

Abstract
It is now widely accepted that compensatory mechanisms are involved during the early phase of Parkinson's disease (PD) to delay the expression of motor symptoms. However, the neurochemical mechanisms underlying this presymptomatic period are still unclear. Here, we measured in vivo longitudinal changes of both the dopaminergic and serotonergic systems in seven asymptomatic 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-intoxicated monkeys (when motor symptoms are less apparent) using PET. We used the progressively MPTP-intoxicated monkey model that expresses recovery from motor symptoms to study the changes in dopamine synthesis ([18F]DOPA), dopamine D2/D3 receptors ([11C]raclopride), and serotonin transporter 11C-N,N-dimethyl-2-(-2-amino-4-cyanophenylthio) benzylamine ([11C]DASB) and serotonin 1A receptor ([18F]MPPF) levels between four different states (baseline, early symptomatic, full symptomatic and recovered). During the early symptomatic state, we observed increases of [18F]DOPA uptake in the anterior putamen, [11C]raclopride binding in the posterior striatum, and 2′-methoxyphenyl-(N-2′-pyridinyl)-p-[18F]fluoro-benzamidoethylpiperazine [18F]MPPF uptake in the orbitofrontal cortex and dorsal ACC. After recovery from motor symptoms, the results mainly showed decreased [11C]raclopride binding in the anterior striatum and limbic ACC. In addition, our findings supported the importance of pallidal dopaminergic neurotransmission in both the early compensatory mechanisms and the functional recovery mechanisms, with reduced aromatic L-amino acid decarboxylase (AAAD) activity closely related to the appearance or perseveration of motor symptoms. In parallel, this study provides preliminary evidence of the role of the serotonergic system in compensatory mechanisms. Nonetheless, future studies are needed to determine whether there are changes in SERT availability in the early symptomatic state and if [18F]MPPF PET imaging might be a promising biomarker of early degenerative changes in PD.
SIGNIFICANCE STATEMENT The present research provides evidence of the potential of combining a multitracer PET imaging technique and a longitudinal protocol applied on a progressively 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-intoxicated monkey model to further elucidate the nature of the compensatory mechanisms involved in the preclinical period of Parkinson's disease (PD). In particular, by investigating the dopaminergic and serotonergic changes both presynaptically and postsynaptically at four different motor states (baseline, early symptomatic, full symptomatic, and recovered), this study has allowed us to identify putative biomarkers for future therapeutic interventions to prevent and/or delay disease expression. For example, our findings suggest that the external pallidum could be a new target for cell-based therapies to reduce PD symptoms.
Keywords: compensatory mechanisms, dopamine, MPTP, Parkinson's disease, PET imaging, serotonin
Introduction
Parkinson's disease (PD) is a neurodegenerative disorder clinically characterized by the progressive emergence of motor symptoms. However, those impairments are observed only when the striatal level of DA has decreased by 60–80% (Bernheimer et al., 1973). During the early phase of the disease, parkinsonian patients do not show motor symptoms despite progressive but strong dopaminergic depletion. This suggests the existence of impressive compensatory mechanisms in this presymptomatic period to counterbalance the degenerative process (Zigmond, 1997). Human PET imaging studies have shown that, early in the disease, the activity of the enzyme aromatic L-amino acid decarboxylase (AAAD) is increased in the preserved striatal dopaminergic terminals (Lee et al., 2000; Ribeiro et al., 2002), as well as in extrastriatal structures such as the pallidum (Whone et al., 2003) and the cingulate cortex (Rakshi et al., 1999). In addition, increased dopamine (DA) release (Snyder et al., 1990; Schneider et al., 1994) or turnover by the remaining axons (Agid et al., 1973; Altar et al., 1987) and increased numbers or sensitivity of the DA receptors (Zigmond et al., 1984; Bezard and Gross, 1998) have been observed in PD animal models. Altogether, these findings have emphasized the role of dopaminergic compensatory mechanisms. Nonetheless, nondopaminergic systems could also play a role in compensation, such as the GABAergic (Schroeder and Schneider, 2002), glutamatergic (Bezard et al., 1998), and serotonergic systems (Gaspar et al., 1993; Mounayar et al., 2007; Boulet et al., 2008). The precise neurochemical mechanisms involved during the early phase of the disease are still unclear.
To gain further understanding of these compensatory mechanisms, we previously used a progressive approach of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-intoxicated monkey model of PD (Mounayar et al., 2007; Boulet et al., 2008; Neumane et al., 2012). With this method, monkeys developed all the motor symptoms and then fully recovered after the final MPTP injection despite a large DA denervation, suggesting that compensatory mechanisms could be involved in the recovery process. Overall, our studies supported the notion that residual striatal DA played a role in compensation in PD, revealed the importance of extrastriatal structures such as the external pallidum, and highlighted the possible involvement of serotonin (5-HT). However, the conclusions were limited because the observed changes were restricted to the subcortical presynaptic level and there is no evidence yet that the recovery process involves the same mechanisms as those involved during the presymptomatic state of PD when the level of dopaminergic lesion is less pronounced.
Accordingly, in the present study, we combined a multitracer PET imaging method and a longitudinal protocol using this progressively MPTP-intoxicated monkey model of PD to investigate the in vivo changes in the DA and 5-HT systems both at the presynaptic and the postsynaptic levels. Because functional compensatory mechanisms might differ depending on the extent of dopaminergic denervation, we successively observed the changes between four different motor states, starting from baseline, followed by an early symptomatic and then a full symptomatic parkinsonian state, and proceeding to the recovered state. We used both the [18F]DOPA (thought to reflect AAAD activity) and [11C]raclopride (a selective dopamine D2/D3 receptors antagonist) radiotracers to identify regions with dopaminergic dysfunctions, as described previously in human studies (for review, see Brooks and Pavese, 2011). Abnormalities in both serotonin transporter (SERT) availability (Politis et al., 2010a, 2010b) and 5-HT1A receptors (Ballanger et al., 2012) have been associated with depression in PD patients, a nonmotor symptom that often appears years before motor disturbances. Therefore, follow-up of the serotonergic system has been addressed using the 11C-N,N-dimethyl-2-(-2-amino-4-cyanophenylthio) benzylamine ([11C]DASB) (a specific marker of SERT availability) and 2′-methoxyphenyl-(N-2′-pyridinyl)-p-[18F]fluoro-benzamidoethylpiperazine ([18F]MPPF) (a selective 5-HT1A receptor antagonist) radiotracers to identify regions with respectively presynaptic and/or postsynaptic 5-HT dysfunctions. We hypothesized that there would be an early increase in presynaptic DA activities in structures less affected by MPTP toxicity, followed by a profound decrease during the full parkinsonian motor state, whereas the recovery process should involve modifications in DA receptors and 5-HT compensatory activities.
Materials and Methods
Animals.
Seven male cynomolgus monkeys (Macaca fascicularis, MF) between 3 and 5 years old (young adults) and weighing 4–6 kg were provided by Tamarinier Ltee, Mauritius Island and used in this study. These monkeys were cared for and treated in strict accordance with National Institutes of Health guidelines (1996) and with the European Community Council Directive of 2010 (2010/63/UE) and the recommendations of the French National Committee (2013/11).
MPTP treatment and experimental schedule.
The timeline of the experimental design is shown in Figure 1A. After collection of baseline PET imaging data, the animals were given intramuscular MPTP injections (0.3–0.6 mg/kg body weight; Sigma-Aldrich) performed under light anesthesia (ketamine 0.5 mg/kg, atropine 0.05 mg/kg) at intervals of 4–5 d until the emergence of most of the motor parkinsonian symptoms. The total MPTP dose given to each monkey varied between 1.2 and 2.2 mg/kg. Once MPTP injections were stopped, almost all the animals (5/7) totally recovered spontaneously from their motor symptoms (Fig. 1C).
Figure 1.

Study design and schedule of PET scan acquisitions. A, Timeline of the experimental design with the number (n) of animals in each state and for each radioligand. B, Mean time (days) ± SD between last MPTP injection and PET acquisition for each radioligand. C, Evolution of parkinsonian score during MPTP intoxication and recovery. Data represent the evolution of appearance and disappearance of motor symptoms during and after MPTP intoxication. Timelines are aligned such that day 0 corresponds to the day on which the maximal parkinsonian score was obtained from an individual animal. The rectangle represents PET scan acquisition. D, Positive correlation between maximal parkinsonian score and the time needed for each animal to recover spontaneously from their motor symptoms once MPTP injections were stopped (n = 7).
Throughout the study, the severity of parkinsonism was evaluated using the rating scale proposed by Schneider and Kovelowski (1990). We have already described the effects of MPTP evaluated using this rating scale in previous studies (Pessiglione et al., 2004; Mounayar et al., 2007; Boulet et al., 2008). Briefly, this scale includes 12 items rated between 0 and 2 or 3, with a total score of 29. It takes into account classical motor symptoms (bradykinesia, rigidity, tremor, freezing, posture and arm posture), but also spontaneous activities (arm movements, spontaneous eye movements, and home cage activity) and other activities (vocalization, triggered eye movements, and feeding). For each monkey, the evolution of these measurements was evaluated daily by one or two experimenters accustomed to handling the animals. Moreover, all of the evaluation sessions were recorded to allow additional evaluations by an experimenter blinded to the state of the animal. During these additional evaluations, the intensity of some items was evaluated by quantitative measurement of specific events (counting of eye and arm movements and home cage activity), which were statistically compared with measurements made during baseline periods before MPTP treatment. For these measurements, a score of 1, 2, or 3 indicated a statistical reduction of >50%, a statistical reduction of >90%, or a complete absence of movement, respectively. Some symptoms were evaluated specifically in either the chair (rigidity, spontaneous and triggered eye movements, and arm movements during tasks) or the cage (home cage activity), whereas the other symptoms were evaluated in both the chair and the cage (bradykinesia/akinesia, posture). The home cage activity was evaluated using closed circuit television and an activity digitalization system (Vigie Primates; for details, see Mounayar et al., 2007).
PET scans were acquired longitudinally at four different times: (1) in the baseline state, (2) in the early symptomatic state (4–5 d after the second MPTP injection), (3) in the full symptomatic state (between 4 and 14 d after the last MPTP injection during the peak of motor symptoms), and (4) in the recovered state (at least 1 week after the parkinsonian score returned to 0). However, it was not possible to undertake PET with all of the radiotracers in all of the animals, which therefore explains the different number of animals within each state (Fig. 1A). The mean time (days) between last MPTP dose and PET scans was (mean ± SD) −11 ± 3 for the early symptomatic state, 9 ± 5 for the full symptomatic state, and 59 ± 22 for the recovered state (Fig. 1B). On the day of each PET scan, the monkeys were pretreated with atropine (0.05 mg/kg) and 15 min later were anesthetized by an intramuscular dose of zoletil (15 mg/kg). The monkeys were then transported to the Imaging Centre (CERMEP, Lyon, France), where they were placed in a PET-compatible stereotaxic apparatus. Anesthesia was maintained during the scan and a lactated Ringer's solution was infused continuously through a saphenous vein catheter. The respiratory frequency, pO2, and heart rate were monitored throughout the experiment.
PET data acquisition.
All PET scans were performed in 3D mode using a Siemens CTI HR + tomograph, with an axial field of view of 15.2 cm, yielding 63 planes and a nominal in-plane resolution of 4.1 mm FWHM according to the NEMA protocol (Brix et al., 1997). PET measurements were performed with four radiotracers: [18F]DOPA, [11C]raclopride, [11C]DASB, and [18F]MPPF. For each experimental state, the mean time (days) between PET scans was (mean ± SD) 20 ± 13 for the baseline state, 5 ± 2 for the early symptomatic state, 7 ± 4 for the full symptomatic state, and 12 ± 6 for the recovered state. It should be noted that the [11C]raclopride and [18F]MPPF PET scans were always acquired in 1 d, with the [11C]raclopride collected first due to the short half-life of carbon-11. Except for those two ligands, the order of PET scans was randomized for each monkey and each state to avoid, as far as possible, the neurochemical effects of anesthesia. Although we cannot exclude that repeated anesthesia may have had an effect on our PET results, it is worth mentioning that treatment with zoletil does not affect the binding of dopaminergic and serotonergic transporters' radioligands (Elfving et al., 2003).
Before the tracer injection, a transmission scan (68 Ge rotating rod sources; 10 min) was acquired to correct for tissular 511 keV gamma attenuation. Dynamic acquisition started with the intravenous injection of the radiotracer and lasted 100 min for [18F]DOPA, 90 min for [11C]DASB, 70 min for [18F]MPPF, and 60 min for [11C]raclopride PET scans. Synthesis of radioligands was performed in the cyclotron unit of the CERMEP. The mean doses (±SD) of injected [18F]DOPA, [11C]raclopride, [11C]DASB, and [18F]MPPF were 139 ± 24, 143.9 ± 29.6, 156.9 ± 20, and 136.2 ± 28.1 MBq, respectively. The 3D emission data were reconstructed with attenuation and scatter correction by a 3D filtered back projection (Hamming filter; cutoff frequency, 0.5 cycles/pixel) algorithm and a zoom factor of three. Reconstructed volumes were 128 × 128 matrices of 0.32 × 0.32 mm2 pixels in 63 2.42 mm spaced planes.
PET data preprocessing.
To obtain parametric images, the PET studies were analyzed by suitable tracer kinetic modeling. The parameters computed were the uptake rate (Ki, 10−3 min−1) for [18F]DOPA and the BPND of [11C]raclopride, [11C]DASB, and [18F]MPPF. The [18F]DOPA Ki was calculated using frames recording for between 30 and 90 min for the linearization and the Patlak graphical analysis, considering the cerebellum as the reference area. Parametric images of BPND were obtained using a simplified reference tissue model (Gunn et al., 1997) with the cerebellum as the reference. Using Statistical Parametric Mapping (SPM8, Wellcome Trust Centre for NeuroImaging, University College London) in MATLAB (version 8.1, R2013a; The MathWorks), the parametric images were transformed into a common space using the brain Macaca fascicularis MRI template (Ballanger et al., 2013) available from http://www.cermep.fr (section download/atlas). Finally, a Gaussian-smoothing kernel of 6 mm was applied to the normalized parametric images to localize significant changes at the voxel level.
ROI-based analysis.
Tissue time–activity concentration curves were extracted using our maximum probability atlas (Ballanger et al., 2013) for eight ROIs within the BG (namely the anterior and posterior parts of the putamen, the caudate nucleus, the ventral striatum, and the external and internal segments of the pallidum), as well as 11 other ROIs within the thalamus; the raphe nucleus; the dorsal and ventral parts of the ACC; the dorsolateral, medial, orbital, and ventral parts of the frontal cortex; the amygdala; the hippocampus; and the insula considering regional distribution of DA and 5-HT.
Histological procedures for dopaminergic cell loss.
At the end of the experiments, the monkeys received a lethal overdose of anesthetic drug (pentobarbital) and were then transcardially perfused with saline, followed by 5 L of 4% paraformaldehyde fixative solution. The brains were removed from the skull, immersed in PBS with 20% sucrose for 48 h, frozen, and cut into 50-μm-thick frontal sections perpendicular to the intercommissural plane, which were stored for immunohistochemical procedures (see details in Jan et al., 2000). Dopaminergic innervation of the SN cell loss was studied by immunocytochemical localization of TH (see details in Mounayar et al., 2007). In the SNc, TH-positive cells were counted in nine regularly spaced sections covering the anteroposterior extent of the structure using an image analysis system (Mercator; ExploraNova). The sections were matched anatomically in each of the animals and the total number of TH-positive cells was estimated after correction using the Abercrombie method. The percentage of neuronal loss in the SNc was evaluated by comparison with baseline values from four intact monkeys (Table 1).
Table 1.
Dopaminergic cell loss
| Monkey | Dopaminergic cell loss (%) |
Total | ||
|---|---|---|---|---|
| A8 | A9 | A10 | ||
| MF3 | 74 | 86.4 | 73.2 | 78 |
| MF4 | 71.6 | 75.4 | 65.1 | 70.8 |
| MF5 | 72 | 80.8 | 68.1 | 73.8 |
| MF6 | 58.1 | 78.2 | 50.4 | 62.5 |
| MF7 | 71 | 68.3 | 61 | 66.9 |
| MF10 | Data not available | |||
| MF11 | 81.4 | 67.8 | 58 | 69.5 |
| Mean | 71.4 | 76.2 | 62.6 | 70.3 |
| SD | 7.5 | 7.2 | 8 | 5.4 |
Data represent the percentage of neuronal loss in the substantia nigra collected for each monkey (MF) at the end of the study (with the exception of MF10).
Statistical analyses.
Statistical differences in ROIs Ki and BPND among the four different states were assessed using two-way repeated-measures ANOVA, followed by the Holm post hoc test (see Table 2 for details). It should be noted that the [11C]DASB PET scan of MF11 collected in the recovered state was excluded for technical reasons. Nonlinear multivariate regressions were used to analyze dependences between the behavioral markers (motor scores and recovery time), the histological markers (SN cell loss), and the different binding parameters. For all of the statistical analyses performed using STATA 10.0/SE (StataCorpLP), a probability level of <5% (p < 0.05) was considered to be significant.
Table 2.
Evolution of [18F]DOPA uptake and [11C]Raclopride, [11C]DASB, and [18F]MPPF bindings among the different states and the different regions of interest
| Region | Part | [18F]Dopa Ki unit × 10−3 min−1 |
[11C]Raclopride BPND |
[11C]DASB BPND |
[18F]MPPF BPND |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Baseline (n = 7) | Early (n = 4) | Symptomatic (n = 7) | Recovered (n = 7) | Baseline (n = 5) | Early (n = 4) | Symptomatic (n = 6) | Recovered (n = 7) | Baseline (n = 5) | Symptomatic (n = 5) | Recovered (n = 4) | Baseline (n = 7) | Early (n = 4) | Symptomatic (n = 7) | Recovered (n = 7) | ||
| Putamen | Anterior | 5 ± 2.2 | 5.3 ± 2.2 | 2.2 ± 2† | 2 ± 0.8‡ | 3.7 ± 0.8 | 3.8 ± 0.4 | 4.2 ± 0.6 | 3.3 ± 0.7†‡ | 1.17 ± 0.2 | 1.3 ± 0.3 | 1.19 ± 0.2 | 0.74 ± 0.2 | 0.89 ± 0.2 | 0.84 ± 0.2 | 0.79 ± 0.1 |
| Posterior | 5.2 ± 2.3 | 6.2 ± 3.6 | 1.9 ± 2.3†/*† | 1.3 ± 0.7‡/*‡ | 4 ± 0.3 | 4.6 ± 0.2 | 4.9 ± 0.4 | 4.1 ± 0.8 | 1.09 ± 0.2 | 1.24 ± 0.2† | 1.13 ± 0.2†‡0.058 | 0.62 ± 0.2 | 0.72 ± 0.2 | 0.7 ± 0.1 | 0.65 ± 0.2 | |
| Caudate | Anterior | 4.3 ± 2.3 | 3.8 ± 1.9 | 1.8 ± 1.7† | 1.9 ± 0.8‡0.053 | 3.6 ± 0.5 | 3.7 ± 0.3 | 4 ± 0.5†0.06 | 3 ± 1.1†‡ | 0.88 ± 0.2 | 1.02 ± 0.3 | 0.92 ± 0.2 | 0.56 ± 0.2 | 0.73 ± 0.2*0.06 | 0.66 ± 0.2 | 0.67 ± 0.1 |
| Posterior | 3.8 ± 2.2 | 3.8 ± 2.8 | 1 ± 1.1†/*† | 1.1 ± 0.6‡/*‡ | 3.1 ± 0.2 | 3.5 ± 0.5 | 3.6 ± 0.7 | 2.8 ± 1.1 | 0.9 ± 0.2 | 1.04 ± 0.3 | 0.94 ± 0.2 | 0.38 ± 0.1 | 0.44 ± 0.1 | 0.42 ± 0.1 | 0.45 ± 0.2 | |
| Ventral striatum | Anterior | 3.4 ± 1.6 | 4.1 ± 1.4 | 2.8 ± 1.5 | 3.2 ± 0.7 | 2 ± 0.6 | 2 ± 0.4 | 2 ± 0.4 | 1.8 ± 0.3 | 1.11 ± 0.3 | 1.18 ± 0.5 | 1.15 ± 0.3 | 0.64 ± 0.2 | 0.81 ± 0.3 | 0.72 ± 0.2 | 0.69 ± 0.2 |
| Posterior | 2.2 ± 1 | 2.7 ± 1.8 | 1.1 ± 0.9 | 1.2 ± 0.5 | 2 ± 0.3 | 2.2 ± 0.2 | 2.2 ± 0.3 | 2.5 ± 1.4 | 1.05 ± 0.2 | 1.15 ± 0.4 | 1.06 ± 0.3 | 0.71 ± 0.1 | 0.70 ± 0.1 | 0.75 ± 0.1 | 0.69 ± 0.2 | |
| Pallidum | External | 4.4 ± 2.7 | 6 ± 4.2 | 2.5 ± 1.6 | 2.3 ± 0.6 | 2.8 ± 0.3 | 3 ± 0.3 | 3.1 ± 0.7 | 3 ± 0.8 | 1.18 ± 0.2 | 1.38 ± 0.3† | 1.26 ± 0.3 | ||||
| Internal | 2.1 ± 2 | 3.1 ± 2.3 | 1.6 ± 1.2 | 2.2 ± 0.8 | 1.4 ± 0.2 | 1.5 ± 0.2 | 1.5 ± 0.4 | 1.7 ± 1 | 1.16 ± 0.2 | 1.37 ± 0.3† | 1.21 ± 0.3 | |||||
| Thalamus | 1.55 ± 0.2 | 1.8 ± 0.4†0.087 | 1.67 ± 0.2 | |||||||||||||
| ACC | Ventral | 0.81 ± 0.3 | 0.9 ± 0.4 | 0.91 ± 0.3 | 1.36 ± 0.4 | 1.64 ± 0.6 | 1.44 ± 0.3 | 1.39 ± 0.3 | ||||||||
| Dorsal | 0.58 ± 0.2 | 0.7 ± 0.2 | 0.67 ± 0.1 | 1.84 ± 0.5 | 2.21 ± 0.6 | 1.99 ± 0.4 | 1.88 ± 0.3 | |||||||||
| Frontal cortex | Dorsolateral | 0.38 ± 0.2 | 0.44 ± 0.3 | 0.41 ± 0.1 | 1.23 ± 0.4 | 1.49 ± 0.4 | 1.37 ± 0.4 | 1.21 ± 0.4 | ||||||||
| Medial | 0.41 ± 0.2 | 0.49 ± 0.3 | 0.45 ± 0.2 | 1.51 ± 0.4 | 1.78 ± 0.5 | 1.63 ± 0.4 | 1.50 ± 0.5 | |||||||||
| Orbital | 0.51 ± 0.3 | 0.62 ± 0.3 | 0.54 ± 0.2 | 1.13 ± 0.3 | 1.34 ± 0.3*0.053 | 1.27 ± 0.2 | 1.10 ± 0.3*‡†‡0.066 | |||||||||
| Ventral | 0.37 ± 0.2 | 0.45 ± 0.3 | 0.39 ± 0.1 | 1.06 ± 0.3 | 1.18 ± 0.3 | 1.18 ± 0.2 | 1.00 ± 0.3 | |||||||||
| Limbic structures | Amygdala | 0.96 ± 0.3 | 0.9 ± 0.3 | 0.91 ± 0.3 | 1.19 ± 0.2 | 1.24 ± 0.2 | 1.26 ± 0.1 | 1.10 ± 0.2 | ||||||||
| Hippocampus | 0.61 ± 0.2 | 0.63 ± 0.2 | 0.6 ± 0.2 | 1.44 ± 0.3 | 1.46 ± 0.2 | 1.44 ± 0.3 | 1.34 ± 0.3 | |||||||||
| Insula | 1.05 ± 0.2 | 1.17 ± 0.3 | 1.09 ± 0.2 | 1.52 ± 0.4 | 1.67 ± 0.4 | 1.62 ± 0.3 | 1.46 ± 0.3 | |||||||||
| Raphe | 1.8 ± 0.2 | 2.17 ± 0.4 | 2.18 ± 0.2 | 0.56 ± 0.2 | 0.58 ± 0.1 | 0.52 ± 0.1 | 0.47 ± 0.3 | |||||||||
Data represent mean Ki or BPND values ± SD. Symbols in italics indicate significant differences. p < 0.05 between states: *baseline versus early symptomatic; †baseline versus full symptomatic; ‡baseline versus; *†early versus full symptomatic; *‡early symptomatic versus recovered; and †‡full symptomatic versus recovered.
In parallel, for an exploratory search involving the entire cortex, we also performed on the normalized parametric images voxel-by-voxel analyses using SPM8 software. Due to the small sample size, statistical maps were thresholded at a level of p < 0.001 uncorrected with an extent threshold of at least 80 contiguous voxels. For visualization, voxelwise levels of significance (t-maps) were projected onto the MF MRI template (Ballanger et al., 2013).
Results
MPTP intoxication, evolution of motor score during intoxication, and recovery
The monkeys received 3–5 injections of MPTP for a cumulative dose of 1.2 to 2.2 mg/kg (mean 1.6 mg/kg). Four monkeys (MF6, MF7, MF10, and MF11) were scanned in an early symptomatic state after two MPTP injections (parkinsonian scores of 8, 6, 5, and 1, respectively, at the time of the [18F]DOPA scan and of 9, 3, 5, and 1, respectively, at the time of the [11C]raclopride and [18F]MPPF scans; Fig. 1C). Among those animals, MF6 already presented overt mild motor symptoms (including bradykinesia, rigidity, freezing, and parkinsonian posture) and a slight reduction in the analysis of home cage activity and spontaneous eye movements associated with a mild parkinsonian posture were reported in the other animals. After full MPTP intoxication, all animals (7/7) developed motor symptoms and exhibited recovery after stopping treatment (Fig. 1C). Recovery was complete for most of the monkeys (5/7), whereas two monkeys (MF3 and MF10) presented only a partial recovery with mild motor symptoms (parkinsonian scores of 9 and 8, respectively, at the end; Fig. 1C). Of particular interest, in both monkeys, bradykinesia, parkinsonian posture, and a reduction in home cage activity were still observed during the recovery period. Interestingly, the maximal parkinsonian score obtained for those 2 monkeys (22.5 ± 0.7) was greater than the maximal score observed in monkeys with total recovery (13.4 ± 4.5). Recovery after the maximal effect and cessation of MPTP injections was gradual over 4–8 weeks (mean 38.8 ± 10.9 d to maximal recovery; Fig. 1C). No correlation was found between the motor score and the cumulative dose of MPTP received by each animal (r = 0.63, p = 0.13, n = 7). A positive correlation was found between the motor score and the recovery time (r = 0.85, p = 0.015, n = 7), indicating that, the greater the maximal effect, the longer the time to recovery (Fig. 1D). Finally, the maximal motor score was positively correlated with the total dopaminergic cell death in the SN (r = 0.87, p = 0.023, n = 6). Specifically, cell death in A9 and A10 were significantly correlated with motor scores (r = 0.85, p = 0.032 and r = 0.84, p = 0.036, respectively, n = 6). It is worth noting that cell death in A8 was not correlated with motor scores (r = 0.19, p = 0.72, n = 6). No significant correlation was reported between cell death and recovery time.
PET imaging at baseline
As expected, the regional distribution of radioactivity was in good agreement with that known from in vivo studies in humans for each radiotracer, with the highest uptake in the striatum for both the [18F]DOPA and [11C]raclopride radiotracers (Ito et al., 1999; Nagano et al., 2000), in the raphe and the thalamus for the [11C]DASB (Houle et al., 2000; Wilson et al., 2002), and in various components of the limbic system (such as the dorsal ACC, the insula, and the hippocampus) for [18F]MPPF ligand (Aznavour and Zimmer, 2007; Fig. 2A, Table 2).
Figure 2.
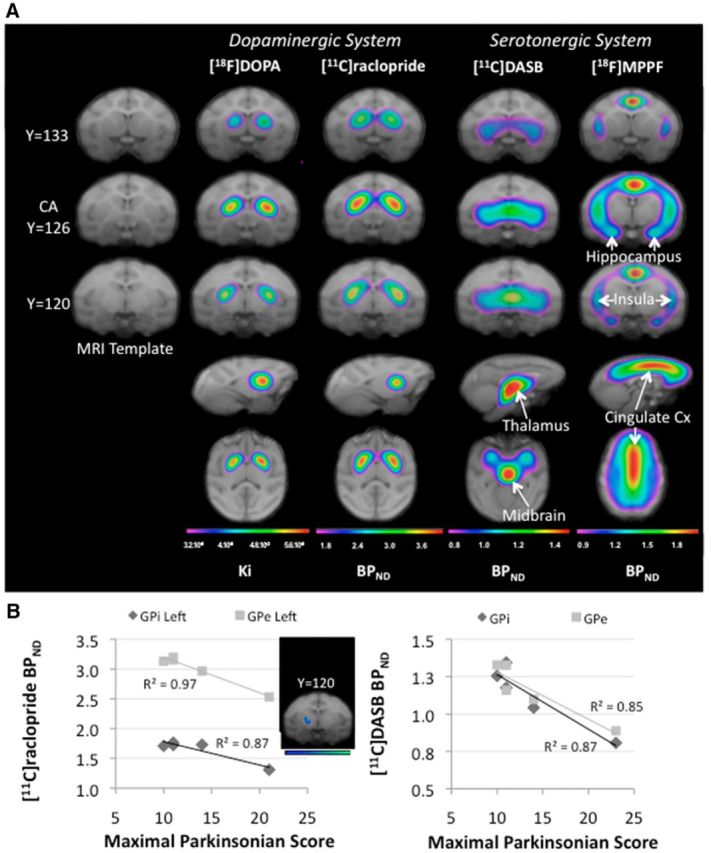
PET imaging at baseline. A, Spatially normalized mean parametric images of each radiotracer collected at baseline superimposed on the brain Macaca fascicularis MRI template (Ballanger et al., 2013). The parametric images are shown in color, whereas the MRIs are shown in grayscale. Colors represent the level of binding expressed as uptake ratio using the cerebellum as a nonspecific reference for each radioligand (Ki for [18F]DOPA and BPND for [11C]raclopride, [11C]DASB, and [18F]MPPF). Red represents high Ki or BPND, whereas blue denotes low Ki or BPND. For each radiotracer, three coronal (first three rows), one sagittal (fourth row), and one transaxial (fifth row) slices are shown. B, Negative correlation reported within the pallidum internal (GPi) and external (GPe) parts between both the [11C]Raclopride and [11C]DASB BPND collected at baseline and the maximal parkinsonian score obtained in the full symptomatic state (n = 5).
Regression analyses
Interestingly, both [11C]raclopride BPND and [11C]DASB BPND collected at baseline were inversely related to the maximal parkinsonian score (corresponding to the full symptomatic motor state) in multiple regions. In particular, negative correlations were reported within both the left GPe and GPi (r = −0.99, p = 0.002 and r = −0.93, p = 0.02 respectively, Fig. 2B, n = 5) for the [11C]raclopride BPND and in the amygdala (r = −0.88, p = 0.049), hippocampus (r = −0.88, p = 0.047), PCC (r = −0.98, p = 0.004), left insula (r = −0.90, p = 0.037), and right dorsal ACC (r = −0.91, p = 0.032), as well as the GPe and GPi (r = −0.92, p = 0.025 and r = −0.93, p = 0.02, respectively, Fig. 2B), the posterior striatum (r = −0.92, p = 0.027 for the caudate, r = −0.98, p = 0.005 for the putamen), and the posterior ventral striatum (r = −0.92, p = 0.026) for the [11C]DASB BPND (n = 5).Moreover, we also found a negative correlation between the total time to recovery and the [11C]DASB BPND collected at baseline in the amygdala (r = −0.94, p = 0.017), hippocampus (r = −0.93, p = 0.02), and the GPe and GPi (r = −0.94, p = 0.019 and r = −0.96, p = 0.009, respectively) and the posterior parts of the putamen and the ventral striatum (r = −0.93, p = 0.023 and r = −0.97, p = 0.005, respectively, n = 5). Finally, cell death in A9 was negatively correlated with [18F]DOPA uptake collected at baseline within the GPe and the putamen posterior (r = −0.90, p = 0.01 and r = −0.85, p = 0.03 respectively, n = 6), whereas cell death in A8 was negatively correlated with [11C]raclopride binding collected at baseline within the anterior caudate (r = −0.92, p = 0.025, n = 5) and the putamen in its anterior (r = −0.97, p = 0.005, n = 5) and posterior parts (r = −0.92, p = 0.026, n = 5). In parallel, there was also a negative relationship between the percentage of cell death in A8 and the level of [11C]DASB binding collected at baseline within the anterior putamen (r = −0.99, p = 0.01, n = 4). It is worth noting that cell death in A10 was negatively correlated with 11C]raclopride binding collected at baseline in both the GPe (r = −0.96, p = 0.008, n = 5) and the putamen posterior ventral (r = −0.96, p = 0.01, n = 5), whereas the percentage of total dopaminergic cell death within the SN was inversely related to the level of [11C]DASB binding collected at baseline within the GPi (r = −0.96, p = 0.04, n = 4).
Changes in the early symptomatic state
Compared with baseline, ROI-based analyses only revealed a trend for an increase in [18F]MPPF BPNDin the early symptomatic state within the anterior caudate (F(1,15) = 8.67, p = 0.06) and the orbitofrontal cortex (F(1,15) = 8.52; p = 0.053; Table 2). In parallel, SPM analyses revealed a cluster (k = 563 voxels) of increased Ki with a peak of activity in the left anterior putamen (T = 7.98; Fig. 3B); two clusters of increased [11C]raclopride BPND in the right and left parts of the posterior putamen (T = 9.11, k = 447 voxels and T = 8.42, k = 102 voxels respectively, Fig. 4B); and three clusters of increased [18F]MPPF BPND in the right orbitofrontal cortex (k = 139 voxels, T = 58.28), right dorsal ACC (k = 660 voxels, T = 55.08), and right orbitofrontal cortex extending to the anterior caudate (k = 119 voxels, T = 47.8; Fig. 5B). Finally, regression analysis showed a negative relationship between [18F]DOPA uptake and motor status (both collected in the early symptomatic state) within the right GPe (r = −0.97, p = 0.047, n = 4; Fig. 3D).
Figure 3.
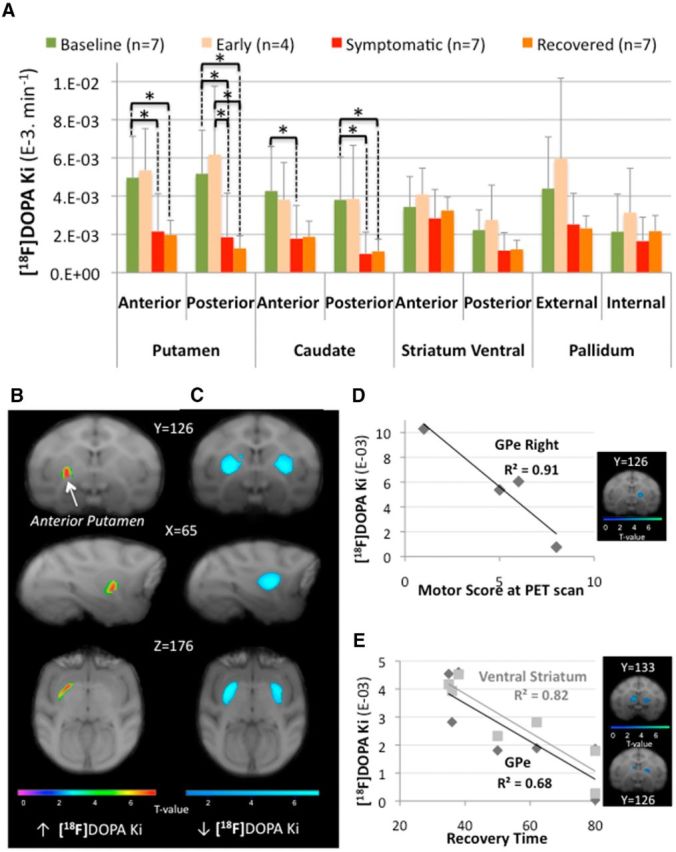
[18F]DOPA uptake modifications. A, ROI assessment of [18F]DOPA uptake (Ki) between the different states (n: number of animals in each state). Asterisks indicate significant differences (p < 0.05). The bar graphs show the mean ± SD. B, C, SPM showing changes in [18F]DOPA uptake (Ki) rendered over coronal (first row), sagittal (second row), and transaxial (third row) sections of the brain Macaca fascicularis MRI template from Ballanger et al. (2013). The statistical threshold of the voxel-based analyses was set at p < 0.001 uncorrected. Color bars represent the t value. Specifically, B displays the significant cluster of increase Ki observed within the left anterior putamen in the early symptomatic state compared with baseline and C displays significant clusters (involving the bilateral putamen) of decreased Ki observed in the recovered state compared with baseline. D, E, Correlation analyses between [18F]DOPA uptake (Ki) and motor score or recovery time during the different states. In particular, motor scores were negatively correlated with Ki values within the external pallidum (GPe) collected in the early symptomatic state (D, n = 4) and recovery time was negatively correlated with Ki values within the GPe and the ventral striatum collected during the full symptomatic state (E, n = 7). For illustration, results from voxelwise regression with SPM are shown at a more liberal threshold (uncorrected p < 0.05, D, and p < 0.01, E). Color bars represent the t value.
Figure 4.
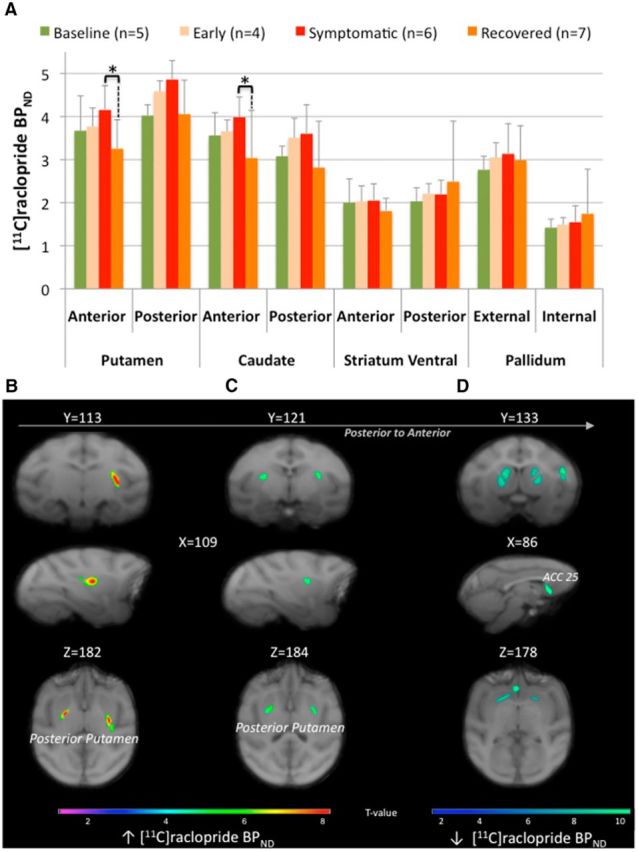
[11C]Raclopride binding potential modifications. A, ROI assessment of [11C]raclopride binding potential (BPND) between the different states (n: number of animals in each state). Asterisks indicate significant differences (p < 0.05). The bar graphs show the mean ± SD. B–D, SPM showing changes in [11C]raclopride binding potential (BPND) rendered over coronal (first row), sagittal (second row), and transaxial (third row) sections of the brain Macaca fascicularis MRI template from Ballanger et al. (2013). The statistical threshold of the voxel-based analyses was set at p < 0.001 uncorrected. Specifically, B and C display the significant clusters of increase BPND within the bilateral posterior putamen observed in both the early symptomatic (B) and full symptomatic (C) states compared with baseline, whereas D displays the significant clusters of decreased BPND within the striatum and the limbic part of the ACC (BA 25) observed in the recovered state compared with the full symptomatic condition. Color bars represent the t value.
Figure 5.

[18F]MPPF binding potential modifications. A, ROI assessment of [18F]MPPF binding potential (BPND) between the different states (n: number of animals in each state). The bar graphs show the mean ± SD. B, SPM showing changes in [18F]MPPF BPND rendered over coronal (first row), sagittal (second row), and transaxial (third row) sections of the brain Macaca fascicularis MRI template from Ballanger et al. (2013). The statistical threshold of the voxel-based analysis was set at p < 0.001 uncorrected. Color bars represent the t value. B, Significant clusters of increased [18F]MPPF BPND observed in the early symptomatic state compared with baseline within the anterior caudate and at the cortical level within the orbitofrontal cortex and the dorsal ACC.
Changes in the full symptomatic state
Compared with baseline, ROI assessment of [18F]DOPA uptake showed a significant decrease in the putamen (in its anterior (F(1,15) = 9.22; p = 0.049) and posterior (F(1,15) = 12.85; p = 0.011) parts) and the caudate nucleus (in its anterior (F(1,15) = 9.35; p = 0.048) and posterior (F(1,15) = 16.21; p = 0.0066) parts; Fig. 3A). Interestingly, the Ki values collected during the full symptomatic state were also significantly reduced compared with the early symptomatic state in the posterior striatum (F(1,15) = 9.88; p = 0.02 and F(1,15) = 8.87; p = 0.037 for the putamen and the caudate, respectively). The SPM analysis was in good agreement with these latter findings by revealing four clusters of decreased Ki values in the full symptomatic compared with the early symptomatic state within the left and right posterior putamen (k = 1282, T = 6.23 and k = 527, T = 4.96), as well as the left and right caudate posterior (k = 490, T = 5.42 and k = 104, T = 4.83, respectively). In the pallidum, the SPM analysis (full symptomatic state vs baseline) revealed a significant decrease of Ki values within the bilateral GPe (k = 547 voxels and T = 8.63 for the left side and k = 439 voxels, T = 6.22 for the right side, data not shown). In parallel, ROI analysis of [11C]raclopride BPND showed a trend for an increase in the anterior caudate nucleus compared with baseline (F(1,12) = 8.62; p = 0.06; Fig. 4A, Table 2), whereas the SPM analysis revealed a significant increase of [11C]raclopride BPND in the posterior putamen [T = 5.3 for the cluster (k = 112) in the left side and T = 5.12 for the cluster (k = 98) in the right side; Figure 4C]. Assessment of 5-HT innervation using PET imaging of the SERT with [11C]DASB revealed a significant increase (compared with baseline) in the posterior putamen (F(1,7) = 22.34; p = 0.0064) and the pallidum in its external (F(1,7) = 13.17; p = 0.025) and internal (F(1,7) = 10.8; p = 0.04) segments (Fig. 6A,B, Table 2). At the postsynaptic level, SPM analysis revealed a cluster of increased [18F]MPPF BPND in the right DLPFC (k = 127 voxels, T = 6.85, data not shown).
Figure 6.
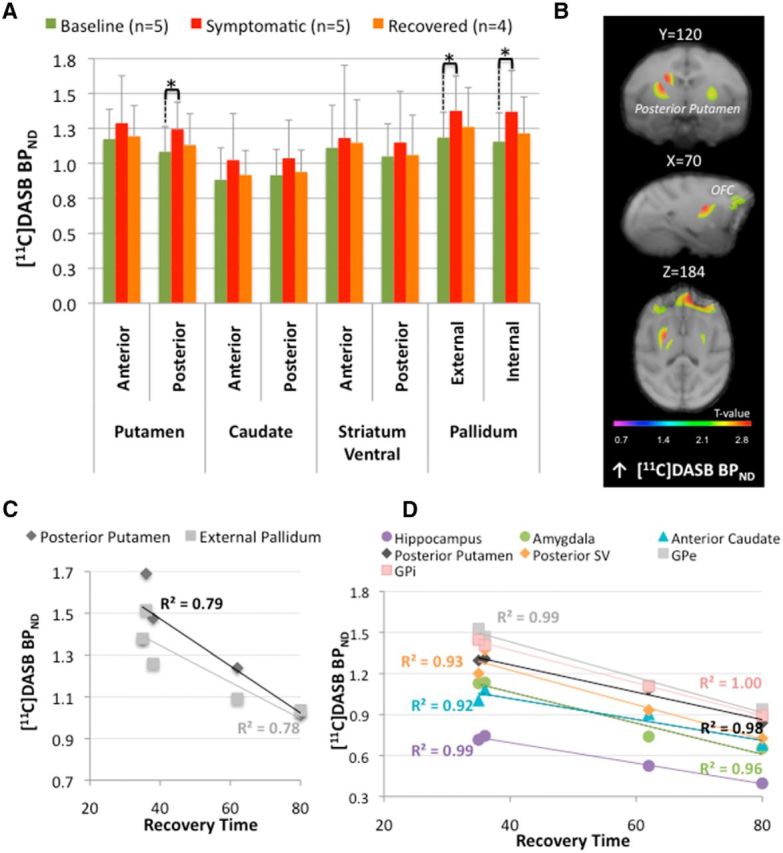
[11C]DASB binding potential modifications. A, ROI assessment within the basal ganglia (BG) of [11C]DASB binding potential (BPND) between the different states (n: number of animals in each state). Asterisks indicate significant differences (p < 0.05). The bar graphs show the mean ± SD. B, SPM showing changes in [11C]DASB BPND rendered over coronal (first row), sagittal (second row), and transaxial (third row) sections of the brain Macaca fascicularis MRI template from Ballanger et al. (2013). The statistical threshold of the voxel-based analysis was set at p < 0.001 uncorrected. Color bars represent the t value. Specifically, the figure illustrates the increase of [11C]DASB BPND observed in the full symptomatic state compared with baseline within the bilateral posterior putamen. C, D, Correlation analyses between [11C]DASB BPND and recovery time during the full symptomatic and recovered states. Overall recovery time was negatively correlated with [11C]DASB BPND within the posterior putamen and the external pallidum during the full symptomatic state (C, n = 5) and within the pallidum, the posterior putamen and ventral striatum, the anterior caudate, and the hippocampus and amygdala during the recovered state (D, n = 4).
Regression analyses showed that [18F]DOPA uptake collected during the full symptomatic state in the anterior ventral striatum was inversely correlated with the motor status of the monkeys (r = −0.79, p = 0.03, n = 7, data not shown). Negative correlations were also detected between Ki values in the full symptomatic state and recovery time in the anterior ventral striatum and the GPe (respectively r = −0.91, p = 0.005 and r = −0.83, p = 0.022, n = 7; Fig. 3E). In addition, we found negative relationships between [11C]DASB BPND collected during the symptomatic state in the posterior putamen and the motor score (r = −0.90, p = 0.036, n = 5) and recovery time (r = −0.88, p = 0.046, n = 5). There was also a negative correlation between [11C]DASB bindingcollected in the symptomatic state and the recovery time in the GPe (r = −0.89, p = 0.043, n = 5; Fig. 6C). Finally, there was a positive relationship between the percentage of cell death in A8 and the amount of [11C]raclopride binding in the symptomatic state compared with baseline within the anterior ventral striatum (r = 0.92, p = 0.03, n = 5). The monkeys with greater neuronal loss in the retrorubral area were those with the larger increase in [11C]raclopride binding in the symptomatic state compared with baseline within this specific region. In parallel, we found a negative relationship between the percentage of cell death in A10 and the difference in [11C]DASB binding between the full symptomatic state and baseline in the caudate nucleus in its anterior (r = −0.97, p = 0.03) and posterior (r = −0.99, p = 0.006) parts (n = 4). The monkeys with less neuronal loss in the ventral tegmental region were those with the larger increase in [11C]DASB binding in the symptomatic state compared with baseline within this structure.
Changes in the recovered state
Interestingly, ROI-based analysis showed no statistical difference in [18F]DOPA uptake between the full symptomatic and recovered states, whereas, compared with the early symptomatic state, [18F]DOPA uptake was greatly decreased in the posterior striatum (putamen (F(1,15) = 13.28; p = 0.012) and caudate (F(1,15) = 7.82; p = 0.04). Compared with baseline, the loss of [18F]DOPA uptake was observed in both the putamen in its anterior (F(1,15) = 10.48; p = 0.033) and posterior (F(1,15) = 17.62; p = 0.0047) parts and the posterior caudate nucleus (F(1,15) = 14.47; p = 0.009), with a trend in the anterior caudate (F(1,15) = 8.52; p = 0.053). In parallel, ROI analysis of [11C]raclopride BPND did not reveal any difference between the recovered state and baseline. However, there was an overall tendency for decreased [11C]raclopride BPND in the recovered state compared with the symptomatic state. This decrease was significant in the anterior striatum caudate (F(1,12) = 14.38; p = 0.015) and putamen (F(1,12) = 14.20; p = 0.016) (Fig. 4A). In addition, SPM analysis confirmed the ROI analysis by revealing two clusters within the right anterior putamen (k = 249, T = 9.22) and the right anterior caudate (k = 80, T = 8.59), as well as a significant cluster of decreased [11C]raclopride BPND within the limbic ACC (k = 877, T = 14.09; Fig. 4D). Finally, assessment of the 5-HT system revealed a significant decrease of [18F]MPPF BPND in the orbitofrontal cortex (compared with the early symptomatic state; F(1,15) = 10.62; p = 0.03) and a trend for decreased [18F]MPPF BPND in the orbitofrontal cortex and decreased [11C]DASB BPND in the posterior putamen (compared with the full symptomatic state; F(1,15) = 7.30; p = 0.066 and F(1,7) = 7.51; p = 0.058, respectively).
Regression analyses showed that recovery time was inversely related to the [18F]DOPA uptake collected in the recovered state in the anterior ventral striatum (r = −0.8; p = 0.034, n = 7, data not shown), as well as to the [18F]MPPF binding collected in the hippocampus during the recovered state (r = −0.76, p = 0.047, n = 7, data not shown) and the [11C]DASB binding (n = 5) in multiple regions: the hippocampus (r = −0.99, p = 0.004), the amygdala (r = −0.98, p = 0.02), the GPe (r = −0.99, p = 0.007), the GPi (r = −0.99, p = 0.001), the posterior parts of the putamen and ventral striatum (r = −0.99, p = 0.009 and r = −0.96, p = 0.036, respectively), and the anterior caudate (r = −0.96, p = 0.043). In parallel, a positive significant relationship between recovery time and [11C]raclopride binding in the posterior putamen collected in the recovered state was found (r = 0.82, p = 0.048, n = 7, data not shown). Finally, cell death in A9 was positively correlated with the delta of [18F]DOPA uptake between baseline and recovered state within the GPe and the putamen posterior, as well as the putamen posterior ventral (r = 0.83, p = 0.04, r = 0.81, p = 0.049, and r = 0.84, p = 0.04 respectively, n = 6). The monkeys with greater neuronal loss in the SNc were those with a larger decrease in [18F]DOPA uptake in the recovered state compared with baseline within these three regions.
Discussion
To our knowledge, this is the first multitracer PET imaging study in nonhuman primates investigating dopaminergic and serotonergic changes both presynaptically and postsynaptically in four different motor states. Two main results emerged. First, different compensatory mechanisms were highlighted, depending on the level of dopaminergic depletion. In particular, our findings supported the importance of dopaminergic neurotransmission in extrastriatal structures (GPe and ACC) for compensation with an early presynaptic regulation followed by more prominent changes in the expression of D2/D3 dopaminergic receptors during recovery. Second, we identified early changes in the expression of 5-HT1A receptors and presynaptic serotonergic modifications strongly related to the recovery time.
Different DA compensatory mechanisms depending on the parkinsonian state
During the early symptomatic state, we found an increase in [18F]DOPA uptake (thought to reflect increased AAAD activity) in the anterior putamen, consistent with the classic hypothesis of dopaminergic striatal compensation in the surviving fibers (Lee et al., 2000; Pifl and Hornykiewicz, 2006). This was associated with an increase in [11C]raclopride binding (thought to reflect an increased number of dopaminergic D2/D3 receptors) specifically in the posterior striatum, similar to PD patients (Nandhagopal et al., 2011). Because parkinsonian motor features mainly depend on the degree of DA depletion in the sensorimotor striatum (Kaasinen et al., 2000; Nandhagopal et al., 2009), it is not surprising that the few remaining fibers in this territory are also involved in the early compensatory phenomenon. A strength of our progressive protocol was the possibility of collecting behavioral and histological markers, which allowed us to perform correlation analyses with the PET data to gain additional information. Such analyses revealed the importance of the relationship between dopaminergic level in the GPe and the appearance and severity of the motor symptoms. In particular, the more the monkeys presented parkinsonian symptoms during the early symptomatic state, the less [18F]DOPA uptake was observed in the GPe, suggesting its implication in early compensatory mechanisms, probably by maintaining the minimum DA concentration required for normal motor functioning. Interestingly, in de novo parkinsonian patients, an increase of [18F]DOPA uptake has been reported in the GPi (Whone et al., 2003; Pavese et al., 2012). It is notable that [18F]DOPA uptake has been shown to reflect the activity of overall brain monoaminergic population. Accordingly, we cannot exclude the possibility that an increase in [18F]DOPA uptake might also reflect the AAAD activity of serotonergic terminals (Pavese et al., 2012). This issue needs further study.
The recovered state is particularly interesting because this probably represents the lesional situation closest to PD patients around the time of diagnosis. Indeed, we showed an SNc cell loss of ∼74%, a reduction of [18F]DOPA uptake by ∼63% in the striatum of recovered monkeys that was higher in the posterior putamen (72%), and a relative sparing of AAAD activity in the anterior ventral striatum throughout the different states (Doudet et al., 2006; Blesa et al., 2012). These findings are consistent with the extensive loss of dopaminergic neurons that has already taken place when patients are diagnosed with PD (Hornykiewicz and Kish, 1987; Morrish et al., 1998); the characteristic caudorostral gradient of DA degeneration and greater vulnerability of the dopaminergic projection onto the posterior putamen region, as reported in PD patients (Snow et al., 2000; De La Fuente-Fernández et al., 2003); and the partial preservation of the mesolimbic DA innervation also observed in PD patients (Farley et al., 1977; Brooks and Piccini, 2006). Also of interest were the relationships reported between dopaminergic activity within the posterior putamen (as revealed by PET), cell loss in the SNc, the maximal motor impairment, and the recovery time. Indeed, recovered monkeys with higher SNc cell loss were those with greater parkinsonian impairment and higher reduction of [18F]DOPA uptake in the posterior putamen compared with baseline. Moreover, the monkeys with longer recovery were those with higher [11C]raclopride binding, suggesting greater DA depletion in this structure. Moreover, we provided further evidence that dopaminergic fibers remaining inside the associative limbic territory of the striatum could also be involved in the recovery process (Mounayar et al., 2007; Boulet et al., 2008). This was reflected within the anterior ventral striatum with the negative correlation between recovery time and [18F]DOPA uptake collected in both the symptomatic and recovered states. The monkeys with high AAAD activity in the anterior ventral striatum had faster recovery. Finally, in both the symptomatic and recovered states, striatal AAAD activity was very low compared with baseline with no significant difference between them, as observed previously (Boulet et al., 2008; Blesa et al., 2012). This lack of difference between these opposite motor states strongly suggests the existence of extrastriatal mechanisms during recovery. Indeed, we demonstrated a significant decrease in the limbic ACC during this period. Moreover, we found that monkeys with a higher level of [18F]DOPA uptake in the GPe presented faster recovery, confirming the contribution of pallidal dopaminergic neurotransmission also in the recovery mechanisms (Mounayar et al., 2007; Neumane et al., 2012).
Serotonin modifications: implications for compensation or indirect markers of competition with the DA system
Previously, we suggested a role for serotonergic neurotransmission in motor recovery by showing an increase of striatal serotonergic fibers in recovered monkeys (Mounayar et al., 2007). Here, we report an increase in [11C]DASB binding within the BG during the symptomatic state, confirming and extending previous results obtained with immunohistological (Mounayar et al., 2007) and biochemical (Boulet et al., 2008) techniques. Specifically, the changes mainly involved the posterior putamen and the GPe, in which negative correlations were reported between [11C]DASB binding and recovery time. These results strongly suggested a positive involvement of serotonergic innervation in compensation within these structures, at least for functional motor recovery. However, we cannot exclude that this SERT upregulation is not directly responsible for the recovery phenomenon, but rather is an artifact related to the competition between the 5-HT and DA systems. A decrease in DA fibers may allow the sprouting process of serotonergic fibers to take place, leading to an increase in density of serotonergic terminals, as observed in the present study with the [11C]DASB radiotracer. Nevertheless, if the 5-HT system is not involved in the recovery process, how can we explain the strong correlations observed in the recovery state between [11C]DASB binding and recovery time in many brain structures (including the BG, hippocampus, and amygdala)? Consistent with this idea, we recently showed that administration of 3,4-methylenedioxy-N-methamphetamine to induce a 5-HT lesion in MPTP-recovered monkeys led to the reappearance of the rigidity only, showing the importance of 5-HT transmission in the posterior putamen and the GPi for this particular symptom (Beaudoin-Gobert et al., 2015).
At the postsynaptic level, the present study revealed early changes in the expression of 5-HT1A receptors in subcortical and cortical structures that were less affected by the MPTP-induced dopaminergic degeneration, such as the anterior caudate, the ACC, and the OFC. Although this does not provide direct evidence for the involvement of 5-HT1A receptors in early compensatory mechanisms, it might be interesting to investigate in humans whether [18F]MPPF could be a promising biomarker of early degenerative changes in PD. This should be explored further, because postmortem studies have found an increase in 5-HT1A receptor levels in both the orbitofrontal and temporal cortex of parkinsonian patients (Chen et al., 1998; Huot et al., 2012).
Finally, the inverse relationship observed in multiple regions between the [11C]DASB binding collected at baseline and the maximal motor impairment obtained in the full symptomatic stage (or the total time for recovery) raises the question of whether baseline density of serotonergic terminals will predict the intensity of motor impairment after MPTP treatment. Indeed, the monkeys with lower levels of [11C]DASB binding at baseline had a higher maximal motor score in the parkinsonian condition. Is it possible that the [11C]DASB radiotracer may be a marker of vulnerability to develop symptom expression? In the same vein, it has been suggested that alterations in availability of serotonergic terminals may be an important factor in the development of anxiety (Reimold et al., 2008; Oler et al., 2009). Accordingly, is there a relationship between the level of anxiety and the vulnerability of some animals to express parkinsonian symptoms and their ability to recover from the parkinsonian state? Further investigations should be performed to elucidate these issues.
In conclusion, the present work highlights the role of pallidal dopaminergic neurotransmission in both the early compensatory mechanisms and the functional recovery mechanisms, with reduced AAAD activity closely related to the appearance or perseveration of motor symptoms. This finding expands possibilities for new treatments (such as cell-based therapies) that could artificially promote dopaminergic levels within the GPe to delay disease expression. In parallel, this study provides preliminary evidence of the role of the serotonergic system in compensatory mechanisms. Nonetheless, future studies are needed to determine whether there are changes in SERT availability in the early symptomatic state and if [18F]MPPF PET imaging might be a promising biomarker of early degenerative changes in PD.
Footnotes
This work was supported by the French National Agency of Research (Grant ANR-09-MNPS-018). We thank Jean-Luc Charrieau and Fabrice Hérant for expert animal care and C. Kennard for proofreading of the manuscript and English corrections.
The authors declare no competing financial interests.
References
- Agid Y, Javoy F, Glowinski J. Hyperactivity of remaining dopaminergic neurones after partial destruction of the nigro-striatal dopaminergic system in the rat. Nat New Biol. 1973;245:150–151. doi: 10.1038/newbio245150a0. [DOI] [PubMed] [Google Scholar]
- Altar CA, Marien MR, Marshall JF. Time course of adaptations in dopamine biosynthesis, metabolism, and release following nigrostriatal lesions: implications for behavioral recovery from brain injury. J Neurochem. 1987;48:390–399. doi: 10.1111/j.1471-4159.1987.tb04106.x. [DOI] [PubMed] [Google Scholar]
- Aznavour N, Zimmer L. [18F]MPPF as a tool for the in vivo imaging of 5-HT1A receptors in animal and human brain. Neuropharmacology. 2007;52:695–707. doi: 10.1016/j.neuropharm.2006.09.023. [DOI] [PubMed] [Google Scholar]
- Ballanger B, Klinger H, Eche J, Lerond J, Vallet AE, Le Bars D, Tremblay L, Sgambato-Faure V, Broussolle E, Thobois S. Role of serotonergic 1A receptor dysfunction in depression associated with Parkinson's disease. Mov Disord. 2012;27:84–89. doi: 10.1002/mds.23895. [DOI] [PubMed] [Google Scholar]
- Ballanger B, Tremblay L, Sgambato-Faure V, Beaudoin-Gobert M, Lavenne F, Le Bars D, Costes N. A multi-atlas based method for automated anatomical Macaca fascicularis brain MRI segmentation and PET kinetic extraction. Neuroimage. 2013;77:26–43. doi: 10.1016/j.neuroimage.2013.03.029. [DOI] [PubMed] [Google Scholar]
- Beaudoin-Gobert M, Epinat J, Météreau E, Duperrier S, Neumane S, Ballanger B, Lavenne F, Liger F, Tourvielle C, Bonnefoi F, Costes N, Bars DL, Broussolle E, Thobois S, Tremblay L, Sgambato-Faure V. Behavioural impact of a double dopaminergic and serotonergic lesion in the non-human primate. Brain. 2015;138:2632–2647. doi: 10.1093/brain/awv183. [DOI] [PubMed] [Google Scholar]
- Bernheimer H, Birkmayer W, Hornykiewicz O, Jellinger K, Seitelberger F. Brain dopamine and the syndromes of Parkinson and Huntington: clinical, morphological and neurochemical correlations. J Neurol Sci. 1973;20:415–455. doi: 10.1016/0022-510X(73)90175-5. [DOI] [PubMed] [Google Scholar]
- Bezard E, Gross CE. Compensatory mechanisms in experimental and human parkinsonism: towards a dynamic approach. Prog Neurobiol. 1998;55:93–116. doi: 10.1016/S0301-0082(98)00006-9. [DOI] [PubMed] [Google Scholar]
- Bezard E, Bioulac B, Gross CE. Glutamatergic compensatory mechanisms in experimental parkinsonism. Prog Neuropsychopharmacol Biol Psychiatry. 1998;22:609–623. doi: 10.1016/S0278-5846(98)00030-X. [DOI] [PubMed] [Google Scholar]
- Blesa J, Pifl C, Sánchez-González MA, Juri C, García-Cabezas MA, Adánez R, Iglesias E, Collantes M, Peñuelas I, Sánchez-Hernández JJ, Rodríguez-Oroz MC, Avendaño C, Hornykiewicz O, Cavada C, Obeso JA. The nigrostriatal system in the presymptomatic and symptomatic stages in the MPTP monkey model: a PET, histological and biochemical study. Neurobiol Dis. 2012;48:79–91. doi: 10.1016/j.nbd.2012.05.018. [DOI] [PubMed] [Google Scholar]
- Boulet S, Mounayar S, Poupard A, Bertrand A, Jan C, Pessiglione M, Hirsch EC, Feuerstein C, François C, Féger J, Savasta M, Tremblay L. Behavioral recovery in MPTP-treated monkeys: neurochemical mechanisms studied by intrastriatal microdialysis. J Neurosci. 2008;28:9575–9584. doi: 10.1523/JNEUROSCI.3465-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brix G, Zaers J, Adam LE, Bellemann ME, Ostertag H, Trojan H, Haberkorn U, Doll J, Oberdorfer F, Lorenz WJ. Performance evaluation of a whole-body PET scanner using the NEMA protocol. National Electrical Manufacturers Association. J Nucl Med. 1997;38:1614–1623. [PubMed] [Google Scholar]
- Brooks DJ, Pavese N. Imaging biomarkers in Parkinson's disease. Prog Neurobiol. 2011;95:614–628. doi: 10.1016/j.pneurobio.2011.08.009. [DOI] [PubMed] [Google Scholar]
- Brooks DJ, Piccini P. Imaging in Parkinson's disease: the role of monoamines in behavior. Biol Psychiatry. 2006;59:908–918. doi: 10.1016/j.biopsych.2005.12.017. [DOI] [PubMed] [Google Scholar]
- Chen CP, Alder JT, Bray L, Kingsbury AE, Francis PT, Foster OJ. Post-synaptic 5-HT1A and 5-HT2A receptors are increased in Parkinson's disease neocortex. Ann N Y Acad Sci. 1998;861:288–289. doi: 10.1111/j.1749-6632.1998.tb10229.x. [DOI] [PubMed] [Google Scholar]
- De La Fuente-Fernández R, Lim AS, Sossi V, Adam MJ, Ruth TJ, Calne DB, Stoessl AJ, Lee CS. Age and severity of nigrostriatal damage at onset of Parkinson's disease. Synapse. 2003;47:152–158. doi: 10.1002/syn.10160. [DOI] [PubMed] [Google Scholar]
- Doudet DJ, Rosa-Neto P, Munk OL, Ruth TJ, Jivan S, Cumming P. Effect of age on markers for monoaminergic neurons of normal and MPTP-lesioned rhesus monkeys: a multi-tracer PET study. Neuroimage. 2006;30:26–35. doi: 10.1016/j.neuroimage.2005.09.044. [DOI] [PubMed] [Google Scholar]
- Elfving B, Bjørnholm B, Knudsen GM. Interference of anaesthetics with radioligand binding in neuroreceptor studies. Eur J Nucl Med Mol Imaging. 2003;30:912–915. doi: 10.1007/s00259-003-1171-8. [DOI] [PubMed] [Google Scholar]
- Farley IJ, Price KS, Hornykiewicz O. Dopamine in thelimbic regions of the human brain: normal and abnormal. Adv Biochem Psychopharmacol. 1977;16:57–64. [PubMed] [Google Scholar]
- Gaspar P, Febvret A, Colombo J. Serotonergic sprouting in primate MTP-induced hemiparkinsonism. Exp Brain Res. 1993;96:100–106. doi: 10.1007/BF00230443. [DOI] [PubMed] [Google Scholar]
- Gunn RN, Lammertsma AA, Hume SP, Cunningham VJ. Parametric imaging of ligand-receptor binding in PET using a simplified reference region model. Neuroimage. 1997;6:279–287. doi: 10.1006/nimg.1997.0303. [DOI] [PubMed] [Google Scholar]
- Hornykiewicz O, Kish SJ. Biochemical pathophysiology of Parkinson's disease. Adv Neurol. 1987;45:19–34. [PubMed] [Google Scholar]
- Houle S, Ginovart N, Hussey D, Meyer JH, Wilson AA. Imaging the serotonin transporter with positron emission tomography: initial human studies with [11C]DAPP and [11C]DASB. Eur J Nucl Med. 2000;27:1719–1722. doi: 10.1007/s002590000365. [DOI] [PubMed] [Google Scholar]
- Huot P, Johnston TH, Visanji NP, Darr T, Pires D, Hazrati LN, Brotchie JM, Fox SH. Increased levels of 5-HT1A receptor binding in ventral visual pathways in Parkinson's disease. Mov Disord. 2012;27:735–742. doi: 10.1002/mds.24964. [DOI] [PubMed] [Google Scholar]
- Ito H, Okubo Y, Halldin C, Farde L. Mapping of central D2 dopamine receptors in man using [11C]raclopride: PET with anatomic standardization technique. Neuroimage. 1999;9:235–242. doi: 10.1006/nimg.1998.0401. [DOI] [PubMed] [Google Scholar]
- Jan C, François C, Tandé D, Yelnik J, Tremblay L, Agid Y, Hirsch E. Dopaminergic innervation of the pallidum in the normal state, in MPTP-treated monkeys and in parkinsonian patients. Eur J Neurosci. 2000;12:4525–4535. [PubMed] [Google Scholar]
- Kaasinen V, Ruottinen HM, Någren K, Lehikoinen P, Oikonen V, Rinne JO. Upregulation of putaminal dopamine D2 receptors in early Parkinson's disease: a comparative PET study with [11C] raclopride and [11C]N-methylspiperone. J Nucl Med. 2000;41:65–70. [PubMed] [Google Scholar]
- Lee CS, Samii A, Sossi V, Ruth TJ, Schulzer M, Holden JE, Wudel J, Pal PK, de la Fuente-Fernandez R, Calne DB, Stoessl AJ. In vivo positron emission tomographic evidence for compensatory changes in presynaptic dopaminergic nerve terminals in Parkinson's disease. Ann Neurol. 2000;47:493–503. [PubMed] [Google Scholar]
- Morrish PK, Rakshi JS, Bailey DL, Sawle GV, Brooks DJ. Measuring the rate of progression and estimating the preclinical period of Parkinson's disease with [18F]dopa PET. J Neurol Neurosurg Psychiatr. 1998;64:314–319. doi: 10.1136/jnnp.64.3.314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mounayar S, Boulet S, Tandé D, Jan C, Pessiglione M, Hirsch EC, Féger J, Savasta M, François C, Tremblay L. A new model to study compensatory mechanisms in MPTP-treated monkeys exhibiting recovery. Brain. 2007;130:2898–2914. doi: 10.1093/brain/awm208. [DOI] [PubMed] [Google Scholar]
- Nagano AS, Ito K, Kato T, Arahata Y, Kachi T, Hatano K, Kawasumi Y, Nakamura A, Yamada T, Abe Y, Ishigaki T. Extrastriatal mean regional uptake of fluorine-18-FDOPA in the normal aged brain–an approach using MRI-aided spatial normalization. Neuroimage. 2000;11:760–766. doi: 10.1006/nimg.2000.0584. [DOI] [PubMed] [Google Scholar]
- Nandhagopal R, Kuramoto L, Schulzer M, Mak E, Cragg J, Lee CS, Mckenzie J, McCormick S, Samii A, Troiano A, Ruth TJ, Sossi V, de la Fuente-Fernandez R, Calne DB, Stoessl AJ. Longitudinal progression of sporadic Parkinson's disease: a multi-tracer positron emission tomography study. Brain. 2009;132:2970–2979. doi: 10.1093/brain/awp209. [DOI] [PubMed] [Google Scholar]
- Nandhagopal R, Kuramoto L, Schulzer M, Mak E, Cragg J, McKenzie J, McCormick S, Ruth TJ, Sossi V, de la Fuente-Fernández R, Stoessl AJ. Longitudinal evolution of compensatory changes in striatal dopamine processing in Parkinson's disease. Brain. 2011;134:3290–3298. doi: 10.1093/brain/awr233. [DOI] [PubMed] [Google Scholar]
- Neumane S, Mounayar S, Jan C, Epinat J, Ballanger B, Costes N, Féger J, Thobois S, François C, Sgambato-Faure V, Tremblay L. Effects of dopamine and serotonin antagonist injections into the striatopallidal complex of asymptomatic MPTP-treated monkeys. Neurobiol Dis. 2012;48:27–39. doi: 10.1016/j.nbd.2012.06.002. [DOI] [PubMed] [Google Scholar]
- Oler JA, Fox AS, Shelton SE, Christian BT, Murali D, Oakes TR, Davidson RJ, Kalin NH. Serotonin transporter availability in the amygdala and bed nucleus of the stria terminalis predicts anxious temperament and brain glucose metabolic activity. J Neurosci. 2009;29:9961–9966. doi: 10.1523/JNEUROSCI.0795-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavese N, Simpson BS, Metta V, Ramlackhansingh A, Chaudhuri KR, Brooks DJ. [18F]FDOPA uptake in the raphe nuclei complex reflects serotonin transporter availability: a combined [18F]FDOPA and [11C]DASB PET study in Parkinson's disease Neuroimage. 2012;59:1080–1084. doi: 10.1016/j.neuroimage.2011.09.034. [DOI] [PubMed] [Google Scholar]
- Pessiglione M, Guehl D, Hirsch EC, Féger J, Tremblay L. Disruption of self-organized actions in monkeys with progressive MPTP-induced parkinsonism. I. Effects of task complexity. Eur J Neurosci. 2004;19:426–436. doi: 10.1111/j.0953-816X.2003.03088.x. [DOI] [PubMed] [Google Scholar]
- Pifl C, Hornykiewicz O. Dopamine turnover is upregulated in the caudate/putamen of asymptomatic MPTP-treated rhesus monkeys. Neurochemistry International. 2006;49:519–524. doi: 10.1016/j.neuint.2006.03.013. [DOI] [PubMed] [Google Scholar]
- Politis M, Wu K, Loane C, Kiferle L, Molloy S, Brooks DJ, Piccini P. Staging of serotonergic dysfunction in Parkinson's disease: an in vivo 11C-DASB PET study. Neurobiol Dis. 2010a;40:216–221. doi: 10.1016/j.nbd.2010.05.028. [DOI] [PubMed] [Google Scholar]
- Politis M, Wu K, Loane C, Turkheimer FE, Molloy S, Brooks DJ, Piccini P. Depressive symptoms in PD correlate with higher 5-HTT binding in raphe and limbic structures. Neurology. 2010b;75:1920–1927. doi: 10.1212/WNL.0b013e3181feb2ab. [DOI] [PubMed] [Google Scholar]
- Rakshi JS, Uema T, Ito K, Bailey DL, Morrish PK, Ashburner J, Dagher A, Jenkins IH, Friston KJ, Brooks DJ. Frontal, midbrain and striatal dopaminergic function in early and advanced Parkinson's disease A 3D [(18)F]dopa-PET study. Brain. 1999;122:1637–1650. doi: 10.1093/brain/122.9.1637. [DOI] [PubMed] [Google Scholar]
- Reimold M, Batra A, Knobel A, Smolka MN, Zimmer A, Mann K, Solbach C, Reischl G, Schwärzler F, Gründer G, Machulla HJ, Bares R, Heinz A. Anxiety is associated with reduced central serotonin transporter availability in unmedicated patients with unipolar major depression: a [11C]DASB PET study. Mol Psychiatry. 2008;13:606–613. 557. doi: 10.1038/sj.mp.4002149. [DOI] [PubMed] [Google Scholar]
- Ribeiro MJ, Vidailhet M, Loc'h C, Dupel C, Nguyen JP, Ponchant M, Dollé F, Peschanski M, Hantraye P, Cesaro P, Samson Y, Remy P. Dopaminergic function and dopamine transporter binding assessed with positron emission tomography in Parkinson disease. Arch Neurol. 2002;59:580–586. doi: 10.1001/archneur.59.4.580. [DOI] [PubMed] [Google Scholar]
- Schneider JS, Kovelowski CJ. Chronic exposure to low doses of MPTP. I. Cognitive deficits in motor asymptomatic monkeys. Brain Res. 1990;519:122–128. doi: 10.1016/0006-8993(90)90069-N. [DOI] [PubMed] [Google Scholar]
- Schneider JS, Rothblat DS, DiStefano L. Volume transmission of dopamine over large distances may contribute to recovery from experimental parkinsonism. Brain Res. 1994;643:86–91. doi: 10.1016/0006-8993(94)90012-4. [DOI] [PubMed] [Google Scholar]
- Schroeder JA, Schneider JS. GABA(A) and mu-opioid receptor binding in the globus pallidus and entopeduncular nucleus of animals symptomatic for and recovered from experimental Parkinsonism. Brain Res. 2002;947:284–289. doi: 10.1016/S0006-8993(02)03010-X. [DOI] [PubMed] [Google Scholar]
- Snow BJ, Vingerhoets FJ, Langston JW, Tetrud JW, Sossi V, Calne DB. Pattern of dopaminergic loss in the striatum of humans with MPTP induced parkinsonism. J Neurol Neurosurg Psychiatr. 2000;68:313–316. doi: 10.1136/jnnp.68.3.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder GL, Keller RW, Jr, Zigmond MJ. Dopamine efflux from striatal slices after intracerebral 6-hydroxydopamine: evidence for compensatory hyperactivity of residual terminals. J Pharmacol Exp Ther. 1990;253:867–876. [PubMed] [Google Scholar]
- Whone AL, Moore RY, Piccini PP, Brooks DJ. Plasticity of the nigropallidal pathway in Parkinson's disease. Ann Neurol. 2003;53:206–213. doi: 10.1002/ana.10427. [DOI] [PubMed] [Google Scholar]
- Wilson AA, Ginovart N, Hussey D, Meyer J, Houle S. In vitro and in vivo characterisation of [11C]-DASB: a probe for in vivo measurements of the serotonin transporter by positron emission tomography. Nucl Med Biol. 2002;29:509–515. doi: 10.1016/S0969-8051(02)00316-5. [DOI] [PubMed] [Google Scholar]
- Zigmond MJ. Do compensatory processes underlie the preclinical phase of neurodegenerative disease? Insights from an animal model of parkinsonism. Neurobiol Dis. 1997;4:247–253. doi: 10.1006/nbdi.1997.0157. [DOI] [PubMed] [Google Scholar]
- Zigmond MJ, Acheson AL, Stachowiak MK, Stricker EM. Neurochemical compensation after nigrostriatal bundle injury in an animal model of preclinical parkinsonism. Arch Neurol. 1984;41:856–861. doi: 10.1001/archneur.1984.04050190062015. [DOI] [PubMed] [Google Scholar]