

Abstract
Although we are beginning to understand the late stage of neurodegenerative diseases, the molecular defects associated with the initiation of impaired cognition are poorly characterized. Here, we demonstrate that in the adult brain, the coxsackievirus and adenovirus receptor (CAR) is located on neuron projections, at the presynapse in mature neurons, and on the soma of immature neurons in the hippocampus. In a proinflammatory or diseased environment, CAR is lost from immature neurons in the hippocampus. Strikingly, in hippocampi of patients at early stages of late-onset Alzheimer's disease (AD), CAR levels are significantly reduced. Similarly, in triple-transgenic AD mice, CAR levels in hippocampi are low and further reduced after systemic inflammation. Genetic deletion of CAR from the mouse brain triggers deficits in adult neurogenesis and synapse homeostasis that lead to impaired hippocampal plasticity and cognitive deficits. We propose that post-translational CAR loss of function contributes to cognitive defects in healthy and diseased-primed brains.
SIGNIFICANCE STATEMENT This study addressed the role of the coxsackievirus and adenovirus receptor (CAR), a single-pass cell adhesion molecule, in the adult brain. Our results demonstrate that CAR is expressed by mature neurons throughout the brain. In addition, we propose divergent roles for CAR in immature neurons, during neurogenesis, and at the mature synapse. Notably, CAR loss of function also affects hippocampal plasticity.
Keywords: adult neurogenesis, Alzheimer's disease, coxsakievirus and adenovirus receptor, inflammation, synapse
Introduction
Cell adhesion molecules (CAMs) are multifunctional proteins that play various roles in all tissues in cell migration, signaling, ion transport, and cell–cell and cell–extracellular matrix attachment. CAMs such as the cadherins, ephrins, neurexins, and NCAM (a member of the Ig superfamily) play critical roles in general brain homeostasis (Togashi et al., 2009). CAMs regulate adult neurogenesis, dendritic spine development, and synapse remodeling, which, when combined, form the basis of neuronal plasticity. During synaptogenesis, CAMs influence axonal growth; path finding and target recognition; the differentiation of presynaptic and postsynaptic specializations; and the regulation of synapse size, stability, strength, and plasticity (Yamagata et al., 2003). When defects in CAM functions occur, they can lead to neurological and psychiatric diseases.
In combination with genetic triggers, a compounding and unifying factor in the physiopathology of many brain diseases is a proinflammatory environment that perturbs synapse homeostasis and adult neurogenesis (Lucas et al., 2006). A proinflammatory environment, whether initiated in the brain or systemically, is responsible for impaired cognition in the healthy brain and amplifies cognitive defects in many neurodegenerative diseases, including Alzheimer's disease (AD) (Holmes et al., 2009). Proinflammatory cytokine-induced cognitive defects can be recapitulated in healthy and AD mice (Valero et al., 2014), highlighting the global repercussions in brain homeostasis. Conspicuously, the mechanistic link is poorly understood.
The coxsackievirus and adenovirus receptor (CAR) is a single-pass transmembrane protein belonging to the Ig superfamily (Freimuth et al., 2008). Its extracellular region contains two globular Ig-like domains and its cytoplasmic tail harbors protein-interacting motifs (Loustalot et al., 2016). CAR functions are best characterized in epithelial cells, where it acts as a CAM and participates in the maintenance of tight junctions (Honda et al., 2000; Coyne and Bergelson, 2005). As the name suggests, CAR was identified as an attachment molecule for group B coxsackieviruses and some adenoviruses (Roelvink et al., 1999), including canine adenovirus type 2 (CAV-2) (Soudais et al., 2000). Because CAV-2 vectors preferentially infect neurons and can be transported efficiently from axon terminals to efferent regions (Soudais et al., 2001, 2004), the vectors have become powerful tools to investigate anatomical organization of neural circuits and higher-order brain functions and to treat brain diseases (Junyent and Kremer, 2015). Existing data indicate that CAR is responsible for CAV-2 neuron binding, entry, and retrograde axonal transport (Salinas et al., 2009, 2014).
Although a role for CAR in brain development has been proposed (Patzke et al., 2010), its cellular and subcellular location and functions in the adult brain are poorly characterized. Here, we demonstrate that, in the healthy brain, CAR is abundant in axon tracks throughout the brain, on the soma of immature neurons in the dentate gyrus (DG), in the mossy fibers of the stratum lucidum (SLu), at the presynapse in some mature neurons, and recruited to activated presynapses. Genetic deletion of CAR in the mouse brain affects adult neurogenesis, synaptic function, and behavior. In addition, we show that a proinflammatory environment induced by AD and/or systemic inflammation is associated with CAR loss in the hippocampus. Together, our data link posttranslational CAR loss in the hippocampus to inflammation, modulation of hippocampal plasticity, and impaired cognition in healthy and diseased brain.
Materials and Methods
Mice, neuron cultures, and human tissue samples.
Nestin-Cre mice and CARflox/flox mice (Tronche et al., 1999; Chen et al., 2006) were crossed to generate CAR-CNSKO mice. Unless specified otherwise, experiments were performed on ∼2-month-old CARflox/flox and CARflox/flox nestin-Cre littermates. Animal from both sexes were used and the numbers of animals of each sex is provided in the figure legends or directly in the figures themselves. The samples from 3xTgAD mice were described previously (Valero et al., 2014). Animals used in histological procedures were anesthetized with intraperitoneal injection of ketamine (100 mg/kg) and xylazine (10 mg/kg) and then perfused with 4% PFA in PBS. All applicable international, national, and/or institutional guidelines for the care and use of animals were followed. Human brain samples were processed in accordance with European bioethics laws regarding patient information: written consent was obtained from participant. AD and control hippocampal extracts have been described previously (Marcello et al., 2012). Primary hippocampal neurons were prepared from OF1 embryonic day 18 (E18) mice embryos. Depolarization was obtained by treating neurons with 90 mm KCl for 5 min in solution containing the following (in mm): 5 HEPES, 10 glucose, 2.5 CaCl2, 1 MgCl2, and 137 NaCl. Assays with CAV-2 fiber knob (FKCAV) were performed as described previously (Salinas et al., 2014).
Immunohistochemistry and immunofluorescence.
For CAR immunohistochemistry (IHC), free-floating coronal sections of brain were rinsed in PBS, pH 7.2, and then treated with 0.5% H2O2 and 10% methanol in PBS for 15 min and washed with PBS. Sections were permeabilized with PBS-T (PBS with 0.5% Triton X-100) and incubated for 1 h in 10% FBS in PBS-T. Sections were incubated overnight at 4°C with goat anti-CAR Ab (R&D Systems). Two rabbit anti-CAR antibodies (Bethyl A302–847A and Atlas HPA003342) gave identical signals during immunofluorescence (IF) assay in wild-type (WT) and CAR-CNSKO mice. Sections were then sequentially incubated for 2 h with biotinylated horse anti-goat Ab (Vector Laboratories) and then incubated with the avidin-biotin-peroxidase complex (Vector Laboratories) before peroxidase reaction. For IF assays, free-floating coronal sections of brain were rinsed in PBS, pH 7.2, permeabilized with PBS-T, and incubated for 1 h in blocking solution (10% FBS in PBS-T). Sections were then incubated overnight at 4°C with goat anti-CAR (R&D Systems), mouse anti-PSA-NCAM (Developmental Studies Hybridoma Bank) or mouse anti-NeuN (Millipore). Sections were then incubated sequentially for 2 h with the corresponding secondary antibodies (Life Technologies). Dissociated primary neurons were washed with PBS and permeabilized with 4% PFA/PBS or −20°C methanol/acetone. Cells were then washed 3 times in PBS, permeabilized, and blocked for 30 min in PBS containing 2% bovine albumin/10% horse serum/PBS. Cells were incubated with the following primary antibodies overnight at 4°C: goat anti-CAR (R&D Systems); mouse anti-VGLUT, rabbit anti-gephyrin, and mouse anti-VGAT (Synaptic Systems); mouse anti-PSD95 (Abcam); mouse anti-synaptophysin (SYP) (Sigma-Aldrich); and mouse anti-MAP2 (Roche), followed by three washes with PBS. Incubation with the appropriate secondary antibodies was performed for 1 h at room temperature, followed by 3 washes with PBS. The coverslips were mounted on slides with fluorescent mounting medium (DAKO) containing DAPI. Images were acquired using a Zeiss LSM 780 confocal microscope with ZEN imaging software.
Analyses of synaptic proteins.
Presynaptic and postsynaptic protein extracts from the hippocampi of control and CAR-CNSKO mice were used in immunoblotting. After behavioral studies, mice were killed by decapitation and hippocampi were removed, frozen, and stored at −80°C. Tissues were sonicated in 5% SDS/PBS and the protein concentration was measured using the BCA Kit (Pierce, France). Five to 15 μg of proteins were loaded, separated by SDS-PAGE (12%), and transferred to a PVDF membrane. Membranes were blocked and incubated overnight at 4°C with mouse anti-VGLUT, mouse anti-VGAT, rabbit anti-gephyrin, rabbit anti-GABA-A receptor γ2, rat anti-NCAM 180 (Synaptic Systems), mouse anti-PSD95 (Abcam), mouse anti-NR1 (Millipore), and mouse anti-SYP and anti-β-tubulin (β-tub) (Sigma-Aldrich). Membranes were then rinsed and incubated for 1 h at room temperature with the appropriate HRP-conjugated secondary antibodies (Sigma-Aldrich). ECL reagents were used to reveal peroxidase activity. Band intensity was normalized to β-tubulin levels and quantified using ImageJ software. Preparation of the synaptosome postsynaptic density (PSD) and presynaptic web (PSW) fractions was performed as described previously (Garside et al., 2009). Electrophysiology on sagittal hippocampal slices was performed as described previously (Chafai et al., 2012). Recordings were conducted in an average of three slices per animal.
In vitro neuron depolarization assays.
Primary hippocampal neurons were prepared from OF1 E18 mice embryos (Charles River Laboratories) as described previously (Salinas et al., 2014). To study CAR involvement in neurotransmission, chemical protocols were used to induce depolarization or LTP on mature hippocampal neurons (days in vitro 21, DIV21) (Kang and Schuman, 1995; Ackermann and Matus, 2003). Depolarization was obtained by treating neurons with the following (in mm): 90 KCl, 5 HEPES, 10 glucose, 2.5 CaCl2, 1 MgCl2, and 137 NaCl for 5 min. LTP was induced by treating neurons with a 20 ng/ml final concentration of recombinant BDNF (Peprotech) with or without boiling for 45 min. Treatments with FKCAV were performed first on ice for 10 min and then the medium was changed and the neurons incubated at 37°C for the indicated times. Cytokines (murine and human TNF and IFN-γ) were purchased from Peprotech.
mRNA quantification.
Total RNA was extracted from brain of 3xTgAD mice and reverse transcribed using random hexamer primers (Roche) and SuperScript III Reverse Transcriptase (Invitrogen). For quantitative analyses, primers that selectively amplify murine CAR were used (forward: TCTTCTGCTGTCACAGGAAAC and reverse: CTGGGGACTTGGTTATACTGC) and real-time PCR was performed using SYBR Green PCR mix and LC 480 machine (Roche).
Adult neurogenesis.
Mice received intraperitoneal injections of BrdU or EdU at a dose of 100 mg/kg body weight and were perfused with 4% PFA/PBS. Brains were removed and processed for IHC or IF. EdU was detected by adding the Click-iT reaction buffer (from the Click-iT EdU imaging kit; Invitrogen).
Behavior assays.
Behavior testing (Maurice et al., 2009) was performed between 10:00 and 14:00 hours. In all experiments, CARflox/flox animals were used as controls. For locomotion measure, mice were placed in the center of an open white Plexiglas box (50 × 50 × 50 cm) equipped with infrared light-emitting diodes and their movements monitored for 10 min using Videotrack software (Viewpoint). For cognitive tests, mice were subjected to a series of behavioral tests to assess anxiety, learning, and memory, finishing with the most stressful procedures: open-field, elevated-plus maze, Y maze, and water maze (spatial reference memory procedure during 5 d, probe test on day 6, and visible platform procedure during 3 d).
Statistical analyses.
Statistical tests and analyses were performed and verified by the SERANAD Complex Biological Data Analysis Service (http://www.igmm.cnrs.fr/spip.php?rubrique168&lang=en). p < 0.05 is reported as significant.
Results
CAR is in axons, at the presynapse, and on the soma of cells in the DG
To characterize CAR cellular and subcellular location, we analyzed the distribution of CAR in the brains of WT mice by IHC and IF. CAR staining is notable in the posterior corpus callosum, between layers IV and V of the cerebral cortex, and in layer I of the cerebral cortex, which is primarily axons from other cortical areas and apical dendrites of local neurons (Fig. 1A). In the hippocampus, CAR staining overlaps with the axons projecting from the entorhinal cortex and in mossy fibers in the SLu (Fig. 1B,C). Despite strong axon labeling in most regions, cell bodies are strikingly devoid of CAR. However, somatic CAR staining was notable in some cells the subgranular zone (SGZ) and their projections in the granular cell layer (GCL) of the DG (Fig. 1B,D). In contrast to the report describing CAR expression in the human brain (Persson et al., 2006), we did not detect CAR in cells with glia-like morphology in the mouse brain.
Figure 1.

CAR in axons and on the soma of cells in the hippocampus. A, Representative image of anti-CAR IHC in a coronal section from the brain of a healthy adult mouse. Solid black arrows show intense CAR staining in layer I and near layer IV/V of the cerebral cortex and posterior corpus callosum. B, Magnification of the black boxed area in A showing CAR labeling in the hippocampus, soma of cells in the DG SGZ and GCL (solid black arrows), in axons projecting from the entorhinal cortex (black arrowhead) and in the SLu (open arrows). C, Magnification of the white boxed area in B showing IHC and IF (black and white open arrows, respectively) staining of CAR in fibers within the SLu. D, Magnification of black boxed area in B showing IHC and IF staining (black and white arrowheads, respectively) of CAR on the soma and projections from cells in the SGZ and GCL. Scale bars: A, 1 mm; B, 100 μm; C, D, 50 μm.
In addition to the preferential transduction of neurons, CAV-2 vector transport from the injections site to cell bodies in efferent regions can be remarkably efficient in some neuron types, suggesting that CAR is likely located at the presynapse (Junyent and Kremer, 2015). To address CAR subcellular distribution, we isolated synaptosomes from mouse brains and showed that CAR is present in the synapotosome fraction (Fig. 2A). In epithelial cells, CAR forms high-affinity intercellular homodimers via its Ig-like D1 and D2 domains. To determine whether the intercellular CAR–CAR interaction is similar at the highly specialized neuron synapse, we separated the PSD from the PSW plus vesicles (PSW+V) component. Whereas the biochemical separation of these two fractions is not without minor cross-contamination, we nonetheless find that CAR is exclusively in the presynapse fraction (Fig. 2A). We also found that CAR is in the presynapse fraction of synaptosomes prepared from human brain (Fig. 2B). To address CAR location using another approach, we stained mature (>DIV14) primary hippocampal neurons for CAR. In these neurons, CAR is present in the somato-dendritic and axonal compartments and colocalizes with the synaptic marker SYP (36.1 ± 4.1% of SYP+ structures contain CAR; Fig. 2C). In addition, CAR puncta overlap with VGLUT, PSD95, and VGAT (Fig. 2D,E), demonstrating that in these conditions, CAR is present at the synapse in excitatory and inhibitory synapses. Consistent with the biochemical assays, at DIV21, when dendritic spines are fully mature, we were unable to detect CAR in dendritic spines (Fig. 2F). These data are consistent with the lack of CAR at or near the postsynapse. Together, these data demonstrate that CAR is preferentially expressed by neurons in the mouse brain, on the soma of cells in the SGZ of the DG, in the mossy fibers in the SLu, and in axons projecting from the entorhinal cortex. At the synapse, CAR is at the presynapse and undetectable at the postsynapse, suggesting that intercellular CAR–CAR interactions that predominate in epithelial cells are not occurring at the synapse.
Figure 2.

CAR is a synaptic protein. A, CAR is present in presynaptic fractions in the mature mouse brain. Synaptosomes from adult mouse brain were screened for CAR. Representative immunoblots showing input, synaptosome, PSD, and PSW+V. SYP was used as a marker for the PSW+V; NR1 and PSD95 as markers for the PSD; and NeuN for soma. B, CAR is present in presynaptic fractions in the mature human brain. Synaptosomes isolated from adult human cortex were screened for CAR levels by immunoblotting. Shown is a representative immunoblot of CAR expression at the presynapse. C, Hippocampal neurons (≥DIV14) were costained for endogenous CAR (in magenta) and with the presynaptic marker SYP (in green). Arrows show double-positive structures (36.1 ± 4.1% of SYP+ structures contain CAR). D, E, DIV14 murine hippocampal neurons were labeled for glutamatergic and GABAergic synapses using VGLUT (D) and VGAT (E), respectively. Colocalization (white arrows) with PSD95 indicates synapses. CAR colocalizes with VGLUT and VGAT, showing that CAR was expressed in excitatory and inhibitory synapses. F, Hippocampal neurons (≥DIV21) were costained for endogenous CAR (in magenta) and MAP2 (in green) to visualize dendritic spines (white arrows). Scale bars: C, F, 10 μm; D, E, 15 μm.
CAR loss of function affects hippocampal GCL organization
To better understand CAR function in the brain, we generated conditional CAR KO (CAR-CNSKO) mice by crossing nestin-Cre and CARflox/flox mice (Fig. 3A; see Materials and Methods). CAR-CNSKO mice thrived, have no obvious phenotypic differences compared with WT mice, express CAR in somatic tissues, but have no detectable CAR expression in the brain (Fig. 3B). IHC analyses in CAR-CNSKO mice confirm the specificity of the CAR antibodies and staining in control mice (Fig. 3C). By comparing cresyl violet staining of WT, CARflox/flox (hereafter referred to as control) and CAR-CNSKO mice, we found that there were no gross morphological anomalies in the CAR-CNSKO mouse brain (Fig. 3D). However, upon detailed examination, we found that the GCL of the DG in CAR-CNSKO mice is less densely packed and occupies a greater area than in control littermates (Fig. 3E,F). This difference in area is significant for the GCL (p < 0.05), but not for the pyramidal layer of CA1 or CA3 (data not shown). These data suggest that CAR loss of function may affect hippocampal neurotransmission.
Figure 3.
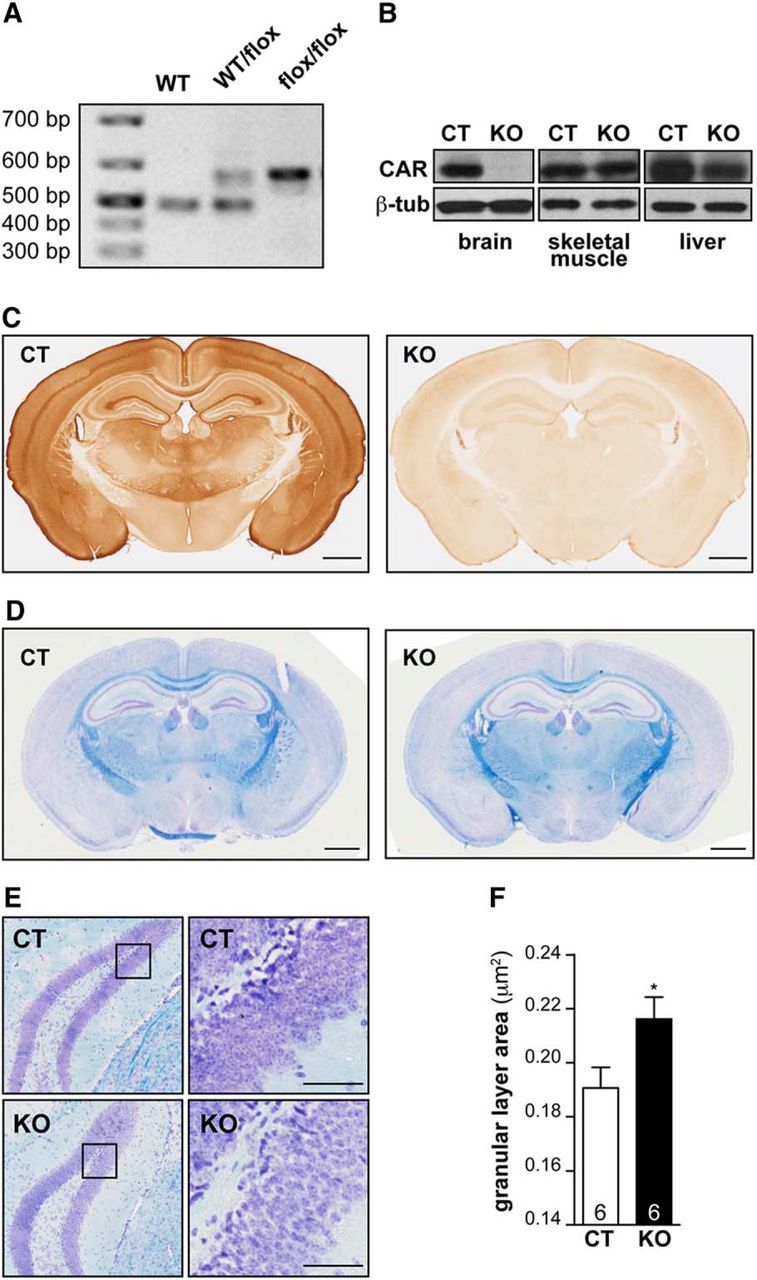
CAR-CNSKO mice have hippocampal defect. A, Deletion of Cxadr in the CNS. Example of genotyping results using primers flanking the Cxadr exon 2 region. B, Immunoblot analyses from control (CT) and CAR-CNSKO mice showed deletion of CAR (∼46 kDa) in the brain, whereas expression in skeletal muscle and liver is similar to controls. β-tubulin was used as a loading control. C, IHC of CAR expression in CT and CAR-CNSKO mice. D, Brain morphology revealed by luxol blue staining of CT and CAR-CNSKO mice. E, Cresyl violet and luxol fast blue coloration of CT and CAR-CNSKO brains. Coronal brain sections of the DG: boxed region is magnified in the panel on the right to show the GCL. F, Average area of the GCL in CT and CAR-CNSKO mice. The number of animals in groups is indicated within the columns. Scale bars: C, 1 mm; D, 1 mm; E, 20 μm.
CAR loss perturbs hippocampal synaptic plasticity
Because CAR loss of function affects the organization of the DG, we investigated whether the global plasticity of hippocampal neurotransmission is affected. We therefore measured short-term plasticity as paired-pulse facilitation (PPF, measured as paired-pulse ratio, PPR) and long-term plasticity as LTP induction and maintenance in organotypic slices from CAR-CNSKO mice. Schaffer collaterals were stimulated and evoked field EPSPs (fEPSPs) were recorded in the stratum radiatum of the CA1 area. After stable recording baseline activity, high-frequency stimulation (HFS) was applied to induce LTP (Fig. 4A). No significant differences were observed between PPRs from male CAR-CNSKO and control male mice before or 60 min after HFS (Fig. 4B). By contrast, PPRs from female CAR-CNSKO mice were significantly lower than PPRs from female controls (Fig. 4C). To visualize LTP, the level of post-HFS potentiation is expressed as a percentage of the mean fEPSP amplitude before LTP induction. Male CAR-CNSKO mice have LTP levels comparable to controls, as observed by the increased fEPSPs peaks after HSF for both groups (Fig. 4D). Conversely, in female CAR-CNSKO mice, the levels of post-HFS potentiation rapidly decreased (Fig. 4E). After 60 min, the amplitude of the fEPSPs in female CAR-CNSKO mice is not different from baseline activity, whereas in controls, increased fEPSPs peaks were observed from 10 to 60 min after HSF. Together, these data demonstrate that CAR loss of function perturbs global hippocampal synaptic plasticity and neurotransmission in a sex-biased manner.
Figure 4.

Effect of CAR loss of function on neurotransmission. A, fEPSPs recordings were performed in hippocampal slices from adult female control (CT) and CAR-CNSKO mice. Representative recordings of the fEPSPs before (black) and after (red) HFS. The LTP (comparison between red and black traces) is shown. B, PPR in male CT and CAR-CNSKO mice. C, PPR in female CT and CAR-CNSKO mice. Results are expressed as the ratio between the fEPSP peaks of response 1 and 2. These measures were performed before (basal activity) and after induction of LTP (60 min after HFS). The number of slices in each group is indicated within the columns. D, LTP induction in male CT and CAR-CNSKO mice. E, LTP induction in female CT and CAR-CNSKO mice. LTP was assessed for 1 h after a 10 min stable baseline response. fEPSP peak data were converted to percentages by setting the baseline fEPSP peak data to 100%. Results are expressed as means ± SEM. The number of animals in groups is indicated within the columns. Data were analyzed using an unpaired Student's t test (B, C) or a two-way ANOVA followed by a Sidak test (D, E). *p < 0.05, **p < 0.01.
Impaired adult hippocampal neurogenesis
Adult neurogenesis influences learning and memory by the generation and integration of neurons into existing networks (Lepousez et al., 2015). In the adult mouse brain, neurogenesis is restricted to the subventricular zone and the SGZ of the DG in the hippocampus. To identify the subpopulation of CAR+ cells in the SGZ, we coincubated sections with anti-CAR and anti-PSA-NCAM Abs (PSA-NCAM is a marker of immature neurons). We found that cells with CAR on the soma and their projections in the inner layer of the DG are also PSA-NCAM+ (Fig. 5A,B). Staining with CAR and NeuN (a marker of mature neurons) shows that some “NeuN-low” cells also have CAR staining on the cell body (Fig. 5C).
Figure 5.

CAR involvement in adult neurogenesis. GCL of the DG (A) and SLu (B) of CT mice were stained with anti-CAR, anti-PSA-NCAM, and DAPI. C, GCL of the DG of CT mice was stained with anti-CAR, anti-NeuN, and DAPI. Boxed regions in the upper left panel in A–C are expanded in the upper right panels. White arrows show overlapping expression. D–I, Adult neurogenesis in CAR-CNSKO mice. Two-month-old mice were injected with thymidine analogs and brains were processed 1 and 28 d after injection by IF. Confocal microscopy images showing cells positive for EdU and NeuN (D) and EdU and PSA-NCAM (E). F, G, Quantification of proliferative cells (incorporation of a thymidine analog) in the SGZ of the DG in CT and CAR-CNSKO mice at 1 and 28 d after injection. H, Percentage of new neurons/DG [(EdU+ + NeuN+ cells/EdU+ cells) × 100] CT and CAR-CNSKO mice 28 d post-injection. I, Percentage of immature neurons/DG [(EdU+ + PSA-NCAM+ cells/EdU+ cells) × 100)] CT and CAR-CNSKO mice 28 d after injection. Results are expressed as means ± SEM and were analyzed using an unpaired t test. *p < 0.05, **p < 0.01, ***p < 0.001. The number of animals in groups is indicated within the columns. Scale bars: A–G, 50 μm; D, E, 10 μm.
We then investigated whether CAR loss of function affects proliferation, survival, and/or differentiation of adult newborn neurons in the DG. To address this question, proliferating cells in the brains of CAR-CNSKO mice were tagged using thymidine analogs that incorporate into the genome of dividing cells. The fate of newborn CAR− cells in the DG was evaluated by comparing EdU+/NeuN+ and EdU+/PSA-NCAM+ cells in CAR-CNSKO versus control mice (Fig. 5D,E). Killing mice 1 d after injection allows one to quantify proliferation, whereas killing mice at 28 d after injection allows one to quantify neuron survival/differentiation. In both groups, the number of EdU+ cells in the DG was similar (Fig. 5F,G). However, there were significantly fewer EdU+ neurons (NeuN+) in both male and female CAR-CNSKO mice compared with control mice 28 d after injection (Fig. 5H). The decrease in mature neurons is mirrored by an increase in the percentage of EdU+ immature (PSA-NCAM+) neurons (Fig. 5I). Together, these data demonstrate that adult NPC proliferation and survival are not significantly affected, whereas NPC differentiation is.
Synapses are perturbed in the CAR-CNSKO mouse hippocampus
Because CAR loss of function affects global hippocampal plasticity and some CAMs can also be targeted to synapses during plasticity (Ackermann and Matus, 2003), we investigated whether activation or chemical induction of LTP affects presynaptic CAR. We therefore compared the location of SYP and CAR after neuronal depolarization. By measuring CAR/SYP colocalization, we found that the percentage of CAR+ synapses increased (Fig. 6A). We then investigated whether CAR levels change at the synapse after the induction of neuronal plasticity. To this end, we incubated hippocampal neurons with BDNF and quantified of CAR/SYP colocalization. Using this assay, we found that CAR can be recruited to synaptic termini (Fig. 6B).
Figure 6.
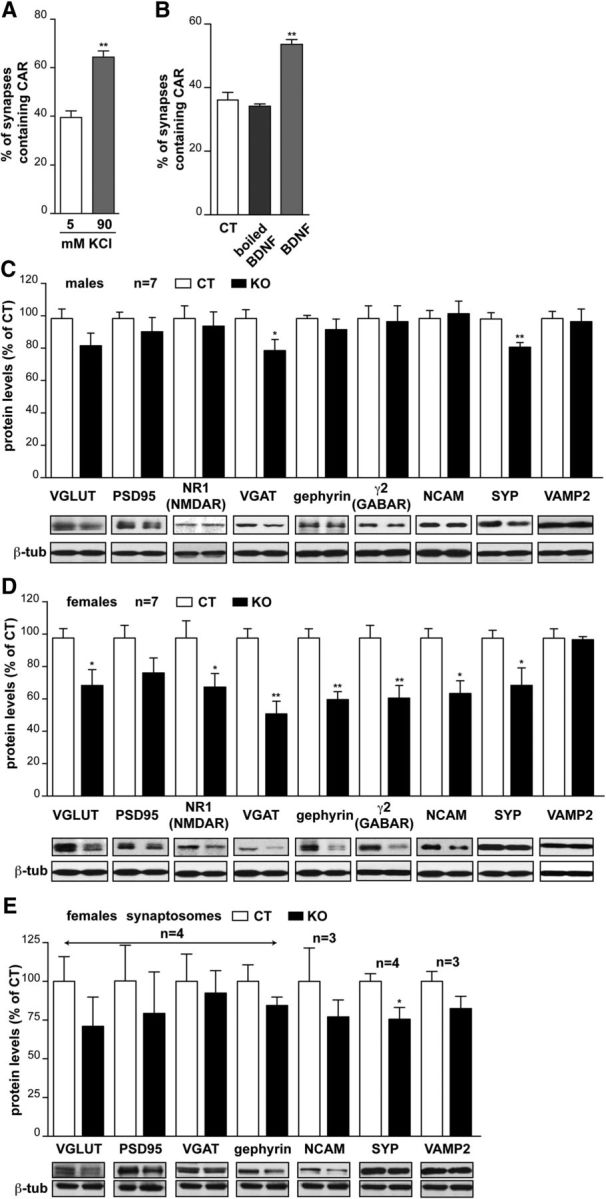
CAR loss of function affects synapse homeostasis. A, Quantification of colocalization between SYP and CAR when neuronal depolarization is induced by 90 mm KCl. B, Quantification of colocalization between SYP and CAR after incubation with BDNF or boiled BDNF. Each condition is the percentage of synapses (SYP+ structures) containing CAR and is expressed as means ± SEM of three independent experiments. Greater than 200 SYP+ structures were analyzed at each condition. C, D, Effect of CAR loss of function on synapse content in the hippocampus. Quantification of immunoblots for synaptic proteins relevant proteins and respective representative immunoblots are shown under the columns. The intensity of the bands was quantified and normalized to β-tubulin levels for each marker. NB, Some membranes were used for more than one protein and therefore the same control β-tubulin bands were used to compare the levels of different synaptic proteins. Results are expressed as a percentage in control mice. Because differences appeared between sexes, we separated males (C) and females (D). n = the number of animals. E, Effect of CAR loss of function on synapse content from synaptosomes preparation. Quantification of immunoblots for synaptic proteins relevant proteins and respective representative immunoblots are shown under the columns. The intensity of the bands was quantified and normalized to β-tubulin levels for each marker. All results are expressed as means ± SEM and were analyzed using an unpaired t test. *p < 0.05, **p < 0.01.
Synaptic plasticity also involves a feedback loop to ensure the production and targeting of actors involved in neurotransmission through recruitment/exclusion of proteins, local mRNA translation, and transcription. We therefore investigated whether CAR loss of function affects hippocampal synapse content and/or genesis. To monitor synaptic protein content in CAR-CNSKO mice, the levels of several synapse proteins were quantified by immunoblotting. In male CAR-CNSKO mice, VGAT and SYP were lower compared with controls (Fig. 6C). Unexpectedly, in female CAR-CNSKO mice, the majority of hippocampal synapse proteins assayed here are present at lower levels compared with control mice (Fig. 6D). To further characterize CAR at the synapse, we quantified the synapse protein content in synaptosomes prepared from the hippocampus (Fig. 6E) and found that the difference in synapse protein levels in CAR-CNSKO did not vary significantly compared with controls, which could be consistent with a synaptic loss occurring globally in the hippocampus that cannot been seen on isolated synaptosomes. Together, these data suggest that CAR loss of function affects global hippocampal synaptic content, but that there is likely a difference in synapse number, type, and/or maturation level in female CAR-CNSKO mice.
CAR loss of function affects behavior
Because adult hippocampal neurogenesis and synapse homeostasis are processes associated with cognition, we subjected CAR-CNSKO mice to a series of behavior tests associated with hippocampal functions. Because CAR could be expressed at the neuromuscular junction (Shaw et al., 2004) and locomotion and exploration deficits can interfere with cognitive tasks, we initially examined the mobility of mice in an open-field paradigm. Neither the distance traveled nor the locomotion speed was significantly different between control and CAR-CNSKO mice (Fig. 7A,B).
Figure 7.
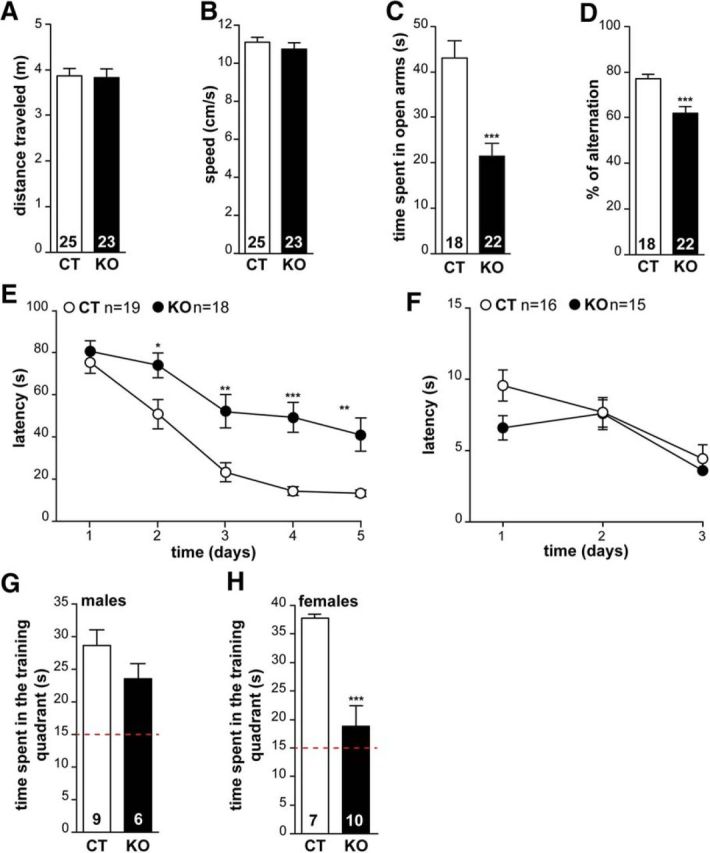
CAR loss of function affects behavior. Locomotion of CT and CAR-CNSKO mice was analyzed in the open field for 10 min. A, Locomotion activity was evaluated in terms of distance traveled (m) and (B) locomotion speed calculated as total distance over time in movement (cm/s). The loss of CAR in the CNS did not alter locomotor behavior. C, Anxiety was measured using their ability to explore the open arms of an elevated plus maze. The apparatus consisted of a plus-shape maze with two opposite open and enclosed arms. The arms extended from a central elevated platform. Each mouse was placed at the center of the maze and could freely explore for 10 min. Results were expressed as total time spent in the open arms (10 min). Results were pooled as we found no difference between males and females. D, Spatial working memory was recorded as described in a three-arm maze converging at an equal angle. Each mouse was placed at the end of arm and allowed to move freely through the maze during an 8 min session. The sequence and number of arm entries were recorded. Results are expressed as the percentage of alternations. Results were pooled as we found no difference between males and females. E, The Morris water maze was a circular pool arbitrarily divided into four quadrants. A hidden platform was immersed beneath the water surface in the training quadrant. Reference memory training consisted of 3 swims/d for 5 d with 20 min intertrial time interval. Animals were allowed to swim for 90 s and to use visual extra-maze cues. Mice were then left on the platform for 20 s. The median latency was calculated for each training day and expressed as mean ± SEM. On day 6, a probe test was performed to measure memory retention. The platform was removed and each animal was allowed to swim freely for 60 s. The time spent in the training quadrant was determined using Videotrack software (Viewpoint). Results were pooled as we found no difference between males and females. F, Swimming time to a visible platform over 3 d did not show differences between CT and CAR-CNSKO animals. G, H, Memory retention in CAR-CNSKO mice. The probe test was performed 1 d after the last training session in a single 60 s swim without the platform. The presence in the training quadrant was analyzed over the chance level (red line at 15 s). There were notable differences between the male and female mice, so the results are presented separately. All results are expressed as means ± SEM and the number of animals in groups is indicated within the columns of results. Data were analyzed using an unpaired Student's t test (A–D, G, H) or a two-way ANOVA followed by a Tukey post hoc test (E). *p < 0.05, **p < 0.01, ***p < 0.001.
In the elevated plus maze, male and female CAR-CNSKO mice spent less time in the open arm compared with control mice (Fig. 7C), reflecting an abnormal level of anxiety. In the Y maze, a nonaversive task based on rodents' natural exploratory instincts, spontaneous alternation performances were impaired in male and female CAR-CNSKO mice (Fig. 7D), suggesting an impact on spatial working memory deficit. In the Morris water maze, male and female CAR-CNSKO mice showed a higher latency to find the hidden platform in the training quadrant compared with control littermates on days 2–5 (Fig. 7E), suggesting a possible altered learning process. CAR-CNSKO mice showed no sign of visual impairment or swimming performance in the visible platform session (Fig. 7F). In the probe trial conducted 24 h after training, control and CAR-CNSKO male mice swam preferentially in the training quadrant during the 60 s session (Fig. 7G). In contrast, the time spent in the training quadrant was near the random values for female CAR-CNSKO mice (Fig. 7H), suggesting that the retention of spatial memory is altered in female CAR-CNSKO mice. Together, our data demonstrate a sex-biased role for CAR in hippocampal plasticity and, together with possible sensorial, affective, or behavioral factors, may underscore a function in spatial memory.
Decreased CAR levels in a proinflammatory environment and in the mouse AD brain
To the best of our knowledge, mutations in the CAR gene (CXADR) have not been linked with brain dysfunction. Because CAR plays primordial roles in other tissue such as the heart, where loss of function is embryonically lethal (Chen et al., 2006), this may preclude an association with brain diseases. However, there are posttranscriptional mechanisms that can lead to the loss of CAR: studies using nonneuronal cells and tissues suggest that CAR levels are indirectly reduced by proinflammatory cytokines (e.g., TNF and IFN-γ) (Vincent et al., 2004). In addition, CAR is a substrate for α, β, and γ-secretases (Zhou et al., 2012), which, when dysregulated, can be implicated in the pathogenesis of AD. These observations led us to assay CAR levels in primary murine hippocampal neurons after ionomycin-induced secretase activation and TNF and IFN-γ treatment. As a positive control for CAR loss, we incubated hippocampal neurons with FKCAV, which disrupts homodimeric CAR interactions and induces CAR internalization and degradation (Salinas et al., 2014). After ionomycin treatment, the levels of the CAR ectodomain increased in the supernatant and full-length CAR decreased in total cell extracts (Fig. 8A). CAR levels were also significantly decreased in primary cultures of murine hippocampal neurons and adult murine NPCs incubated with TNF and IFN-γ in a dose-dependent response (Fig. 8B–D). These data demonstrate that TNF and IFN-γ play a direct role on CAR processing in neurons.
Figure 8.

CAR levels are reduced by secretases and proinflammatory cytokines. A, Primary murine hippocampal neurons were mock or ionomycin treated. Supernatant and cells were collected separately and assayed for the presence of CAR using an anti-CAR that recognizes the extracellular domains. A representative immunoblot from three independent experiments is shown. β-tubulin was used as a loading control. B, Primary cultures of murine hippocampal neurons were incubated with FKCAV or increasing concentrations of TNF and IFN-γ for 72 h. Representative immunoblot showing CAR levels, with β-tubulin used as a loading control, are shown. C, Primary cultures of adult NPCs were incubated with FKCAV or increasing concentrations of TNF and IFN-γ for 72 h. Representative immunoblot showing CAR levels with β-tubulin used as a loading control is shown. D, Quantitative analyses of the data in B. Results are expressed as means ± SEM of three independent experiments and were analyzed using an unpaired Student's t test. *p < 0.05, **p < 0.01 vs mock condition. E, Effect of systemic inflammation triggered by intraperitoneal injection of LPS in healthy mice. LPS injections (bottom) triggers CAR loss from SGZ and GCL (left) and SLu of the DG (middle) compared with PBS-injected mice (top row). Representative IHC analyses of PSA-NCAM in the SLu in LPS-injected mice compared with PBS-injected mice 1 week after injection (right). Scale bars, 50 μm.
Systemic inflammation perturbs brain function via overlapping mechanisms, including proinflammatory cytokines entering the brain, and via microglia activation and/or cytokine secretion within the brain (Habbas et al., 2015; Heneka et al., 2015). We therefore investigated whether global CAR levels in the brain are affected by systemic inflammation. To address this possibility, we injected lipopolysaccharides (LPS) from Gram-negative bacteria, which induce a rapid and strong proinflammatory immune response, into the peritoneal cavity of healthy mice. We then quantified global CAR levels by immunoblotting. We found that at 1 or 7 weeks after injection, no significant change in CAR levels, as assayed by immunoblotting, was found in any region (data not shown).
By contrast to the diffuse CAR staining throughout the axon tracks in most of the brain (Fig. 1A), immature neurons in the DG have high levels of somal CAR. We therefore investigated whether somal CAR is affected and found that, at 1 week after LPS injection, CAR staining was strikingly reduced in immature neurons in the DG and their axons that project to CA3. In contrast, PSA-NCAM staining was unaffected (Fig. 8E), demonstrating that the lack of CAR immunoreactivity is not due to the loss of immature neurons. These data suggest that, during acute induction of proinflammatory cytokines by systemic stimuli, CAR levels on immature neurons and in the SLu are reduced.
Activated secretases and a proinflammatory environment are closely linked hallmarks of several neurodegenerative diseases and create a feedforward loop (Holmes et al., 2009). In AD, an inflammatory environment can lead to increased β-secretase (BACE1: β-site amyloid precursor protein-cleaving enzyme) expression, Aβ overproduction, and senile plaque accumulation (Yamamoto et al., 2007), which in turn promote an NF-κB proinflammatory cytokine response. Because inflammation, synapse homeostasis, and perturbation of CAM functions can be associated with neurologic and psychiatric diseases (Togashi et al., 2009)—in particular late-onset AD, which is inexorably linked with inflammation—we investigated whether CAR levels are perturbed in the AD hippocampi. 3xTgAD mice recapitulate some AD-related characteristics, such as an increased level of proinflammatory cytokines, age-related formation of senile plaques, neurofibrillary tangles, impaired synapse homeostasis, and cognitive functions (Oddo et al., 2003). We therefore compared CAR levels in total protein extracts from hippocampi from age-matched controls and 5- to 8-month-old (transitional stage of AD progression) and 16- to 20-month-old (late stage of AD progression) 3xTgAD mice. We found that CAR was reduced in both age groups, with a greater decrease in the older cohort compared with age-matched controls (Fig. 9A,B). To determine whether the decrease is due to a pretranslational or posttranslational mechanism, we quantified Cxadr mRNA levels using the same samples (Fig. 9C) and did not detect a difference of Cxadr mRNA levels between WT and 3xTgAD mice, suggesting posttranslational CAR loss.
Figure 9.
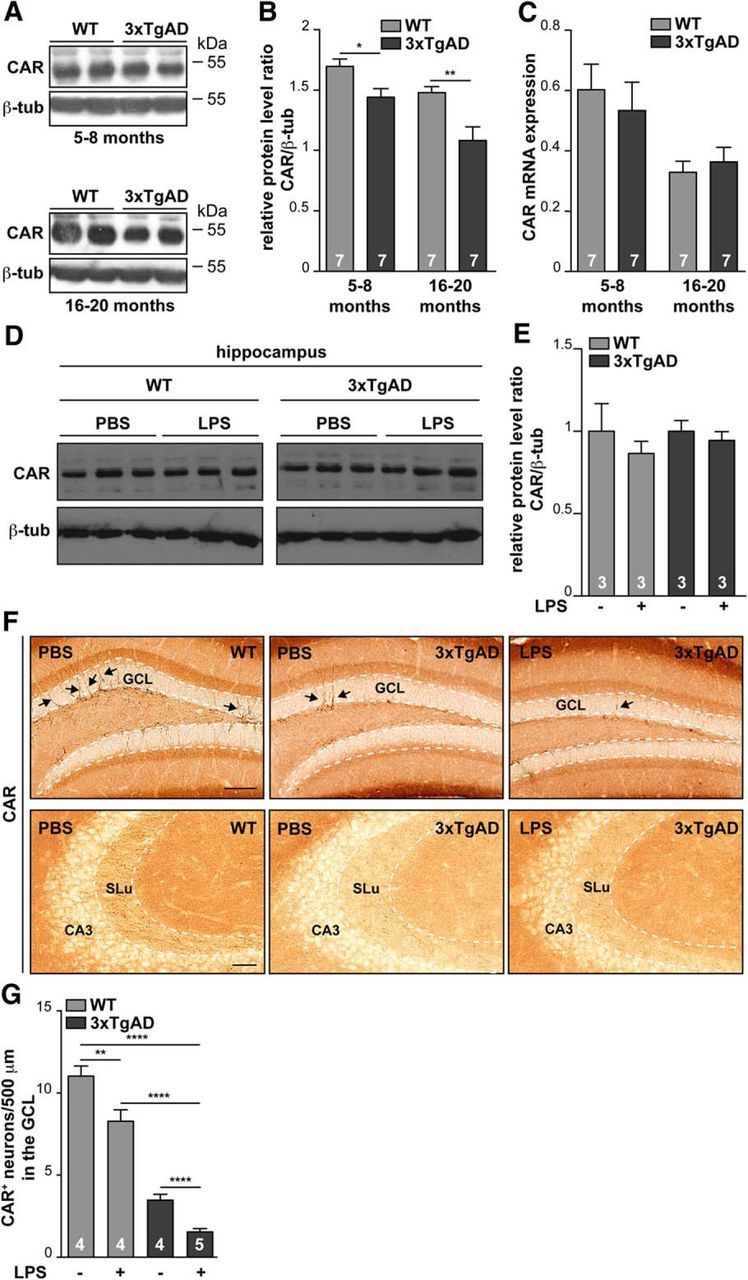
CAR loss in murine AD hippocampus. Representative immunoblots of CAR levels in proteins extracted from the hippocampi from 5- to 8-month-old (A, top) and 16- to 20-month-old (A, bottom) WT and 3xTgAD mice. B, Quantification of CAR levels in the immunoblot from A in 3xTgAD mice. CAR levels were normalized to β-tubulin. Number of animals is indicated within the columns. C, Relative Cxadr mRNA levels, normalized to GAPDH mRNA, in hippocampi of WT and 3xTgAD mice. D–G, Wild-type and 3xTgAD mice were injected in the peritoneal cavity with LPS and killed 7 weeks after injection. D, Representative immunoblot of CAR levels from hippocampi extract. E, Quantification of the immunoblot shown in D. F, Representative images of CAR staining (black arrows) in coronal sections containing the GCL (top row) and SLu (bottom row) from mice ± LPS injections. G, Quantification of CAR+ cells in the GCL from WT and 3xTgAD mice injected in the peritoneal cavity with LPS and killed 7 weeks after injection. Quantification was performed using ImageJ. A 500-μm-long segmented line (10 pixel width) was drawn through the middle of the GCL and signal intensity along that line was plotted. Each peak above the threshold (80 in the grayscale 0–255) corresponds to a neuron (≥6 sections from each of the four mice). Results are expressed as mean ± SEM and were analyzed using an unpaired Student's t test. *p < 0.05, **p < 0.01, ****p < 0.0001. Scale bar, 50 μm.
Systemic inflammation induced by LPS injections impairs long-term spatial memory and neurogenesis in healthy and 3xTgAD mice (Valero et al., 2014). LPS-induced defect in spatial memory is exacerbated in 3xTgAD mice and is associated with a significant reduction in the number of synaptic puncta. We therefore investigated whether systemic LPS-induced systemic inflammation affected CAR levels in the 3xTgAD mouse brain. To address this possibility, we injected LPS into the peritoneal cavity of control and 3xTgAD mice. Control and 3xTgAD mice that had previously shown memory defects (Valero et al., 2014) were killed 7 weeks after LPS injection and global CAR levels were quantified by immunoblotting. We found that global CAR levels in LPS-injected WT and 3xTgAD mice were not significantly different from PBS-injected controls (Fig. 9D,E). In contrast, IHC analyses showed that CAR is strikingly absent from the soma and neurites of cells in the SGZ of the DG and in the mossy fibers of the SLu after LPS injections (Fig. 9F). Of note, there are fewer CAR+ neurons in 3xTgAD mice versus WT healthy mice (Fig. 9G). Moreover, LPS injections significantly reduced CAR levels on immature neurons in healthy and 3xTgAD mice versus PBS-injected controls. Together, these data demonstrate that CAR can be lost from the hippocampus in healthy and AD mice via a posttranslational mechanism induced by systemic inflammation.
CAR levels in the hippocampus of human AD patients
For neurodegenerative diseases, extrapolating results from mice to humans has been historically challenging. Therefore, we assayed protein extracts from hippocampi from the Braak IV stage of patients diagnosed with AD and age-matched controls. In the hippocampi of these well characterized AD patients, the levels of several synapse proteins, including SYP and SNAP25, were not altered, demonstrating that significant synapse loss had not yet occurred (Marcello et al., 2012). Remarkably though, global CAR levels were significantly decreased already at Braak IV stage of late-onset AD (Fig. 10A,B). Together, these data demonstrate that CAR levels are significantly reduced in the human AD brain when impaired cognition starts, but dementia is not yet declared. Moreover, these results demonstrate that the CAR-CNSKO and 3xTgAD mice reproduce a key aspect of CAR loss and strengthen the clinical relevance of rodent models.
Figure 10.

CAR loss in human AD brain. A, Protein extracts from the hippocampus from AD patients and age-matched controls were subjected to immunoblotting and CAR level were normalized to β-tubulin. B, Quantitative analyses of CAR and VGLUT levels in A. Results are expressed as means ± SEM and the number of samples is indicated within the columns. Data were analyzed using an unpaired Student's t test. **p < 0.01.
Discussion
Before this study, there were no known roles for CAR in the adult brain. Here, we demonstrate in the healthy adult brain that CAR has cell-type-specific functions. CAR is predominantly located on neuron projections and present at the presynapse, where it can be recruited upon stimulation. In the in the GCL of the DG, CAR staining is notable on the soma and projections of immature PSA-NCAM+ neurons. In the CAR-CNSKO mouse brain, CAR loss of function perturbs synapse content, LTP, adult neurogenesis, and hippocampal-associated behavior. When healthy mice are challenged with LPS-induced systemic inflammation, CAR levels notably decrease in the DG. In the diseased-primed brain, CAR levels are significantly decreased in the hippocampus of humans during the early phase of late-onset AD. CAR loss also occurs in the brain of 3xTgAD mice. CAR levels are further reduced in 3xTgAD mice challenged with LPS.
CAR in the healthy brain
Neuronal plasticity arises from processes including adult neurogenesis, dendritic spine development, and synapse remodeling. In the hippocampus, we found an overlapping pattern of expression/localization between CAR and PSA-NCAM. The similarities between CAR and PSA-NCAM are notable: both are widely expressed in the embryonic and early postnatal brain (Cremer et al., 2000; Durbec and Cremer, 2001; Loustalot et al., 2016). In the adult brain, PSA-NCAM is found in the DG, where its expression corresponds to the period when postmitotic neuroblasts extend their processes and migrate (Cremer et al., 2000; Durbec and Cremer, 2001). Moreover, CAR and PSA-NCAM are also readily detected in the mossy fibers in the SLu. After newborn neurons reach the GCL and develop into mature granule cells, PSA-NCAM (Seki et al., 2007) and CAR expression are downregulated and restricted to specific compartments. These immature neurons expressing CAR and PSA-NCAM have unique functional properties, including enhanced synaptic plasticity and a lower threshold for the induction of glutamatergic potentiation (Schmidt-Hieber et al., 2004). These properties make them susceptible to be recruited upon hippocampal activation. Synaptic plasticity is associated with synapse formation, remodeling, and elimination. In primary cultures of hippocampal neurons, which resemble immature neurons in the adult hippocampus, CAR is recruited to the synapse. These observations underscore the potential role of CAR in neuronal circuit remodeling regulating hippocampal plasticity. It will be important to determine whether hippocampal CAR, like NCAM, is modulated posttranslationally after behavior tasks, contextual fear conditioning, or passive avoidance (Sandi, 2004).
In the adult brain, NCAM stabilizes neural circuits. The polysialylation of NCAM induces anti-adhesion properties, allowing structural plasticity of neuronal network including activity-dependent synaptic plasticity and formation of long-term memory (Bonfanti, 2006). Abolishing glycosylation of the extracellular domains of CAR may reduce intercellular adhesion (Excoffon et al., 2007), but how this occurs is unclear. Could glycosylation limit intracellular cis-CAR–CAR interactions and influence CAR function in newborn or mature neurons? Like nonpolysialylated NCAM, CAR may be involved in interactions that induce neuronal and neurite outgrowth in some adult neurons (Seidenfaden et al., 2006). Although our data in the brain of CAR-CNSKO mice poorly dovetail with a primordial role for CAR during axon growth (Patzke et al., 2010), they do not exclude the possibility that CAR plays a role in axon guidance in a subset of neurons.
Assuming that CAR will behave, at least in some respects, as a prototypical CAM with noncanonical functions, it would not be surprising that, once recruited to synapses, CAR interacts with different intracellular and extracellular partners than those on the somal plasma membrane. Among synaptic CAMs, integrins play a role in LTP by controlling actin reorganization and spine remodeling in an NMDA-dependent mechanism (Shi and Ethell, 2006). Of particular relevance here is that fibronectin, the extracellular matrix protein mediating integrin effects, also binds CAR to promote neurite extension in vitro (Patzke et al., 2010).
CAR loss of function and neurogenesis
Both intrinsic and extrinsic mechanisms regulate adult neurogenesis. Like some NCAM isoforms (Burgess et al., 2008), our data are consistent with a role for CAR in newborn neuron differentiation and integration in existing circuitries. Based on IHC staining, a significant amount of CAR is in axon tracks and the majority of CAR in the brain is not lost after acute LPS-induced inflammation in mice. A striking exception is CAR on the cell body of immature neurons. CAM processing by secretases regulates neuron proliferation, migration, and plasticity. The juxtaposition of these observations merit further investigations as to whether CAR processing on immature neurons is a fundamental event during neurogenesis. In addition to BACE1, CAR is also a substrate for α and γ-secretases, which can liberate the CAR ectodomain and intracellular domain. Protease cleavage of CAR could switch its role to a signaling molecule by the generation of its intracellular domain because a phosphorylated fragment of the CAR intracellular domain can be found in the nucleus (Dephoure et al., 2008). The CAR intracellular domain therefore may influence transcriptional regulation and affect neurogenesis and/or synapse homeostasis.
Adult human neurogenesis creates ∼700 neurons/d in the DG (Spalding et al., 2013), which, over an average life span, corresponds to approximately one-third of the total numbers of neurons in the entire hippocampus (or to the equivalent of the renewal of the DG). In CAR-CNSKO mice, the disorganization of the GCL in the DG is consistent with abnormal neuron integration. A similar phenotype is found in mice lacking Reelin, which modulates CAM-dependent migration (D'Arcangelo et al., 1995), and therefore CAR loss of function may impair similar guidance cues.
CAR, hippocampal plasticity, and AD
As the decade-long progression to AD symptoms occurs, synapse function and adult neurogenesis are battered by recurring acute and/or chronic proinflammatory insults. A growing body of evidence supports the link between neuroinflammation and cognitive impairment (Peterson and Toborek, 2014). Similar to the AD brain, adult neurogenesis and synapse function are perturbed in CAR-CNSKO mice. In 3xTgAD mice, a decrease in adult NPC proliferation and integration correlates with the presence of senile plaques and of Aβ-containing neurons (Rodríguez et al., 2008). These stimuli increase TNF and IFN-γ production, which can increase γ-secretase activity directly through a JNK-dependent MAPK pathway (Liao et al., 2004). Because CAR is also processed by α, β, and γ-secretase (Houri et al., 2013), a link between systemic inflammation and CAR loss in the hippocampus is correlative. Habbas et al. (2015) proposed a mechanistic explanation for TNF-induced memory and spatial impairment involving astrocyte-stimulated secretion of glutamate, which increases the frequency of presynaptic vesicular release. However, how, or if, this is related to CAR loss is unclear and will need further analyses.
CAR at the synapse
In some murine models of brain disorders, synapse content is altered in an age-dependent process and parallels the onset of cognitive deficits (Greten-Harrison et al., 2010). Many components of AD pathophysiology affect neurogenesis and synapse homeostasis and the onset of impaired cognition in AD is an enigma: histological analyses of brains from suspected early-stage AD patients have no striking differences compared with many age-matched controls. However, synapse dysfunction occurs early in AD, followed by a gradual presynapse and spine loss. Notably, at the Braak IV stage, severe synapse loss has not yet occurred. In a previous study, it was surprising to find that, after acute CAR depletion in the 6-week-old mouse striatum, a return to predepletion levels takes at least 1 month (Salinas et al., 2014). It would not be unexpected to find that CAR replacement is significantly slower in the aged human brain and, therefore, repetitive CAR depletion, combined with other insults, would have a detrimental effect on hippocampal plasticity. Our data demonstrating that CAR levels are significantly reduced at early stages of late-onset AD, before most synaptic protein levels are changed, underscore CAR's role in hippocampal plasticity and its likely impact in AD-related cognitive impairment.
CAR and the sexes
It has been noted repeatedly that male and female brains do not respond identically to biological insults. Conspicuously, female CAR-CNSKO mice are more affected in synapse content, short- and long-term synaptic plasticity, and memory retention. This stronger phenotype in female CAR-CNSKO mice might be due to less neuroprotective mechanisms. One plausible explanation for sex-biased difference is sex steroid concentrations because the hippocampus synthesizes estrogen and androgen. Estradiol levels in the hippocampus are 8 nm in male rodents, but ≤2 nm in females (Ooishi et al., 2012). Numerous studies show that estrogens have neuroprotective properties and improve plasticity and memory processes (Roepke et al., 2011). However, as noted previously, translational studies from rodents to humans, especially for neurodegenerative diseases, can be challenging to interpret. In humans, there is a link between estrogen levels and CAR: the CXADR promoter possesses an estrogen-responsive element (Vindrieux et al., 2011). More efficient replacement of inflammation-depleted CAR in the male brain via estrogen-induced transcription would fit attractively into the existing data.
In addition, the interplay between sex steroids and the immune system likely plays an important role. Several neurological conditions show striking gender imbalances in prevalence and outcome. Interestingly, autoimmune diseases also have a stronger prevalence in females (Ngo et al., 2014) and a gender link exists between inflammation states and the propensity to develop certain diseases. In some mammals, females tend to show enhanced immune responses and resistance to infections, which is likely due to immunomodulatory actions of sex steroid hormones. In general, estradiol has immune-enhancing effects on humoral immunity, but a divergent impact on cellular immunity depending on the dose. Testosterone generally depresses both humoral and cell-mediated immunity and increases susceptibility to bacterial and viral infections (for review, see Hanamsagar and Bilbo, 2016). In addition, estrogen can regulate cytokine expression by microglia at the basal level and in the presence of an inflammatory challenge (Mor et al., 1999; Vegeto et al., 2001; Barreto et al., 2007), whereas testosterone is known to have an inhibitory effect on glial activation (Barreto et al., 2007). In a pathological context such as AD, in which women are more susceptible, accumulating evidence has suggested that sex hormones might contribute to the etiology of cognitive changes or plasticity alteration acting through inflammatory reactions (Au et al., 2016). The sexual dimorphism in hippocampal plasticity observed in CAR-CNSKO mice might also involve differential inflammatory mechanisms.
Conclusion
We characterized CAR expression in the healthy and diseased brain and show that it is a multifunctional CAM in immature and mature neurons. Using transgenic mice depleted in CAR expression in the brain, we identify roles for CAR during adult neurogenesis and synapse biology. We delineate a pathway in which secretases and proinflammatory cytokines perturb CAR levels and affect adult neurogenesis, synapse homeostasis and hippocampal plasticity. We propose that the consequence of recurrent or chronic proinflammatory insults, combined with the etiological origin of the neurodegenerative diseases, affects CAR function and ultimately reaches a threshold in aged and diseased brain and exacerbates cognitive decline. Clearly, the connections between CAR-linked defects in adult neurogenesis, synaptic plasticity, and cognitive tasks and the reduction of synaptic protein levels in the hippocampus will require focused and extended analyses in each area.
Footnotes
Work in the Kremer laboratory was funded by the EC European Commission FP7 (BrainCAV #226992, BrainVectors ##286071), the L'Agence Nationale de la Recherche ANR, E-Rare, the La Fondation de France, and L'Association de Recherche pour la Sclérose Latérale Amyotrophique. E.J.K. and S.S. are Inserm fellows. We thank the staff of the IGMM animal facilities MRI for help with microscopy studies; T. Gostan for help with statistical analyses; S. Musardo and S. Pelucchi for technical help; F. Tronche for the nestin-Cre mice; T. Maurice for help with the behavioral studies; R. G. Kelly for help with LPS injections; and J. Zabner and K. Excoffin for anti-CAR antibodies.
References
- Ackermann M, Matus A. Activity-induced targeting of profilin and stabilization of dendritic spine morphology. Nat Neurosci. 2003;6:1194–1200. doi: 10.1038/nn1135. [DOI] [PubMed] [Google Scholar]
- Au A, Feher A, McPhee L, Jessa A, Oh S, Einstein G. Estrogens, inflammation and cognition. Front Neuroendocrinol. 2016;40:87–100. doi: 10.1016/j.yfrne.2016.01.002. [DOI] [PubMed] [Google Scholar]
- Barreto G, Veiga S, Azcoitia I, Garcia-Segura LM, Garcia-Ovejero D. Testosterone decreases reactive astroglia and reactive microglia after brain injury in male rats: role of its metabolites, oestradiol and dihydrotestosterone. Eur J Neurosci. 2007;25:3039–3046. doi: 10.1111/j.1460-9568.2007.05563.x. [DOI] [PubMed] [Google Scholar]
- Bonfanti L. PSA-NCAM in mammalian structural plasticity and neurogenesis. Prog Neurobiol. 2006;80:129–164. doi: 10.1016/j.pneurobio.2006.08.003. [DOI] [PubMed] [Google Scholar]
- Burgess A, Wainwright SR, Shihabuddin LS, Rutishauser U, Seki T, Aubert I. Polysialic acid regulates the clustering, migration, and neuronal differentiation of progenitor cells in the adult hippocampus. Dev Neurobiol. 2008;68:1580–1590. doi: 10.1002/dneu.20681. [DOI] [PubMed] [Google Scholar]
- Chafai M, Corbani M, Guillon G, Desarménien MG. Vasopressin inhibits LTP in the CA2 mouse hippocampal area. PLoS One. 2012;7:e49708. doi: 10.1371/journal.pone.0049708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JW, Zhou B, Yu QC, Shin SJ, Jiao K, Schneider MD, Baldwin HS, Bergelson JM. Cardiomyocyte-specific deletion of the coxsackievirus and adenovirus receptor results in hyperplasia of the embryonic left ventricle and abnormalities of sinuatrial valves. Circ Res. 2006;98:923–930. doi: 10.1161/01.RES.0000218041.41932.e3. [DOI] [PubMed] [Google Scholar]
- Coyne CB, Bergelson JM. CAR: a virus receptor within the tight junction. Adv Drug Deliv Rev. 2005;57:869–882. doi: 10.1016/j.addr.2005.01.007. [DOI] [PubMed] [Google Scholar]
- Cremer H, Chazal G, Lledo PM, Rougon G, Montaron MF, Mayo W, Le Moal M, Abrous DN. PSA-NCAM: an important regulator of hippocampal plasticity. Int J Dev Neurosci. 2000;18:213–220. doi: 10.1016/S0736-5748(99)00090-8. [DOI] [PubMed] [Google Scholar]
- D'Arcangelo G, Miao GG, Chen SC, Soares HD, Morgan JI, Curran T. A protein related to extracellular matrix proteins deleted in the mouse mutant reeler. Nature. 1995;374:719–723. doi: 10.1038/374719a0. [DOI] [PubMed] [Google Scholar]
- Dephoure N, Zhou C, Villén J, Beausoleil SA, Bakalarski CE, Elledge SJ, Gygi SP. A quantitative atlas of mitotic phosphorylation. Proc Natl Acad Sci U S A. 2008;105:10762–10767. doi: 10.1073/pnas.0805139105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durbec P, Cremer H. Revisiting the function of PSA-NCAM in the nervous system. Mol Neurobiol. 2001;24:53–64. doi: 10.1385/MN:24:1-3:053. [DOI] [PubMed] [Google Scholar]
- Excoffon KJ, Gansemer N, Traver G, Zabner J. Functional effects of coxsackievirus and adenovirus receptor glycosylation on homophilic adhesion and adenoviral infection. J Virol. 2007;81:5573–5578. doi: 10.1128/JVI.02562-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freimuth P, Philipson L, Carson SD. The coxsackievirus and adenovirus receptor. Curr Top Microbiol Immunol. 2008;323:67–87. doi: 10.1007/978-3-540-75546-3_4. [DOI] [PubMed] [Google Scholar]
- Garside D, Hargrove RL, Winecker RE. Concentration of oxymorphone in postmortem fluids and tissue. J Anal Toxicol. 2009;33:121–128. doi: 10.1093/jat/33.3.121. [DOI] [PubMed] [Google Scholar]
- Greten-Harrison B, Polydoro M, Morimoto-Tomita M, Diao L, Williams AM, Nie EH, Makani S, Tian N, Castillo PE, Buchman VL, Chandra SS. alphabetagamma-Synuclein triple knockout mice reveal age-dependent neuronal dysfunction. Proc Natl Acad Sci U S A. 2010;107:19573–19578. doi: 10.1073/pnas.1005005107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habbas S, Santello M, Becker D, Stubbe H, Zappia G, Liaudet N, Klaus FR, Kollias G, Fontana A, Pryce CR, Suter T, Volterra A. Neuroinflammatory TNF impairs memory via astrocyte signaling. Cell. 2015;163:1730–1741. doi: 10.1016/j.cell.2015.11.023. [DOI] [PubMed] [Google Scholar]
- Hanamsagar R, Bilbo SD. Sex differences in neurodevelopmental and neurodegenerative disorders: focus on microglial function and neuroinflammation during development. J Steroid Biochem Mol Biol. 2016;160:127–133. doi: 10.1016/j.jsbmb.2015.09.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heneka MT, et al. Neuroinflammation in Alzheimer's disease. Lancet Neurol. 2015;14:388–405. doi: 10.1016/S1474-4422(15)70016-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes C, Cunningham C, Zotova E, Woolford J, Dean C, Kerr S, Culliford D, Perry VH. Systemic inflammation and disease progression in Alzheimer disease. Neurology. 2009;73:768–774. doi: 10.1212/WNL.0b013e3181b6bb95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honda T, Saitoh H, Masuko M, Katagiri-Abe T, Tominaga K, Kozakai I, Kobayashi K, Kumanishi T, Watanabe YG, Odani S, Kuwano R. The coxsackievirus-adenovirus receptor protein as a cell adhesion molecule in the developing mouse brain. Mol Brain Res. 2000;77:19–28. doi: 10.1016/S0169-328X(00)00036-X. [DOI] [PubMed] [Google Scholar]
- Houri N, Huang KC, Nalbantoglu J. The Coxsackievirus and Adenovirus Receptor (CAR) undergoes ectodomain shedding and regulated intramembrane proteolysis (RIP) PLoS One. 2013;8:e73296. doi: 10.1371/journal.pone.0073296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Junyent F, Kremer EJ. CAV-2-why a canine virus is a neurobiologist's best friend. Curr Opin Pharmacol. 2015;24:86–93. doi: 10.1016/j.coph.2015.08.004. [DOI] [PubMed] [Google Scholar]
- Kang H, Schuman EM. Long-lasting neurotrophin-induced enhancement of synaptic transmission in the adult hippocampus. Science. 1995;267:1658–1662. doi: 10.1126/science.7886457. [DOI] [PubMed] [Google Scholar]
- Lepousez G, Nissant A, Lledo PM. Adult neurogenesis and the future of the rejuvenating brain circuits. Neuron. 2015;86:387–401. doi: 10.1016/j.neuron.2015.01.002. [DOI] [PubMed] [Google Scholar]
- Liao YF, Wang BJ, Cheng HT, Kuo LH, Wolfe MS. Tumor necrosis factor-alpha, interleukin-1beta, and interferon-gamma stimulate gamma-secretase-mediated cleavage of amyloid precursor protein through a JNK-dependent MAPK pathway. J Biol Chem. 2004;279:49523–49532. doi: 10.1074/jbc.M402034200. [DOI] [PubMed] [Google Scholar]
- Loustalot F, Kremer EJ, Salinas S. Membrane dynamics and signaling of coxsackievirus and adenovirus receptor. Int Rev Cell Mol Biol. 2016;322:331–362. doi: 10.1016/bs.ircmb.2015.10.006. [DOI] [PubMed] [Google Scholar]
- Lucas SM, Rothwell NJ, Gibson RM. The role of inflammation in CNS injury and disease. Br J Pharmacol. 2006;147:S232–S240. doi: 10.1038/sj.bjp.0706400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcello E, Epis R, Saraceno C, Gardoni F, Borroni B, Cattabeni F, Padovani A, Di Luca M. SAP97-mediated local trafficking is altered in Alzheimer disease patients' hippocampus. Neurobiol Aging. 2012;33:422.e1–10. doi: 10.1016/j.neurobiolaging.2010.09.015. [DOI] [PubMed] [Google Scholar]
- Maurice T, Hippert C, Serratrice N, Dubois G, Jacquet C, Antignac C, Kremer EJ, Kalatzis V. Cystine accumulation in the CNS results in severe age-related memory deficits. Neurobiol Aging. 2009;30:987–1000. doi: 10.1016/j.neurobiolaging.2007.09.006. [DOI] [PubMed] [Google Scholar]
- Mor G, Nilsen J, Horvath T, Bechmann I, Brown S, Garcia-Segura LM, Naftolin F. Estrogen and microglia: a regulatory system that affects the brain. J Neurobiol. 1999;40:484–496. doi: 10.1002/(SICI)1097-4695(19990915)40:4<484::AID-NEU6>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- Ngo ST, Steyn FJ, McCombe PA. Gender differences in autoimmune disease. Front Neuroendocrinol. 2014;35:347–369. doi: 10.1016/j.yfrne.2014.04.004. [DOI] [PubMed] [Google Scholar]
- Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, Metherate R, Mattson MP, Akbari Y, LaFerla FM. Triple-transgenic model of Alzheimer's disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron. 2003;39:409–421. doi: 10.1016/S0896-6273(03)00434-3. [DOI] [PubMed] [Google Scholar]
- Ooishi Y, Kawato S, Hojo Y, Hatanaka Y, Higo S, Murakami G, Komatsuzaki Y, Ogiue-Ikeda M, Kimoto T, Mukai H. Modulation of synaptic plasticity in the hippocampus by hippocampus-derived estrogen and androgen. J Steroid Biochem Mol Biol. 2012;131:37–51. doi: 10.1016/j.jsbmb.2011.10.004. [DOI] [PubMed] [Google Scholar]
- Patzke C, Max KE, Behlke J, Schreiber J, Schmidt H, Dorner AA, Kröger S, Henning M, Otto A, Heinemann U, Rathjen FG. The coxsackievirus-adenovirus receptor reveals complex homophilic and heterophilic interactions on neural cells. J Neurosci. 2010;30:2897–2910. doi: 10.1523/JNEUROSCI.5725-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persson A, Fan X, Widegren B, Englund E. Cell type- and region-dependent coxsackie adenovirus receptor expression in the central nervous system. J Neurooncol. 2006;78:1–6. doi: 10.1007/s11060-005-9055-3. [DOI] [PubMed] [Google Scholar]
- Peterson PK, Toborek M. Neuroinflammation and neurodegeneration. Springer Publishing. 2014 [Google Scholar]
- Rodríguez JJ, Jones VC, Tabuchi M, Allan SM, Knight EM, LaFerla FM, Oddo S, Verkhratsky A. Impaired adult neurogenesis in the dentate gyrus of a triple transgenic mouse model of Alzheimer's disease. PLoS One. 2008;3:e2935. doi: 10.1371/journal.pone.0002935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roelvink PW, Mi Lee G, Einfeld DA, Kovesdi I, Wickham TJ. Identification of a conserved receptor-binding site on the fiber proteins of CAR-recognizing adenovirus. Science. 1999;286:1568–1571. doi: 10.1126/science.286.5444.1568. [DOI] [PubMed] [Google Scholar]
- Roepke TA, Ronnekleiv OK, Kelly MJ. Physiological consequences of membrane-initiated estrogen signaling in the brain. Front Biosci (Landmark Ed) 2011;16:1560–1573. doi: 10.2741/3805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salinas S, Bilsland LG, Henaff D, Weston AE, Keriel A, Schiavo G, Kremer EJ. CAR-associated vesicular transport of an adenovirus in motor neuron axons. PLoS Pathog. 2009;5:e1000442. doi: 10.1371/journal.ppat.1000442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salinas S, Zussy C, Loustalot F, Henaff D, Menendez G, Morton PE, Parsons M, Schiavo G, Kremer EJ. Disruption of the coxsackievirus and adenovirus receptor-homodimeric interaction triggers lipid microdomain- and dynamin-dependent endocytosis and lysosomal targeting. J Biol Chem. 2014;289:680–695. doi: 10.1074/jbc.M113.518365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandi C. Stress, cognitive impairment and cell adhesion molecules. Nat Rev Neurosci. 2004;5:917–930. doi: 10.1038/nrn1555. [DOI] [PubMed] [Google Scholar]
- Schmidt-Hieber C, Jonas P, Bischofberger J. Enhanced synaptic plasticity in newly generated granule cells of the adult hippocampus. Nature. 2004;429:184–187. doi: 10.1038/nature02553. [DOI] [PubMed] [Google Scholar]
- Seidenfaden R, Krauter A, Hildebrandt H. The neural cell adhesion molecule NCAM regulates neuritogenesis by multiple mechanisms of interaction. Neurochem Int. 2006;49:1–11. doi: 10.1016/j.neuint.2005.12.011. [DOI] [PubMed] [Google Scholar]
- Seki T, Namba T, Mochizuki H, Onodera M. Clustering, migration, and neurite formation of neural precursor cells in the adult rat hippocampus. J Comp Neurol. 2007;502:275–290. doi: 10.1002/cne.21301. [DOI] [PubMed] [Google Scholar]
- Shaw CA, Holland PC, Sinnreich M, Allen C, Sollerbrant K, Karpati G, Nalbantoglu J. Isoform-specific expression of the Coxsackie and adenovirus receptor (CAR) in neuromuscular junction and cardiac intercalated discs. BMC Cell Biol. 2004;5:42. doi: 10.1186/1471-2121-5-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Ethell IM. Integrins control dendritic spine plasticity in hippocampal neurons through NMDA receptor and Ca2+/calmodulin-dependent protein kinase II-mediated actin reorganization. J Neurosci. 2006;26:1813–1822. doi: 10.1523/JNEUROSCI.4091-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soudais C, Boutin S, Hong SS, Chillon M, Danos O, Bergelson JM, Boulanger P, Kremer EJ. Canine adenovirus type 2 attachment and internalization: Coxsackievirus-adenovirus receptor, alternative receptors, and an RGD-independent pathway. J Virol. 2000;74:10639–10649. doi: 10.1128/JVI.74.22.10639-10649.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soudais C, Laplace-Builhe C, Kissa K, Kremer EJ. Preferential transduction of neurons by canine adenovirus vectors and their efficient retrograde transport in vivo. FASEB J. 2001;15:2283–2285. doi: 10.1096/fj.01-0321fje. [DOI] [PubMed] [Google Scholar]
- Soudais C, Skander N, Kremer EJ. Long-term in vivo transduction of neurons throughout the rat central nervous system using novel helper-dependent CAV-2 vectors. FASEB J. 2004;18:391–393. doi: 10.1096/fj.03-0438fje. [DOI] [PubMed] [Google Scholar]
- Spalding KL, Bergmann O, Alkass K, Bernard S, Salehpour M, Huttner HB, Boström E, Westerlund I, Vial C, Buchholz BA, Possnert G, Mash DC, Druid H, Frisén J. Dynamics of hippocampal neurogenesis in adult humans. Cell. 2013;153:1219–1227. doi: 10.1016/j.cell.2013.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Togashi H, Sakisaka T, Takai Y. Cell adhesion molecules in the central nervous system. Cell Adh Migr. 2009;3:29–35. doi: 10.4161/cam.3.1.6773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tronche F, Kellendonk C, Kretz O, Gass P, Anlag K, Orban PC, Bock R, Klein R, Schütz G. Disruption of the glucocorticoid receptor gene in the nervous system results in reduced anxiety. Nat Genet. 1999;23(1):99–103. doi: 10.1038/12703. [DOI] [PubMed] [Google Scholar]
- Valero J, Mastrella G, Neiva I, Sánchez S, Malva JO. Long-term effects of an acute and systemic administration of LPS on adult neurogenesis and spatial memory. Front Neurosci. 2014;8:83. doi: 10.3389/fnins.2014.00083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vegeto E, Bonincontro C, Pollio G, Sala A, Viappiani S, Nardi F, Brusadelli A, Viviani B, Ciana P, Maggi A. Estrogen prevents the lipopolysaccharide-induced inflammatory response in microglia. J Neurosci. 2001;21:1809–1818. doi: 10.1523/JNEUROSCI.21-06-01809.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent T, Pettersson RF, Crystal RG, Leopold PL. Cytokine-mediated downregulation of coxsackievirus-adenovirus receptor in endothelial cells. J Virol. 2004;78:8047–8058. doi: 10.1128/JVI.78.15.8047-8058.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vindrieux D, Le Corre L, Hsieh JT, Métivier R, Escobar P, Caicedo A, Brigitte M, Lazennec G. Coxsackie and adenovirus receptor is a target and a mediator of estrogen action in breast cancer. Endocr Relat Cancer. 2011;18:311–321. doi: 10.1530/ERC-10-0230. [DOI] [PubMed] [Google Scholar]
- Yamagata M, Sanes JR, Weiner JA. Synaptic adhesion molecules. Curr Opin Cell Biol. 2003;15:621–632. doi: 10.1016/S0955-0674(03)00107-8. [DOI] [PubMed] [Google Scholar]
- Yamamoto M, Kiyota T, Horiba M, Buescher JL, Walsh SM, Gendelman HE, Ikezu T. Interferon-gamma and tumor necrosis factor-alpha regulate amyloid-beta plaque deposition and beta-secretase expression in Swedish mutant APP transgenic mice. Am J Pathol. 2007;170:680–692. doi: 10.2353/ajpath.2007.060378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L, Barão S, Laga M, Bockstael K, Borgers M, Gijsen H, Annaert W, Moechars D, Mercken M, Gevaert K, De Strooper B. The neural cell adhesion molecules L1 and CHL1 are cleaved by BACE1 protease in vivo. J Biol Chem. 2012;287:25927–25940. doi: 10.1074/jbc.M112.377465. [DOI] [PMC free article] [PubMed] [Google Scholar]