

Abstract
Passive immunization with anti-tau monoclonal antibodies has been shown by several laboratories to reduce age-dependent tau pathology and neurodegeneration in mutant tau transgenic mice. These studies have used repeated high weekly doses of various tau antibodies administered systemically for several months and have reported reduced tau pathology of ∼40–50% in various brain regions. Here we show that direct intrahippocampal administration of the adeno-associated virus (AAV)-vectored anti-phospho-tau antibody PHF1 to P301S tau transgenic mice results in high and durable antibody expression, primarily in neurons. Hippocampal antibody levels achieved after AAV delivery were ∼50-fold more than those reported following repeated systemic administration. In contrast to systemic passive immunization, we observed markedly reduced (≥80–90%) hippocampal insoluble pathological tau species and neurofibrillary tangles following a single dose of AAV-vectored PHF1 compared with mice treated with an AAV-IgG control vector. Moreover, the hippocampal atrophy observed in untreated P301S mice was fully rescued by treatment with the AAV-vectored PHF1 antibody. Vectored passive immunotherapy with an anti-tau monoclonal antibody may represent a viable therapeutic strategy for treating or preventing such tauopathies as frontotemporal dementia, progressive supranuclear palsy, or Alzheimer's disease.
SIGNIFICANCE STATEMENT We have used an adeno-associated viral (AAV) vector to deliver the genes encoding an anti-phospho-tau monoclonal antibody, PHF1, directly to the brain of mice that develop neurodegeneration due to a tau mutation that causes frontotemporal dementia (FTD). When administered systemically, PHF1 has been shown to modestly reduce tau pathology and neurodegeneration. Since such antibodies do not readily cross the blood–brain barrier, we used an AAV vector to deliver antibody directly to the hippocampus and observed much higher antibody levels and a much greater reduction in tau pathology. Using AAV vectors to deliver antibodies like PHF1 directly to brain may constitute a novel approach to treating various neurodegenerative disorders, such as FTD and Alzheimer's disease.
Keywords: AAV vector, Alzheimer's disease, anti-tau antibody, passive immunization, PHF-tau, tauopathy
Introduction
In Alzheimer's disease (AD), the early and progressive development of amyloid-containing plaques and tau-containing neurofibrillary tangles (NFTs) eventually lead to neurodegeneration in such brain regions as the hippocampus and cerebral cortex, resulting in memory loss and other behavioral symptoms (Selkoe, 2001). Under normal conditions, tau is an abundant soluble cytoplasmic protein that binds to microtubules to promote microtubule stability and function (Drechsel et al., 1992; Jeganathan et al., 2008). In AD and other tauopathies, tau aggregates, becomes hyperphosphorylated, and forms insoluble NFTs (Greenberg and Davies, 1990; Mandelkow and Mandelkow, 2012). Although the exact toxic tau species is unknown, there is abundant evidence that one or more pathological tau species cause synaptic dysfunction and ultimately neuronal cell death (Iqbal and Grundke-Iqbal, 2002; Patterson et al., 2011; Lasagna-Reeves et al., 2012a,b; Tai et al., 2014; Usenovic et al., 2015; Meier et al., 2016). In many respects, the progressive spread of tau pathology along distinct anatomical pathways in the brain closely correlate with disease progression and severity in a number of tauopathies, including AD, frontotemporal lobar degeneration (FTD), Pick's disease, progressive supranuclear palsy (PSP), and corticobasal degeneration. In several of these disorders, specific mutations in the tau gene (MAPT) itself have been shown to cosegregate with disease, providing strong direct evidence that tau pathology itself is sufficient to cause neurodegeneration (Ghetti et al., 2015). Because the extent of tau pathology in AD and other tauopathies so closely correlates with the severity of neurodegeneration, synapse loss, and cognitive deficits (Braak and Braak, 1991; Arriagada et al., 1992), attempts to prevent, reduce, or slow the development of tau pathology have become prominent therapeutic strategies for AD and related tauopathies (Giacobini and Gold, 2013).
Immunotherapy by either active vaccination or passive immunization has emerged as one of the more promising approaches for reducing or preventing tau pathology (Pedersen and Sigurdsson, 2015; Sigurdsson, 2016). Active vaccination directed against phospho-tau (p-tau) peptides or passive immunization with specific high-affinity anti-tau monoclonal antibodies have both been shown to reduce tau pathology and to reduce behavioral deficits in mutant tau transgenic mice (Boutajangout et al., 2010; Bi et al., 2011; Chai et al., 2011; Yanamandra et al., 2013, 2015; d'Abramo et al., 2013, 2015; Collin et al., 2014; Sankaranarayanan et al., 2015). Exactly how passive immunization reduces tau pathology in these mouse models is poorly understood and both cell autonomous (Congdon et al., 2013; Gu et al., 2013; Collin et al., 2014) and nonautonomous (d'Abramo et al., 2013; Sankaranarayanan et al., 2015) mechanisms have been postulated.
While reductions in tau pathology following passive immunization with tau monoclonal antibodies have been observed by several laboratories, the absolute reductions have been rather modest (≤40–50%) and quite variable (Chai et al., 2011; d'Abramo et al., 2013, 2015). Moreover, traditional active and passive immunization approaches have several limitations, notably the very high doses or serum concentrations of antibody required since only very low levels of antibody reach the brain parenchyma from the systemic circulation (Paul, 2011). Effective passive immunization of mutant tau transgenic mice also requires frequent repeated antibody dosing to achieve sufficient serum and brain antibody levels. To circumvent these limitations, we have used an adeno-associated virus (AAV) vector to administer a tau monoclonal antibody, PHF1, previously shown to reduce tau pathology following passive immunization (Chai et al., 2011; d'Abramo et al., 2015), directly to the hippocampus of mutant tau transgenic mice. We now report that vectored intracerebral immunization with this tau monoclonal antibody results in high and durable hippocampal antibody levels (≥50-fold more than that measured following systemic antibody administration), as well as abundant intraneuronal antibody expression. In contrast to previous studies using passive immunization with this same antibody, we observed marked reductions (≥80–90%) in hippocampal tau pathology, including marked reductions in insoluble pathological p-tau species and NFTs following a single intracerebral dose of the AAV-vectored PHF1 antibody.
Materials and Methods
Cloning of antibody sequences and vector production.
PHF1 cDNA sequences were amplified from PHF1 hybridoma cells (a generous gift from Dr. Peter Davies) using a rapid amplification of cDNA ends method (Thermo Fisher Scientific). Total RNA was extracted from the PHF1 hybridoma cell lysates and cDNA was synthesized in two independent reactions by using primers annealing conserved regions of the constant heavy and light chains or by using random hexamers. Heavy and light chain sequences were then amplified from the cDNA using nested primers, cloned into a TOPO vector (Thermo Fisher Scientific) and fully sequenced. Subsequently, the full antibody constructs were assembled by overlapping PCR and cloned into a pAAV plasmid under the control of the CAG promoter, with the heavy and light chains separated by the Tav 2A sequence downstream of a furin cleavage recognition site (Furin 2A cleavage sequence). The pAAV plasmid expressing the anti-tau antibody PHF1 was packaged into an AAV serotype rh.10 capsid to produce the AAVrh.10PHF1 vector. The AAVrh.10 vector was subsequently produced by cotransfection of HEK 293T cells with the pAAV expression plasmids and a helper plasmid carrying the AAVrh.10 cap gene and with AAV and adenovirus genes necessary for capsid production and expression cassette encapsidation. The AAVrh.10PHF1 vector was purified as previously described (Sondhi et al., 2012) and the vector genome titer determined by TaqMan quantitative PCR.
PHF1 expression.
PHF1 antibody expression was assessed in vitro by a Western assay. HEK293T cells were transfected with the pAAVrh.10PHF1 plasmids and, 48 h after transfection, cell culture supernatants were assayed by SDS-PAGE using a goat anti-murine IgG1-specific antibody conjugated to horseradish peroxidase (HRP; Santa Cruz Biotechnology). Antibodies present in the cell culture supernatants were further evaluated for specific binding to p-tau protein. Briefly, brain lysates from age-matched control (non-AD) subjects and AD patients were separated by SDS-PAGE and probed by Western analysis using the supernatant from the pAAVPFH1 transfected cells as a primary antibody and a goat anti-mouse IgG antibody conjugated to HRP as a secondary antibody (Santa Cruz Biotechnology).
Animal and human tissue.
Homozygous P301S mutant tau transgenic mice (kindly provided by Dr. Michel Goedert, Cambridge, United Kingdom) expressing the P301S mutant human 0N4R isoform (383aa) cDNA under control of the Thy1.2 promoter were maintained and characterized as previously described (Allen et al., 2002). These mice were intercrossed and maintained on a C57BL/6J background and developed abundant age-dependent tau pathology (Allen et al., 2002). Human frontal cortical tissue samples from AD and non-AD individuals were obtained from the Banner Sun Health Research Institute (Sun City, AZ), the Kathleen Price Bryan Brain Bank at Duke University (Durham, NC), and Dr. Bernardino Ghetti of Indiana University (Indianapolis, IN).
Isolation of sarkosyl-insoluble fraction from human brain.
The sarkosyl-insoluble fraction of AD or non-AD frontal cortical tissue was prepared using the method described by Greenberg and Davies (1990) with minor modification (Fig. 1E). Briefly, brain tissue was homogenized with cold homogenization buffer (H buffer: 10 mm Tris/1 mm ethylene glycol-bis(2-aminoethylether)-N,N,N′,N′-tetraacetic acid/0.8 m NaCl/10% sucrose, pH 7.4) in a Teflon glass homogenizer. The resulting brain homogenate was then centrifuged at 27,000 × g for 20 min at 4°C. The resulting supernatant was subjected to extraction with 1% (w/v) N-lauroylsarcosine in the presence of 1% (v/v) 2-mercaptoethanol at 37°C for 2.5 h followed by centrifugation at 108,000 × g for 30 min at room temperature. The pellet recovered from this centrifugation was quickly rinsed three times with PBS and dissolved in PBS and stored at −80°C. This preparation is called the sarkosyl-insoluble fraction (SI-tau).
Figure 1.

Diagram of the PHF1 vector, expression cassette, and in vitro expression of PHF1. A, Schematic of the full-length antibody, including light and heavy chains. B, Diagram of the AAVrh.10PHF1 genome. The full-length antibody expression cassette is flanked by the two inverted terminal repeats of AAV serotype 2 (ITR) and encapsidation signal (Ψ). The expression cassette comprises the following: the human cytomegalovirus (CMV) enhancer, the chicken β-actin promoter/splice donor and 5′ end of intron, the 3′ end of the rabbit β-globin intron and splice acceptor, the full-length PHF1 antibody sequence expressed in a single open reading frame (ORF) with an optimized Kozak sequence, and the polyadenylation/transcription stop signal from rabbit β-globin. The full-length antibody ORF includes the IgG1 leader peptide and variable and constant regions (heavy chain) in frame with the Igκ leader peptide, variable and constant regions (light chain), by inclusion of a furin cleavage recognition sequence upstream of a Tav 2A sequence (furin 2A). Expression of PHF1 was examined in the supernatant of 293T cells 48 h after transfection with pAAVPHF1. C, pAAVPHF1-transfected cells produce PHF1 full-length antibody secreted into the extracellular media. Western analysis lanes show supernatants from pAAVPHF1, pAAVαPCRV antibody control, and mock transfected cells. Arrows indicate antibody heavy and light chains. PHF1 and control antibodies were detected using a goat anti-mouse IgG antibody conjugated with HRP. D, PHF1 from the supernatant of transfected cells binds to pathogenic tau from an AD brain S1 fraction. Brain S1 fractions from non-AD and AD patients were separated by SDS PAGE (NuPAGE 4–12% Bis-Tris protein gels, Life Technologies) and assayed by Western blot using cell culture supernatants from pAAVPHF1-transfected 293T cells as a primary antibody and a goat anti-mouse IgG antibody conjugated to HRP as secondary antibody. Arrows indicated the three expected bands (55, 61, and 66 kDa proteins) for hyperphosphorylated/pathogenic Tau (p-Tau). A cortical S1 fraction from a non-AD subject was used as a negative control. M.W., Molecular weight standards. E, A schematic of biochemical procedures for obtaining human AD and P301S brain tissue fractions (details in Materials and Methods).
Stereotaxic injections into the hippocampus of adult P301S mice.
AAVrh.10 vectors expressing either a control IgG (anti-nicotine monoclonal antibody) or PHF1 were stereotactically injected bilaterally into the hippocampus (1 × 1010 vg per injection) of P301S male or female mice (N = 15 each group). We and others have observed gender differences in tau pathology that develop in P301S mice as they age (Allen et al., 2002; Chai et al., 2011). For this study, mice were treated at 2–3 months of age and the animals were killed and brain tissue analyzed at 6 months of age. Intrahippocampal injections of AAV vectors were performed by stereotaxic surgery as previously described (Cetin et al., 2006) with modifications. Briefly, under sterile conditions, mice were anesthetized with isofluorane and secured on a stereotaxic frame (David Kopf Instruments). An incision was made over the skull, the periosteum removed, and burr holes the size of the injection needle were subsequently drilled using a high-speed drill. The AAV preparation (2 μl) was bilaterally injected into the hippocampus of mice using a 33 gauge needle (Hamilton) affixed to a syringe pump (KD Scientific) at a rate of 0.2 μl/min. The injection was performed using the following coordinates: −2.1 mm anteroposterior (AP) from bregma, ±2.0 mm mediolateral (ML) from bregma, and −1.7 mm dorsoventral (DV) below the dura. After injection, the needle was left in place for 5 min to minimize backflow and then slowly withdrawn. Animals were individually housed and monitored until they regained full consciousness. Three to 4 months after injection, the mice were deeply anesthetized with avertin and perfused via a cardiac syringe with 0.3% heparinized saline (2500 IU/L). Brains were rapidly collected and each brain was divided along the sagittal plane with one hemisphere processed for histological and immunohistochemical (IHC) analyses and the other hemisphere microdissected into the hippocampus, cerebral cortex, and brainstem; then rapidly frozen on dry ice; and stored at −80°C for biochemical analyses (see below).
Mouse tissue preparation.
Tissues were prepared as described by Chai et al. (2011). For biochemical analyses (Fig. 1E), brain tissue was quickly thawed and homogenized in cold H buffer, and the lysates centrifuged at 21,000 × g for 20 min at 4°C. The supernatants were collected as S1 fractions. Half of the S1 fractions were further centrifuged at 100,000 × g for 1 h at 4°C. The resulting pallet (P1 fraction), which contains insoluble p-tau, was collected and resuspended in ELISA buffer (Cell Signaling Technology).
ELISAs.
Human total tau, pS202/T205-tau (AT8), or pT212/pS214-tau (AT100) were measured by specific sandwich ELISAs as described by Chai et al. (2011). Briefly, 96-well plates were precoated with 5 μg/ml AT8, 2 μg/ml AT100 (Thermo Fisher Scientific), or 2 μg/ml JRF/htau/10 targeting an N-terminal epitope (a gift from Drs. Marc Mercken and Tom Malia, Johnson & Johnson) overnight at 4°C followed by blocking with Starting Block blocking buffer (Thermo Fisher Scientific). Samples (S1 or P1) were diluted in Superblock buffer (Thermo Fisher Scientific) and loaded onto the plates together with biotinylated-HT7 antibody (1:300; catalog #MN 1000B, Thermo Fisher Scientific). After incubation for 1 h at room temperature, samples were then washed nine times with TBS/0.5% Tween 20 wash buffer followed by incubation with streptavidin-HRP (catalog #016-030-084, Jackson Immunoresearch) for 30 min. Following this, the plates were developed by incubating with one-step 3,3,5,5-tetramethylbenzidine (TMB) substrate (Thermo Fisher Scientific) for 30 min and stopped with 2N H2SO4 and then read using a BioTek Synergy H1 Hybrid Reader at 450 nm. The amount of AT8, AT100, or Tau 10-immunoreactive tau was determined using a standard curve derived from human AD brain homogenates and plotted as a relative amount of S1 brain homogenate equivalents as described previously (Chai et al., 2011).
Brain PHF1 levels after intraparenchymal injection of AAVrh.10PHF1.
Since PHF1 is a mouse IgG1, HRP-labeled and biotin-labeled anti-mouse IgG1 was used to detect PHF1 in the hippocampal fractions or tissue sections using an ELISA or IHC, respectively. For the ELISA, 96-well plates were coated with SI-tau overnight, followed by incubation with PHF1-present or PHF1-absent samples for 1 h. Next, HRP-labeled anti-mouse IgG (Jackson Immunoresearch) was added to each well and the PHF1-HRP-conjugated IgG complexes were then detected by one-step TMB substrate, stopped with 2N H2SO2, and read at OD450. For IHC, 40 μm brain sections were first incubated with a solution containing 3% H2O2 in 1 × PBS with 0.05% Triton for 30 min to quench endogenous peroxidase activity. Sections were then blocked sequentially with superblock solution (5% powdered milk, 5% BSA, 5% goat serum, in 1 × PBS with 0.05% Triton), as well as mouse Ig blocking reagent (M.O.M. Kit, catalog #BMK-2202, Vector Laboratories) for 1 h each at room temperature. Next, sections were immersed in biotin-labeled anti-mouse IgG (catalog #BMK-2202, Vector Laboratories) for 1 h, which allows the formation of an anti-tau antibody and biotinylated mouse IgG complex. Following this, sections were incubated with HRP-labeled streptavidin using a Vectastatin ABC kit (Vector Laboratories). Finally, the sections were stained with ImmPACT DAB (catalog #CK-4105, Vector Laboratories) for 30 s and PHF1 was visualized using a Nikon Eclipse 80i microscope.
Immunofluorescence.
To investigate the specific cellular localization of the PHF1 antibody delivered by AAVrh.10, 40 μm brain sections containing the hippocampus were first blocked as described above, then double-stained with Alexa Fluor 488-conjugated goat anti-mouse (1:500; catalog #A11029, Life Technologies) and cell-type-specific antibodies, including either a rabbit anti-GFAP antibody (a specific astrocyte marker; 1:500; catalog #AB7260, Abcam) or a guinea pig anti-NeuN antibody (a specific neuronal marker; 1:1000; catalog #ABN90, Millipore). Alexa Fluor 568-conjugated goat anti-guinea pig (catalog #A11075, Life Technologies) and Alexa Fluor 594 goat anti-rabbit (catalog #A11072, Life Technologies) secondary antibodies were used for detecting NeuN and GFAP, respectively. Sections were covered with coverslips using VectaShield (catalog #H-1000, Vector Laboratories). To examine the degree of tau pathology in the PHF1-expressing area, brain sections were double-stained with Alexa Fluor 488-conjugated goat anti-mouse (1:500; catalog #A11029, Life Technologies) as well as biotinylated AT100 antibody (Thermo Fisher Scientific). Sections were prepared as described above with minor modification. After being treated with the superblock/mouse IgG blocking steps, sections were subsequently incubated with avidinD/biotin solution (Avidin/Biotin Blocking Kit, catalog #SP-2001, Vector Laboratories) for 30 min. Sections were then rinsed three times in 1 × PBS solution containing 0.05% Triton and 0.5% BSA, 5 min each, followed by incubation with biotinylated AT100 and Alexa Fluor 488 goat anti-mouse (1:500; diluted in 1 × PBS plus 0.05% Triton with 0.5% BSA) at 4°C overnight. AT100 immunoreactivity was detected by immersing brain sections in Alexa Fluor 568 streptavidin-containing solution (1:500; diluted in 1 × PBS containing 0.05% Triton and 0.5% BSA) for 1 h. Sections were covered with coverslips using VectaShield. Fluorescent imaging was performed on a Nikon Eclipse 80i microscope.
Thioflavin-S staining.
Thioflavin-S staining to quantify NFTs was performed as described previously (Yanamandra et al., 2015). Three randomly selected hippocampal brain sections from each mouse were stained in the dark with thioflavin-S in 50% ethanol (0.05%, w/v) for 8 min, washed in 80% ethanol twice, and then washed in distilled water three times. Thioflavin-S-positive cells were quantified in the dentate gyrus (DG) of the hippocampus by an investigator (F.Y.) blind to the treatments.
Image analysis.
All image analyses were performed using NIS-Elements software (Nikon). To count AT100-positive immunoreactivity and NFT-positive cells, three and four sections, respectively, through the hippocampus of each animal were used and the data averaged. All measurements were performed by an investigator blinded to the treatment condition. Neurons and astrocytes were counted in ≥2 random sections from the subiculum region of each mouse (Gu et al., 2013). For volumetric analysis of the hippocampus, ≥3 coronal brain sections corresponding to bregma coordinates −1.7 to −2.3 mm (Franklin and Paxinos, 2008) were used. All of these mounted tissue sections were stained with AT8-labeled or biotin-labeled anti-mouse IgG antibody (catalog #BMK-2202, Vector Laboratories). The stained tissues were scanned and hippocampal volumes were measured blindly with the NIS-Elements software as previously described with modification (Yanamandra et al., 2015).
Statistical analysis.
Data were statistically analyzed using GraphPad Prism 6 (GraphPad Software). A p value of <0.05 was considered significant. Error bars represent the SEM. Statistical analyses between two groups were performed by Student's t test.
Results
Construction of an AAVrh.10PHF1 vector and in vitro confirmation of PHF1 expression
We first identified PHF1 cDNA sequences by amplification from PHF1 hybridoma cells and full-length antibody (with both heavy and light chains) constructs were cloned into a pAAV plasmid under the control of the CAG promoter (Fig. 1A). The pAAV plasmid-expressing PHF1 was packaged into an AAV serotype rh.10 capsid to yield the AAVrh.10PHF1 vector (Fig. 1A,B). AAVrh.10 vectors are known to predominantly transduce neurons (Sondhi et al., 2012; see below). The expression of PHF1 by pAAVPHF1 was confirmed by first transfecting HEK293 cells with pAAVPHF1, pAAVαPCRV IgG antibody control, and mock control. Supernatants from pAAVPHF1, pAAVαPCRV antibody control, and mock transfected cells were collected for Western analyses to demonstrate the presence of PHF1 antibody. Figure 1C shows (1) that the supernatant from pAAVPHF1-treated cells contains antibody heavy and light chains and (2) PHF1 from the supernatant of transfected cells readily binds to pathogenic tau isolated from AD brain S1 fractions (Fig. 1D). Brain S1 fractions prepared identically from non-AD cases were used as a negative control and did not react with PHF1 (Fig. 1D). Antibody expressed by the pAAVαPCRV controls did not recognize PHF tau (data not shown).
Expression, tissue distribution, and cellular localization of PHF1 anti-tau antibody delivered by an AAVrh.10 vector
AAVrh.10 vectors expressing either control IgG or PHF1 were stereotactically injected bilaterally into the hippocampus of homozygous P301S male mice, a tauopathy mouse model that expresses a familial tau mutation (P301S) that causes frontal temporal dementia with parkinsonism-17 (Allen et al., 2002). These mice develop abundant age-dependent tau pathology (Allen et al., 2002). We first examined the expression and regional distribution of PHF1 delivered by AAVrh.10 directly to the hippocampus of P301S mice. Using a specific ELISA, we found that PHF1 is abundantly expressed in the hippocampus of mice treated with AAVrh.10PHF1 (18.66 ± 2.39 μg/mg protein; Fig. 2A) and at levels ∼50-fold greater than the brain levels reported after passive immunization with PHF1 (Chai et al., 2011). Using this same ELISA, there was no detectable PHF1 in the hippocampus of the control group of mice (Fig. 2A). However, we also observed some expression of PHF1 in the cerebral cortex of AAVrh.10PHF1-treated P301S mice, albeit at much lower levels (0.24 ± 0.004 μg/mg protein). We did not detect expression of PHF1 in brain regions distal to the hippocampal injection site (e.g., brainstem), indicating that the expression of PHF1 was mostly confined to the hippocampus (dorsal to rostral) and, to a much lesser extent, the neighboring cerebral cortex. Using IHC we confirmed that PHF1 is abundantly expressed throughout the hippocampus, including in the subiculum and DG (Figs. 2B–D) and with a distinct pattern of primarily neuronal expression. Next, we investigated exactly what cell type PHF1 is expressed in following treatment with AAVrh.10PHF1. The latter was achieved by double-staining hippocampal sections from mice treated with AAVrh.10PHF1 using either a neuron-specific or astrocyte-specific marker (Fig. 3). PHF1 is colocalized primarily to NeuN-positive cells (neurons; Fig. 3A–D) but not GFAP-positive cells (astrocytes; Fig. 3E–H). Quantitative analysis in the subiculum of mice treated with AAVrh.10PHF1 (Fig. 3A, boxed area) revealed that ≥95% of PHF1-expressing cells were NeuN-positive neurons (Fig. 3I), and only ∼2.0% of PHF1-expressing cells were GFAP-positive astrocytes (Fig. 3I).
Figure 2.
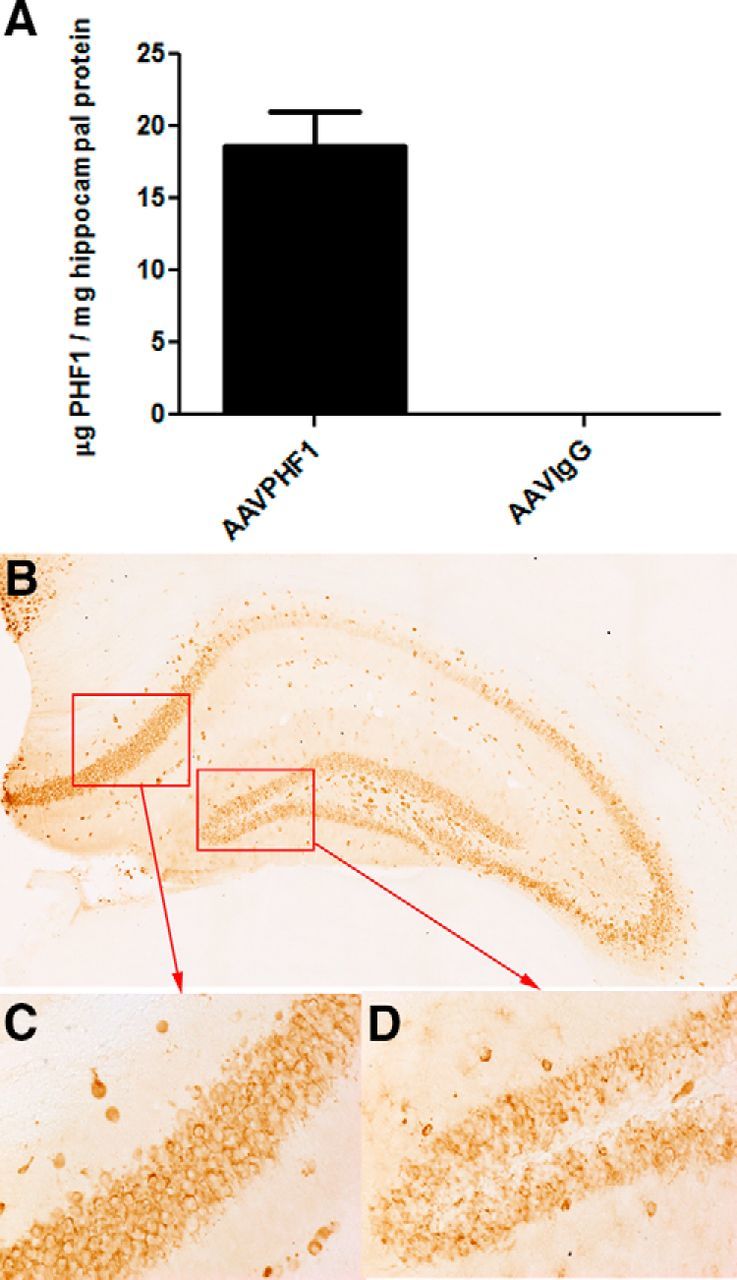
The presence of PHF1 in the hippocampus of P301S male mice treated with AAVrh.10PHF1 vector. A, PHF1, present in the soluble fraction of the hippocampus, is detected by binding to paired helical filamentous tau coated onto an ELISA plate. The antibody-antigen complex is visualized and quantified using HRP-labeled anti-mouse antibody. The complex and its TMB substrate were read at OD450 on a plate reader. PHF1 is not found in mice treated with the AAVrh.10IgG (anti-nicotine antibody) control vector. B, PHF1 antibody is detected in the hippocampus of P301S male mice treated with the AAVrh.10PHF1 vector by IHC using an anti-mouse IgG1 antibody. C, D, Higher-power images of the subiculum and DG regions (red boxes) of B. Note that PHF1 is a mouse IgG1.
Figure 3.

PHF1 is expressed primarily in hippocampal neurons of P301S male mice treated with AAVrh.10PHF1 vector. A–H, PHF1 is primarily colocalized to NeuN-positive neurons (A–D) but not GFAP-positive astrocytes (D–H). A, A representative photomicrograph of double-staining IHC using an anti-mouse antibody (green) and NeuN antibody (red) and hippocampal sections from P301S mice treated with AAVrh.10PHF1. B, C, Higher-power images of PHF1 (B) and NeuN (C) expression from the boxed area of A. D, A merged image of B and C. E, A representative photomicrograph of double-staining IHC using an anti-mouse antibody (green) and GFAP antibody (red) and of hippocampal sections of P301S mice treated with AAVrh.10PHF1. F, G, Higher-power images of PHF1 (F) and GFAP (G) expression in the boxed area of E. H, A merged image of F and G. I, Quantitative analysis of the subiculum of AAVrh.10PHF1-treated P301S mice (n = 12) demonstrated that 95.5 ± 0.009% of PHF1-expressing cells are NeuN-positive neurons, whereas only 2.12 ± 0.32% of PHF1-expressing cells are GFAP-positive astrocytes.
Insoluble p-tau is markedly reduced in the hippocampus of AAVrh.10PHF1-treated P301S mice
Next we examined whether the level of total tau in the hippocampus of P301S mice was altered following treatment with AAVrh.10PHF1 or AAVrh.10IgG. As shown in Figure 4A, total tau levels of the AAVrh.10PHF1-treated, theAAVrh.10IgG-treated, and the untreated P301S mice remained unchanged, suggesting that treatment with either vector fails to non-specifically reduce total tau levels and does not result in significant neurotoxicity. We then used ELISAs that specifically measure pS202/pT205 tau (AT8) or pT212 /pS215 tau (AT100) to quantify pathological insoluble p-tau levels in the hippocampus of 2-month-old and 6-month-old untreated P301S mice as well as from P301S mice treated with either AAVrh.10PHF1 or AAVrh.10IgG. P301S mice did not display detectable insoluble p-tau at 2 months of age whereas very high levels of AT8 and AT100 immunoreactivity were present at 6 months of age (Fig. 4). Insoluble p-tau levels in the hippocampus of 6-month-old P301S mice treated with the AAVrh.10IgG control vector were comparable to those of untreated P301S mice of the same age (Fig. 4B,C). Importantly, we observed a rather marked decrease of AT8 immunoreactivity (Fig. 4B; ∼84% reduction, p < 0.0001) and AT100 immunoreactivity (Fig. 4C; ∼93% reduction, p < 0.0001) in the hippocampus of P301S mice treated with AAVrh.10PHF1 compared with mice treated with AAVrh.10IgG or untreated P301S mice. We also measured insoluble p-tau levels in the brainstem of AAVrh.10PHF1-treated P301S mice and found that insoluble p-tau levels in the brainstem remain unchanged compared with both groups of control mice (data not shown). This is consistent with our finding that no PHF1 antibody was detectable in the brainstem of these mice following injection of AAVrh.10PHF1 to the hippocampus, suggesting that the reduction of insoluble p-tau in the hippocampus of these mice was due to expression of PHF1 in this brain region.
Figure 4.

Marked reduction of insoluble p-tau in the hippocampus of AAVrh.10PHF1-treated P301S male mice. S1 and P1 fractions from wild-type and various groups of P301S mice were prepared to measure total tau and p-tau, respectively. A, Total tau levels are not changed in AAVrh.10-treated P301S versus untreated male mice. B, P301S male mice do not display detectable AT8 immunoreactivity (IR) in the hippocampal insoluble fraction at 2 months of age, whereas a very high level of AT8 IR is present at 6 months of age. Insoluble p-tau levels in the hippocampus of 6-month-old P301S mice treated with the AAVrh.10IgG control vector are comparable to those of untreated P301S mice of the same age, but markedly reduced following treatment with AAVPHF1 (84%; p < 0.0001). C, Similar to the results found in the AT8 ELISA analyses in B, there is a significant reduction (93.8% reduction; p < 0.0001) in AT100 IR in the hippocampal insoluble fraction of AAVrh.10PHF1-treated P301S mice. Relative IR represents the total IR or p-tau IR detected in the indicated amount of S1 protein (in nanograms) from the brain homogenates of cases of AD (see Materials and Methods for details). **** p ≤ 0.0001.
Insoluble p-tau and NFTs are reduced in the subiculum and DG of the hippocampus of AAVrh.10PHF1-treated P301S mice
We next quantified insoluble p-tau immunoreactivity in the subiculum and DG using AT100 IHC, an antibody that has previously been shown to detect only insoluble p-tau (Allen et al., 2002). We found a highly significant reduction in AT100-positive immunoreactivity in the subiculum (Fig. 5A,B; ∼72% reduction, p = 0.0025) and the DG (Fig. 5C,D, ∼84% reduction, p < 0.0001) of AAVrh.10PHF1 vector-treated compared with AAVrh.10IgG-treated mice. Finally, using thioflavin-S, we also quantified NFTs, comprising abnormal hyperphosphorylated and aggregated intracellular tau, in the hippocampus of P301S mice treated with either the AAVrh.10PHF1 or AAVrh.10IgG vector. Consistent with the IHC data, we found a marked reduction in the number of NFTs in the DG of mice treated with AAVrh.10PHF1 (Fig. 5E,F; ∼83% reduction, p < 0.0001 vs controls).
Figure 5.
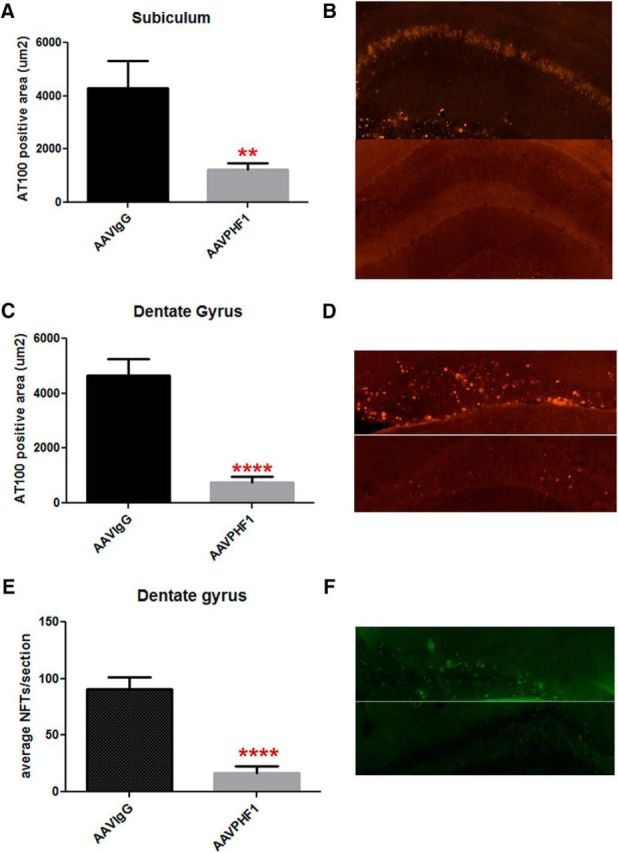
Marked decrease of insoluble p-tau and NFTs in the subiculum and DG of AAVrh.10PHF1-treated P301S male mice. A, C, The area of AT100-positive immunoreactivity (IR) is significantly reduced in the subiculum (A, ∼72% reduction, p = 0.0025) and DG (C, ∼84% reduction, p < 0.0001) of AAVrh.10PHF1-treated P301S male mice. B, D, Representative photomicrographs for A (B) and C (D). Top, AT100 immunofluorescence in the subiculum and DG, respectively, of P301S mice treated with the AAVrh.10IgG control vector. Bottom, AT100 IR of P301S mice treated with AAV rh.10PHF1 vector. E, Thioflavin-S staining was used to visualize and quantify NFTs, comprising abnormally misfolded and hyperphosphorylated tau in the DG of AAVrh.10IgG control and AAVrh10PHF1-treated P301S mice. Quantitative analysis demonstrates that the number of NFTs is significantly reduced in the DG of AAVrh.10PHF1-treated mice compared with control mice (∼83% reduction, p < 0.0001). F, A representative image of E. Top, Thioflavin-S staining of the DG from mice treated with the control vector. Bottom, Staining of the DG from AAVrh.10PHF1-treated P301S mice. All quantitative analyses were performed blind to treatment (A, C, and E). ** 0.001 < p ≤ 0.01; **** p ≤ 0.0001.
To further replicate and extend these findings, we performed an independent experiment to examine the effects of the AAV vector-delivered PHF1 antibody in P301S female mice. Three-month-old P301S female mice were administered AAVrh.10PHF1 (1 × 1010 vg) or AAVrh.10mCherry as a control bilaterally into the hippocampus. After 3 months, tissues, including brains, were collected and processed as described above. Similar to the results shown for males in Figures 2 and 3, we detected high levels of PHF1 in the hippocampus, predominantly in hippocampal neurons, in the AAVrh.10PHF1-treated P301S female mice. Some PHF1 was also detected to a lesser extent in the cortex but none could be measured in the brainstem. Consistent with the findings in male P301S mice, total tau levels were not altered in P301S female mice following treatment with either AAVrh.10PHF1 or AAVrh.10mCherry (data not shown). There was, however, a marked reduction (70–90%) of insoluble p-tau in the hippocampus of AAVrh.10PHF1-treated P301S female mice compared with that of AAVrh.10mCherry-treated control mice (Fig. 6A,B). Furthermore, we observed a significant decrease in p-tau immunoreactivity in the cerebral cortex of AAVrh.10PHF1-treated versus AAVrh.10mcherry-treated female P301S mice (Fig. 6C,D; ∼47% reduction in AT8 immunoreactivity, p = 0.05; 70% reduction in AT100 immunoreactivity, p = 0.04). As anticipated, there was no change in insoluble p-tau immunoreactivity (IR) in the brainstem (data not shown) of AAVrh.10PHF1-treated P301S female mice (data not shown). We next examined the number of NFTs present in the hippocampus of AAVrh.10-treated P301S female mice and also observed a marked reduction in NFTs in the DG compared with control mice (Fig. 6E,F). Together, these data demonstrate rather marked reductions in tau pathology (insoluble p-tau and NFTs) in P301S mice of either gender with a single intrahippocampal injection of the AAVrh.10PHF1 vector and especially in hippocampal subfields expressing high levels of PHF1, predominantly in neurons.
Figure 6.

Marked reduction in insoluble p-tau and NFTs in the DG of AAVrh.10-treated P301S female mice. A, B, The levels of AT8 IR (A; ∼70% reduction; p = 0.05) and AT100 IR (B; 84% reduction; p = 0.003) are markedly reduced in the hippocampal insoluble fraction of AAVrh.10PHF1-treated P301S female mice (n = 7) compared with the AAVrh.10mCherry control mice (n = 13). C, D, We also observed a reduction of AT8 immunoreactivity (IR; C: ∼47%; p = 0.05) and AT100 IR (D: ∼70%; p = 0.04) in the cortical insoluble fraction from AAVPHF1-treated P301S compared with those of mCherry-treated P301S mice. E, Thioflavin-S staining was used to quantify the number of NFTs. Top, A representative photomicrograph of NFTs in the DG of P301S female mice treated with the AAVrh.10mCherry control vector. Bottom, A photomicrograph of the DG from AAVrh.10PHF1 vector-treated mice. F, Quantitative analysis performed blind to treatment demonstrates that the number of NFTs is significantly reduced in the DG of AAVrh.10PHF1-treated P301S female mice (∼88% reduction; n = 13; p < 0.0001). * 0.01 < p ≤ 0.05; ** 0.001 < p ≤ 0.01; **** p ≤ 0.0001.
Hippocampal atrophy observed in homozygous P301S female mice is rescued by AAVrh.10PHF1 treatment
Decreased hippocampal volume has been observed in another strain of P301S mice (PS19; Yanamandra et al., 2015) and an increase in hippocampal volume has been demonstrated in these mice following anti-tau antibody treatment via passive immunization (Yanamandra et al., 2015). We also observed a modest but significant reduction (∼8%; p = 0.01) of hippocampal volume in 6-month-old homozygous P301S female mice (Fig. 7) compared with age-matched wild-type (C57/B16) female mice. A more marked reduction in hippocampal volume was also observed in AAVrh.10mCherry-treated P301S mice (∼21%; p = 0.0006) but not in AAVrh.10PHF1-treated mice compared with wild-type mice. Importantly, compared with the hippocampal volume of untreated P301S mice, we observed no reduction in hippocampal volume in P301S mice treated with the AAV-vectored PHF1 antibody (Fig. 7B). These data suggest that the hippocampal atrophy characteristic of P301S homozygous female mice can be rescued by treatment with the vectored PHF1 antibody. Moreover, delivery of PHF1 to hippocampal neurons using AAVrh.10 over the time frame of our study does not appear to cause overt toxicity or changes in hippocampal volume. However, we did observe a mild astrocytosis but no microgliosis in hippocampal sections from AAVrh.10PHF1-treated P301S mice compared with untreated wild-type C57B1/6 or P301S mice (data not shown). We also observed some apparent toxicity (i.e., loss of hippocampal volume) with the mCherry control vector as has been reported with AAV delivery of other foreign reporter genes/proteins (Fig. 7A,B; Klein et al., 2006).
Figure 7.

Hippocampal atrophy in P301S mice is rescued by treatment with AAVrh.10PHF1. Hippocampal volumes are not changed between AAVrh.10PHF1-treated P301S female mice and wild-type mice, indicating that there is no obvious neurotoxicity following AAVrh.10PHF1 treatment. Hippocampal volumes were measured using three coronal brain sections from each animal at the following level relative to bregma: −1.7, −1.94, and −2.30. A, A representative Nissl-stained hippocampal section from a wild-type female mouse as well as from untreated, AAVrh.10mCherry-treated, and AAVrh.10PHF1-treated P301S female mice. B, Statistical analysis demonstrates that there is a decrease in hippocampal volume (∼8% reduction; p = 0.01) in P301S female mice at 6 months of age compared with that of aged-matched wild-type female control mice. A more significant reduction in hippocampal volume (∼21%) was observed in AAVrh.10mCherry-treated P301S mice (p = 0.0006) compared with that of wild-type mice. No significant difference in hippocampal volume was observed between AAVrh.10PHF1-treated and wild-type female mice. Importantly, compared with the hippocampal volume of untreated P301S female mice, treatment with the AAVrh.10PHF1 vector fully rescued this loss of hippocampal volume. Hippocampal volumes were measured using NIS software (Nikon). * 0.01 < p ≤ 0.05; *** 0.0001 < p ≤ 0.001.
Discussion
It is now well established that passive immunization with repeated systemic doses of various monoclonal antibodies directed against pathological tau epitopes can reduce the development and progression of tau pathology and neurodegeneration in mutant P301S or P301L tauopathy mice (Chai et al., 2011; Pedersen and Sigurdsson, 2015). In general, rather high weekly or biweekly doses of antibody (20–45 mg/kg weekly cumulative dose) have been administered for 3–6 months and tau pathology, measured by quantifying soluble or insoluble p-tau immunoreactivity or NFTs, is reduced by ≤40–50% compared with treatment with an IgG control antibody or that observed in untreated mice (Chai et al., 2011; d'Abramo et al., 2013). These reductions in tau pathology have been accompanied by reduced functional behavioral impairment in some (Boutajangout et al., 2010; Chai et al., 2011) but not all (Bi et al., 2011) studies. Collectively though these data provide “proof of principle” that passive immunization with repeated high doses of certain anti-tau monoclonal antibodies can reduce pathological tau, especially if these antibodies are administered before the development of significant tau pathology. However, the incomplete and modest reduction in tau pathology observed following treatment with very high systemic doses of these antibodies and the requirement for repeated frequent dosing may pose considerable therapeutic challenges in humans with various tauopathies.
In the present study, we have used a single administration of an AAV vector to persistently express the anti-tau monoclonal antibody PHF1 in the hippocampus of P301S mice, a mouse tauopathy model that develops severe age-dependent tau pathology and neurodegeneration (Allen et al., 2002). Following intrahippocampal administration of a single dose of the vectored anti-tau antibody PHF1 (AAVrh.10PHF1), we observed high levels of antibody expression (measured 4 months following administration) that were primarily confined to the hippocampus and to a much lesser extent the surrounding cerebral cortex, with antibody levels of ∼18 μg/mg protein in the hippocampus. This level of PHF1 antibody measured in the hippocampus following intracerebral administration of the AAV-vectored PHF1 antibody cDNA is ≥50-fold more than that measured in the brain following a single systemic dose of 45 mg/kg (Chai et al., 2011), and in line with previous studies demonstrating that only ∼0.1% of a systemically administered IgG antibody reaches brain parenchyma (Paul, 2011). However, it is important to underscore that when using AAVrh.10 to deliver PHF1 to the hippocampus, antibody expression was primarily confined to neurons (Sondhi et al., 2012) and presumably much of the expressed antibody is retained in the cytoplasm of hippocampal neurons (Fig. 2). In general, only very low levels of antibody are taken up by neurons following systemic administration (Atwal et al., 2011). Consequently, the intracellular levels of antibody observed in our study are likely several orders of magnitude higher than those that occur after traditional passive immunization. It will also be important to ascertain the level of antibody present in the interstitial fluid or CSF following intracerebral administration of AAV-vectored antibody, as it is unclear whether neurons have the capacity to secrete antibody when expressed via an AAV vector (see below). Nonetheless, the high hippocampal levels of PHF1 are associated with a marked (≥80–90%) reduction in insoluble pathological p-tau levels in the subiculum and the DG, measured both by ELISA and IHC, as well as a marked reduction (≥80%) in NFTs.
We also observed significant reductions in p-tau immunoreactivity in the cerebral cortex of P301S mice even though the AAVrh.10PHF1 vector was administered directly to the hippocampus and despite much lower levels of cortical PHF1 antibody. Such reductions in tau pathology are rather striking compared with previous passive immunization studies with this same antibody, i.e., when PHF1 is administered systematically at very high weekly doses (Chai et al., 2011; d'Abramo et al., 2015). We underscore, however, that vectored antibody expression in our study was purposely confined to the hippocampus, the site of vector administration, and not surprisingly tau pathology in other brain regions (except in the cerebral cortex) was relatively unaffected. It is also well known that tau pathology in the P301S mice used in our study develops most robustly in the spinal cord and brainstem (Allen et al., 2002), areas with negligible antibody expression following intrahippocampal AAVrh.10PHF1 administration. In our study, we also initiated treatment before the development of significant tau pathology (Allen et al., 2002). It will therefore be interesting in future experiments to test our vectored immunotherapy approach in mice that have already developed substantial tau pathology. Importantly, there was no evidence of neurotoxicity after 4 months of hippocampal antibody expression, as total tau levels and hippocampal volume were grossly unchanged in treated versus untreated mice. In fact, we observed a small (∼8%) but significant reduction in hippocampal volume in untreated 6-month-old female P301S mice compared with age-matched wild-type controls, which was not observed in P301S mice treated with our AAV-vectored PHF1 antibody. These data suggest that hippocampal atrophy, which has been reported in P301S mice, can also be rescued by treatment with the AAV-vectored PHF1 antibody.
What could account for the very marked reduction in hippocampal tau pathology we observed following AAV-vectored anti-tau antibody delivery? There are several possibilities, including the relatively high and sustained levels of antibody expressed in the hippocampus after AAV-vectored delivery (levels 50-fold greater than those measured following systemic administration) and (or) the very high levels of intraneuronal antibody expression. Currently it is unknown exactly how passive immunization with anti-tau monoclonal antibodies effectively reduces age-dependent tau pathology in mutant tau transgenic mice. It has been postulated however that these systemically administered antibodies enter the brain, albeit to a limited extent, and bind to pathological tau species to either prevent their further spread to adjacent neurons (Clavaguera et al., 2009) or to promote their clearance either extracellularly by microglia (Luo et al., 2015) or intracellularly through degradation via lysosomal enzymes or another degradative pathway (for review, see Pedersen and Sigurdsson, 2015). In this regard, Sigurdsson and colleagues have reported that certain tau antibodies are taken up by neurons mainly via low-affinity Fc receptors, where they appear to bind to and sequester pathological tau species, which prevents their spread to adjacent neurons and (or) promotes their clearance intracellularly (Congdon et al., 2013). These authors have reported that antibody uptake into neurons is a prerequisite for tau clearance (Congdon et al., 2013). Similarly, Collin and coworkers (2014) have used high-resolution confocal microscopy to demonstrate internalization of the p-tau antibody MAb86 by neurons following systemic administration to transgenic mice. This anti-tau antibody, which also reduces the accumulation of tau pathology in these mice following passive immunization, binds to tau species associated with lipid rafts, its presumed cellular entry mechanism (Collin et al., 2014). These authors postulate that the internalized anti-tau antibody promotes lysosomal clearance of pathological tau while simultaneously protecting lysosomes from the damaging effects of these same toxic tau species. The high intraneuronal levels of anti-tau antibody we observe following AAV delivery may also prevent secretion and transneuronal propagation of misfolded tau species (Clavaguera et al., 2009; Yanamandra et al., 2013; Ahmed et al., 2014). Whether the effect of passive immunization with anti-tau monoclonal antibodies to reduce tau pathology in mutant tau transgenic mice is due primarily to a cell-autonomous or cell-nonautonomous mechanism of action is unclear, but it is likely that AAV-vectored delivery of these anti-tau antibodies will facilitate either (or both) mechanisms. However, if in the case of human tauopathies due to germline mutations in MAPT a cell-autonomous mechanism predominates, then AAV-vectored delivery of an anti-tau antibody like PHF1 directly to neurons could be much more effective than traditional passive immunization.
Together, our results provide support for a potential strategy of vectored intracerebral immunotherapy whereby an anti-tau monoclonal antibody is directly delivered to brain parenchyma to substantially reduce or prevent the development of tau pathology and thus to slow the progression of tau-dependent neurodegeneration, which characterizes such tauopathies as FTD, PSP, and AD. The advantages of this approach include the relatively high and durable levels of antibody expression achieved in the brain with only a single dose of vector and the marked reduction in tau pathology observed compared with traditional passive immunization. However, for such an approach to be effective, relatively widespread delivery of antibody to affected or vulnerable brain regions will likely need to be achieved. In this regard, widespread brain delivery of AAV-vectored genes has recently been demonstrated in mice and in nonhuman primates following intrathalamic administration (Kells et al., 2009; Baek et al., 2010; Zhao et al., 2016) as well as following intrathecal (Yoon et al., 2016) and even systemic (Zincarelli et al., 2008; Deverman et al., 2016) vector administration. Further studies on alternative routes of administration with various vector capsids are thus warranted and could result in an optimized vector and delivery protocol suitable for eventual clinical studies.
Footnotes
This work was supported by Appel Alzheimer's Research Institute and Alzheimer's Drug Discovery Foundation. We thank Maria Chiuchiolo, Caroline Ferreira and Yihharn Hwang for help with the studies. We thank Todd Carter and Dinah Sah for constructive comments and suggestions on the manuscript.
The authors declare no competing financial interests. However, the authors note that work was performed at Weill Cornell Medical College and a patent application for the use of AAV-vectored anti-tau monoclonal antibodies has been submitted by Weill Cornell. W.L. and S.M.P. are currently employees of Voyager Therapeutics, a CNS gene therapy company working on AAV vectors to deliver monoclonal antibodies to the CNS.
References
- Ahmed Z, Cooper J, Murray TK, Garn K, McNaughton E, Clark H, Parhizkar S, Ward MA, Cavallini A, Jackson S, Bose S, Clavaguera F, Tolnay M, Lavenir I, Goedert M, Hutton ML, O'Neill MJ. A novel in vivo model of tau propagation with rapid and progressive neurofibrillary tangle pathology: the pattern of spread is determined by connectivity, not proximity. Acta Neuropathol. 2014;127:667–683. doi: 10.1007/s00401-014-1254-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen B, Ingram E, Takao M, Smith MJ, Jakes R, Virdee K, Yoshida H, Holzer M, Craxton M, Emson PC, Atzori C, Migheli A, Crowther RA, Ghetti B, Spillantini MG, Goedert M. Abundant tau filaments and nonapoptotic neurodegeneration in transgenic mice expressing human P301S tau protein. J Neurosci. 2002;22:9340–9351. doi: 10.1523/JNEUROSCI.22-21-09340.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arriagada PV, Growdon JH, Hedley-Whyte ET, Hyman BT. Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer's disease. Neurology. 1992;42:631–639. doi: 10.1212/WNL.42.3.631. [DOI] [PubMed] [Google Scholar]
- Atwal JK, Chen Y, Chiu C, Mortensen DL, Meilandt WJ, Liu Y, Heise CE, Hoyte K, Luk W, Lu Y, Peng K, Wu P, Rouge L, Zhang Y, Lazarus RA, Scearce-Levie K, Wang W, Wu Y, Tessier-Lavigne M, Watts RJ. A therapeutic antibody targeting BACE1 inhibits amyloid-beta production in vivo. Sci Transl Med. 2011;3:84ra43. doi: 10.1126/scitranslmed.3002254. [DOI] [PubMed] [Google Scholar]
- Baek RC, Broekman ML, Leroy SG, Tierney LA, Sandberg MA, D'Azzo A, et al. AAV-mediated gene delivery in adult GM1-gangliosidase mice corrects lysosomal storage in CNS and improves survival. PloS One. 2010 doi: 10.1371/journal.pone.0013468. 5.e13468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi M, Ittner A, Ke YD, Götz J, Ittner LM. Tau-targeted immunization impedes progression of neurofibrillary histopathology in aged P301L tau transgenic mice. PLoS One. 2011;6:e26860. doi: 10.1371/journal.pone.0026860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boutajangout A, Quartermain D, Sigurdsson EM. Immunotherapy targeting pathological tau prevents cognitive decline in a new tangle mouse model. J Neurosci. 2010;30:16559–16566. doi: 10.1523/JNEUROSCI.4363-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991;82:239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- Cetin A, Komai S, Eliava M, Seeburg PH, Osten P. Stereotaxic gene delivery in the rodent brain. Nat Protoc. 2006;1:3166–3173. doi: 10.1038/mprot.2006.450. [DOI] [PubMed] [Google Scholar]
- Chai X, Wu S, Murray TK, Kinley R, Cella CV, Sims H, Buckner N, Hanmer J, Davies P, O'Neill MJ, Hutton ML, Citron M. Passive immunization with anti-tau antibodies in two transgenic models: reduction of tau pathology and delay of disease progression. J Biol Chem. 2011;286:34457–34467. doi: 10.1074/jbc.M111.229633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clavaguera F, Bolmont T, Crowther RA, Abramowski D, Frank S, Probst A, Fraser G, Stalder AK, Beibel M, Staufenbiel M, Jucker M, Goedert M, Tolnay M. Transmission and spreading of tauopathy in transgenic mouse brain. Nat Cell Biol. 2009;11:909–913. doi: 10.1038/ncb1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collin L, Bohrmann B, Göpfert U, Oroszlan-Szovik K, Ozmen L, Grüninger F. Neuronal uptake of tau/pS422 antibody and reduced progression of tau pathology in a mouse model of Alzheimer's disease. Brain. 2014;137:2834–2846. doi: 10.1093/brain/awu213. [DOI] [PubMed] [Google Scholar]
- Congdon EE, Gu J, Sait HB, Sigurdsson EM. Antibody uptake into neurons occurs primarily via clathrin-dependent Fc gamma receptor endocytosis and is a prerequisite for acute tau protein clearance. J Biol Chem. 2013;288:35452–35465. doi: 10.1074/jbc.M113.491001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- d'Abramo C, Acker CM, Jimenez HT, Davies P. Tau passive immunotherapy in mutant P301L mice: antibody affinity versus specificity. PLoS One. 2013;8:e62402. doi: 10.1371/journal.pone.0062402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- d'Abramo C, Acker CM, Jimenez H, Davies P. Passive immunization in JNPL3 transgenic mice using an array of phospho-tau specific antibodies. PLoS One. 2015;10:e0135774. doi: 10.1371/journal.pone.0135774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deverman BE, Pravdo PL, Simpson BP, Kumar SR, Chan KY, Banerjee A, Wu WL, Yang B, Huber N, Pasca SP, Gradinaru V. Cre-dependent selection yields AAV variants for widespread gene transfer to the adult brain. Nat Biotechnol. 2016;34:204–209. doi: 10.1038/nbt.3440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drechsel DN, Hyman AA, Cobb MH, Kirschner MW. Modulation of the dynamic instability of tubulin assembly by the microtubule-associated protein tau. Mol Biol Cell. 1992;3:1141–1154. doi: 10.1091/mbc.3.10.1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franklin KBJ, Paxinos G. The mouse brain in stereotaxic coordinates. San Diego: Academic; 2008. [Google Scholar]
- Ghetti B, Oblak AL, Boeve BF, Johnson KA, Dickerson BC, Goedert M. Invited review: frontotemporal dementia caused by microtubule-associated protein tau gene (MAPT) mutations: a chameleon for neuropathology and neuroimaging. Neuropathol Appl Neurobiol. 2015;41:24–46. doi: 10.1111/nan.12213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giacobini E, Gold G. Alzheimer disease therapy—moving from amyloid-beta to tau. Nat Rev Neurol. 2013;9:677–686. doi: 10.1038/nrneurol.2013.223. [DOI] [PubMed] [Google Scholar]
- Greenberg SG, Davies P. A preparation of Alzheimer paired helical filaments that displays distinct tau proteins by polyacrylamide gel electrophoresis. Proc Natl Acad Sci U S A. 1990;87:5827–5831. doi: 10.1073/pnas.87.15.5827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu J, Congdon EE, Sigurdsson EM. Two novel tau antibodies targeting the 396/404 region are primarily taken up by neurons and reduce Tau protein pathology. J Biol Chem. 2013;288:33081–33095. doi: 10.1074/jbc.M113.494922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iqbal K, Grundke-Iqbal I. Neurofibrillary pathology leads to synaptic loss and not the other way around in Alzheimer disease. J Alzheimers Dis. 2002;4:235–238. doi: 10.3233/jad-2002-4313. [DOI] [PubMed] [Google Scholar]
- Jeganathan S, von Bergen M, Mandelkow EM, Mandelkow E. The natively unfolded character of tau and its aggregation to Alzheimer-like paired helical filaments. Biochemistry. 2008;47:10526–10539. doi: 10.1021/bi800783d. [DOI] [PubMed] [Google Scholar]
- Kells AP, Hadaczek P, Yin D, Bringas J, Varenika V, Forsayeth J, Bankiewicz KS. Efficient gene therapy-based method for the delivery of therapeutics to primate cortex. Proc Natl Acad Sci U S A. 2009;106:2407–2411. doi: 10.1073/pnas.0810682106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein RL, Dayton RD, Leidenheimer NJ, Jansen K, Golde TE, Zweig RM. Efficient neuronal gene transfer with AAV8 leads to neurotoxic levels of tau or green fluorescent proteins. Mol Ther. 2006;13:517–527. doi: 10.1016/j.ymthe.2005.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasagna-Reeves CA, Castillo-Carranza DL, Sengupta U, Sarmiento J, Troncoso J, Jackson GR, Kayed R. Identification of oligomers at early stages of tau aggregation in Alzheimer's disease. FASEB J. 2012a;26:1946–1959. doi: 10.1096/fj.11-199851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasagna-Reeves CA, Castillo-Carranza DL, Sengupta U, Guerrero-Munoz MJ, Kiritoshi T, Neugebauer V, Jackson GR, Kayed R. Alzheimer brain-derived tau oligomers propagate pathology from endogenous tau. Sci Rep. 2012b;2:700. doi: 10.1038/srep00700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo W, Liu W, Hu X, Hanna M, Caravaca A, Paul SM. Microglial internalization and degradation of pathological tau is enhanced by an anti-tau monoclonal antibody. Sci Rep. 2015;5:11161. doi: 10.1038/srep11161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandelkow EM, Mandelkow E. Biochemistry and cell biology of tau protein in neurofibrillary degeneration. Cold Spring Harb Perspect Med. 2012;2:a006247. doi: 10.1101/cshperspect.a006247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meier S, Bell M, Lyons DN, Rodriguez-Rivera J, Ingram A, Fontaine SN, Mechas E, Chen J, Wolozin B, LeVine H, 3rd, Zhu H, Abisambra JF. Pathological tau promotes neuronal damage by impairing ribosomal function and decreasing protein synthesis. J Neurosci. 2016;36:1001–1007. doi: 10.1523/JNEUROSCI.3029-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson KR, Remmers C, Fu Y, Brooker S, Kanaan NM, Vana L, Ward S, Reyes JF, Philibert K, Glucksman MJ, Binder LI. Characterization of prefibrillar Tau oligomers in vitro and in Alzheimer disease. J Biol Chem. 2011;286:23063–23076. doi: 10.1074/jbc.M111.237974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul SM. Therapeutic antibodies for brain disorders. Sci Transl Med. 2011;3:84ps20. doi: 10.1126/scitranslmed.3002620. [DOI] [PubMed] [Google Scholar]
- Pedersen JT, Sigurdsson EM. Tau immunotherapy for Alzheimer's disease. Trends Mol Med. 2015;21:394–402. doi: 10.1016/j.molmed.2015.03.003. [DOI] [PubMed] [Google Scholar]
- Sankaranarayanan S, Barten DM, Vana L, Devidze N, Yang L, Cadelina G, Hoque N, DeCarr L, Keenan S, Lin A, Cao Y, Snyder B, Zhang B, Nitla M, Hirschfeld G, Barrezueta N, Polson C, Wes P, Rangan VS, Cacace A, et al. Passive immunization with phospho-tau antibodies reduces tau pathology and functional deficits in two distinct mouse tauopathy models. PLoS One. 2015;10:e0125614. doi: 10.1371/journal.pone.0125614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe DJ. Alzheimer's disease: genes, proteins, and therapy. Physiol Rev. 2001;81:741–766. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- Sigurdsson EM. Tau immunotherapy. Neurodegener Dis. 2016;16:34–38. doi: 10.1159/000440842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sondhi D, Johnson L, Purpura K, Monette S, Souweidane MM, Kaplitt MG, Kosofsky B, Yohay K, Ballon D, Dyke J, Kaminksy SM, Hackett NR, Crystal RG. Long-term expression and safety of administration of AAVrh.10hCLN2 to the brain of rats and nonhuman primates for the treatment of late infantile neuronal ceroid lipofuscinosis. Hum Gene Ther Methods. 2012;23:324–335. doi: 10.1089/hgtb.2012.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tai HC, Wang BY, Serrano-Pozo A, Frosch MP, Spires-Jones TL, Hyman BT. Frequent and symmetric deposition of misfolded tau oligomers within presynaptic and postsynaptic terminals in Alzheimer's disease. Acta Neuropathol Commun. 2014;2:146. doi: 10.1186/s40478-014-0146-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usenovic M, Niroomand S, Drolet RE, Yao L, Gaspar RC, Hatcher NG, Schachter J, Renger JJ, Parmentier-Batteur S. Internalized tau oligomers cause neurodegeneration by inducing accumulation of pathogenic tau in human neurons derived from induced pluripotent stem cells. J Neurosci. 2015;35:14234–14250. doi: 10.1523/JNEUROSCI.1523-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanamandra K, Kfoury N, Jiang H, Mahan TE, Ma S, Maloney SE, Wozniak DF, Diamond MI, Holtzman DM. Anti-tau antibodies that block tau aggregate seeding in vitro markedly decrease pathology and improve cognition in vivo. Neuron. 2013;80:402–414. doi: 10.1016/j.neuron.2013.07.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanamandra K, Jiang H, Mahan TE, Maloney SE, Wozniak DF, Diamond MI, Holtzman DM. Anti-tau antibody reduces insoluble tau and decreases brain atrophy. Ann Clin Transl Neurol. 2015;2:278–288. doi: 10.1002/acn3.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon SY, Bagel JH, O'Donnell PA, Vite CH, Wolfe JH. Clinical Improvement of alpha-mannosidosis cat following a single cisterna magna infusion of AAV. Mol Ther. 2016;24:26–33. doi: 10.1038/mt.2015.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao L, Gottesdiener AJ, Parmar M, Li M, Kaminsky SM, Chiuchiolo MJ, Sondhi D, Sullivan PM, Holtzman DM, Crystal RG, Paul SM. Intracerebral adeno-associated virus gene delivery of APOE2 markedly reduces brain amyloid pathology in Alzheimer's disease mouse models. Neurobiol Aging. 2016;44:159–172. doi: 10.1016/j.neurobiolaging.2016.04.020. [DOI] [PubMed] [Google Scholar]
- Zincarelli C, Soltys S, Rengo G, Rabinowitz JE. Analysis of AAV serotypes 1–9 mediated gene expression and tropism in mice after systemic injection. Mol Ther. 2008;16:1073–1080. doi: 10.1038/mt.2008.76. [DOI] [PubMed] [Google Scholar]