

Abstract
Shank2 is a multidomain scaffolding protein implicated in the structural and functional coordination of multiprotein complexes at excitatory postsynaptic sites as well as in psychiatric disorders, including autism spectrum disorders. While Shank2 is strongly expressed in the cerebellum, whether Shank2 regulates cerebellar excitatory synapses, or contributes to the behavioral abnormalities observed in Shank2−/− mice, remains unexplored. Here we show that Shank2−/− mice show reduced excitatory synapse density in cerebellar Purkinje cells in association with reduced levels of excitatory postsynaptic proteins, including GluD2 and PSD-93, and impaired motor coordination in the Erasmus test. Shank2 deletion restricted to Purkinje cells (Pcp2-Cre;Shank2fl/fl mice) leads to similar reductions in excitatory synapse density, synaptic protein levels, and motor coordination. Pcp2-Cre;Shank2fl/fl mice do not recapitulate autistic-like behaviors observed in Shank2−/− mice, such as social interaction deficits, altered ultrasonic vocalizations, repetitive behaviors, and hyperactivity. However, Pcp2-Cre;Shank2fl/fl mice display enhanced repetitive behavior in the hole-board test and anxiety-like behavior in the light-dark test, which are not observed in Shank2−/− mice. These results implicate Shank2 in the regulation of cerebellar excitatory synapse density, motor coordination, and specific repetitive and anxiety-like behaviors.
SIGNIFICANCE STATEMENT The postsynaptic side of excitatory synapses contains multiprotein complexes, termed the postsynaptic density, which contains receptors, scaffolding/adaptor proteins, and signaling molecules. Shank2 is an excitatory postsynaptic scaffolding protein implicated in the formation and functional coordination of the postsynaptic density and has been linked to autism spectrum disorders. Using Shank2-null mice and Shank2-conditional knock-out mice with a gene deletion restricted to cerebellar Purkinje cells, we explored functions of Shank2 in the cerebellum. We found that Shank2 regulates excitatory synapse density, motor coordination, and specific repetitive and anxiety-like behaviors, but is not associated with autistic-like social deficits or repetitive behaviors.
Keywords: autism, cerebellum, excitatory synapse, GluD2, Purkinje cell, Shank2
Introduction
The Shank/ProSAP family of excitatory postsynaptic scaffolding proteins contains three known members, Shank1, Shank2, and Shank3, which are thought to play important roles in the structural assembly of the multiprotein complexes in the postsynaptic density (PSD) as well as in the functional coordination of excitatory synaptic signaling (Sheng and Sala, 2001; Boeckers et al., 2002; Sheng and Hoogenraad, 2007; Feng and Zhang, 2009; Grabrucker et al., 2011; Sheng and Kim, 2011; Sala et al., 2015).
Shank2 (also known as ProSAP1) contains several domains for protein–protein interactions (Du et al., 1998; Boeckers et al., 1999b; Lim et al., 1999; Naisbitt et al., 1999; Sheng and Kim, 2000), which mediate the interactions of Shank2 with diverse PSD proteins, including GKAP/SAPAP, Homer, and GluD2 (Boeckers et al., 1999a, b; Naisbitt et al., 1999; Tu et al., 1999; Uemura et al., 2004).
Human and mouse genetic studies have also linked Shank2 to several psychiatric disorders (Grabrucker et al., 2011, 2014; Jiang and Ehlers, 2013; Guilmatre et al., 2014; Yoo et al., 2014; Sala et al., 2015; Schmeisser, 2015). In humans, diverse genetic variations of the human SHANK2 gene have been identified in individuals with autism spectrum disorders (ASDs), intellectual disability, developmental delay, and schizophrenia (Pinto et al., 2010; Wischmeijer et al., 2011; Berkel et al., 2012; Leblond et al., 2012, 2014; Prasad et al., 2012; Rauch et al., 2012; Sanders et al., 2012; Chilian et al., 2013; Schluth-Bolard et al., 2013; Costas, 2015; Peykov et al., 2015a, b; Homann et al., 2016).
In mice, viral-mediated delivery of Shank2-mutant proteins results in reduced excitatory synaptic transmission and impaired cognition (Berkel et al., 2012). In addition, transgenic mice carrying deletions of Shank2 gene exons display autistic-like behaviors in association with dysregulated NMDA receptor (NMDAR) function (Schmeisser et al., 2012; Won et al., 2012), behaviors that are rescued by pharmacological normalization of NMDAR function (Won et al., 2012; Lee et al., 2015).
Shank2 is expressed in diverse regions of rodent brain, including the cerebellum (Du et al., 1998; Boeckers et al., 1999a, b; Lim et al., 1999). In the cerebellum, a brain region that has increasingly demonstrated associations with ASDs and other psychiatric disorders (Baudouin et al., 2012; Tsai et al., 2012; Wang et al., 2014), Shank2 mRNAs are more abundant in the Purkinje cell (PC) layer and, to a lesser extent, in the molecular layer but are relatively rare in the granule cell layer, a pattern similar to that of Shank1 mRNAs but unlike that of Shank3 mRNAs, which are more abundant in the granule cell layer (Boeckers et al., 1999b; Böckers et al., 2004; Peça et al., 2011). However, whether cerebellar Shank2 protein plays a significant role in the regulation of excitatory synapses and animal behaviors has not been explored.
Here, using Shank2-null (Shank2−/−) mice as well as Shank2 conditional knock-out (KO) mice in which Shank2 gene deletion is restricted to PCs, we found that Shank2 in cerebellar PCs is important for the regulation of excitatory synapse density, motor coordination, and specific repetitive and anxiety-like behaviors.
Materials and Methods
Shank2−/− mice.
Shank2 conventional KO mice (Shank2−/− or Shank2 KO mice) under the genetic background of C57BL/6N have been described (Won et al., 2012). All mice were housed and bred at the mouse facility of Korea Advanced Institute of Science and Technology (KAIST) and maintained according to the Animal Research Requirements of KAIST. All animals were fed ad libitum and housed under 12 h light/dark cycle (light phase during 1:00 A.M. to 1:00 P.M.). We crossed male and female heterozygous Shank2+/− mice to produce littermate pairs of wild-type (WT) and KO mice. Mice were weaned at the age of postnatal day 21, and mixed-genotype littermate mice with same gender were housed together until experiments. All procedures were approved by the Committee of Animal Research at KAIST.
Generation of Pcp2-Cre;Shank2fl/fl mice.
Floxed Shank2 mice carrying exons 6 and 7 of the Shank2 gene flanked with loxP sites under the genetic background of C57BL/6J were designed and generated by Biocytogen (see Fig. 5A). To remove the cassette, these mice were crossed with Protamine-Flp mice, which yielded heterozygous floxed Shank2 mice (Shank2fl/+). Shank2fl/+ mice were then crossed with Pcp2-Cre transgenic mice under the genetic background of C57BL/6J (Zhang et al., 2004) to yield double heterozygous progenies (Pcp2cre;Shank2fl/+). We crossed female Pcp2cre;Shank2fl/+ mice and male Shank2fl/fl animals to generate Pcp2-Cre;Shank2fl/fl mice (used as conditional KO [cKO]) and Shank2fl/fl mice (used as control mice). Genotypes of Pcp2-Cre;Shank2fl/fl mice were determined by PCR using the following primers: for floxed (367 bp) or WT allele (253 bp): 5′-CGC ACT GTG GGC TCA TCA GAT G-3′ and 5′-CAG ACG CAT CTT CCA GGG AAG C-3′; for Pcp2-cre allele (272 bp): 5′-GTG TTG CCG CGC CAT CTG C-3′ and 5′-CAC CAT TGC CCC TGT TTC ACT ATC-3′. Only male mice were used for behavioral experiments, except for pup ultrasonic vocalization (USV) test. Both male and female were used for electrophysiological and biochemical experiments. Animal housing conditions were identical to Shank2 conventional KO mice.
Figure 5.

Generation and characterization of PC-specific Shank2−/− mice. A, B, cKO strategy for the production of Pcp2-Cre;Shank2fl/fl mice. We used Shank2fl/fl mice without Cre expression as controls (indicated as WT) in our experiments. C, Reduced levels of Shank2 protein in the cerebellum of Pcp2-Cre;Shank2fl/fl mice (6–8 weeks), as shown by immunoblotting of total lysates of the cerebrum and cerebellum in Pcp2-Cre;Shank2fl/fl mice. n = 3 female mice for WT and Pcp2-Cre;Shank2fl/fl (cKO). ***p < 0.001 (Student's t test). D, Reduced levels of Shank2 proteins in the Pcp2-Cre;Shank2fl/fl cerebellum (P56), as shown by immunofluorescence staining. Images shown are examples from experiments performed on three WT and KO pairs. The residual Shank2 signals in the cKO panel may represent those from non-PCs.
Immunohistochemistry.
Mice were anesthetized and perfused transcardially with 4% PFA, followed by postfixation for 3 h at 4°C, and vibratome sectioning (40 μm; Leica VT 1200S). Sections were blocked in 5% normal goat serum in PBS, incubated in primary antibodies overnight at 4°C, washed several times, and then incubated in secondary antibodies for 3 h at room temperature. Single optical sections of confocal images were acquired by an A1R laser scanning confocal microscope (Nikon).
Electrophysiology.
We used PCs in cerebellar lobules III–V for whole-cell recordings. Chemicals used were obtained from Sigma or Wako Pure Chemical Industries, unless otherwise specified. Sagittal slices (200 μm) of cerebellar vermis were bathed in extracellular solution (ACSF) containing the following (in mm): 125 NaCl, 2.5 KCl, 1.3 MgCl2, 2 CaCl2, 1.25 NaH2PO4, 26 NaHCO3, 20 glucose, and 0.01 bicuculline methocloride (Tocris Bioscience). Patch pipettes (resistance 5–6 mΩ) were filled with the following (in mm): 130 potassium gluconate, 2 NaCl, 4 MgCl2, 4 Na2-ATP, 0.4 Na-GTP, 20 HEPES, pH 7.2, and 0.25 EGTA. EPSCs were evoked in PCs (holding potential of −70 mV) by activating parallel fibers (PFs) with a glass-stimulating electrode on the surface of the molecular layer (PF-EPSCs). PF-EPSCs were acquired and analyzed using pClamp software (Molecular Devices). To evoke LTD by electrical stimulation (PF and ΔV), PF stimuli were paired with PC depolarization (0 mV, 200 ms), 300 times at 1 Hz. Data were accepted if the series resistance changed by <20%, the input resistance was >80 mΩ, and the holding current changed by <20%. Miniature EPSCs (mEPSCs) were recorded as described previously (Yamamoto et al., 2012). Patch pipettes were filled with the solution containing the following (in mm): 60 CsCl, 10 Cs d-gluconate, 20 tetraethylammonium-Cl, 20 BAPTA, 2 CaCl2, 4 MgCl2, 4 ATP, 0.4 GTP, and 30 HEPES, pH 7.2, adjusted with CsOH. The extracellular solution included tetrodotoxin (1 μm; Tocris Bioscience). mEPSCs were recorded for 2 min and analyzed offline using Clampfit 10.5 (Molecular Devices).
Electron microscopy.
WT and Shank2−/− mice were deeply anesthetized with sodium pentobarbital (80 mg/kg, i.p.) and were intracardially perfused with 10 ml of heparinized normal saline, followed by 50 ml of a freshly prepared fixative of 2.5% glutaraldehyde and 1% PFA in 0.1 m phosphate buffer (PB, pH 7.4). Cerebellum was removed from the whole brain and sectioned with vibratome at 100 μm thickness. The sections were postfixed in the same fixative for 2 h, osmicated with 1% osmium tetroxide in 0.1 m phosphate buffer for 1 h, dehydrated in graded alcohols, flat embedded in Durcupan ACM (Fluka), and cured for 48 h at 60°C. Small pieces containing cerebellar molecular layer were cut out of the wafers and glued onto the plastic block by cyanoacrylate. Ultrathin sections were cut and mounted on Formvar-coated single slot grids, stained with uranyl acetate and lead citrate, and examined with an electron microscope (Hitachi H-7500; Hitachi) at 80 kV accelerating voltage. Twenty-four electron micrographs representing 655.49 μm2 neuropil regions in each mouse were taken at a 30,000× and used for quantification. The density, length, and thickness of the PSD, as well as the number of mismatched excitatory synapses and free dendritic spines were quantified from five WT and Shank2−/− mouse pairs. The measurements were all performed by an experimenter blind to the genotype. Digital images were captured with GATAN Digital Micrograph software driving a CCD camera (SC1000 Orius; Gatan) and saved as TIFF files. Brightness and contrast of the images were adjusted in Adobe Photoshop 7.0 (Adobe Systems).
Antibodies.
The following antibodies have been described: Homer1 (#1133) (Won et al., 2012), PSD-93 (#1634), GluA1 (#1193), and GluA2 (#1195) (Kim et al., 2009). Antibodies against Shank1 were generated using a synthetic peptide of Shank1 (RSPSRGRHPEDAKRQPRGRPSSSGT; amino acids 498–522 of mouse Shank1) as an immunogen (#2100). Commercial antibodies were purchased from the following sources: Shank2, neuroligin 1, neuroligin 2, neuroligin 3, VGluT1, VGAT, gephyrin (Synaptic Systems); Gluδ2/GluD2, GluN1 (BD Transduction Laboratories); GluA2/3, Grin2A, mGluR1, mGluR5, VGluT2 (Millipore); Grin2C, α-tubulin (Sigma); PSD-95 (NeuroMab); IP3R, CaMKIIα/β (Cell Signaling Technology); Shank3 (Santa Cruz Biotechnology); and IRSp53, GAD65 (Abcam). For immunohistochemistry, following antibodies were used: Shank2 (Synaptic Systems); parvalbumin, calbindin, VGAT (Sigma); and VGluT1 (Millipore).
Cerebellar whole lysates and crude synaptosomes.
Brains from Shank2−/− mice and their WT littermates, and those from Pcp2-Cre;Shank2fl/fl mice and their WT littermates, were extracted on ice at postnatal day 21. Cerebella were dissected from the cerebrum and briefly homogenized with ice-cold homogenization buffer (0.32 m sucrose, 10 mm HEPES, pH 7.4, 2 mm EDTA, 2 mm EGTA, protease inhibitors, phosphatase inhibitors). Whole lysates were prepared by boiling with β-mercaptoethanol directly after homogenization. For crude synaptosomes, each homogenate was centrifuged at 1200 × g for 10 min, and the supernatants were further centrifuged at 12,000 × g for 10 min. The pellet from each brain was resuspended in brain homogenization buffer and boiled with β-mercaptoethanol.
The order of behavioral experiments.
Experienced researchers performed mouse handling for 5 min per day for 3 d, for all mice before behavioral experiments. For conventional Shank2−/− mice, three independent cohorts were used. Cohort 1 underwent light-dark test and hole-board test, sequentially. Cohort 2 was used in the Erasmus ladder test. Cohort 3 was used in the visual cliff test. For Pcp2-Cre;Shank2fl/fl mice, eight independent cohorts were used. Cohort 1 underwent the following experiments sequentially: long-term measurement of movements in the Laboras test (isolated for 4 d during measurement), 100 lux open field test, repetitive behaviors, low-speed rotarod test, and small-configuration 3-chamber test (4 d of isolation before experiment). Cohort 2 underwent the following experiments sequentially: 0 lux open field test, high-speed rotarod test, hole-board test, and large-configuration 3-chamber test (4 d of isolation before the experiment). Cohort 3 sequentially went through direct social interaction test (4 d of isolation before the experiment) and adult courtship USV (4 d of isolation before the experiment). Cohort 4 was used in the olfactory test and the visual cliff test. Other four cohorts were independently used in the following tests: pup USV, hole-board test, small-configuration 3-chamber test, and large-configuration 3-chamber test. Animals were group housed unless otherwise indicated, and each cohort had at least 2 d of break between experiments.
Erasmus ladder test.
Erasmus ladder (Noldus) was previously described (Van Der Giessen et al., 2008). The test consisted of 6 daily sessions: two unperturbed sessions, one unperturbed session with a fixed obstacle, and three perturbed sessions. In first three sessions, each mouse was trained to cross the ladder from one side to the other side, which is counted as one trial. One session was over when a mouse successfully completed 50 trials. The last three perturbed sessions were designed to measure learning ability of mice, using 2 kHz tone sound as a conditioned stimulus (CS) and sudden rising of an obstacle rung as an unconditioned stimulus (US). Fifty trials of each perturbed session were composed of two-thirds of CS-US paired trials and one-third of CS-only trials, which was given in pseudorandom order. There were three types of rungs in Erasmus ladder: upper rungs, lower rungs, and obstacle rungs. Steps on upper rungs were only considered as normal steps, whereas steps on lower rungs were counted as missteps. A step skipping three upper rungs at a time was counted as a jump.
Hole-board test.
Each mouse was allowed to explore hole-board apparatus (40 × 40 × 35 cm) for 20 min. The hole-board apparatus was made of white acrylic plate with 16 holes (3 cm in diameter, arranged in 4 × 4). Video recordings were made from the bottom-side view. The frequency of nose-poking into a hole was counted manually in a blind manner.
Light-dark test.
Animals were placed in the light chamber with their heads toward the opposite wall from the dark chamber, and allowed to explore the light-dark apparatus (20 × 30 × 20 cm, 800 lux for light chamber, 20 × 13 × 20 cm, 5 lux for dark chamber), which has a 5-cm-wide entrance between the two chambers. The latency to enter the dark chamber and time spent in light and dark chambers were analyzed using Ethovision 10.0 software (Noldus).
Visual cliff test.
We used a box (33 × 33 × 22 cm) with gray acrylic walls and transparent acrylic bottom. The box was placed between two tables, elevated to a height of 1 m above the floor. One half of the box was located on a checkerboard-patterned paper (referred as “safe” area), whereas the other half of the box was kept transparent (referred as “cliff” area). Mice were gently placed in the corner of the safe area and were recorded of movements for 10 min. Time spent in the safe or cliff areas was evaluated using Ethovision 10.0 software (Noldus).
Olfactory habituation/dishabituation test.
The test procedure was previously described (Won et al., 2012). Briefly, we exposed mice to Kim-wipes soaked with five different odors: water, banana, coffee, stranger mice's cage 1, and stranger mice's cage 2. The pieces of Kim-wipes were contained in 60-φ Petri dishes with nine small holes in the cover. A single mouse was placed in a new home cage and introduced to a mock Petri dish for 30 min before the experiment. Each odor was sequentially placed into the cage for 6 min. This six-odor set was presented to the mouse three times with an intersession interval of 1 min. A skilled experimenter measured the sniffing time toward Petri dishes in a blind manner.
Three-chamber social interaction test.
The 3-chamber social interaction test measures sniffing time and time spent in chamber of each test mouse. It consists of three phases and the duration of each phase is 10 min or 5 min. A test mouse, either WT or Shank2−/− mice, was isolated for 4 d before the experiment, and the experiment was conducted at the dark phase, after 30 min habituation in a dark experiment booth. The white acrylic 3-chambered apparatus was placed of two small containers for an object or stranger mouse in the upper corner of the right and left chambers. In the first phase, a test mouse was placed in the center area of the 3-chambered apparatus and allowed to freely explore the whole apparatus. In the second phase, the test mouse was gently guided to the center chamber, and the gates between center and side chambers were transiently blocked using an opaque acryl plate, while a stranger mouse (Stranger 1) and an inanimate blue cylindrical object (Object) were placed in the two corner containers. Stranger was randomly positioned in the left or right chamber. After the two gate ways were reopened, the test mouse was allowed to explore the Stranger1 or the Object freely. In the third phase, the test mouse was confined in the center chamber again, while the Object was replaced with a new stranger (Stranger 2). Mouse was allowed to freely explore and interact with Strangers 1 and 2. Stranger mice were congenic 129S1/SvlmJ strain, which were handled for 5 min during 3 d before experiments and habituated in containers for 10 min 1 d before the experiment. We used two different protocols as described below: (1) Protocol 1: this protocol consisted of three 10 min phases. The apparatus was smaller (40 cm × 20 cm × 25 cm), with plastic cylindrical containers. In the second phase, Stranger 1 and Object were placed in each container; and (2) Protocol 2: this protocol consisted of three 5 min phases. The apparatus was larger (60 cm × 40 cm × 20 cm), with stainless steel fan-shaped containers. In the second phase, Stranger 1 was placed in a container, and the other container was left empty. For quantification, time spent sniffing was measured when the nose part of test mouse was positioned within 2 cm from each container. Time spent in chamber was measured when the central body part of test mouse was positioned in each chamber. Ethovision 10.0 software (Noldus) was used to automatically measure the exploration time. The preference index was calculated by dividing the subtraction of times spent exploring targets (Stranger 1 vs Object/Empty, or Stranger 2 vs Stranger 1) by total exploration time.
Direct social interaction test.
The subject mice isolated in their home cage at least for 4 d were used in this assay. At habituation day, the mice were individually habituated in white acryl box (30 × 30 × 40 cm) for 10 min. At the recording day, two same genotype mice that encounter each other for the first time were put into the habituated box and allowed to interact each other for 10 min while all behavioral information was recorded. Interaction duration was measured when the subject mice physically interact each other.
Adult USVs during courtship behavior.
Male adult subject mice were socially isolated in their home cages at least for 4 d to let them recognize the cage as their own territory. Female adult stranger mice group-caged were prepared for this experiment. A subject male mouse was habituated in the experimental booth for 5 min while recording basal vocalization. Next, a stranger female mouse was introduced into the subject cage and allowed to interact each other freely while recording courtship USVs of the subject mouse for 5 min. Avisoft SASLab Pro was used to analyze USVs.
Pup USV.
Ultrasound Microphone (Avisoft) was used to record pup USVs. The microphone was placed ∼20 cm above the testing chamber. Pups at postnatal day 5–13 were recorded daily. Each pup was separated from its mother and placed into a glass container in a Styrofoam box, and USVs were recorded for 180 s. For the analysis of USV calls, spectrograms were generated with 75% overlap of temporal resolution and 25 kHz cutoff frequency using Avisoft SASLab Pro software.
Repetitive behaviors.
Each mouse was placed in a new home cage with beddings for 20 min. The first 10 min was considered as a habituation period. A skilled experimenter scored the duration of certain stereotypic behaviors during the last 10 min in a blind manner. Grooming was defined as a mouse stroking or scratching its face or body area, or licking its body. Jumping was defined as both two hind limbs simultaneously pushing against the ground. Digging was defined as a mouse uses its head or fore limbs to dig out beddings.
Marble burying test.
Mice were placed in the corner of a cage, containing 5-cm-thick beddings and 21 marbles (metal, 2 cm in diameter, ordered 3 by 7). Mice were allowed to freely explore the cage for 30 min. Afterward, experimenter counted the number of marbles buried in a blind manner. A marble was considered to be buried when the two-thirds of its height was hidden in beddings.
Rotarod test.
Rotarod test was performed using a five lane rotarod treadmill (Ugo Basile). Mice were trained for 5 consecutive days, one trial per day. One trial lasted for 300 s while the rotarod was accelerated from 4 to 40 rpm, or from 8 to 80 rpm. Mice were carefully placed on each lane of the rotarod and acclimated for 30 s. Experimenter checked the latency to fall of each mouse. Latency was regarded as 300 s when a mouse withstood full 300 s.
Open field test.
White acrylic boxes were used as the open field arenas (40 × 40 × 40 cm). Each mouse was placed into the center of the box and allowed to freely explore for 60 min. The illumination was either 100 lux or 0 lux at the center of the open field. Locomotor activity of each mouse was recorded from a camera above and analyzed using EthoVision 10.0 software (Noldus). The center zone was defined as the center 2 × 2 squares when the arena was divided into 4 × 4 squares.
Elevated plus maze test.
Mice were singly placed on the center of the elevated plus maze and allowed to explore for 8 min. The apparatus was made of gray acrylic plates and elevated 75 cm from the floor, with two open arms (30 × 5 × 0.5 cm, 550 lux) and two closed arms (30 × 5 × 30 cm, 30 lux). Time spent in open or closed arms and the frequency of entries to each arm were automatically measured using Ethovision 10.0 software (Noldus).
Long-term measurement of movements.
Long-term (4 d) locomotor, climbing, rearing, grooming, eating, and drinking activities were recorded and automatically analyzed using the Laboratory Animal Behavior Observation Registration and Analysis System (LABORAS; Metris). Mice were individually caged in a specialized LABORAS recording environment for 96 consecutive hours and fed ad libitum.
Statistical analysis.
All the behavioral and electrophysiological experiments were performed by researchers who are blind to the experimental conditions. Statistical analyses were performed using GraphPad Prism 6 software. Statistical test results are described in each figure legends.
Results
Shank2 proteins at cerebellar excitatory synapses
To study cerebellar functions of Shank2, we first characterized the distribution patterns of Shank2 protein in the cerebellum by immunofluorescence staining. Shank2 signals were strong in the molecular layer of the cerebellar cortex, where PC dendrites are localized, but were nearly absent in the granule cell and PC layers (Fig. 1A). Shank2 signals in Purkinje and molecular layers were absent in Shank2−/− mice. In the molecular layer, Shank2 fluorescence signals were located in protrusive spine-like structures on PC dendrites (Fig. 1B). Notably, discrete Shank2 clusters were also detected on dendrites of parvalbumin-positive interneurons (Fig. 1C). These punctate Shank2 clusters colocalized extensively with VGluT1 (an excitatory synaptic marker) but minimally with VGAT (an inhibitory synaptic marker) (Fig. 1D), indicative of PF excitatory synaptic localization of Shank2 in cerebellar PCs.
Figure 1.

Cerebellar Shank2 protein is localized at excitatory synapses. A, Strong signals of Shank2 protein in the molecular, but not granule, cell layer of the cerebellum, as revealed by immunofluorescence staining. Calbindin was used to label PCs. Stars represent PC bodies. ML, Molecular layer; GC, granule cell layer. B, Shank2 clusters on the dendrites of PCs, as revealed by immunofluorescence staining. C, Shank2 clusters on dendrites of parvalbumin-positive GABAergic interneurons. Arrowheads indicate sites of colocalization. D, Shank2 largely colocalizes with excitatory, but not inhibitory, synaptic markers. VGluT1 and VGAT were used as excitatory and inhibitory synaptic markers, respectively. Arrowheads indicate sites of colocalization.
Shank2−/− PCs show reduced mEPSC frequency, PSD density, and trans-synaptic adhesion
We next tested whether Shank2 exerts any influence on excitatory synapse density and/or function. PCs from Shank2−/− mice at postnatal day 19–23 (P19-P23) showed a significantly reduced (∼40%) frequency of mEPSCs, which are largely derived from PF-PC synapses, compared with that of WT mice, whereas the amplitude of mEPSCs was not affected (Fig. 2A–C).
Figure 2.

Reduced mEPSC frequency, PSD density, and synaptic contact and alignment in the Shank2−/− cerebellum. A–C, Reduced frequency but normal amplitude of mEPSCs in Shank2−/− PCs (P19–P23). n = 23 cells from 8 mice (4 male and 4 female) for WT and 22 cells from 7 mice (3 male, 4 female) for KO. *p < 0.05 (Student's t test). D–G, Reduced density but normal length and thickness of PSDs apposed to presynaptic axon terminals (n), and increased frequency of misaligned PSDs (m) and free spines (f) in the molecular layer of the Shank2−/− cerebellum, as shown by EM analysis. White arrowheads indicate boundaries of active zones. Black arrowheads indicate boundaries of the PSD. Scale bar, 500 nm. n = 5 (3 male, 2 female) mice for WT and KO. *p < 0.05 (Student's t test). ***p < 0.001 (Student's t test).
An electron microscopic (EM) analysis of the molecular layer of Shank2−/− mice revealed a marked reduction (∼40%) in the density of excitatory synapses (mainly representing PF-PC synapses), as shown by the reduced number of PSDs apposed to presynaptic axon terminals (Fig. 2D,E). The length and thickness of PSDs were unaltered in Shank2−/− mice, consistent with the normal amplitude of mEPSCs.
Notably, brain slices from Shank2−/− mice displayed significant increases in the number of mismatched excitatory synapses, in which the PSD was misaligned with the presynaptic nerve terminal, as well as free spines that were not in close contact with the presynaptic nerve terminal (Fig. 2F,G). Together, these results suggest that Shank2 deletion reduces cerebellar excitatory synapse density, and this change involves decreases in the density of the PSD as well as altered contact and alignment of the PSD with presynaptic axon terminals.
Reduced levels of excitatory synaptic proteins in the Shank2−/− cerebellum
To explore biochemical mechanisms that may underlie the reduced excitatory synapse density in the Shank2−/− cerebellum, we measured total and crude synaptosomal levels of excitatory synaptic proteins from WT and Shank2−/− mice at P19-P22. We found significant reductions in the levels of excitatory synaptic membrane proteins, including GluD2, the NMDA receptor subunit GluN2C, and AMPA receptor subunits (GluA1 and GluA2) (Fig. 3A). In addition to these membrane proteins, levels of cytoplasmic proteins such as PSD-95, PSD-93, and Homer were reduced. Many of these proteins showed reduced total levels, whereas some (GluA1, GluN2C, and PSD-95) did not (Fig. 3B). Levels of inhibitory synaptic proteins (gephyrin, VGAT, and GAD65) were unchanged, except for a small reduction in gephyrin in crude synaptosomes of Shank2−/− mice. These results suggest that Shank2−/− deletion leads to reductions in the synaptic levels of many excitatory synaptic proteins.
Figure 3.

Reduced synaptic and total levels of excitatory synaptic proteins in the Shank2−/− cerebellum. A, B, Analysis of crude synaptosomal (A) and total (B) levels of excitatory synaptic proteins in the Shank2−/− cerebellum (P19–P22) by immunoblotting. In all bar graphs, amounts of proteins in KO samples were normalized to those in WT samples. n = 6 (3 male, 3 female) mice for WT and KO. *p < 0.05 (Student's t test). **p < 0.01 (Student's t test). ***p < 0.001 (Student's t test).
Suppressed motor coordination in Shank2−/− mice
We next tested whether Shank2 deletion affects cerebellum-dependent motor coordination, using the Erasmus ladder test (Van Der Giessen et al., 2008). Shank2−/− mice displayed a significantly increased number of missteps and fewer normal steps compared with WT mice (Fig. 4A,B). In addition, Shank2−/− mice showed frequent back steps and jumps (Fig. 4C,D). These results suggest that Shank2 is important for cerebellum-dependent motor coordination in mice.
Figure 4.

Suppressed motor coordination in Shank2−/− mice. A–D, Suppressed motor coordination of Shank2−/− mice (5–6 months) in the Erasmus ladder test. n = 6 male mice for WT and 7 male mice for KO. *p < 0.05 (repeated-measures ANOVA). **p < 0.01 (repeated-measures ANOVA). ***p < 0.001 (repeated-measures ANOVA).
Generation and characterization of PC-specific Shank2−/− mice
Because Shank2 protein is highly expressed in cerebellar PCs (Fig. 1), we attempted PC-specific deletion of Shank2 by crossing Pcp2-Cre mice (L7-Cre) with mice in which exons 6 and 7 were flanked by LoxP sites (Shank2fl/fl) (Fig. 5A,B), resulting in deletion of exons 6 and 7 in PCs (Pcp2-Cre;Shank2fl/fl). This deletion strategy was chosen to recapitulate the same exon deletion used for the conventional Shank2−/− mice (Won et al., 2012).
Pcp2-Cre;Shank2fl/fl mice showed significantly reduced (∼90%) levels of Shank2 protein in the cerebellum compared with Shank2fl/fl control mice (defined hereafter as WT for Pcp2-Cre;Shank2fl/fl mice), as shown by immunoblotting and immunofluorescence staining (Fig. 5C,D), suggesting that PCs are the main source of cerebellar Shank2 expression. The weak Shank2 signals detected in the molecular layer of the Pcp2-Cre;Shank2fl/fl cerebellum (Fig. 5D) may represent Shank2 protein expressed in other cell types.
Reduced mEPSC frequency and excitatory synaptic protein levels in the cerebellum of Pcp2-Cre;Shank2fl/fl mice
To determine the impact of specific Shank2 deletion in PCs on synaptic function and protein composition, we tested whether Pcp2-Cre;Shank2fl/fl mice display altered excitatory synapse density and/or synaptic protein levels. Pcp2-Cre;Shank2fl/fl PCs displayed mEPSCs with reduced frequency but normal amplitude (Fig. 6A–C), similar to the results obtained with conventional Shank2−/− mice (Fig. 2A–C). This result suggests that postsynaptic deletion of Shank2 in PCs is sufficient to recapitulate the reduced mEPSC frequency observed in conventional Shank2−/− mice.
Figure 6.

Reduced mEPSC frequency and synaptic levels of GluD2 and PSD-93 in the cerebellum of Pcp2-Cre;Shank2fl/fl mice. A–C, Reduced frequency but normal amplitude of mEPSCs in Pcp2-Cre;Shank2fl/fl (cKO) PCs (P19–P23). n = 16 cells from 7 (3 male, 4 female) mice for WT and 16 cells from 6 (3 male, 3 female) mice for cKO. *p < 0.05 (Student's t test). D, E, Reduced synaptic levels of GluD2 and PSD-93 in the Pcp2-Cre;Shank2fl/fl cerebellum (P21–P22), as determined by immunoblotting of crude synaptosomes (D). The total level of GluD2, but not PSD-93, was reduced, as determined by immunoblotting of total lysates (E). n = 6 (2 male, 4 female) for WT and cKO. *p < 0.05 (Student's t test). **p < 0.01 (Student's t test). ***p < 0.001 (Student's t test). F, Side-by-side comparison of synaptic protein levels in crude synaptosomes and total lysates from Shank2−/− (red) and Pcp2-Cre;Shank2fl/fl (blue) mice. The data presented in Figures 3A, B and 6D, E were reused here for comparison.
An examination of excitatory synaptic protein levels using Pcp2-Cre;Shank2fl/fl cerebellar crude synaptosomes showed significant decreases in two specific synaptic proteins, GluD2 and PSD-93 (Fig. 6D). An analysis of total cerebellar proteins showed that only GluD2, and not PSD-93, was reduced (Fig. 6E), indicating that the reduced synaptic levels of GluD2 were caused by a reduction in its total protein levels. These results contrast with the reductions in a large number of proteins in conventional Shank2−/− mice (Fig. 6F).
Normal LTD at PF-PC synapses in Shank2−/− and Pcp2-Cre;Shank2fl/fl mice
Because Shank2 has been implicated in the regulation of synaptic plasticity at hippocampal synapses (Schmeisser et al., 2012; Won et al., 2012), we examined LTD at PF-PC synapses in the cerebellum of Pcp2-Cre;Shank2fl/fl mice. We found normal LTD at Pcp2-Cre;Shank2fl/fl PF-PC synapses (Fig. 7A). Similarly, conventional Shank2−/− mice showed normal LTD at PF-PC synapses (Fig. 7B), although there was a decreasing tendency. These results suggest that Shank2 deletion in PCs does not affect LTD.
Figure 7.
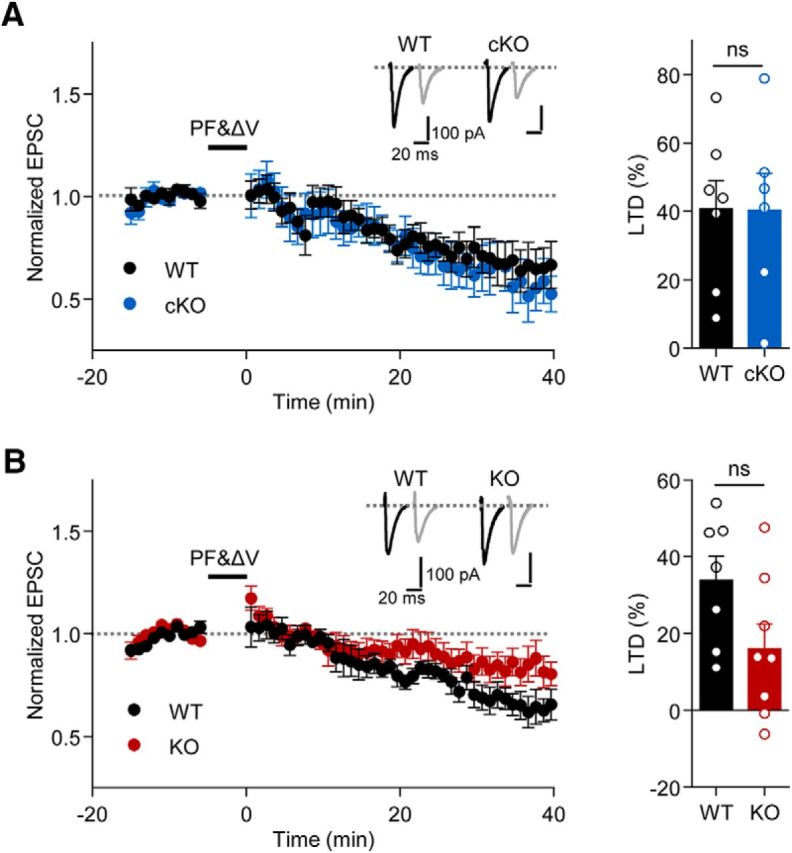
Normal LTD at PF-PC synapses in Shank2−/− and Pcp2-Cre;Shank2fl/fl mice. A, B, Normal LTD at cerebellar PF-PC synapses in Pcp2-Cre;Shank2fl/fl (A) and Shank2−/− (B) mice (P19–P23). Bar graphs represent the average of normalized EPSCs during the last 10 min of the recordings. n = 7 cells from 7 (4 male, 3 female) mice for WT and 6 cells from 4 (1 male, 3 female) mice for cKO; n = 7 cells from 5 (3 male, 2 female) mice for WT and 8 cells from 6 (1 male, 5 female) mice for KO. Student's t test.
Pcp2-Cre;Shank2fl/fl mice display impaired motor coordination but no autistic-like behaviors
We next determined whether Pcp2-Cre;Shank2fl/fl mice recapitulate the behavioral abnormalities observed in Shank2−/− mice. Pcp2-Cre;Shank2fl/fl mice showed impaired motor coordination in the Erasmus test (Fig. 8), similar to Shank2−/− mice (Fig. 4), suggesting that the impaired motor coordination observed in Shank2−/− mice is largely attributable to Shank2 deletion in PCs.
Figure 8.

Pcp2-Cre;Shank2fl/fl mice display impaired motor coordination. A–D, Suppressed motor coordination in Pcp2-Cre;Shank2fl/fl mice (5–6 months) in the Erasmus ladder test. n = 13 male mice for WT and 12 male mice for cKO. *p < 0.05 (repeated-measures ANOVA). **p < 0.01 (repeated-measures ANOVA). ***p < 0.001 (repeated-measures ANOVA).
Pcp2-Cre;Shank2fl/fl mice were then subjected to a battery of behavioral tests that have revealed autistic-like behaviors in Shank2−/− mice (Won et al., 2012). In contrast to Shank2−/− mice, Pcp2-Cre;Shank2fl/fl mice did not display any detectable abnormalities in these behaviors, including social interaction, social novelty recognition, USVs, repetitive behaviors (self-grooming, jumping, digging, and marble burying), and open-field behavior (Figs. 9–11). In addition, Pcp2-Cre;Shank2fl/fl mice displayed normal visual and olfactory functions (Fig. 9A,B).
Figure 9.

Normal social interaction in Pcp2-Cre;Shank2fl/fl mice. A, Normal visual function in Pcp2-Cre;Shank2fl/fl mice (2–3 months) in the visual cliff test. n = 10 male mice for WT and 8 male mice for cKO. ***p < 0.001 (Student's t test). B, Normal olfactory function in Pcp2-Cre;Shank2fl/fl mice (2–3 months). n = 10 male mice for WT and 8 male mice for cKO. Repeated-measures ANOVA. C–F, Normal social interaction (C, D) and social novelty recognition (E, F) in Pcp2-Cre;Shank2fl/fl mice (2–5 months) in two different configurations of the 3-chamber social interaction test: small chambers combined with three 10 min phases (C, E), and large chambers combined with three 5 min phases (D, F). n = 22 male mice for WT and 20 male for cKO for small chamber setup, and 21 male for WT and 22 male for cKO for large chamber setup. *p < 0.05 (Student's t test). ***p < 0.001 (Student's t test). G, Normal social interactions of Pcp2-Cre;Shank2fl/fl mice (2 months) in the direct social interaction test. n = 9 male for WT and 7 male for cKO. Student's t test.
Figure 10.

Normal USVs and repetitive behaviors in Pcp2-Cre;Shank2fl/fl mice. A, B, Normal levels of USVs induced by a stranger female in male adult Pcp2-Cre;Shank2fl/fl mice (3–5 months). n = 8 male mice for WT and cKO. *p < 0.05 (Student's t test). C–E, Normal levels of pup USVs induced by separation of Pcp2-Cre;Shank2fl/fl pups from their mothers. n = 12 (9 male, 3 female) for WT and 9 (6 male, 3 female) for cKO. Student's t test. F, G, Normal self-grooming, jumping, digging, and marble burying in Pcp2-Cre;Shank2fl/fl mice (2 months). n = 22 male for WT and 20 male for cKO. Student's t test. H, I, Normal motor function of Pcp2-Cre;Shank2fl/fl mice (2 months) in two different configurations of the rotarod test (4–40 rpm and 8–80 rpm). 4–40 rpm: n = 14 male for WT and 12 male for cKO; 8–80 rpm: n = 14 male for WT and 17 male for cKO. Repeated-measures ANOVA.
Figure 11.
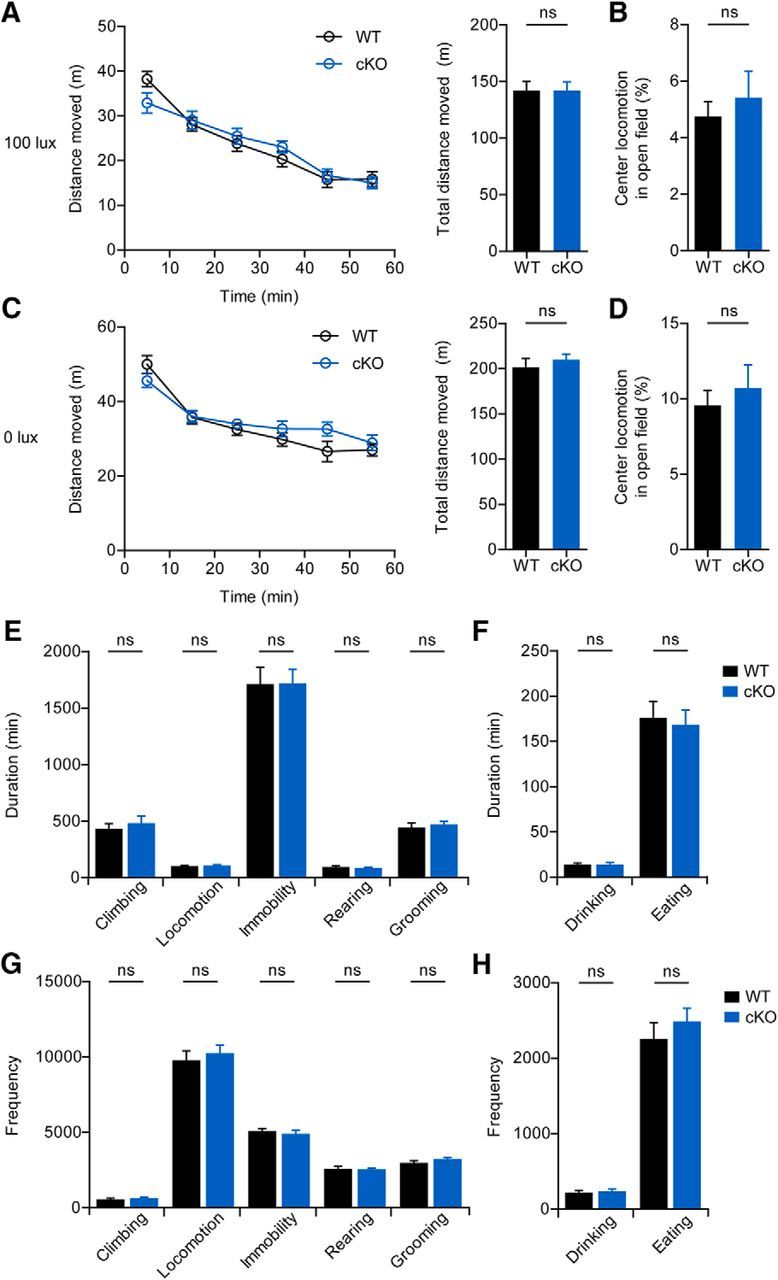
Normal open-field behavior and long-term movements in Pcp2-Cre;Shank2fl/fl mice. A–D, Normal locomotor activity (A, C) and time spent in the center (B, D) by Pcp2-Cre;Shank2fl/fl mice (2 months) in the open field test performed under two different light intensities: A, B, 100 lux. C, D, 0 lux. n = 16 male mice for WT and 18 male for cKO (100 lux); n = 14 for WT and 12 for cKO (0 lux). Repeated-measures ANOVA and Student's t test. E–H, Normal duration (E, F) and frequency (G, H) of long-term movements (climbing, locomotion, immobility, rearing, self-grooming, drinking, and eating) of Pcp2-Cre;Shank2fl/fl mice (2 months) in the Laboras test. n = 16 male for WT and cKO. Student's t test.
Pcp2-Cre;Shank2fl/fl mice showed normal social interaction, even when rigorously tested using two different formats of the 3-chamber social interaction test (small chambers combined with three 10 min phases, and large chambers combined with three 5 min phases) as well as direct social interaction assays (Fig. 9).
In contrast to their impaired motor coordination in Erasmus tests, Pcp2-Cre;Shank2fl/fl mice performed normally in the rotarod test (Fig. 10H,I), suggesting that Shank2 deletion has distinct influences on different motor functions.
Pcp2-Cre;Shank2fl/fl mice display enhanced repetitive and anxiety-like behaviors that are distinct from those in Shank2−/− mice
Although Pcp2-Cre;Shank2fl/fl mice showed normal repetitive behaviors (self-grooming, jumping, digging, and marble burying) (Fig. 10F,G), they showed increased repetitive behavior in the hole-board test (Fig. 12A,B), another measure of repetitive behavior (Takeda et al., 1998; Moy et al., 2008; Wang et al., 2011). In contrast, this enhanced hole-board repetitive behavior was not observed in conventional Shank2−/− mice (Fig. 12C,D). Therefore, Shank2−/− and Pcp2-Cre;Shank2fl/fl mice show distinct repetitive behaviors, with Shank2−/− mice exhibiting enhanced jumping (Won et al., 2012) and Pcp2-Cre;Shank2fl/fl mice showing enhanced hole-board repetitive behavior.
Figure 12.

Enhanced hole-board repetitive behavior and light-dark anxiety-like behavior in Pcp2-Cre;Shank2fl/fl mice. A–D, Increased repetitive behavior of Pcp2-Cre;Shank2fl/fl mice (3–5 months) in the hole-board test (A, B), but normal behavior in Shank2−/− mice (C, D). n = 17 male mice for WT and cKO; n = 12 male for WT and 14 male for KO. *p < 0.05 (Student's t test). **p < 0.01 (Student's t test). E, F, Enhanced anxiety-like behavior of Pcp2-Cre;Shank2fl/fl mice (3–5 months) in the light-dark test (E) but normal behavior in Shank2−/− mice (F). n = 22 male for WT and 20 male for cKO; n = 8 male for WT and 10 male for KO. **p < 0.01 (Student's t test). G, Normal anxiety-like behavior of Pcp2-Cre;Shank2fl/fl mice (3–5 months) in the elevated plus maze test. n = 24 male for WT and cKO. Student's t test.
The two mouse models also exhibited differences in anxiety-like behavior. Whereas Pcp2-Cre;Shank2fl/fl mice showed normal behavior in the elevated plus maze (Fig. 12G) and in the center region of the open-field arena (Fig. 11B,D), they exhibited mildly increased anxiety-like behavior in the light-dark test (Fig. 12E). This latter finding contrasts with that of Shank2−/− mice, which showed normal behavior in the light-dark test (Fig. 12F). These results, together with the previously reported anxiety-like behavior of Shank2−/− mice in an elevated plus maze but normal behavior in the center region of an open-field arena (Won et al., 2012), suggest that Shank2−/− and Pcp2-Cre;Shank2fl/fl mice show distinct anxiety-like behaviors.
Discussion
Our data suggest that Shank2 is important for regulating the density of PF-PC synapses in the cerebellum. In support of this hypothesis, we found that (1) Shank2−/− mice showed not only a reduction in the number of PSDs but also increases in the number of mismatched excitatory synapses and free dendritic spines; and (2) both Shank2−/− and Pcp2-Cre;Shank2fl/fl PCs showed reduced mEPSC frequency. Although the decreased mESPC frequency suggests potential involvement of presynaptic changes, the significant decrease in the number of excitatory synapses supported by the EM results suggests that it is unlikely.
In terms of molecular mechanisms that may underlie Shank2-dependent regulation of PF-PC synapses, our data suggest the involvement of two interesting candidate proteins: GluD2 and PSD-93/Chapsyn-110. GluD2 is predominantly detected in the postsynaptic side of PF-PC synapses in PCs (Landsend et al., 1997) and directly interacts with Shank2 (Uemura et al., 2004) and extracellularly with Cbln1 and presynaptic neurexins to promote synapse development (Matsuda et al., 2010; Uemura et al., 2010; Yuzaki, 2013). In addition, results from GluD2 KO mice indicate that GluD2 is important for the regulation of PF-PC synapses and motor coordination (Kashiwabuchi et al., 1995; Kurihara et al., 1997; Takeuchi et al., 2005; Kakegawa et al., 2009).
The phenotypes observed in Shank2−/− and Pcp2-Cre;Shank2fl/fl mice are similar to those of mice lacking GluD2. Yet, whether the 30%–40% reduction in GluD2 observed in Shank2−/− and Pcp2-Cre;Shank2fl/fl mice is sufficient to cause the same phenotypes as whole deletion of GluD2 is questionable. Another protein reduced in Shank2−/− and Pcp2-Cre;Shank2fl/fl mice is PSD-93/Chapsyn-110 (Brenman et al., 1996; Kim et al., 1996), a member of the PSD-95 family that is predominantly expressed in PCs (Fukaya et al., 1999; Roche et al., 1999; McGee et al., 2001) and is present at both PF-PC and climbing fiber-PC synapses (Roche et al., 1999).
PSD-93, like Shank2, interacts with GluD2 at PF-PC synapses (Roche et al., 1999) and indirectly associates with Shank2 through GKAP/SAPAP (Kim et al., 1997; Takeuchi et al., 1997; Boeckers et al., 1999a; Naisbitt et al., 1999; Tu et al., 1999). Notably, however, it has been shown that deletion of PSD-93 in mice does not cause detectable changes in the synaptic localization of GluD2, short-term plasticity of PF-EPSCs, or motor functions measured by gait and rotarod tests (McGee et al., 2001). However, it still remains to be determined whether PSD-93-mutant mice display alterations in mEPSCs, synaptic structures at the EM level, and motor coordination in the Erasmus test. In addition, given the marked increase (∼9-fold) in the cerebellar levels of PSD-93 in GluD2 KO mice determined by quantitative EM analysis (Yamasaki et al., 2011), an increase that likely represents a compensatory change, it is possible that GluD2 and PSD-93 act synergistically with Shank2 to regulate PF-PC synapses.
Behaviorally, Pcp2-Cre;Shank2fl/fl mice do not recapitulate the majority of autistic-like behaviors observed in Shank2−/− mice, including social interaction, USVs, repetitive behavior, and hyperactivity. These results suggest that Shank2 expressed in cerebellar PCs is unlikely to be associated with autistic-like behaviors. We doubt that the differences between Shank2−/− and Pcp2-Cre;Shank2fl/fl mice are attributable to differential time courses of Pcp2 and Shank2 expressions because both proteins are mainly detected at postnatal stages and reach peak levels at ∼P14-P21 (Boeckers et al., 1999b; Zhang et al., 2004).
Notably, however, Pcp2-Cre;Shank2fl/fl mice showed hole-board repetitive behavior that was not observed in Shank2−/− mice. Moreover, Pcp2-Cre;Shank2fl/fl mice showed mild anxiety-like behavior in the light-dark test, but not in the elevated plus maze test, whereas Shank2−/− mice showed essentially opposite phenotypes. These results suggest that Shank2 deletion in PCs has distinct influences on specific repetitive and anxiety-like behaviors compared with whole cerebellum deletion of Shank2.
These differences, despite the same Shank2 deletion in PCs in the two mouse lines, could be due to that Shank2 is additionally deleted in other cell types, such as interneurons in the cerebellum of conventional Shank2−/− mice. Specifically, in Pcp2-Cre;Shank2fl/fl mice, only excitatory synapses on PCs would be altered. In contrast, in Shank2−/− mice, excitatory synapses on interneuronal dendrites may be additionally altered, modifying inhibitory synaptic input onto PCs, in addition to excitatory synaptic input, although this possibility needs to be addressed in future studies.
Recently, a similar study on the cerebellar functions of Shank2 has been reported, which used mice carrying the deletion of Shank2 exon 7 globally and in PCs (Peter et al., 2016). Notably, these mice displayed normal density of excitatory synapses (Shank2−/− mice) and impaired social interaction (Pcp2-Cre;Shank2fl/fl). These differences might be attributable to that our mice carry a different exon deletion (exons 6 and 7). Alternatively, it could be the different genetic backgrounds of Shank2−/− mice (C57BL/6N in our study and mixed C57BL/6N and C57BL/6J in the other study).
In conclusion, our results suggest that cerebellar Shank2 is important for the regulation of excitatory synapse density, motor coordination, and repetitive and anxiety-like behaviors.
Footnotes
This work was supported by the National Research Foundation (NRF) Global PhD Fellowship Program NRF-2013H1A2A1032785 250 to S.H., NRF-2013-Fostering Core Leaders of the Future Basic 249 Science Program to C.C., MSIP Grant 2008–0062282 to Y.C.B., Korea Institute of Science and Technology Institutional Program 2E26190 to K.T.-Y., and Institute for Basic Science IBS-R002-D1 to E.K.
The authors declare no competing financial interests.
References
- Baudouin SJ, Gaudias J, Gerharz S, Hatstatt L, Zhou K, Punnakkal P, Tanaka KF, Spooren W, Hen R, De Zeeuw CI, Vogt K, Scheiffele P. Shared synaptic pathophysiology in syndromic and nonsyndromic rodent models of autism. Science. 2012;338:128–132. doi: 10.1126/science.1224159. [DOI] [PubMed] [Google Scholar]
- Berkel S, Tang W, Treviño M, Vogt M, Obenhaus HA, Gass P, Scherer SW, Sprengel R, Schratt G, Rappold GA. Inherited and de novo SHANK2 variants associated with autism spectrum disorder impair neuronal morphogenesis and physiology. Hum Mol Genet. 2012;21:344–357. doi: 10.1093/hmg/ddr470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Böckers TM, Segger-Junius M, Iglauer P, Bockmann J, Gundelfinger ED, Kreutz MR, Richter D, Kindler S, Kreienkamp HJ. Differential expression and dendritic transcript localization of Shank family members: identification of a dendritic targeting element in the 3′ untranslated region of Shank1 mRNA. Mol Cell Neurosci. 2004;26:182–190. doi: 10.1016/j.mcn.2004.01.009. [DOI] [PubMed] [Google Scholar]
- Boeckers TM, Winter C, Smalla KH, Kreutz MR, Bockmann J, Seidenbecher C, Garner CC, Gundelfinger ED. Proline-rich synapse-associated proteins ProSAP1 and ProSAP2 interact with synaptic proteins of the SAPAP/GKAP family. Biochem Biophys Res Commun. 1999a;264:247–252. doi: 10.1006/bbrc.1999.1489. [DOI] [PubMed] [Google Scholar]
- Boeckers TM, Kreutz MR, Winter C, Zuschratter W, Smalla KH, Sanmarti-Vila L, Wex H, Langnaese K, Bockmann J, Garner CC, Gundelfinger ED. Proline-rich synapse-associated protein-1/cortactin binding protein 1 (ProSAP1/CortBP1) is a PDZ-domain protein highly enriched in the postsynaptic density. J Neurosci. 1999b;19:6506–6518. doi: 10.1523/JNEUROSCI.19-15-06506.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boeckers TM, Bockmann J, Kreutz MR, Gundelfinger ED. ProSAP/Shank proteins: a family of higher order organizing molecules of the postsynaptic density with an emerging role in human neurological disease. J Neurochem. 2002;81:903–910. doi: 10.1046/j.1471-4159.2002.00931.x. [DOI] [PubMed] [Google Scholar]
- Brenman JE, Christopherson KS, Craven SE, McGee AW, Bredt DS. Cloning and characterization of postsynaptic density 93, a nitric oxide synthase interacting protein. J Neurosci. 1996;16:7407–7415. doi: 10.1523/JNEUROSCI.16-23-07407.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chilian B, Abdollahpour H, Bierhals T, Haltrich I, Fekete G, Nagel I, Rosenberger G, Kutsche K. Dysfunction of SHANK2 and CHRNA7 in a patient with intellectual disability and language impairment supports genetic epistasis of the two loci. Clin Genet. 2013;84:560–565. doi: 10.1111/cge.12105. [DOI] [PubMed] [Google Scholar]
- Costas J. The role of SHANK2 rare variants in schizophrenia susceptibility. Mol Psychiatry. 2015;20:1486. doi: 10.1038/mp.2015.119. [DOI] [PubMed] [Google Scholar]
- Du Y, Weed SA, Xiong WC, Marshall TD, Parsons JT. Identification of a novel cortactin SH3 domain-binding protein and its localization to growth cones of cultured neurons. Mol Cell Biol. 1998;18:5838–5851. doi: 10.1128/MCB.18.10.5838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng W, Zhang M. Organization and dynamics of PDZ-domain-related supramodules in the postsynaptic density. Nat Rev Neurosci. 2009;10:87–99. doi: 10.1038/nrn2540. [DOI] [PubMed] [Google Scholar]
- Fukaya M, Ueda H, Yamauchi K, Inoue Y, Watanabe M. Distinct spatiotemporal expression of mRNAs for the PSD-95/SAP90 protein family in the mouse brain. Neurosci Res. 1999;33:111–118. doi: 10.1016/S0168-0102(98)00120-5. [DOI] [PubMed] [Google Scholar]
- Grabrucker AM, Schmeisser MJ, Schoen M, Boeckers TM. Postsynaptic ProSAP/Shank scaffolds in the cross-hair of synaptopathies. Trends Cell Biol. 2011;21:594–603. doi: 10.1016/j.tcb.2011.07.003. [DOI] [PubMed] [Google Scholar]
- Grabrucker AM. A role for synaptic zinc in ProSAP/Shank PSD scaffold malformation in autism spectrum disorders. Dev Neurobiol. 2014;74:136–146. doi: 10.1002/dneu.22089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guilmatre A, Huguet G, Delorme R, Bourgeron T. The emerging role of SHANK genes in neuropsychiatric disorders. Dev Neurobiol. 2014;74:113–122. doi: 10.1002/dneu.22128. [DOI] [PubMed] [Google Scholar]
- Homann OR, Misura K, Lamas E, Sandrock RW, Nelson P, McDonough SI, DeLisi LE. Whole-genome sequencing in multiplex families with psychoses reveals mutations in the SHANK2 and SMARCA1 genes segregating with illness. Mol Psychiatry. 2016 doi: 10.1038/mp.2016.24. doi: 10.1038/mp.2016.24. Advance online publication. Retrieved March 22, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang YH, Ehlers MD. Modeling autism by SHANK gene mutations in mice. Neuron. 2013;78:8–27. doi: 10.1016/j.neuron.2013.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakegawa W, Miyazaki T, Kohda K, Matsuda K, Emi K, Motohashi J, Watanabe M, Yuzaki M. The N-terminal domain of GluD2 (GluRδ2) recruits presynaptic terminals and regulates synaptogenesis in the cerebellum in vivo. J Neurosci. 2009;29:5738–5748. doi: 10.1523/JNEUROSCI.6013-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashiwabuchi N, Ikeda K, Araki K, Hirano T, Shibuki K, Takayama C, Inoue Y, Kutsuwada T, Yagi T, Kang Y. Impairment of motor coordination, Purkinje cell synapse formation, and cerebellar long-term depression in GluR δ2 mutant mice. Cell. 1995;81:245–252. doi: 10.1016/0092-8674(95)90334-8. [DOI] [PubMed] [Google Scholar]
- Kim E, Cho KO, Rothschild A, Sheng M. Heteromultimerization and NMDA receptor-clustering activity of Chapsyn-110, a member of the PSD-95 family of proteins. Neuron. 1996;17:103–113. doi: 10.1016/S0896-6273(00)80284-6. [DOI] [PubMed] [Google Scholar]
- Kim E, Naisbitt S, Hsueh YP, Rao A, Rothschild A, Craig AM, Sheng M. GKAP, a novel synaptic protein that interacts with the guanylate kinase-like domain of the PSD-95/SAP90 family of channel clustering molecules. J Cell Biol. 1997;136:669–678. doi: 10.1083/jcb.136.3.669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MH, Choi J, Yang J, Chung W, Kim JH, Paik SK, Kim K, Han S, Won H, Bae YS, Cho SH, Seo J, Bae YC, Choi SY, Kim E. Enhanced NMDA receptor-mediated synaptic transmission, enhanced long-term potentiation, and impaired learning and memory in mice lacking IRSp53. J Neurosci. 2009;29:1586–1595. doi: 10.1523/JNEUROSCI.4306-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurihara H, Hashimoto K, Kano M, Takayama C, Sakimura K, Mishina M, Inoue Y, Watanabe M. Impaired parallel fiber->Purkinje cell synapse stabilization during cerebellar development of mutant mice lacking the glutamate receptor δ2 subunit. J Neurosci. 1997;17:9613–9623. doi: 10.1523/JNEUROSCI.17-24-09613.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landsend AS, Amiry-Moghaddam M, Matsubara A, Bergersen L, Usami S, Wenthold RJ, Ottersen OP. Differential localization of δglutamate receptors in the rat cerebellum: coexpression with AMPA receptors in parallel fiber-spine synapses and absence from climbing fiber-spine synapses. J Neurosci. 1997;17:834–842. doi: 10.1523/JNEUROSCI.17-02-00834.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leblond CS, Heinrich J, Delorme R, Proepper C, Betancur C, Huguet G, Konyukh M, Chaste P, Ey E, Rastam M, Anckarsäter H, Nygren G, Gillberg IC, Melke J, Toro R, Regnault B, Fauchereau F, Mercati O, Lemière N, Skuse D, et al. Genetic and functional analyses of SHANK2 mutations suggest a multiple hit model of autism spectrum disorders. PLoS Genet. 2012;8:e1002521. doi: 10.1371/journal.pgen.1002521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leblond CS, Nava C, Polge A, Gauthier J, Huguet G, Lumbroso S, Giuliano F, Stordeur C, Depienne C, Mouzat K, Pinto D, Howe J, Lemière N, Durand CM, Guibert J, Ey E, Toro R, Peyre H, Mathieu A, Amsellem F, et al. Meta-analysis of SHANK mutations in autism spectrum disorders: a gradient of severity in cognitive impairments. PLoS Genet. 2014;10:e1004580. doi: 10.1371/journal.pgen.1004580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee EJ, Lee H, Huang TN, Chung C, Shin W, Kim K, Koh JY, Hsueh YP, Kim E. Trans-synaptic zinc mobilization improves social interaction in two mouse models of autism through NMDAR activation. Nat Commun. 2015;6:7168. doi: 10.1038/ncomms8168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim S, Naisbitt S, Yoon J, Hwang JI, Suh PG, Sheng M, Kim E. Characterization of the shank family of synaptic proteins: multiple genes, alternative splicing, and differential expression in brain and development. J Biol Chem. 1999;274:29510–29518. doi: 10.1074/jbc.274.41.29510. [DOI] [PubMed] [Google Scholar]
- Matsuda K, Miura E, Miyazaki T, Kakegawa W, Emi K, Narumi S, Fukazawa Y, Ito-Ishida A, Kondo T, Shigemoto R, Watanabe M, Yuzaki M. Cbln1 is a ligand for an orphan glutamate receptor δ2, a bidirectional synapse organizer. Science. 2010;328:363–368. doi: 10.1126/science.1185152. [DOI] [PubMed] [Google Scholar]
- McGee AW, Topinka JR, Hashimoto K, Petralia RS, Kakizawa S, Kauer FW, Aguilera-Moreno A, Wenthold RJ, Kano M, Bredt DS, Kauer F. PSD-93 knock-out mice reveal that neuronal MAGUKs are not required for development or function of parallel fiber synapses in cerebellum. J Neurosci. 2001;21:3085–3091. doi: 10.1523/JNEUROSCI.21-09-03085.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moy SS, Nadler JJ, Poe MD, Nonneman RJ, Young NB, Koller BH, Crawley JN, Duncan GE, Bodfish JW. Development of a mouse test for repetitive, restricted behaviors: relevance to autism. Behav Brain Res. 2008;188:178–194. doi: 10.1016/j.bbr.2007.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naisbitt S, Kim E, Tu JC, Xiao B, Sala C, Valtschanoff J, Weinberg RJ, Worley PF, Sheng M. Shank, a novel family of postsynaptic density proteins that binds to the NMDA receptor/PSD-95/GKAP complex and cortactin. Neuron. 1999;23:569–582. doi: 10.1016/S0896-6273(00)80809-0. [DOI] [PubMed] [Google Scholar]
- Peça J, Feliciano C, Ting JT, Wang W, Wells MF, Venkatraman TN, Lascola CD, Fu Z, Feng G. Shank3 mutant mice display autistic-like behaviours and striatal dysfunction. Nature. 2011;472:437–442. doi: 10.1038/nature09965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peter S, Ten Brinke MM, Stedehouder J, Reinelt CM, Wu B, Zhou H, Zhou K, Boele HJ, Kushner SA, Lee MG, Schmeisser MJ, Boeckers TM, Schonewille M, Hoebeek FE, De Zeeuw CI. Dysfunctional cerebellar Purkinje cells contribute to autism-like behaviour in Shank2-deficient mice. Nat Commun. 2016;7:12627. doi: 10.1038/ncomms12627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peykov S, Berkel S, Degenhardt F, Rietschel M, Nöthen MM, Rappold GA. Rare SHANK2 variants in schizophrenia. Mol Psychiatry. 2015a;20:1487–1488. doi: 10.1038/mp.2015.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peykov S, Berkel S, Schoen M, Weiss K, Degenhardt F, Strohmaier J, Weiss B, Proepper C, Schratt G, Nöthen MM, Boeckers TM, Rietschel M, Rappold GA. Identification and functional characterization of rare SHANK2 variants in schizophrenia. Mol Psychiatry. 2015b;20:1489–1498. doi: 10.1038/mp.2014.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto D, Pagnamenta AT, Klei L, Anney R, Merico D, Regan R, Conroy J, Magalhaes TR, Correia C, Abrahams BS, Almeida J, Bacchelli E, Bader GD, Bailey AJ, Baird G, Battaglia A, Berney T, Bolshakova N, Bölte S, Bolton PF, et al. Functional impact of global rare copy number variation in autism spectrum disorders. Nature. 2010;466:368–372. doi: 10.1038/nature09146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasad A, Merico D, Thiruvahindrapuram B, Wei J, Lionel AC, Sato D, Rickaby J, Lu C, Szatmari P, Roberts W, Fernandez BA, Marshall CR, Hatchwell E, Eis PS, Scherer SW. A discovery resource of rare copy number variations in individuals with autism spectrum disorder. G3 (Bethesda) 2012;2:1665–1685. doi: 10.1534/g3.112.004689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauch A, Wieczorek D, Graf E, Wieland T, Endele S, Schwarzmayr T, Albrecht B, Bartholdi D, Beygo J, Di Donato N, Dufke A, Cremer K, Hempel M, Horn D, Hoyer J, Joset P, Röpke A, Moog U, Riess A, Thiel CT, et al. Range of genetic mutations associated with severe non-syndromic sporadic intellectual disability: an exome sequencing study. Lancet. 2012;380:1674–1682. doi: 10.1016/S0140-6736(12)61480-9. [DOI] [PubMed] [Google Scholar]
- Roche KW, Ly CD, Petralia RS, Wang YX, McGee AW, Bredt DS, Wenthold RJ. Postsynaptic density-93 interacts with the δ2 glutamate receptor subunit at parallel fiber synapses. J Neurosci. 1999;19:3926–3934. doi: 10.1523/JNEUROSCI.19-10-03926.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sala C, Vicidomini C, Bigi I, Mossa A, Verpelli C. Shank synaptic scaffold proteins: keys to understanding the pathogenesis of autism and other synaptic disorders. J Neurochem. 2015;135:849–858. doi: 10.1111/jnc.13232. [DOI] [PubMed] [Google Scholar]
- Sanders SJ, Murtha MT, Gupta AR, Murdoch JD, Raubeson MJ, Willsey AJ, Ercan-Sencicek AG, DiLullo NM, Parikshak NN, Stein JL, Walker MF, Ober GT, Teran NA, Song Y, El-Fishawy P, Murtha RC, Choi M, Overton JD, Bjornson RD, Carriero NJ, et al. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature. 2012;485:237–241. doi: 10.1038/nature10945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schluth-Bolard C, Labalme A, Cordier MP, Till M, Nadeau G, Tevissen H, Lesca G, Boutry-Kryza N, Rossignol S, Rocas D, Dubruc E, Edery P, Sanlaville D. Breakpoint mapping by next generation sequencing reveals causative gene disruption in patients carrying apparently balanced chromosome rearrangements with intellectual deficiency and/or congenital malformations. J Med Genet. 2013;50:144–150. doi: 10.1136/jmedgenet-2012-101351. [DOI] [PubMed] [Google Scholar]
- Schmeisser MJ. Translational neurobiology in Shank mutant mice-model systems for neuropsychiatric disorders. Ann Anat. 2015;200:115–117. doi: 10.1016/j.aanat.2015.03.006. [DOI] [PubMed] [Google Scholar]
- Schmeisser MJ, Ey E, Wegener S, Bockmann J, Stempel AV, Kuebler A, Janssen AL, Udvardi PT, Shiban E, Spilker C, Balschun D, Skryabin BV, Dieck St, Smalla KH, Montag D, Leblond CS, Faure P, Torquet N, Le Sourd AM, Toro R, et al. Autistic-like behaviours and hyperactivity in mice lacking ProSAP1/Shank2. Nature. 2012;486:256–260. doi: 10.1038/nature11015. [DOI] [PubMed] [Google Scholar]
- Sheng M, Hoogenraad CC. The postsynaptic architecture of excitatory synapses: a more quantitative view. Annu Rev Biochem. 2007;76:823–847. doi: 10.1146/annurev.biochem.76.060805.160029. [DOI] [PubMed] [Google Scholar]
- Sheng M, Kim E. The shank family of scaffold proteins. J Cell Sci. 2000;113:1851–1856. doi: 10.1242/jcs.113.11.1851. [DOI] [PubMed] [Google Scholar]
- Sheng M, Kim E. The postsynaptic organization of synapses. Cold Spring Harb Perspect Biol. 2011;3:a005678. doi: 10.1101/cshperspect.a005678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng M, Sala C. PDZ domains and the organization of supramolecular complexes. Annu Rev Neurosci. 2001;24:1–29. doi: 10.1146/annurev.neuro.24.1.1. [DOI] [PubMed] [Google Scholar]
- Takeda H, Tsuji M, Matsumiya T. Changes in head-dipping behavior in the hole-board test reflect the anxiogenic and/or anxiolytic state in mice. Eur J Pharmacol. 1998;350:21–29. doi: 10.1016/S0014-2999(98)00223-4. [DOI] [PubMed] [Google Scholar]
- Takeuchi M, Hata Y, Hirao K, Toyoda A, Irie M, Takai Y. SAPAPs: a family of PSD-95/SAP90-associated proteins localized at postsynaptic density. J Biol Chem. 1997;272:11943–11951. doi: 10.1074/jbc.272.18.11943. [DOI] [PubMed] [Google Scholar]
- Takeuchi T, Miyazaki T, Watanabe M, Mori H, Sakimura K, Mishina M. Control of synaptic connection by glutamate receptor δ2 in the adult cerebellum. J Neurosci. 2005;25:2146–2156. doi: 10.1523/JNEUROSCI.4740-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai PT, Hull C, Chu Y, Greene-Colozzi E, Sadowski AR, Leech JM, Steinberg J, Crawley JN, Regehr WG, Sahin M. Autistic-like behaviour and cerebellar dysfunction in Purkinje cell Tsc1 mutant mice. Nature. 2012;488:647–651. doi: 10.1038/nature11310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu JC, Xiao B, Naisbitt S, Yuan JP, Petralia RS, Brakeman P, Doan A, Aakalu VK, Lanahan AA, Sheng M, Worley PF. Coupling of mGluR/Homer and PSD-95 complexes by the Shank family of postsynaptic density proteins. Neuron. 1999;23:583–592. doi: 10.1016/S0896-6273(00)80810-7. [DOI] [PubMed] [Google Scholar]
- Uemura T, Mori H, Mishina M. Direct interaction of GluRδ2 with Shank scaffold proteins in cerebellar Purkinje cells. Mol Cell Neurosci. 2004;26:330–341. doi: 10.1016/j.mcn.2004.02.007. [DOI] [PubMed] [Google Scholar]
- Uemura T, Lee SJ, Yasumura M, Takeuchi T, Yoshida T, Ra M, Taguchi R, Sakimura K, Mishina M. Trans-synaptic interaction of GluRδ2 and Neurexin through Cbln1 mediates synapse formation in the cerebellum. Cell. 2010;141:1068–1079. doi: 10.1016/j.cell.2010.04.035. [DOI] [PubMed] [Google Scholar]
- Van Der Giessen RS, Koekkoek SK, van Dorp S, De Gruijl JR, Cupido A, Khosrovani S, Dortland B, Wellershaus K, Degen J, Deuchars J, Fuchs EC, Monyer H, Willecke K, De Jeu MT, De Zeeuw CI. Role of olivary electrical coupling in cerebellar motor learning. Neuron. 2008;58:599–612. doi: 10.1016/j.neuron.2008.03.016. [DOI] [PubMed] [Google Scholar]
- Wang SS, Kloth AD, Badura A. The cerebellum, sensitive periods, and autism. Neuron. 2014;83:518–532. doi: 10.1016/j.neuron.2014.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, McCoy PA, Rodriguiz RM, Pan Y, Je HS, Roberts AC, Kim CJ, Berrios J, Colvin JS, Bousquet-Moore D, Lorenzo I, Wu G, Weinberg RJ, Ehlers MD, Philpot BD, Beaudet AL, Wetsel WC, Jiang YH. Synaptic dysfunction and abnormal behaviors in mice lacking major isoforms of Shank3. Hum Mol Genet. 2011;20:3093–3108. doi: 10.1093/hmg/ddr212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wischmeijer A, Magini P, Giorda R, Gnoli M, Ciccone R, Cecconi L, Franzoni E, Mazzanti L, Romeo G, Zuffardi O, Seri M. Olfactory receptor-related duplicons mediate a microdeletion at 11q13.2q13.4 associated with a syndromic phenotype. Mol Syndromol. 2011;1:176–184. doi: 10.1259/000322054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Won H, Lee HR, Gee HY, Mah W, Kim JI, Lee J, Ha S, Chung C, Jung ES, Cho YS, Park SG, Lee JS, Lee K, Kim D, Bae YC, Kaang BK, Lee MG, Kim E. Autistic-like social behaviour in Shank2-mutant mice improved by restoring NMDA receptor function. Nature. 2012;486:261–265. doi: 10.1038/nature11208. [DOI] [PubMed] [Google Scholar]
- Yamamoto Y, Lee D, Kim Y, Lee B, Seo C, Kawasaki H, Kuroda S, Tanaka-Yamamoto K. Raf kinase inhibitory protein is required for cerebellar long-term synaptic depression by mediating PKC-dependent MAPK activation. J Neurosci. 2012;32:14254–14264. doi: 10.1523/JNEUROSCI.2812-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamasaki M, Miyazaki T, Azechi H, Abe M, Natsume R, Hagiwara T, Aiba A, Mishina M, Sakimura K, Watanabe M. Glutamate receptor δ2 is essential for input pathway-dependent regulation of synaptic AMPAR contents in cerebellar Purkinje cells. J Neurosci. 2011;31:3362–3374. doi: 10.1523/JNEUROSCI.5601-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo J, Bakes J, Bradley C, Collingridge GL, Kaang BK. Shank mutant mice as an animal model of autism. Philos Trans R Soc Lond B Biol Sci. 2014;369:20130143. doi: 10.1098/rstb.2013.0143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuzaki M. Cerebellar LTD vs motor learning: lessons learned from studying GluD2. Neural Netw. 2013;47:36–41. doi: 10.1016/j.neunet.2012.07.001. [DOI] [PubMed] [Google Scholar]
- Zhang XM, Ng AH, Tanner JA, Wu WT, Copeland NG, Jenkins NA, Huang JD. Highly restricted expression of Cre recombinase in cerebellar Purkinje cells. Genesis. 2004;40:45–51. doi: 10.1002/gene.20062. [DOI] [PubMed] [Google Scholar]