarkably, in the dorsal hippocampus of Ndel1 CKO mice, CA1 cells are misaligned in two layers compared with WT CA1 (Fig. 1C). CA1 subfields from heterozygous KO littermates and WT mice are indistinguishable from each other (data not shown). The presence of a CA1 pyramidal neurons bilayer in 7-week-old Ndel1 CKO mice was confirmed by immunofluorescent staining with the neuronal marker NeuN (Fig. 1D) commonly used to detect pyramidal cells. We extend our characterization with Wfs1 antibodies that are specific to several brain regions, including CA1 pyramidal neuron layer (Lein et al., 2007; Kohara et al., 2014), on serial sections of Ndel1 CKO brain (bregma ∼−1.7 mm to bregma ∼−2.7 mm; see Fig. 1E,F). Approximately 40% of Wfs1-positive pyramidal neurons disperse in 7-week-old Ndel1 CKO mice: dispersion in WT (+/+) vs Ndel1 CKO (−/−): 0% versus 39.8 ± 5.4%; mean ± SD (n = 3 animals); p < 0.001 (Student's t test).
To determine the course of the dispersion, we analyzed the architecture of the CA1 from P4 to P70 using immunostaining with Wfs1 (Fig. 1E,G,H). In P4 Cre transgenic animals with intact Neo alleles that are indistinguishable from WT animals, all CA1 neurons were aligned on a single layer. In mice with LoxP-Ndel1 alleles removed at P17 (gene excision occurring at P15 and decreased levels of Ndel1 being detectable by Western blot at P19), a few CA1 pyramidal neurons start to disperse (Fig. 1G,H, white asterisks). The dispersion becomes more obvious as the animals age (Fig. 1G,H for P32 and P51; Fig. 1E for P70). Thus, consistent with the postnatal Ndel1 protein knockdown (Fig. 1A), the bilayer phenotype is not due to a neuronal migration defect during embryonic development (as reported in previous models lacking Lis1, Doublecortin, Reeler, or Dab1) but is the result of alterations in the maintenance of positioning once the neurons have reached their proper location in the developing brain and have integrated within the network. This also contrasts strikingly with the absence of cell dispersion in postnatal KO for the MAP Lis1 (see Hunt et al., 2012) despite the genetic and biochemical association between Ndel1 and Lis1. The Ndel1 CKO CA1 dispersion phenotype is reminiscent of the tectonic hippocampal malformation found in human diseased patients (Sloviter et al., 2004).
Dendritic pathology of Ndel1 CKO CA1 pyramidal cells is associated with fragmentation of microtubules
To gain further insights into the morphology of CA1 pyramidal cells from Ndel1 CKO mice, we performed Golgi staining. From ∼4 weeks of age, misaligned CA1 neurons in the Ndel1 CKO mice display primitive dendritic trees (Fig. 2A) reminiscent of neurons derived from neurologically impaired patients with cytoskeletal abnormalities (for morphology of these neurons, see Lavenex and Banta Lavenex, 2013). Reconstruction of neuronal morphology with the Imaris tracer software followed by quantification revealed an overall ∼40% decrease in total dendritic length and a significant reduction in the number of neuronal processes (for representative neurons, see Fig. 2B; for quantifications, see Fig. 2C) for CA1 cells in Ndel1 CKO mice. This dendritic anomaly was not observed in mice with LoxP-Ndel1 alleles deleted at P17 (Deleted P17; Figs. 1A, 2B, quantifications not shown) but with intact levels of Ndel1 (Fig. 1B). The reduced dendritic arborization detected with Golgi staining and Imaris reconstruction was also detected by immunostaining with the dendritic marker MAP2 (data not shown).
Figure 2.

Dendritic pathology of CA1 pyramidal neurons in Ndel1 CKO mice. A, Dispersed CA1 pyramidal neurons from 7-week-old Ndel1 CKO mice display dendritic pathology as revealed by Golgi staining. B, Imaris software-enabled reconstruction of individual CA1 hippocampal neurons from 9-week-old Ndel1 CKO mice and WT littermates, as well as from control Cre transgenic mice and mice with both Ndel1 LoxP alleles deleted at P17 (Deleted) but with no protein knockdown (Fig. 1B). C, Reduced total neuronal process length and reduced number of dendrites in CA1 pyramidal cells from Ndel1 CKO mice. Total neuronal process length: WT: 2522 ± 376 μm versus Ndel1 CKO: 1607 ± 387 μm; mean ± SD (n = 6 animals). **p < 0.005 (Student's t test). Total number of dendrites per pyramidal cell: WT: 44.0 ± 4.8 versus Ndel1 CKO: 27 ± 5.0; mean ± SD (n = 5 or 6 animals). ***p < 0.001 (Student's t test).
Ultrastructural analysis using electron microscopy further revealed that MTs in dendrites of mutant animals are fragmented and much shorter than those in WT siblings (Fig. 3A). There was a ∼55% decrease in the number of MTs with a length superior to 1 μm and, conversely, a significant increase in the proportion of MTs ≤1 μm in CA1 Ndel1 CKO pyramidal neurons (Fig. 3B). Our results are in agreement with in vitro studies assigning an MT-associated role for Ndel1 during development (for review, see Chansard et al., 2011) and, most importantly, provide the first unequivocal proof that Ndel1 regulates MT structure in adult brain neurons in vivo.
Figure 3.

CA1 pyramidal dendrites in Ndel1 CKO mice exhibit fragmented and shortened microtubules. A, Representative electron microscopic photographs of CA1 hippocampal dendrites from 9-week-old Ndel1 CKO mice and WT littermates. Note the fragmentation and shortening of dendritic microtubules in CA1 pyramidal cells from Ndel1 CKO versus WT mice. B, Bar graphs represent the percentage of microtubules longer, equal to, or shorter than 1 μm length in Ndel1 CKO (−/−) and WT mice (+/+). Percentage of microtubules >1 μm: WT: 27 ± 5.6% versus Ndel1 CKO: 13 ± 4.5%; mean ± SD (n = 3 animals). *p < 0.03. Percentage of microtubules ≤ 1 μm: WT: 73 ± 5.6% versus Ndel1 CKO: 87 ± 4.5%; mean ± SD (n = 3 animals). *p < 0.03. C, Levels of Lis1 and Dynein in hippocampus of Ndel1 CKO mice. Lis1 in WT: 1.00 ± 0.49 versus Ndel1 CKO: 2.25 ± 0.34; mean ± SE (n = 5 animals). *p < 0.05. Dynein: WT: 1.00 ± 0.16 versus Ndel1 CKO: 1.53 ± 0.19; mean ± SE (n = 5 animals). *p < 0.05. Student's t test was used for all statistical analyses in this figure.
To support our electron microscopy data, we quantified the levels of Lis1 and Dynein in Ndel1 CKO hippocampus by Western blot. Ndel1, Lis1, and Dynein form a complex that is critical for MTs organization and extension, as well as transport on MTs (Niethammer et al., 2000; Sasaki et al., 2000; Smith et al., 2000; Malikov et al., 2004; Li et al., 2005; Grabham et al., 2007; Yamada et al., 2010; Moon et al., 2014). In cell cultures, overexpression of Lis1 in the presence of the MT destabilization agent nocodazole washed out for 2 min generates fragments of MTs detached from the centrosomes in a manner reminiscent of Ndel1 CKO dendritic microtubules (Smith et al., 2000, their Fig. 6). By probing the levels of Lis1 and Dynein, we looked for possible mechanism underlying the microtubule fragmentation phenotype. Importantly, we found that the levels of Lis1 and Dynein are abnormally high in Nde1l CKO samples versus WT (Fig. 3C). On the basis of the cell culture experiments performed by Smith et al. (2000), and numerous studies documenting roles for Ndel1/Lis1/Dynein in MT organization and extension, our results suggest that MT fragmentation in Ndel1 CKO neurons involves MT destabilization and Lis1 increased dosage. In sum, our results on Lis1 and Dynein levels substantiate and provide a potential explanation for the MT fragmentation phenotype in Ndel1 CKO CA1 neurons. As MTs are highways for intracellular transport and trafficking, we reason that loss of Ndel1 likely causes dendritic pathology through failed delivery of cellular components required for neuronal maintenance and communication.
Figure 6.
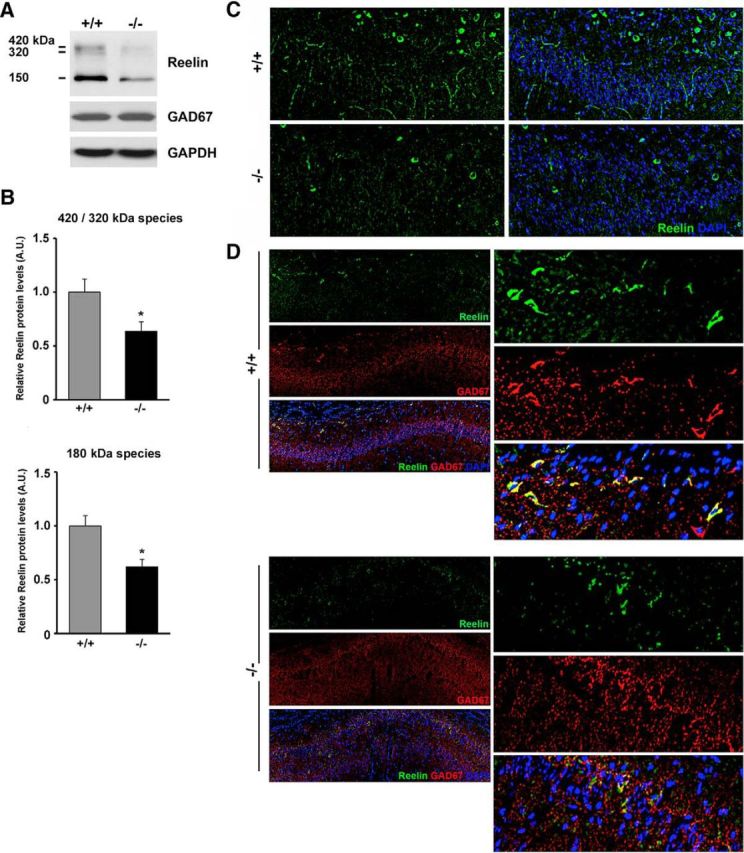
Reduced expression of Reelin in the hippocampus and stratum oriens of Ndel1 CKO mice. A, Expression of all Reelin species (180, 320, 420 kDa) in the hippocampus of 10-week-old Ndel1 CKO mice as detected by Western blots. Note the unchanged levels of the interneuronal marker GAD67 in the mutant animals compared with WT littermates. B, Bar graphs for the arbitrary levels of Reelin in the hippocampus of 10-week-old animals. 420/320 kDa species for WT (+/+): 1.00 ± 0.12 versus Ndel1 CKO (−/−): 0.64 ± 0.09; mean ± SE (n = 3 animals). 150 kDa species for WT (+/+): 1.00 +/− 0.09 versus Ndel1 CKO (−/−): 0.62 ± 0.07; mean ± SE (n = 3 animals), *p < 0.05 by Student's t test. C, Representative confocal images from a 7-week-old Ndel1 CKO mouse and WT littermate depicting the reduced number of Reelin-positive cell bodies in stratum oriens and reduced immunoreactivity for Reelin in CA1. D, Representative confocal images of the CA1 region from a 7-week-old Ndel1 CKO mouse and WT littermate confirming the reduced levels of Reelin in GAD67-positive interneurons of stratum oriens above the CA1 subfield.
Intrinsic hyperexcitability of CA1 pyramidal neurons in Ndel1 CKO mice
To understand the potential impact of MT destabilization and dendritic pathology on the functionality of CA1 Ndel1 CKO neurons, we measured several parameters associated with intrinsic excitability using patch-clamp electrophysiological methods (Fig. 4). Although resting membrane potential was not changed in Ndel1 CKO group, the remaining parameters were significantly altered (Fig. 4A). The input resistance was significantly higher in Ndel1 CKO pyramidal cells compared with control (285.9 ± 16.7 vs 211.8 ± 14.1 mΩ, p < 0.01; Fig. 4A). The higher Ri was correlated with a smaller current needed to elicit spiking in Ndel1 CKO pyramidal cells (threshold current, 61.5 ± 7.8 pA in KO vs 94.5 ± 10.2 pA in control mice; p < 0.05; Fig. 4A). We also found that the number of spikes elicited by current injection was increased in CKO animals compared with the control group (3.2 ± 0.4 vs 1.7 ± 0.2 spikes were elicited by threshold current and 8.7 ± 1.0 vs 4.2 ± 0.6 spikes when threshold current was doubled, p < 0.001; Fig. 4A). The increased input resistance may be linked to the reduced dendritic arborization of Ndel1 CKO pyramidal cells (Fig. 2), as suggested by a recent study emphasizing a similar correlation in CA3 neurons (Narayanan and Chattarji, 2010). A negative correlation between total dendritic length (as found in Figs. 2, 4C) and neuronal firing frequency was also found in other studies (Mainen and Sejnowski, 1996; van Ooyen et al., 2002). Together, our results associate MT fragmentation and dendritic pathology of Ndel1 CKO pyramidal cells with an intrinsic hyperexcitability.
Figure 4.
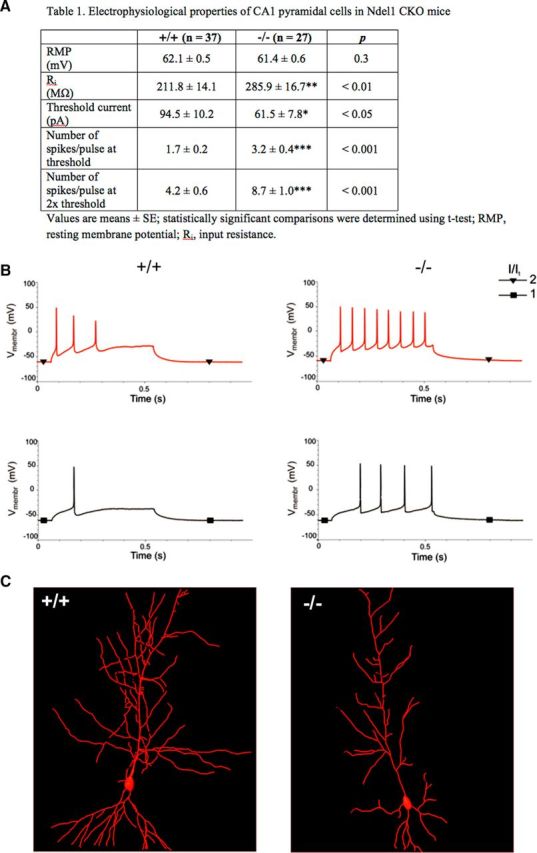
Intrinsic hyperexcitability of CA1 pyramidal neurons in Ndel1 CKO mice. A, Electrophysiological properties of CA1 hippocampal neurons in 7-week-old WT and Ndel1 CKO mice. While the resting membrane potential (RMP) was unaltered in Ndel1 CKO mice, the remaining parameters were significantly changed. Input resistance (Ri) was significantly higher in Ndel1 CKO pyramidal cells compared with control neurons. The higher Ri correlated with the smaller currents required to elicit spiking in Ndel1 CKO pyramidal cells. The number of spikes induced by current injection was increased in the CKO mice compared with control group. A total of 37 control and 27 Ndel1 KO recordings were used in the experiments. B, Representative recordings showing the response of a WT (left) and CKO (right) pyramidal cell to injection of depolarizing current steps at threshold (I/It = 1) and to twice the firing threshold (I/It = 2) levels. C, Representative reconstructions of biocytin-filled neurons show typical morphologies of pyramidal cells from WT (top) and Ndel1 CKO (bottom) littermates. *p < 0.05; **p < 0.01; ***p < 0.001.
Decreased number of synapses in CA1 hippocampus of Ndel1 CKO mice
The dendritic pathology of Ndel1 CKO pyramidal neurons (Fig. 2) raised the question as to whether connectivity within the CA1 is altered. We first evaluated the concentration of dendritic spines on CA1 pyramidal neurons using Golgi-stained tissues from Ndel1 CKO mice. A significant (∼35%) decrease in spine density per dendrite was found on these neurons compared with controls (Fig. 5A). Using electron microscopy, we detected an overall 50% decrease in the total number of synapses in the CA1 (Fig. 5B). Further, we found that abundance of both symmetric (inhibitory) and asymmetric (excitatory) types of synapses per 100 μm2 decreased by ∼50% compared with their control neurons (Fig. 5C), although no neuronal loss was detected using unbiased stereological counts at that age (data not shown). Thus, upon deletion of Ndel1 in principal cells, both inhibitory and excitatory synapses are lost in CA1 hippocampus. These results suggest that connectivity within the CA1 is substantially altered.
Figure 5.
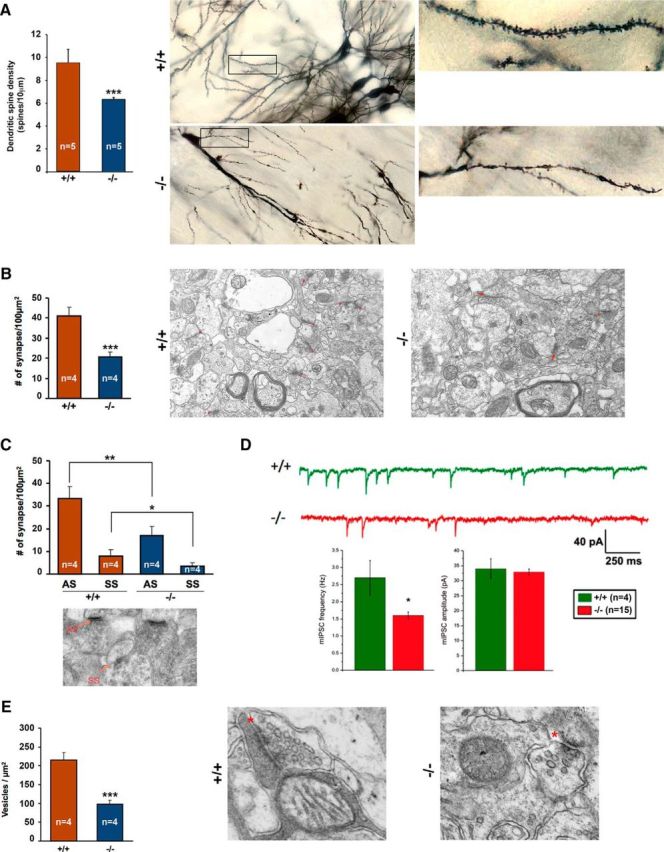
Ndel1 CKO mice show reduced number of synapses, decreased frequency of mIPSCs, and lower number of synaptic vesicles at inhibitory (symmetric) synapses in CA1 hippocampus. A, Approximately 35% decrease in the number of dendritic spines on CA1 pyramidal neurons of 9-week-old Ndel1 CKO mice (−/−) compared with WT littermates (+/+), and representative Golgi-stained CA1 pyramidal neurons. WT: 9.53 ± 1.18 versus Ndel1 CKO: 6.33 ± 0.17; mean ± SD (n = 5 animals). ***p < 0.0005. B, A ∼50% decrease in the total number of synapses in CA1 of 9-week-old Ndel1 CKO mice as detected by electron microscopy, and representative electron microscopic photographs of CA1 hippocampus. WT: 41.0 ± 4.4% versus Ndel1 CKO: 20.6 ± 2.7%; mean ± SD (n = 4 animals). ***p < 0.001. Arrows indicate synapses. C, A ∼50% decrease in the number of asymmetric (AS) and symmetric synapses (SS) in CA1 of 9-week-old Ndel1 CKO mice as counted under electron microscopy. AS: WT: 33.3 ± 5.3% versus Ndel1 CKO: 17.1 ± 4.0%; mean ± SD (n = 4). **p < 0.01. SS: WT: 7.8 ± 3.3% versus Ndel1 CKO: 3.5 ± 1.5%; mean ± SD (n = 4 animals). *p < 0.05. D, Representative traces of mIPSCs recorded in the presence of AP5, DNQX, and TTX on CA1 pyramidal neurons in 7-week-old Ndel1 CKO mice (red trace, n = 14) and WT littermates (green trace, n = 5). Note the decreased mIPSCs frequency (from 2.7 ± 0.5 Hz in control to 1.6 ± 0.1 Hz CKO; *p = 0.01) but no significant change in amplitude (34.0 ± 3.3 pA in control vs 32.9 ± 1.0 pA CKO; p = 0.69). E, A ∼50% decrease in the number of synaptic vesicles at inhibitory (symmetric) synapses per μm2 of CA1 in 9-week-old Ndel1 CKO mice (−/−) compared with WT littermates (+/+), and representative electron microscopic photographs of flattened, ellipsoidal, or pleomorphic vesicles found in symmetric synapses of CA1. WT: 215.6 ± 18.7 versus Ndel1 CKO: 98.5 ± 9.8; mean ± SD (n = 4 animals). ***p < 0.0001. Student's t test was used for all statistical analyses in this figure. Two blinded independent investigators completed all counts in these experiments.
Decreased inhibitory drive onto CA1 pyramidal cells in Ndel1 CKO mice
Next, we sought to investigate functional connectivity within the Ndel1 CKO hippocampus. We focused on the interactions between pyramidal cells and interneurons that are critical for hippocampal function and integrity. We first recorded mIPSCs in CA1 neurons of Ndel1 CKO (n = 14) and control mice (n = 4) in the presence of APV, DNQX, and TTX. A significant decrease in mIPSCs frequency (from 2.7 ± 0.5 Hz in control to 1.6 ± 0.1 Hz CKO; p = 0.01) was detected for CA1 pyramidal cells from Ndel1 CKO mice (Fig. 5D). In contrast, no significant change in amplitude (34.0 ± 3.3 pA in control vs 32.9 ± 1.0 pA CKO; p = 0.69) was observed between the two groups. The unchanged amplitude indicated that postsynaptic GABAA receptors appear relatively intact. In contrast, a change in frequency most likely suggests presynaptic alterations (i.e., a reduced number of GABAA synapses, as found in Fig. 5C) and/or decreased presynaptic release probability. To investigate the latter phenomenon, we counted the number of presynaptic flattened, ellipsoidal, or pleomorphic vesicles in CA1 symmetric synapses. We found a substantially lower number of presynaptic vesicles at these inhibitory CA1 synapses in Ndel1 CKO mice compared with controls (Fig. 5E). Together, our results indicate an overall decrease in inhibitory drive onto CA1 principal cells in Ndel1 CKO mice.
Decreased levels of Reelin in the hippocampus of Ndel1 CKO mice
The above findings on inhibitory synapses and inhibitory drive suggest that CA1 interneurons might be dysfunctional in Ndel1 CKO mice. To further investigate interneuron biology, we analyzed the expression of established interneuronal markers by Western blots and staining. A candidate molecule to which we devoted particular attention was Reelin, a multifunctional glycoprotein secreted by interneurons. Our rationale was based on the following facts. Like Ndel1, Reelin signaling regulates MTs during development (Hiesberger et al., 1999; Niu et al., 2004; González-Billault et al., 2005; Meseke et al., 2013), and perhaps a similar function for Reelin exists in the adult brain. Further, Reelin controls neuronal alignment in the mature brain and inhibition of Reelin signaling by injection of CR-50 Reelin antibodies in the dentate gyrus or the cerebellum causes a “widening” of granule and Purkinje cell layers (D'Arcangelo et al., 1997; Miyata et al., 1997; Nakajima et al., 1997; Heinrich et al., 2006), although to a significantly lesser extent than the dispersion found in the CA1 of Ndel1 CKO mice (Fig. 1).
We first determined the expression of Reelin in the hippocampus of Ndel1 CKO mice by Western blot. As expected, Reelin was expressed in three different forms with approximate molecular weights of 420, 320, and 180 kDa (Fig. 6A). We found a ∼30% decrease in Reelin species in Ndel1 CKO hippocampus compared with WT samples (Fig. 6B). To better localize these changes, we analyzed the distribution of Reelin in the hippocampus using confocal microscopy. Approximately 90% of interneurons in the stratum oriens heavily express Reelin compared with other layers with half percentage of Reelin-expressing neurons (Pesold et al., 1998; Ramos-Moreno et al., 2006). An intense Reelin-positive signal was found in cell bodies above the CA1 in the stratum oriens of WT animals (Fig. 6C). Secreted Reelin-immunoreactive signals were also found “invading” CA1 layer in WT animals (Fig. 6C). Importantly, these Reelin-immunoreactive signals were significantly reduced in stratum oriens and CA1 of mutant mice. The decreased expression of Reelin in stratum oriens interneurons of Ndel1 CKO hippocampus was confirmed by costaining of Reelin and the interneuronal marker GAD-67 (Fig. 6D). Together, we conclude that changes in GABAergic inhibitory synapses in Ndel1 CKO mice (Fig. 5C–E) are associated with decreased Reelin levels in stratum oriens and hippocampus. As Ndel1 is primarily depleted in excitatory neurons, we reasoned that these interneuronal alterations are secondarily associated with pyramidal cell defects.
A single injection of Reelin ameliorates structural, cellular, and anatomical abnormalities
Reelin regulates MTs, dendritic development (Niu et al., 2004; González-Billault et al., 2005; Meseke et al., 2013), neuronal plasticity (Weeber et al., 2002), and it has a certain role in preventing granule and Purkinje cell dispersion in the adult brain (D'Arcangelo et al., 1997; Miyata et al., 1997; Nakajima et al., 1997; Heinrich et al., 2006). Therefore, decreased levels of Reelin in the postnatal brain might contribute to CA1 defects observed in Ndel1 CKO mice. In support of this idea, treatment of P14 primary hippocampal neuron cultures (that contain both pyramidal cells and Reelin-secreting interneurons; data not shown) with CR-50 Reelin blocking antibodies destabilizes neuronal processes (Fig. 7A) in a manner reminiscent of crooked dendrites with fragmented MTs in Ndel1 CKO CA1 (Fig. 2). On these bases, we hypothesized that supplementation of Reelin would prevent CA1 pathologies of Ndel1 CKO mice. To test this hypothesis, we purified full-length Reelin proteins (Fig. 7B) and performed a single unilateral stereotaxic injection in the hippocampus of 15 Ndel1 CKO mice with a mean age of 7 weeks that developed deficits. As variability in Ndel1 CKO phenotypes is possible, for direct comparison, the contralateral side was injected with control vehicle. Electron microscopic analysis of the Reelin-injected hemisphere and contralateral control-injected side revealed that, 1 week after injection, Reelin supplementation improves the fragmentation of dendritic MTs: Reelin-treated dendrites contained twice the number of MTs >1 μm compared with control-treated dendrites (Fig. 7C,D). These results indicate that the MT-associated function of Reelin signaling during embryonic development could be harnessed to counteract the loss of Ndel1's MAP function in mature CA1 pyramidal neurons in vivo.
Figure 7.
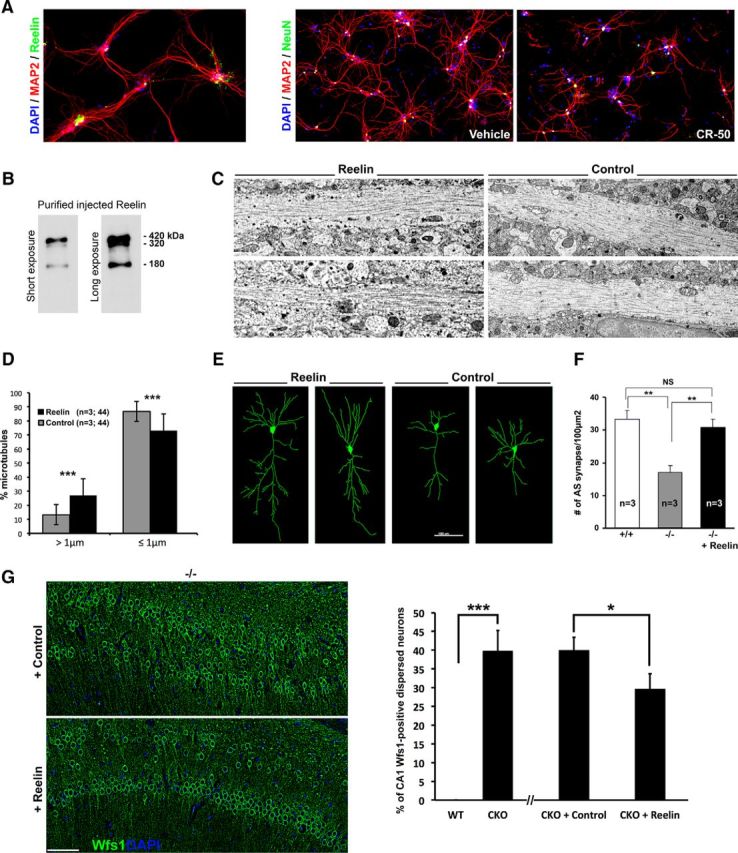
A single injection of Reelin protein ameliorates microtubular fragmentation, dendritic/synaptic pathologies, and CA1 dispersion. A, Primary hippocampal neuron cultures expressing Reelin (right) undergo dendritic attrition when treated with the CR-50 Reelin blocking peptides. B, Western blots showing the three Reelin species purified from a stable cell line. C, Representative electron microscopic images of CA1 hippocampal dendrites in 7-week-old Ndel1 CKO hippocampus after unilateral injection with purified Reelin (at concentration of 0.1 μg/μl); the contralateral side was injected with vehicle control (supernatant from HEK 293 cells, see Materials and Methods). D, Bar graphs represent the number of MTs longer, shorter, or equal to 1 μm in pyramidal dendrites in CA1 injected with either Reelin or control vehicle. Percentage of MTs >1 μm: Ndel1 CKO + control: 13.2 ± 7.1% versus Ndel1 CKO + Reelin: 26.8 ± 7.1%; mean ± SD (n = 3 animals, 44 dendrites per condition). ***p < 0.001. Percentage of MTs ≤ 1 μm: Ndel1 CKO + control: 86.8 ± 12.0% versus Ndel1 CKO + Reelin: 73.2 ± 12.0%; mean ± SD (n = 3 animals, 44 dendrites per condition). ***p < 0.001. E, Representative images of Imaris software-enabled reconstruction of individual CA1 pyramidal neurons from 3 7-week-old Ndel1 CKO mice injected unilaterally with Reelin and contralaterally with PBS. Reelin rescues the dendritic pathology in the Ndel1 CKO mice. Total neuronal process length: Ndel1 CKO + control: 1386 ± 109 μm versus Ndel1 CKO + Reelin: 1879 ± 145 μm; mean ± SEM (n = 10 neurons from 3 animals). *p < 0.05. Total number of dendrites per pyramidal cell: Ndel1 CKO + control: 21.9 ± 1.6 versus Ndel1 CKO + Reelin: 31.7 ± 3.2; mean ± SEM (n = 10 neurons from 3 animals). *p < 0.05. F, Bar graphs represent the number of asymmetric synapses in CA1 of control WT (+/+; 33.3 ± 2.6), untreated Ndel1 CKO (−/−; 17.1 ± 2.0), or Reelin-injected Ndel1 CKO mice (−/− + Reelin; 29.0 ± 2.1); mean ± SD (n = 3 animals per condition). NS, Nonsignificant. **p < 0.01. G, Dispersion of Wfs1-positive CA1 pyramidal neurons is ameliorated by 25% in Ndel1 CKO mice injected in the hippocampus with Reelin at 7 week of age and analyzed at 8-week-old: dispersion in Ndel1 CKO (−/−) injected with control versus Ndel1 CKO (−/−) injected with Reelin: 40.0 ± 3.4% versus 29.7 ± 4.0%; mean ± SD (n = 3 animals; 4 serial sections per animals). *p < 0.05 (Student's t test). Dispersion in WT (+/+) versus Ndel1 CKO (−/−): 0% versus 39.8 ± 5.4%; mean ± SD (n = 3 animals, 4 serial sections per animals). ***p < 0.001 (Student's t test).
Compatible with the amelioration in MT structural integrity, Reelin injection improved dendritic pathology, as evidenced by reconstruction of Golgi-stained neurons (Fig. 7E). Further, Reelin-treated CA1 exhibited as many asymmetric synapses as CA1 from control animals, suggesting functional recovery as well; this corresponds to a ∼100% increase compared with untreated Ndel1 CKO CA1 (Fig. 7F). The injection of Reelin also ameliorated the dispersion of CA1 pyramidal cells by 25% (dispersion in Ndel1 CKO (−/−) injected with control versus Ndel1 CKO (−/−) injected with Reelin: 40.0 ± 3.4% versus 29.7 ± 4.0%; mean ± SD (n = 3 animals; p < 0.05, Student's t test), as demonstrated by immunostaining with the CA1 pyramidal neuron marker Wfs1 (Fig. 7G). From these experiments, we conclude that Reelin rescues the ultrastructural, cellular, and, to a lesser extent, the anatomical CA1 defects observed in Ndel1 CKO mice.
Discussion
Here we report that postnatal disruption of Ndel1 in CA1 excitatory pyramidal cells impairs maintenance of their positioning and integrity, as well as connectivity with interneurons. Consistent with the long suspected but yet unproven MT-associated function of Ndel1 in vivo, the primary lack of Ndel1 in excitatory forebrain pyramidal neurons causes fragmentation of dendritic MTs associated with abnormally elevated levels of Lis1/Dynein, leading eventually to dendritic/synaptic pathologies associated with cell hyperexcitability (Figs. 1–4). Associated with these pyramidal cell changes, the interface between pyramidal cells and GABAergic interneurons becomes disrupted (Fig. 5). Specifically, there are fewer inhibitory synapses, a decreased frequency of mIPSCs, and a substantially lower number of presynaptic vesicles at CA1 inhibitory synapses, all consistent with an overall decrease in inhibitory drive onto principal cells (Fig. 5). Moreover, depletion of Reelin in interneurons of hippocampus of Ndel1 CKO mice contributes to the pathologies, as evidenced by the experiment with Reelin injection (Figs. 6, 7). Thus, Ndel1 and Reelin both contribute to the maintenance of postnatal CA1 integrity and function.
Whether the two proteins cooperate or work independently in postnatal CA1 remains to be determined. Considering that they both regulate MTs during neurodevelopment, the most straightforward explanation for the rescue is that, upon Reelin injection, the MT stabilization and plasticity functions of Reelin are harnessed to primarily counteract the fragmentation of MTs and dendritic/synaptic pathology of Ndel1-deficient CA1 principal cells (Fig. 7). However, it is important to note that Ndel1 CKO mice are intrinsically different from Reeler and Dab1 (a downstream component of Reelin signaling pathway) homozygote and heterozygote-null mice in many aspects (from protein functions to straight KO phenotypes, to gene KO strategy, to mechanisms underlying the bilayer phenotype). Thus, it is not surprising that our Ndel1 CKO mice do not phenocopy these mutant animals.
Our initial studies focused on the CA1 region of the hippocampus because our data indicated that this region is most affected in Ndel1 CKO animals. However, it may be that other regions within the hippocampus or the trisynaptic hippocampal circuitry per se, and in other remote parts of the brain, such as the insular cortex (given that the animals die suddenly), may be significantly impacted. Abnormalities in the structure and function of the insular cortex can lead to cardiac arrhythmias that may account for unexpected mortality seen in the model. Indeed, the average lifespan of the Ndel1 CKO animals is ∼11 weeks with only a few subjects (∼5%) surpassing 20 weeks of age (data not shown). Of course, this early mortality precludes any experiments beyond 10–11 weeks. How Ndel1 and Reelin maintain postnatally other brain regions also remains to be determined.
Our findings support and extend the cortical GABAergic hypothesis (particularly in autism and schizophrenia) that posits reduced inhibitory input onto pyramidal cells in the cortex (Ross et al., 2006; Marín, 2012), to the hippocampus. That Reelin and Ndel1 levels decreased in humans with mental disorders is known (Impagnatiello et al., 1998; Guidotti et al., 2000; Fatemi, 2005; Fatemi et al., 2005; Lipska et al., 2006). There is also evidence that human patients with neurological deficits have dispersed CA1 (Sloviter et al., 2004). Thus, our study may help unravel the basic mechanisms of postnatal hippocampal maintenance and function, and serve as base to understand the significance of decreased levels of Ndel1 and Reelin observed in patients with neurological disorders.
Footnotes
This work was supported by the Canadian Institutes of Health Research to M.D.N. and G.C.T., Alberta Innovates Health Solutions to M.D.N. and G.C.T., the Alberta Children's Hospital Research Institute to C.G. and J.M.R., and the framework of international cooperation program managed by NRF of Korea (2012K2A1A2033117) between the laboratories of M.D.N. and S.K.P., M.D.N. received a Human Frontier Science Program Organization Career Development Award, Canadian Institutes of Health Research New Investigator Award, and Alberta Innovates Health Solutions Scholar Award. This work was also supported in part by the Grants NRF-2012R1A4A1028200 and NRF-2015M3C7A1030964 from the Korean government (MSIP) to S.K.P. K.P. received an Alberta Innovates Health Solutions scholarship. We thank Dr. Tom Curran and Dr. Joachim Herz for the Reelin-expressing cell line and methods of purification.
The authors declare no competing financial interests.
References
- Applegate MD, Landfield PW. Synaptic vesicle redistribution during hippocampal frequency potentiation and depression in young and aged rats. J Neurosci. 1988;8:1096–1111. doi: 10.1523/JNEUROSCI.08-04-01096.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradshaw NJ, Ogawa F, Antolin-Fontes B, Chubb JE, Carlyle BC, Christie S, Claessens A, Porteous DJ, Millar JK. DISC1, PDE4B, and NDE1 at the centrosome and synapse. Biochem Biophys Res Commun. 2008;377:1091–1096. doi: 10.1016/j.bbrc.2008.10.120. [DOI] [PubMed] [Google Scholar]
- Brandon NJ. Dissecting DISC1 function through protein-protein interactions. Biochem Soc Trans. 2007;35:1283–1286. doi: 10.1042/BST0351283. [DOI] [PubMed] [Google Scholar]
- Burdick KE, Kamiya A, Hodgkinson CA, Lencz T, DeRosse P, Ishizuka K, Elashvili S, Arai H, Goldman D, Sawa A, Malhotra AK. Elucidating the relationship between DISC1, NDEL1 and NDE1 and the risk for schizophrenia: evidence of epistasis and competitive binding. Hum Mol Genet. 2008;17:2462–2473. doi: 10.1093/hmg/ddn146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chansard M, Hong JH, Park YU, Park SK, Nguyen MD. Ndel1, Nudel (Noodle): flexible in the cell? Cytoskeleton (Hoboken) 2011;68:540–554. doi: 10.1002/cm.20532. [DOI] [PubMed] [Google Scholar]
- D'Arcangelo G, Miao GG, Chen SC, Soares HD, Morgan JI, Curran T. A protein related to extracellular matrix proteins deleted in the mouse mutant reeler. Nature. 1995;374:719–723. doi: 10.1038/374719a0. [DOI] [PubMed] [Google Scholar]
- D'Arcangelo G, Nakajima K, Miyata T, Ogawa M, Mikoshiba K, Curran T. Reelin is a secreted glycoprotein recognized by the CR-50 monoclonal antibody. J Neurosci. 1997;17:23–31. doi: 10.1523/JNEUROSCI.17-01-00023.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fatemi SH. Reelin glycoprotein: structure, biology and roles in health and disease. Mol Psychiatry. 2005;10:251–257. doi: 10.1038/sj.mp.4001613. [DOI] [PubMed] [Google Scholar]
- Fatemi SH, Hossein Fatemi S, Stary JM, Earle JA, Araghi-Niknam M, Eagan E. GABAergic dysfunction in schizophrenia and mood disorders as reflected by decreased levels of glutamic acid decarboxylase 65 and 67 kDa and Reelin proteins in cerebellum. Schizophr Res. 2005;72:109–122. doi: 10.1016/j.schres.2004.02.017. [DOI] [PubMed] [Google Scholar]
- Förster E, Tielsch A, Saum B, Weiss KH, Johanssen C, Graus-Porta D, Müller U, Frotscher M. Reelin, Disabled 1, and beta 1 integrins are required for the formation of the radial glial scaffold in the hippocampus. Proc Natl Acad Sci U S A. 2002;99:13178–13183. doi: 10.1073/pnas.202035899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Förster E, Zhao S, Frotscher M. Laminating the hippocampus. Nat Rev Neurosci. 2006;7:259–267. doi: 10.1038/nrn1882. [DOI] [PubMed] [Google Scholar]
- Gavrilovici C, D'Alfonso S, Dann M, Poulter MO. Kindling-induced alterations in GABAA receptor-mediated inhibition and neurosteroid activity in the rat piriform cortex. Eur J Neurosci. 2006;24:1373–1384. doi: 10.1111/j.1460-9568.2006.05012.x. [DOI] [PubMed] [Google Scholar]
- Gavrilovici C, Pollock E, Everest M, Poulter MO. The loss of interneuron functional diversity in the piriform cortex after induction of experimental epilepsy. Neurobiol Dis. 2012;48:317–328. doi: 10.1016/j.nbd.2012.07.002. [DOI] [PubMed] [Google Scholar]
- González-Billault C, Del Río JA, Ureña JM, Jiménez-Mateos EM, Barallobre MJ, Pascual M, Pujadas L, Simó S, Torre AL, Gavin R, Wandosell F, Soriano E, Avila J. A role of MAP1B in Reelin-dependent neuronal migration. Cereb Cortex. 2005;15:1134–1145. doi: 10.1093/cercor/bhh213. [DOI] [PubMed] [Google Scholar]
- Grabham PW, Seale GE, Bennecib M, Goldberg DJ, Vallee RB. Cytoplasmic dynein and LIS1 are required for microtubule advance during growth cone remodeling and fast axonal outgrowth. J Neurosci. 2007;27:5823–5834. doi: 10.1523/JNEUROSCI.1135-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guidotti A, Auta J, Davis JM, Di-Giorgi-Gerevini V, Dwivedi Y, Grayson DR, Impagnatiello F, Pandey G, Pesold C, Sharma R, Uzunov D, Costa E. Decrease in reelin and glutamic acid decarboxylase67 (GAD67) expression in schizophrenia and bipolar disorder: a postmortem brain study. Arch Gen Psychiatry. 2000;57:1061–1069. doi: 10.1001/archpsyc.57.11.1061. [DOI] [PubMed] [Google Scholar]
- Heinrich C, Nitta N, Flubacher A, Müller M, Fahrner A, Kirsch M, Freiman T, Suzuki F, Depaulis A, Frotscher M, Haas CA. Reelin deficiency and displacement of mature neurons, but not neurogenesis, underlie the formation of granule cell dispersion in the epileptic hippocampus. J Neurosci. 2006;26:4701–4713. doi: 10.1523/JNEUROSCI.5516-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herz J, Chen Y. Reelin, lipoprotein receptors and synaptic plasticity. Nat Rev Neurosci. 2006;7:850–859. doi: 10.1038/nrn2009. [DOI] [PubMed] [Google Scholar]
- Herz J, Chen Y, Masiulis I, Zhou L. Expanding functions of lipoprotein receptors. J Lipid Res. 2009;50(Suppl):S287–S292. doi: 10.1194/jlr.R800077-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiesberger T, Trommsdorff M, Howell BW, Goffinet A, Mumby MC, Cooper JA, Herz J. Direct binding of Reelin to VLDL receptor and ApoE receptor 2 induces tyrosine phosphorylation of disabled-1 and modulates tau phosphorylation. Neuron. 1999;24:481–489. doi: 10.1016/S0896-6273(00)80861-2. [DOI] [PubMed] [Google Scholar]
- Hunt RF, Dinday MT, Hindle-Katel W, Baraban SC. LIS1 deficiency promotes dysfunctional synaptic integration of granule cells generated in the developing and adult dentate gyrus. J Neurosci. 2012;32:12862–12875. doi: 10.1523/JNEUROSCI.1286-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Impagnatiello F, Guidotti AR, Pesold C, Dwivedi Y, Caruncho H, Pisu MG, Uzunov DP, Smalheiser NR, Davis JM, Pandey GN, Pappas GD, Tueting P, Sharma RP, Costa E. A decrease of reelin expression as a putative vulnerability factor in schizophrenia. Proc Natl Acad Sci U S A. 1998;95:15718–15723. doi: 10.1073/pnas.95.26.15718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Impey S, McCorkle SR, Cha-Molstad H, Dwyer JM, Yochum GS, Boss JM, McWeeney S, Dunn JJ, Mandel G, Goodman RH. Defining the CREB regulon: a genome-wide analysis of transcription factor regulatory regions. Cell. 2004;119:1041–1054. doi: 10.1016/j.cell.2004.10.032. [DOI] [PubMed] [Google Scholar]
- Kerjan G, Gleeson JG. Genetic mechanisms underlying abnormal neuronal migration in classical lissencephaly. Trends Genet. 2007;23:623–630. doi: 10.1016/j.tig.2007.09.003. [DOI] [PubMed] [Google Scholar]
- Kohara K, Pignatelli M, Rivest AJ, Jung HY, Kitamura T, Suh J, Frank D, Kajikawa K, Mise N, Obata Y, Wickersham IR, Tonegawa S. Cell type-specific genetic and optogenetic tools reveal hippocampal CA2 circuits. Nat Neurosci. 2014;17:269–279. doi: 10.1038/nn.3614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakatosova S, Ostatnikova D. Reelin and its complex involvement in brain development and function. Int J Biochem Cell Biol. 2012;44:1501–1504. doi: 10.1016/j.biocel.2012.06.002. [DOI] [PubMed] [Google Scholar]
- Lavenex P, Banta Lavenex P. Building hippocampal circuits to learn and remember: insights into the development of human memory. Behav Brain Res. 2013;254:8–21. doi: 10.1016/j.bbr.2013.02.007. [DOI] [PubMed] [Google Scholar]
- Lein ES, Hawrylycz MJ, Ao N, Ayres M, Bensinger A, Bernard A, Boe AF, Boguski MS, Brockway KS, Byrnes EJ, Chen L, Chen L, Chen TM, Chin MC, Chong J, Crook BE, Czaplinska A, Dang CN, Datta S, Dee NR, et al. Genome-wide atlas of gene expression in the adult mouse brain. Nature. 2007;445:168–176. doi: 10.1038/nature05453. [DOI] [PubMed] [Google Scholar]
- Li J, Lee WL, Cooper JA. NudEL targets dynein to microtubule ends through LIS1. Nat Cell Biol. 2005;7:686–690. doi: 10.1038/ncb1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipska BK, Peters T, Hyde TM, Halim N, Horowitz C, Mitkus S, Weickert CS, Matsumoto M, Sawa A, Straub RE, Vakkalanka R, Herman MM, Weinberger DR, Kleinman JE. Expression of DISC1 binding partners is reduced in schizophrenia and associated with DISC1 SNPs. Hum Mol Genet. 2006;15:1245–1258. doi: 10.1093/hmg/ddl040. [DOI] [PubMed] [Google Scholar]
- Mackie S, Millar JK, Porteous DJ. Role of DISC1 in neural development and schizophrenia. Curr Opin Neurobiol. 2007;17:95–102. doi: 10.1016/j.conb.2007.01.007. [DOI] [PubMed] [Google Scholar]
- Mainen ZF, Sejnowski TJ. Influence of dendritic structure on firing pattern in model neocortical neurons. Nature. 1996;382:363–366. doi: 10.1038/382363a0. [DOI] [PubMed] [Google Scholar]
- Malikov V, Kashina A, Rodionov V. Cytoplasmic dynein nucleates microtubules to organize them into radial arrays in vivo. Mol Biol Cell. 2004;15:2742–2749. doi: 10.1091/mbc.E03-10-0770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marín O. Interneuron dysfunction in psychiatric disorders. Nat Rev Neurosci. 2012;13:107–120. doi: 10.1038/nrn3155. [DOI] [PubMed] [Google Scholar]
- Meseke M, Cavus E, Förster E. Reelin promotes microtubule dynamics in processes of developing neurons. Histochem Cell Biol. 2013;139:283–297. doi: 10.1007/s00418-012-1025-1. [DOI] [PubMed] [Google Scholar]
- Miyata T, Nakajima K, Mikoshiba K, Ogawa M. Regulation of Purkinje cell alignment by reelin as revealed with CR-50 antibody. J Neurosci. 1997;17:3599–3609. doi: 10.1523/JNEUROSCI.17-10-03599.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon HM, Youn YH, Pemble H, Yingling J, Wittmann T, Wynshaw-Boris A. LIS1 controls mitosis and mitotic spindle organization via the LIS1-NDEL1-dynein complex. Hum Mol Genet. 2014;23:449–466. doi: 10.1093/hmg/ddt436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajima K, Mikoshiba K, Miyata T, Kudo C, Ogawa M. Disruption of hippocampal development in vivo by CR-50 mAb against reelin. Proc Natl Acad Sci U S A. 1997;94:8196–8201. doi: 10.1073/pnas.94.15.8196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayanan R, Chattarji S. Computational analysis of the impact of chronic stress on intrinsic and synaptic excitability in the hippocampus. J Neurophysiol. 2010;103:3070–3083. doi: 10.1152/jn.00913.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicodemus KK, Callicott JH, Higier RG, Luna A, Nixon DC, Lipska BK, Vakkalanka R, Giegling I, Rujescu D, St Clair D, Muglia P, Shugart YY, Weinberger DR. Evidence of statistical epistasis between DISC1, CIT and NDEL1 impacting risk for schizophrenia: biological validation with functional neuroimaging. Hum Genet. 2010;127:441–452. doi: 10.1007/s00439-009-0782-y. [DOI] [PubMed] [Google Scholar]
- Niethammer M, Smith DS, Ayala R, Peng J, Ko J, Lee MS, Morabito M, Tsai LH. NUDEL is a novel Cdk5 substrate that associates with LIS1 and cytoplasmic dynein. Neuron. 2000;28:697–711. doi: 10.1016/S0896-6273(00)00147-1. [DOI] [PubMed] [Google Scholar]
- Niu S, Renfro A, Quattrocchi CC, Sheldon M, D'Arcangelo G. Reelin promotes hippocampal dendrite development through the VLDLR/ApoER2-Dab1 pathway. Neuron. 2004;41:71–84. doi: 10.1016/S0896-6273(03)00819-5. [DOI] [PubMed] [Google Scholar]
- Pesold C, Impagnatiello F, Pisu MG, Uzunov DP, Costa E, Guidotti A, Caruncho HJ. Reelin is preferentially expressed in neurons synthesizing gamma-aminobutyric acid in cortex and hippocampus of adult rats. Proc Natl Acad Sci U S A. 1998;95:3221–3226. doi: 10.1073/pnas.95.6.3221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porteous DJ, Thomson P, Brandon NJ, Millar JK. The genetics and biology of DISC1: an emerging role in psychosis and cognition. Biol Psychiatry. 2006;60:123–131. doi: 10.1016/j.biopsych.2006.04.008. [DOI] [PubMed] [Google Scholar]
- Ramos-Moreno T, Galazo MJ, Porrero C, Martínez-Cerdeño V, Clascá F. Extracellular matrix molecules and synaptic plasticity: immunomapping of intracellular and secreted Reelin in the adult rat brain. Eur J Neurosci. 2006;23:401–422. doi: 10.1111/j.1460-9568.2005.04567.x. [DOI] [PubMed] [Google Scholar]
- Ross CA, Margolis RL, Reading SA, Pletnikov M, Coyle JT. Neurobiology of schizophrenia. Neuron. 2006;52:139–153. doi: 10.1016/j.neuron.2006.09.015. [DOI] [PubMed] [Google Scholar]
- Sasaki S, Shionoya A, Ishida M, Gambello MJ, Yingling J, Wynshaw-Boris A, Hirotsune S. A LIS1/NUDEL/cytoplasmic dynein heavy chain complex in the developing and adult nervous system. Neuron. 2000;28:681–696. doi: 10.1016/S0896-6273(00)00146-X. [DOI] [PubMed] [Google Scholar]
- Sasaki S, Mori D, Toyo-oka K, Chen A, Garrett-Beal L, Muramatsu M, Miyagawa S, Hiraiwa N, Yoshiki A, Wynshaw-Boris A, Hirotsune S. Complete loss of Ndel1 results in neuronal migration defects and early embryonic lethality. Mol Cell Biol. 2005;25:7812–7827. doi: 10.1128/MCB.25.17.7812-7827.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sloviter RS, Kudrimoti HS, Laxer KD, Barbaro NM, Chan S, Hirsch LJ, Goodman RR, Pedley TA. “Tectonic” hippocampal malformations in patients with temporal lobe epilepsy. Epilepsy Res. 2004;59:123–153. doi: 10.1016/j.eplepsyres.2004.04.001. [DOI] [PubMed] [Google Scholar]
- Smith DS, Niethammer M, Ayala R, Zhou Y, Gambello MJ, Wynshaw-Boris A, Tsai LH. Regulation of cytoplasmic dynein behaviour and microtubule organization by mammalian Lis1. Nat Cell Biol. 2000;2:767–775. doi: 10.1038/35041000. [DOI] [PubMed] [Google Scholar]
- Stranahan AM, Erion JR, Wosiski-Kuhn M. Reelin signaling in development, maintenance, and plasticity of neural networks. Ageing Res Rev. 2013;12:815–822. doi: 10.1016/j.arr.2013.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsien JZ, Chen DF, Gerber D, Tom C, Mercer EH, Anderson DJ, Mayford M, Kandel ER, Tonegawa S. Subregion- and cell type-restricted gene knockout in mouse brain. Cell. 1996;87:1317–1326. doi: 10.1016/S0092-8674(00)81826-7. [DOI] [PubMed] [Google Scholar]
- van Ooyen A, Duijnhouwer J, Remme MW, van Pelt J. The effect of dendritic topology on firing patterns in model neurons. Network. 2002;13:311–325. doi: 10.1088/0954-898X_13_3_304. [DOI] [PubMed] [Google Scholar]
- Weeber EJ, Beffert U, Jones C, Christian JM, Förster E, Sweatt JD, Herz J. Reelin and ApoE receptors cooperate to enhance hippocampal synaptic plasticity and learning. J Biol Chem. 2002;277:39944–39952. doi: 10.1074/jbc.M205147200. [DOI] [PubMed] [Google Scholar]
- Wynshaw-Boris A, Pramparo T, Youn YH, Hirotsune S. Lissencephaly: mechanistic insights from animal models and potential therapeutic strategies. Semin Cell Dev Biol. 2010;21:823–830. doi: 10.1016/j.semcdb.2010.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada M, Hirotsune S, Wynshaw-Boris A. The essential role of LIS1, NDEL1 and Aurora-A in polarity formation and microtubule organization during neurogensis. Cell Adh Migr. 2010;4:180–184. doi: 10.4161/cam.4.2.10715. [DOI] [PMC free article] [PubMed] [Google Scholar]