

Abstract
While reactive microgliosis is a hallmark of advanced stages of amyotrophic lateral sclerosis (ALS), the role of microglial cells in events initiating and/or precipitating disease onset is largely unknown. Here we provide novel in vivo evidence of a distinct adaptive shift in functional microglial phenotypes in preclinical stages of superoxide dismutase 1 (SOD1)-mutant-mediated disease. Using a mouse model for live imaging of microglial activation crossed with SOD1G93A and SOD1G37R mouse models, we discovered that the preonset phase of SOD1-mediated disease is characterized by development of distinct anti-inflammatory profile and attenuated innate immune/TLR2 responses to lipopolysaccharide (LPS) challenge. This microglial phenotype was associated with a 16-fold overexpression of anti-inflammatory cytokine IL-10 in baseline conditions followed by a 4.5-fold increase following LPS challenge. While infusion of IL-10R blocking antibody, initiated at day 60, caused a significant increase in markers of microglial activation and precipitated clinical onset of disease, a targeted overexpression of IL-10 in microglial cells, delivered via viral vectors expressed under CD11b promoter, significantly delayed disease onset and increased survival of SOD1G93A mice. We propose that the high IL-10 levels in resident microglia in early ALS represent a homeostatic and compensatory “adaptive immune escape” mechanism acting as a nonneuronal determinant of clinical onset of disease.
SIGNIFICANCE STATEMENT We report here for the first time that changing the immune profile of brain microglia may significantly affect clinical onset and duration of disease in ALS models. We discovered that in presymptomatic disease microglial cells overexpress anti-inflammatory cytokine IL-10. Given that IL-10 is major homeostatic cytokine and its production becomes deregulated with aging, this may suggest that the capacity of microglia to adequately produce IL-10 may be compromised in ALS. We show that blocking IL-10 increased inflammation and precipitated clinical disease onset, whereas overexpression of IL-10 in microglia using a gene therapy approach significantly delayed disease onset and increased survival of ALS mice. Based on our results, we propose that targeted overexpression of IL-10 in microglia may have therapeutic potential in ALS.
Keywords: ALS, biophotonics, innate immunity, microglia, TLR2, transgenic mice
Introduction
Amyotrophic lateral sclerosis (ALS) is a late-onset neurodegenerative disorder characterized by progressive muscle weakness leading to a lethal paralysis. The majority of ALS cases are sporadic with no known genetic components, whereas ∼10% of ALS cases are familial (FALS). Approximately one-fifth of these familial cases are caused by mutations in superoxide dismutase 1 (SOD1; Rosen et al., 1993). In the past decades, much effort has been focused on elucidating the mechanisms of disease induced by SOD1. However, despite the extensive research, the exact nature of the underlying toxicity of mutant SOD1 remains unknown. Increasing evidence suggests that misfolded SOD1 (mSOD1) species common for all mutations may be secreted and involved in cytotoxicity (Urushitani et al., 2006; Zhao et al., 2010; Zetterström et al., 2011; Grad et al., 2014). Furthermore, using conformation-specific antibodies, the misfolded SOD1 species have been detected in CSF and spinal cords of sporadic ALS patients, potentially suggesting a common SOD1-dependent pathogenic mechanism in ALS (Bosco et al., 2010; Zetterström et al., 2011; Synofzik et al., 2012).
As an adult-onset neurodegenerative disorder, ALS may have an asymptomatic phase spanning over several decades; however, despite intensive research efforts, the pathological events precipitating disease onset and the mechanisms of early disease spreading remain unclear. Transgenic mice overexpressing superoxide dismutase 1 mutants linked to familial ALS develop progressive motor neuron disease with many pathological features resembling the human disease (Gurney, 1994). In addition, in SOD1 mutant mice the sequence of disease related pathological events evolve in a predictable timely manner; therefore, this disease model system provides a powerful tool to study early disease mechanisms. While previous evidence revealed expression of the pathogenic misfolded SOD1 species in subset of large motor neurons as early as postnatal day 7 (Saxena et al., 2013), the role of microglial cells in the mechanisms influencing early disease pathogenesis is largely unknown.
In the current study, using the sensitive biophotonic ALS models for in vivo imaging of microglial activation/innate immune response developed in our laboratory, we were able to visualized early alterations in microglial phenotypes observed in presymptomatic SOD1G93A and SOD1G37R mice. Our results revealed that, contrary to our expectations, the preonset phase of SOD1-mediated disease is characterized by the development of a distinct anti-inflammatory profile of microglial cells, attenuated Toll-like receptor 2 (TLR2) responses to controlled immune challenge, and a 16-fold overexpression of anti-inflammatory cytokine IL-10. IL-10 is a key immunoregulatory/anti-inflammatory cytokine that mediates a feedback inhibition loop and limits excessive production of proinflammatory cytokines such as TNFα, IL-1β, and IL-6 (Howard and O'Garra, 1992; Moore et al., 2001). To investigate, whether early induction of IL-10 in presymptomatic SOD1 mutant microglia represents an adaptive or maladaptive microglia polarization phenotype, we initiated treatment of ALS mice with specific IL-10 receptor blocking antibody. Here we show that treatment with specific IL-10 receptor blocking antibody, initiated in presymptomatic mice, causes a significant increase in markers of microglial activation and precipitates the clinical onset of disease. On the other hand, a targeted overexpression of IL-10 in lumbar spinal cord microglia delivered via intrathecal injection of AAV2/9 vector expressed under the CD11b promoter significantly delayed the clinical onset of disease and increased survival in SOD1G93A mice. Together, our results suggest that early induction of IL-10 represents a homeostatic and adaptive microglial mechanism that may act as an endogenous, nonneuronal determinant of clinical onset of disease. Finally, we show that therapies aiming to induce the state of microglial immunological homeostasis, such as overexpression of IL-10 in microglial cells, may have therapeutic potential in ALS.
Materials and Methods
Generation of TLR2-Fluc-AcGFP;SOD1G93A and TLR2-Fluc-AcGFP;SOD1G37R transgenic mice
The transgenic TLR2-Fluc-AcGFP reporter mice were generated as described previously (Lalancette-Hébert et al., 2009) and maintained heterozygous in the C57BL/6 background. Transgenic mice overexpressing the SOD1G93A mutation (B6SJL-TgN_[SOD1-G93A]_1 Gur) were purchased from The Jackson Laboratory. Transgenic mice expressing the SOD1G37R mutation (line 29) were a gift from Drs. P. Wong and D. Price from John Hopkins University (Baltimore, MD). The TLR2-Fluc-AcGFP reporter mice were crossed with the SOD1 mutants to generate TLR2-Fluc-AcGFP;SOD1G93A and TLR2-Fluc-AcGFP;SOD1G37R double-transgenic mice. To avoid the effect of genetic background on disease progression and survival, all experiments were performed on age-matched littermates. All genotypes were assessed by PCR. The TLR2-Fluc transgenic mice were detected by the amplification of the luciferase transgene as described previously (Lalancette-Hébert et al., 2009), the SOD1G93A transgene was detected according to the Jackson Laboratory protocols (Keller et al., 2009, 2011), and the SOD1G37R transgene was detected as described previously (Gowing et al., 2009). In our experiments, we used animals of either sex randomly divided into different experimental groups.
Virus construction and preparation
Self-complementary adenoassociated virus (scAAV) serotype 2/9 viral vector CD11b-IL10, encoding the protein IL-10, was prepared using the inverted terminal repeat (ITR)-CMV vector. Mouse IL-10 cDNA tagged with Myc-DDK (OriGene Technologies) was amplified by PCR to generate BamH1 and Not1 restriction sites and then cloned into the initial vector. For the scAAV2/9 viral vector CD11b-IL10 used to express the IL-10 protein specifically under the control of the human CD11b promoter, the CD11b promoter was obtained by PCR from pBluescriptSK+-CD11b-TKmt-30 as described previously (Gowing et al., 2006) and excised to generate a Kpn1/Mlu1 fragment. It was then cloned inside the scAAV2/9-CMV-IL10 after removing the CMV promoter with the same restriction enzymes. Cloning was performed in SURE Competent Cells (Stratagene) to preserve the two ITR sites of the vectors. As described previously (Patel et al., 2014), scAAV recombinant viruses were produced in the 293T cell line cultured in DMEM (Invitrogen) supplemented with 10% normal bovine serum, 100 U/ml penicillin G, and 100 μg/ml streptomycin.
In vivo bioluminescence imaging
As described previously (Maysinger et al., 2007; Cordeau et al., 2008), the images were gathered using an IVIS 200 Imaging System (PerkinElmer). Twenty-five minutes before the imaging session, the mice received intraperitoneal injection of the luciferase substrate d-luciferine (150 mg/kg, dissolved in 0.9% saline; PerkinElmer). Mice were anesthetized with 2% isoflurane in 100% oxygen at a flow rate of 2 L/min and placed in the heated, light-tight imaging chamber. Images were collected using a high-sensitivity charge-coupled device camera with wavelengths ranging from 300 to 600 nm. Exposition time for imaging was 1 min using different fields of view and a F/1 lens aperture. The bioluminescence emission was normalized and displayed in photons per seconds per centimeter squared per steradian (Keller et al., 2009, 2011).
Lipopolysaccharide treatment
Lipopolysaccharide (LPS; serotype O55:B5) from Escherichia coli was dissolved in saline and administered by intraperitoneal injection at 5 mg/kg. Mice were maintained on heating pads between the LPS injection and the various imaging sessions (Lalancette-Hébert et al., 2009).
Surgical procedures
Stereotaxic brain injection.
Mice were anesthetized with 2% isoflurane in 100% oxygen at a flow rate of 2 L/min and placed in a stereotaxic apparatus (David Kopf Instruments). Transgenic and wild-type (WT) mice received intracerebroventricular injection of recombinant mutant SOD1 (1 μg/μl) into the right ventricle. A volume of 2 μl was infused over 4 min using a microinjection pump (model A-99; Razel Scientific Instruments).
Intracerebroventricular brain infusion.
Mouse IL-10 receptor (mIL-10R) blocking antibodies (CD210; BD Pharminogen) were infused through a brain infusion cannula connected with osmotic minipumps (model 2006; pumping rate, 0.15 μl/h; duration of treatment, 42 d; Alzet). The osmotic pumps were filled with either 200 μl of antibody (mIL-10R, 0.5 mg/ml) or saline and preconditioned overnight in 37°C saline. SOD1G93A mice (60 d old) were anesthetized with 2% isoflurane in 100% oxygen at a flow rate of 2 L/min and placed in a stereotaxic apparatus. The sterile brain infusion cannula was implanted into the right ventricle and was fixed with dental cement. The osmotic pumps were implanted under the skin. Mice received continuous 3.6 μg/d of antibody for 42 d. In total, 16 SOD1G93A mice received antibodies, and 13 mice were implanted with saline filled pumps as controls. Measurements of body weight and hindlimb reflex were used to score the clinical effect on SOD1G93A mice. The extensibility and postural reflex of the hindlimbs when the mice were held up by their tails were scored as described previously (Urushitani et al., 2006). Reflex score were gathered twice a week in a blind manner by animal technicians who had no information of the treated mice, but had experience in grading SOD1G93A mice. The survival was defined as the age when the animal could not stand on its feet within 30 s when placed on its side.
Intrathecal injection of AAV-CD11b-IL10.
At 33 d of age, WT control mice and SOD1G93A mice were injected intrathecally with 5.75 × 1010 viral particles of scAAV2/9-Cd11b-IL10 vector or scAAV-scFvD1.3 control vector by a 25 μl Hamilton syringe slipped under the dura, where the vector was slowly released into the CSF. To confirm the position of the syringe between the L4 and L5, the tail reflex of each mice was assessed. Mice were all anesthetized with 2% isoflurane during surgical procedures. To confirm the correct expression of IL-10 in lumbar spinal cord following intrathecal injections, tissues from 90-d-old WT mice and from end-point SODG93A mice were collected for immunohistochemistry labeling Iba1 (1:500, rabbit polyclonal anti-Iba1; Wako Chemicals) and cMyc (1:10, homemade monoclonal mouse anti-cMyc raised against the 9E10 epitope). The sections were washed in PBS and then incubated in corresponding fluorescent secondary antibody (Invitrogen).
Tissue collection.
Animals were anesthetized by an intraperitoneal injection of ketamine/xylazine (100/10 mg/kg) and transcardially perfused with 0.9% saline, followed by ice-cold 4% paraformaldehyde (PFA) at pH 7.4 dissolved in PBS. Tissue samples were then postfixed overnight in 4% PFA and equilibrated in PBS/30% sucrose for 48 h. Tissue were cut in 25 μm transverse sections on a Leica frozen microtome and kept in a cryoprotective solution at −20°C.
Immunofluorescence
As described previously (Lalancette-Hébert et al., 2007), the sections were then incubated overnight at room temperature using primary antibodies, 1:500 rabbit polyclonal anti-Iba1 (Wako Chemicals) and 1:250 mouse anti-TLR2 (eBioscience). After wash in PBS, the sections were incubated in a corresponding fluorescent goat secondary antiserum (Invitrogen).
In situ hybridization
The expression and localization of mRNA encoding for TLR2 and TNF-α were detected on olfactory bulb (OB) and lumbar spinal cord sections using a 35S-labeled riboprobes. Protocols for probe synthesis and in situ hybridization were described previously by Lalancette-Hébert et al. (2007). Slides were dried out under a vacuum overnight, postfixed in 4% paraformaldehyde, and digested by proteinase K (10 μg/ml in 0.1 m Tris HCl, pH 8.0, and 50 mm EDTA, pH 8.0, at 37°C for 25 min), after which the sections were rinsed in water and by a solution of 0.1 m triethanolamine (TEA, pH 8.0), acetylated in 0.25% acetic anhydride in 0.1 m TEA, and dehydrated. The hybridization involves 107 cpm/ml/slide of hybridization mixture and incubation at 60°C overnight in a slide warmer. Slides were rinsed in standard saline citrate (SSC; 1×; 0.15 m NaCl, 15 mm trisodium citrate buffer, pH 7.0) and digested by RNase A at 37°C (20 μg/ml), rinsed in descending concentrations of SSC, washed in 0.1× SSC, and dehydrated through graded concentrations of ethanol. The sections were exposed to X-ray film (Kodak) for 18–24 h and dipped in NTB2 nuclear emulsion (diluted 1:1 with distilled water; Kodak). Slides were kept at 4°C for 2 weeks safe from light, developed in D19 developer (Kodak) at 14°C, washed in water, and fixed in rapid fixer (Kodak). After exposition, tissues were rinsed in distilled water for 45 min and counterstained with thionin (0.25%). Slides were analyzed under dark-field illumination (Nguyen et al., 2001). Counts were made of clusters of silver grain hybridization signal in olfactory bulbs and lumbar spinal cord sections.
Western blots
Following LPS and mSOD1 stimulation in vivo, tissues were collected, and whole protein lysates were extracted by sonication in urea lysis buffer (1% SDS, 6 m urea, Tris HCl 6.8). Protein concentrations were determined by the Bradford method. Protein lysates were fractioned by SDS-PAGE, YM1 was detected with 1/1000 antibody dilution (Stem Cell Technologies, anti-mouse Ym1), and actin was used as protein load control (1/30,000 dilution; Millipore).
Primary adult microglia cultures.
WT and SOD1G93A adult mice (8–9 weeks old) were transcardially perfused with ice-cold saline supplemented with heparine (2 U/ml; Sigma-Aldrich). Olfactory bulb, cerebral cortex, and lumbar spinal cord, from which meninges were removed, were collected in ice-cold hibernate medium (Hibernate A medium; BrainBits) supplemented with B-27 (1×) and 0.5 mm GlutaMax-I, l-ananyl-l-glutamine (Invitrogen). Tissues were dissociated in Earle's balanced salt solution (EBSS; 2 ml) containing 2 mg/ml papain and 100 U/ml DNase (Worthington). The enzymatic reaction was quenched by adding EBSS (0.2 ml) containing 10 mg/ml ovomucoid protease inhibitor and 10 mg/ml albumin (Worthington), and 2 ml culture medium (DMEM with GlutaMax-I) supplemented with 15% heat-inactivated fetal bovine serum and 1% penicillin–streptomycin (Invitrogen). The cells were filtered through a 40 μm cell strainer (BD Bioscience) and separated using a discontinuous Percoll (GE Healthcare) density gradient centrifugation. Cells collected from the Percoll gradient interphase were washed in culture medium and plated on culture-treated glass slides (Becton Dickinson) at a 1.25 × 106 cells/ml density and incubated at 37°C in 95% air/5% CO2 in a humidified culture incubator. After 24 h in cultures, cells were challenged with 1 μg/ml LPS in culture medium for 30 min. Cells were then either homogenized in QIAzol for RNA extraction or fixed with 4% paraformaldehyde and processed for immunocytochemical assays.
Primary cultures: IL-10 treatments.
Primary cell cultures were obtained from the brains of PN1-PN4 SOD1G93A mutant pups. Brains were collected and placed in ice-cold PBS. Following mechanical dissociation, five or six brains were incubated in a 0.25% Trypsine-EDTA solution (Sigma) containing 250K U/ml DNase I (Sigma). After centrifugation, the cell pellets were placed in T-75 cm2 flasks (Sarstedt) for 10 d at 37°C, 5% CO2, in DMEM high-glucose media with 10% fetal bovine serum and antibiotic solution (Sigma). At confluence, the cells were plated onto coverslips in 24 well plates at a concentration of 10,000 cells/well in media. Cells were incubated with G-CSF 24 h later to allow adhesion. At 4 d in vitro, wells were divided into three groups: group 1 received mutant SOD1 (0.5 μg/ml) for 48 h; group 2 received IL-10 (30 ng/ml) from 24 h before SOD1 (0.5 μg/ml) challenge and were fixed 48 h after the SOD1 treatment; group 3 received CD210 (IL-10 receptor blocking antibody, 5 μg/ml and 15 min before adding IL-10) plus IL-10 (30 ng/ml) from 24 h before SOD1 (0.5 μg/ml) challenge and were fixed 48 h after the SOD1 stimulation. Cells were fixed in 4% paraformaldehyde, pH 7.4, for 20 min. Following one-step wash, cells were incubated in cold methanol for 10 min, washed, permeabilized in 0.25% Triton-X for 10 min, blocked in 3% normal goat serum for 1 h, and then were incubated overnight at 4°C using primary antibodies (TNFα, 1:500; Ym1, 1:500; Cd11b, 1:500).
Reverse transcriptase-PCR
Cells from the microglia cultures were homogenized in QIAzol buffer (Qiagen), and total RNA was extracted using the RNeasy Micro Kit (Qiagen) on-column treatment following the manufacturer's instructions. RNA concentrations were measured using a NanoDrop spectrophotometer (Thermo Fisher Scientific), and quality was assessed with an Agilent 2100 Bioanalyser (Agilent Technologies). First-strand cDNA synthesis was accomplished with Superscript III RNase H reverse transcriptase (Invitrogen) and random hexamer oligonucleotides. Subsequent PCR amplifications were performed using oligonucleotides targeting IL-1β, IL-6, TNF-α, IGF-1, TGF-β1, and IL-10. All reactions were normalized using the following reference genes: ATP synthase O subunit (ATP50), hypoxanthine guanine phosphoribosyl transferase 1 (HPRT1), and 18S ribosomal RNA (18S). Quantitative real-time PCR measurements were performed by the qRT-PCR platform of the Centre de Génomique de Québec.
Cytokine array
The inflammatory cytokines expression profile was analyzed with a mouse cytokine antibody array (RayBio Mouse Inflammation Antibody Array 1; RayBiotech). Protein lysates were obtained from WT and SOD1G93A transgenic mice 24 h after LPS intraperitoneal injection by homogenization of olfactory bulbs and lumbar spinal cords segments in 1× cell lysis buffer (included in the RayBiotech kit) with a protease inhibitor cocktail (Sigma, catalog #P8340). The protein concentration was determined for each sample and diluted at 300 μg in 1× blocking buffer. Samples for each group (four mice/group) were pooled and incubated with the array membrane overnight at 4°C. After washing in the supplied washing buffer, membranes were incubated with biotin-conjugated antibodies overnight. Signal detection was performed according to RayBiotech protocols, and membranes were exposed to X-ray film (Biomax MR1; Kodak). The film was analyzed using the Agfa Arcus II system and ImageJ software to measure the optical density of each spot on the membrane.
Flow cytometry
Blood samples were collected from WT and SOD1G93A, with and without 24 h LPS stimulation, by submandibular cheek puncture (100 μl) in EDTA-coated vials (Sarstedt) and maintained under continuous agitation. The blood samples were incubated with Fc Block CD16/32 antibodies (BD Pharmingen) to minimize nonspecific fluorescent labeling. Red blood cells were lysed using Pharm Lyse buffer (BD Bioscience). Samples were then labeled with CD45-V500, CD4-APC, CD8-PE-CF594 and CD11b-PE, or CD45-V500, CD4-APC, and CD25-FITC (BD Bioscience). The panel labeled for CD45, CD4, and CD25 was fixed and permeabilized using the Mouse FoxP3 Buffer set (BD Pharmingen) and cells were then labeled with FoxP3-V450, Il-4-PECy7, and Il-10-PE (BD Bioscience). After labeling, cells were resuspended in stain buffer (FBS; BD Pharmingen), and FACS analysis was performed on an LSRII flow cytometer (BD Bioscience) using FACSDiva software (BD Bioscience).
Results
TLR2-luc-GFP;SOD1G93A mice, a mouse model for live analysis of early microglia phenotypes in ALS
To investigate preonset microglia phenotypes and to evaluate the role of innate immune responses in early disease mechanisms, we took advantage of the TLR2 reporter mouse model generated previously in our laboratory (Lalancette-Hébert et al., 2009). In this model, a TLR2 induction (luciferase expression detectable as bioluminescence photon emission) can be visualized longitudinally from the brain and/or spinal cord of the living mice using bioluminescence/biophotonic imaging and a high-sensitivity/high-resolution CCD camera. It is noteworthy that previous work revealed a role of TLR2 in the pathogenesis of FALS (Liu et al., 2009; Zhao et al., 2010). The double-transgenic TLR2-luc-GFP;SOD1G93A mice were generated by crossing heterozygous mice carrying the SOD1G93A transgene with the heterozygous TLR2-luc-GFP mice coexpressing a dual nonpathogenic reporter transgenes luciferase (luc) and green fluorescent protein (GFP) under transcriptional control of the murine TLR2 gene promoter (Fig. 1A–J). The rationale for using this mouse model emerged from the fact that TLR2 expression is very low or undetectable in resting, nonactivated microglia, but is strongly induced in microglial cells in response to infection or brain injury (Lalancette-Hébert et al., 2009, 2012). Moreover, previous analysis of the TLR2-luc-GFP mouse model clearly demonstrated expression of the TLR2-driven transgenes luc and GFP in activated microglia, revealing that this in vivo model system can be used to measure TLR2-mediated microglial activation (Lalancette-Hébert et al., 2009, 2012).
Figure 1.
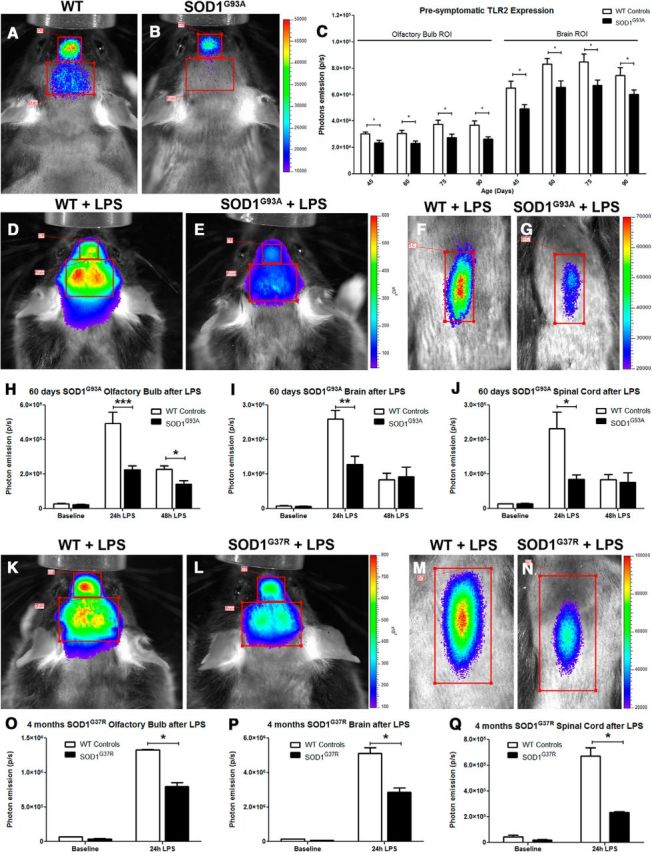
In vivo biophotonic/bioluminescence imaging of TLR2-SOD1 mice. A, B, Representative real-time imaging of the TLR2 induction in TLR2-WT controls (A) and TLR2-SOD1G93A double-transgenic mice (B) at 20 weeks of age (color scales represent photons/second). C, The longitudinal quantitative analysis of the total photon emissions from olfactory bulb and brain ROIs at various time points shows a reduced TLR2 activation in presymptomatic SOD1G93A mice (n between 10 and 20 mice per group per time point; p < 0.05 for all time points). D–G, Real-time imaging of the TLR2 response following LPS stimulation in presymptomatic (60 d) SOD1G93A mice shows a significant reduction in TLR2 activation in the SOD1G93A mice compared to WT littermates. H–J, Photonic quantification from the olfactory bulb ROI 24 h after LPS stimulation (WT, n = 4; SOD1G93A, n = 8; p = 0.0006) and 48 h after LPS (WT, n = 5; SOD1G93A, n = 4; p = 0.0269; H), from the brain ROI 24 h after LPS (WT, n = 4; SOD1G93A, n = 8; p = 0.0065) and 48 h after LPS (WT, n = 4; SOD1G93A, n = 5; I), and from lumbar spinal cord ROI 24 h after LPS (WT, n = 5; SOD1G93A, n = 5; p = 0.0181) and 48 h after LPS (WT, n = 4; SOD1G93A, n = 5; J). K–N, Real-time imaging of the TLR2 response following LPS stimulation in presymptomatic (4 month) SOD1G37R mice show a similarly significant reduction of TLR2 activation. O–Q, Photonic quantification from the olfactory bulb ROI 24 h after LPS stimulation (WT, n = 4; SOD1G37R, n = 2; p = 0.0114; O), from the brain ROI 24 h after LPS (WT, n = 4; SOD1G37R, n = 4; p = 0.0347; P), and from the lumbar spinal cord ROI 24 h after LPS (WT; n = 4, SOD1G37R; n = 4; p = 0.0217; Q). Error bars indicate SEM.
Preonset ALS is associated with marked downregulation of innate immune response and anti-inflammatory microglia profiles
To determine in vivo functional properties of microglial cells in presymptomatic disease, we analyzed the pattern of microglial activation. As a readout, we used biophotonic/bioluminescent TLR2 signals from the live TLR2-luc-GFP;SOD1G93A mice. In the first set of experiments, we investigated microglial activation profile in baseline conditions and in response to LPS challenge. LPS is an activator of innate immunity, and it is noteworthy that LPS-related genes are indeed upregulated in ALS patients (Zhang et al., 2011). Importantly, LPS induces orchestrated transcriptional response in macrophages and microglia that can be divided into relatively limited number of distinct patterns of gene induction and/or repression (Medzhitov and Horng, 2009). We hypothesized that analyses of known patterns of LPS-induced gene expression may serve as a valid readout of the functional response phenotypes of microglial cells in presymptomatic disease. In our experiments, we focused on two major regions of interest: (1) the olfactory bulb/brain area and (2) the spinal cord area. The rationale for analyses of microglial cells from the OB region came from our previous studies where we discovered that the microglia from OB, even in control conditions, express increased levels of the TLR2 signal and may react as early sensors of brain injury (Lalancette-Hébert et al., 2009, 2010). In addition, growing evidence suggests OB pathology in preclinical Parkinson's and Alzheimer's diseases, in human patients as well as in animal models (Lerner and Bagic, 2008; Beach et al., 2009; Wesson et al., 2010).
To analyze early temporal dynamics and activation patterns of microglial cells, we performed a series of live imaging experiments, starting at 35–40 d of age. Because disease progression in ALS models has been associated with the development of inflammatory and neurotoxic microglia phenotypes, we hypothesized that presymptomatic disease will be characterized by aberrant increases in TLR2 signals. To our surprise, analysis of the TLR2 signals revealed no visible signal from the spinal cord of the TLR2-luc-GFP;SOD1G93A mice, whereas the bioluminescence/biophotonic imaging from the OB/brain region of interest area revealed a significant decrease of the baseline TLR2 signal in presymptomatic ALS mice (Fig. 1A–C), thus suggesting a less activated microglial profiles. Next, we investigated how the observed decrease in the baseline TLR2 signal may affect functional response of resident microglial cells; namely, we demonstrated previously that intraperitoneal injection of LPS induces controlled and synchronized increase in resident microglial activation (visualized from live animals as TLR2 signal induction) peaking 24 h after initial challenge (Lalancette-Hébert et al., 2009). The TLR2 responses were evaluated before and 24–48 h after initial injection (5 mg/kg, i.p.). As expected, in control mice, the intraperitoneal injection of LPS was associated with a robust induction of the TLR2 signal in the OB/brain region as well as in the region of the lumbar spinal cord (Figure 1D,F). Quantitative analysis of the biophotonic signals (Figure 1H–J) revealed significantly lower TLR2 signal intensities in the SOD1G93A mutant mice compared to their age-matched TLR2 reporter control littermates (olfactory bulb ROI, baseline, WT, 2.7 × 104 ± 0.3 × 104, n = 4; SOD1G93A, 2.14 × 104 ± 0.2 × 104, n = 8; 24 h after LPS stimulation, WT, 4.94 × 105 ± 0.6 × 105, n = 4; SOD1G93A, 2.25 × 105 ± 0.2 × 105, n = 8; p = 0.0006; 48 h after LPS, WT, 2.26 × 105 ± 0.2 × 105, n = 5; SOD1G93A, 1.41 × 105 ± 0.2 × 105, n = 4; p = 0.0269; brain ROI, baseline, WT, 8.1 × 104 ± 0.7 × 104, n = 4; SOD1G93A, 6.8 × 104 ± 0.7 × 104, n = 8; 24 h after LPS stimulation, WT, 2.59 × 106 ± 0.24 × 106, n = 4; SOD1G93A, 1.28 × 106 ± 0.24 × 106, n = 8; p = 0.0065; 48 h after LPS, WT, 8.36 × 105 ± 1.8 × 105, n = 4; SOD1G93A, 9.21 × 105 ± 2.9 × 105, n = 5; lumbar spinal cord ROI, baseline, WT, 1.34 × 104 ± 0.9 × 104, n = 5; SOD1G93A, 1.42 × 104 ± 0.1 × 104, n = 7; 24 h after LPS stimulation, WT, 2.32 × 105 ± 0.48 × 105, n = 5; SOD1G93A, 0.85 × 105 ± 0.13 × 105, n = 5; p = 0.0181; 48 h after LPS, WT, 8.35 × 104 ± 1.5 × 104, n = 4; SOD1G93A, 7.6 × 104 ± 2.7 × 104, n = 5). Importantly, as revealed in Figure 1H–J, the decrease in signal intensities was observed in all region of interest, including the OB/brain area as well as spinal cord region, suggesting marked differences in the functional phenotypes of presymptomatic microglia carrying SOD1 mutation (Figure 1E,G,H–J).
Because the SOD1G93A mouse model carries a high copy number of the mutated gene and its disease phenotype is characterized by a fast disease progression (onset of clinical symptoms at ∼90 d of age with a median survival of 150 d), we next asked whether observed functional microglia profiles are relevant to SOD1-mutant-mediated disease and/or are linked to a specific mouse model. To answer this question, we crossed the TLR2-luc-GFP reporter mouse with the slow disease progressing SOD1G37R mice [onset of disease at 10 months of age; survival, ∼52 weeks (1 year)]. The same experimental protocol was repeated with presymptomatic 4-month-old double-transgenic TLR2-luc-GFP;SOD1G37R mice. As revealed in Figure 1K–N, the systemic injection of LPS induced a robust increase in the TLR2 signals in control mice, whereas the signal is markedly attenuated in presymptomatic SOD1G37R mice. The quantitative analysis of the TLR2 signal revealed significantly lower signal intensities in the OB/brain region and the spinal cords of presymptomatic SOD1G37R mice compared with age-matched control littermates (olfactory bulb ROI, 24 h after LPS stimulation, WT, 1.33 × 106 ± 0.1 × 105, n = 4; SOD1G93A, 0.80 × 106 ± 0.6 × 105, n = 4; p = 0.0114; brain ROI, 24 h after LPS stimulation, WT, 5.11 × 106 ± 0.34 × 106, n = 4; SOD1G93A, 2.85 × 106 ± 0.27 × 106, n = 4; p = 0.0347; lumbar spinal cord ROI, 24 h after LPS stimulation, WT, 6.69 × 105 ± 0.64 × 105, n = 4; SOD1G93A, 2.36 × 105 ± 0.05 × 105, n = 4; p = 0.0217). Together, our data revealed significant changes in early presymptomatic functional microglia phenotypes in two different SOD1-mutant-mediated disease models. The observed changes strongly suggest a disease-specific downregulation of microglial activation and innate immune responses following standard proinflammatory LPS challenge.
Importantly, in keeping with our in vivo imaging results, the immunofluorescence and in situ hybridization analysis of the OB and the lumbar spinal cord sections revealed attenuated microglia activation patterns in presymptomatic SOD1G93A mice. As shown in Figure 2, A–C and F, the immunohistological analysis of the OB sections revealed more activated “amoeboid-like” morphology of microglial cells in controls as compared to age-matched SOD1G93A mice. Furthermore, a double-immunofluorescence analysis revealed a colocalization of TLR2 immunostaining with the Iba1 microglial marker, confirming that in standard LPS proinflammatory stimuli, TLR2 is indeed induced in activated microglial cells. Next, the quantitative analysis of in situ mRNA signal for TLR2 further confirmed significant downregulation of the TLR2 response in the olfactory bulb and spinal cord of 60-d-old SOD1G93A mice, suggesting that in vivo imaging data paralleled immunohistochemistry and in situ hybridization results (Fig. 2I–N,O,Q; in situ labeled TLR2 cells/tissue section in the olfactory bulb, WT, 2.87 ± 0.63, n = 5; SOD1G93A, 2.50 ± 0.48, n = 5; WT + LPS, 130.4 ± 13.3, n = 5; SOD1G93A + LPS, 79.3 ± 11.6, n = 5; p = 0.0052; in situ labeled TLR2 cells/tissue section in the spinal cord, WT, 0.58 ± 0.23, n = 5; SOD1G93A, 2.21 ± 0.34, n = 5; p = 0.0067; WT + LPS, 23.94 ± 2.47, n = 5; SOD1G93A + LPS, 14.89 ± 1.66, n = 5; p = 0.0043). Because the downstream effects and/or targets of the TLR2 signaling pathway are transcriptional activation of nuclear factor κB (NF-κB) and synthesis of the proinflammatory cytokines such as TNFα, to confirm that the downstream pathway is indeed affected, we also analyzed the levels of TNFα.
Figure 2.
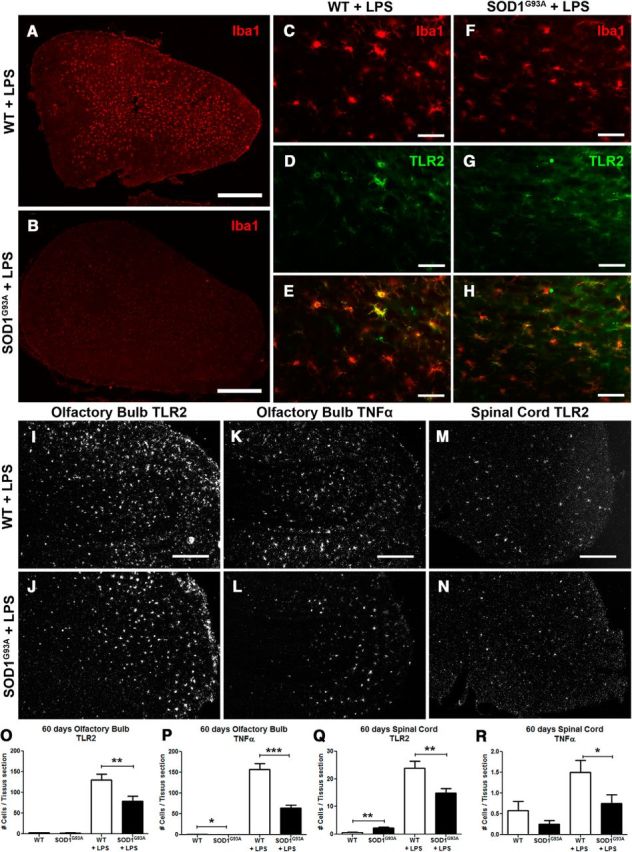
Immunofluorescence and in situ hybridization analysis. A, B, Immunofluorescence of Iba1 staining in the olfactory bulb of WT littermates controls (A) and presymptomatic 60-d-old SOD1G93A mice (B) 24 h after an LPS stimulation. C–H, Higher magnification of Iba1 staining in the olfactory bulb of WT controls (C) and SOD1G93A mice (F), TLR2 staining (D, G), and a merge showing the colocalization of both stainings (E, H). K–N, Representative photomicrographs of the in situ hybridization of the TLR2 expression 24 h after LPS stimulation in the olfactory bulb of WT controls (I) and presymptomatic 60-d-old SOD1G93A mice (J), of TNFα expression in the olfactory bulb (K, L), and TLR2 expression in the lumbar spinal cord (M, N). O–R, Quantification of in situ labeled cells per tissue section of 60-d-old mice, from baseline and 24 h following LPS stimulation: TLR2-labeled cells in the olfactory bulb (WT, n = 5; SOD1G93A, n = 5; WT + LPS, n = 5; SOD1G93A + LPS, n = 5; p = 0.0052; O), TNFα-labeled cells in the olfactory bulb (WT, n = 5; SOD1G93A, n = 5; p = 0.0221, WT + LPS, n = 5; SOD1G93A + LPS, n = 5; p < 0.0001; P), TLR2-labeled cells in the lumbar spinal cord (WT, n = 5; SOD1G93A, n = 5; p = 0.0067, WT + LPS, n = 5; SOD1G93A + LPS, n = 5; p = 0.0043; Q), and TNFα-labeled cells in the lumbar spinal cord (WT, n = 5; SOD1G93A, n = 5; WT + LPS, n = 5; SOD1G93A + LPS, n = 5; p = 0.0376; R). Scale bars: A, B, 500 μm; C–H, 50 μm; K–N, 250 μm. Error bars indicate SEM.
As shown in Figure 2, K, L, P, and R, and in accordance with observed downregulation of the TLR2 pathway, the quantitative analysis of in situ signal revealed significant decrease in TNFα mRNA levels in OB and spinal cord tissue sections of the 60-d-old SOD1G93A mice treated with LPS (in situ labeled TNFα cells/tissue section in the olfactory bulb, WT, 0.92 ± 0.26, n = 5; SOD1G93A, 0.31 ± 0.10, n = 5; p = 0.0221; WT + LPS, 157.6 ± 13.3, n = 5; SOD1G93A + LPS, 64.1 ± 6.79, n = 5; p < 0.0001; in situ labeled TNFα cells/tissue section in the spinal cord, WT, 0.58 ± 0.22, n = 5; SOD1G93A, 0.25 ± 0.09, n = 5; WT + LPS, 1.5 ± 0.29, n = 5; SOD1G93A + LPS, 0.75 ± 0.21, n = 5; p = 0.0376). Together, our results identify early alterations of innate immune response and microglial phenotypes in presymptomatic disease. The downregulation of the TLR2 as well as TNFα pathways suggest that microglial cells in presymptomatic SOD1-mutant-mediated disease may acquire anti-inflammatory polarization phenotypes.
Enhanced IL-10 production by presymptomatic resident SOD1G93A microglia
To gain more insights into these intriguing findings, we next asked whether observed changes in microglial phenotypes are caused by affected environment (i.e., stressed neurons) and/or represent a cell-type-specific change in phenotype caused by inherited disease. To address this issue, we established primary murine microglial cultures derived from adult (P60, the age of observed imaging phenotype; Figs. 1, 2) SOD1G93A mice and WT type age-matched littermates. The primary microglial cultures were derived from OB and lumbar spinal cord microglia since in vivo both of the regions of interest showed changes in phenotype. The cells were treated with (1 μg/ml) LPS for 20 min. As in in vivo experiments, the microglia activation profile was analyzed 24 h after initial stimuli. In accordance with the results obtained in our in vivo imaging studies, in normal culture conditions, the SOD1G93A microglia expressed lower levels of TLR2 compared to WT cells. The similar results were obtained following LPS stimuli. As shown in Figure 3E, the quantitative analysis of the activation profiles after LPS stimuli revealed significantly lower levels of TLR2 immunoreactivity in the SOD1-mutant-expressing cells (WT, 17.30 ± 0.47, n = 5; SOD1G93A, 13.51 ± 0.21, n = 5; p < 0.0001; WT + LPS, 20.59 ± 0.55, n = 5; SOD1G93A + LPS, 12.95 ± 0.21, n = 5; p < 0.0001; values expressed in arbitrary units). Hence, initial in vitro analysis confirmed deregulation of innate immune responses observed in presymptomatic SOD1G93A-expressing microglia in vivo. Next, we hypothesized that observed changes in microglia polarization phenotypes are caused by and/or associated with changes in cytokine profiles. Therefore, using the same experimental paradigm, we performed an unbiased analysis of proinflammatory and anti-inflammatory cytokine profiles. We investigated and compared the cytokine levels of primary microglial cells derived from OB and the lumbar spinal cord region of 60-d-old WT and SOD1 mutant mice. Interestingly, we did not observe major changes in the levels of proinflammatory cytokines following LPS challenge. The quantitative real-time PCR revealed a modest increase in IL-1β levels in SOD1G93A-expressing microglia after LPS stimuli (Fig. 3F; IL-1β, olfactory bulb culture, WT, 34.3 ± 6.4; SOD1G93A, 43.9 ± 5.3; WT + LPS, 1178 ± 0.0005; SOD1G93A + LPS, 1234 ± 16.6; spinal cord culture, WT, 41.0 ± 1.1; SOD1G93A, 109.7 ± 12.5; p = 0.0319; WT + LPS, 1355 ± 9.1; SOD1G93A + LPS, 2639 ± 0.0005; p < 0.0001), whereas no significant difference was observed in mRNA levels of proinflammatory cytokines (IL-6 and TNF-α) in the spinal cord microglial cells after LPS stimuli (Fig. 3 G,H; IL-6, olfactory bulb culture, WT, 5.6 ± 3.7; SOD1G93A, 6.2 ± 1.4; WT + LPS, 115.2 ± 2.2; SOD1G93A + LPS, 102.5 ± 1.3; p = 0.0388; spinal cord culture, WT, 4.7 ± 1.3; SOD1G93A, 13.0 ± 0.9; p = 0.0346; WT + LPS, 219.9 ± 4.3; SOD1G93A + LPS, 256.9 ± 28.9; TNFα, olfactory bulb culture, WT, 225.1 ± 10.3; SOD1G93A, 228.3 ± 2.2; WT + LPS, 1752 ± 17.1; SOD1G93A + LPS, 1551 ± 20.2, p = 0.0170; spinal cord culture, WT, 183.1 ± 7.8; SOD1G93A, 316.0 ± 11.3; p = 0.0105; WT + LPS, 1608 ± 99.5; SOD1G93A + LPS, 1682 ± 21.9). However, the most striking changes were observed in levels of anti-inflammatory cytokines. In addition, the changes were more pronounced in primary microglial cells derived from spinal cords of presymptomatic SOD1G93A mice; namely, in addition to the modest increase in the TGF-β mRNA levels, we detected a sixfold increase in the mRNA levels of anti-inflammatory cytokine IL-10 in baseline conditions followed by a 4.5-fold increase in IL-10 levels following LPS challenge (Fig. 3 I,J; TGF-β1, olfactory bulb culture, WT, 57.4 ± 0.0; SOD1G93A, 58.4 ± 3.1; WT + LPS, 50.8 ± 3.8; SOD1G93A + LPS, 50.0 ± 6.1; spinal cord culture, WT, 42.7 ± 2.9; SOD1G93A, 45.3 ± 2.1; WT + LPS, 34.2 ± 0.8; SOD1G93A + LPS, 60.6 ± 2.6; p = 0.0103; IL-10, olfactory bulb culture, WT, 0.62 ± 0.29; SOD1G93A, 1.7 ± 0.26; WT + LPS, 21.5 ± 0.1; SOD1G93A + LPS, 31.5 ± 0.1; p = 0.0002; spinal cord culture, WT, 0.19 ± 0.29; SOD1G93A, 3.0 ± 0.9; p = 0.0479; WT + LPS, 9.1 ± 0.03; SOD1G93A + LPS, 35.8 ± 3.9; p = 0.0206). As revealed in Figure 3K, an increase in IL-10 levels was further confirmed at the protein level (IL-10 cytokine array from in vivo olfactory bulb homogenates, 24 h after LPS, WT + LPS, 6.8 × 10−3 ± 0.6 × 10−3, n = 6; SOD1G93A + LPS, 19.4 × 10−3 ± 0.7 × 10−3, n = 6; p = 0.0054; lumbar spinal cord homogenates, 24 h after LPS stimulation, WT + LPS, 3.6 × 10−3 ± 0.0 × 10−3, n = 6; SOD1G93A + LPS, 8.1 × 10−3 ± 0.2 × 10−3, n = 6; p = 0.0014). IL-10 is a key homeostatic, anti-inflammatory cytokine that plays a critical role in modulation of inflammatory processes (Saraiva and O'Garra, 2010). Although our results revealed that in presymptomatic SOD1G93A mice IL-10 is overexpressed and produced by resident microglia, evidence suggests that the major source of IL-10 is T regulatory lymphocytes (Tregs) (Kleinewietfeld and Hafler, 2014).
Figure 3.
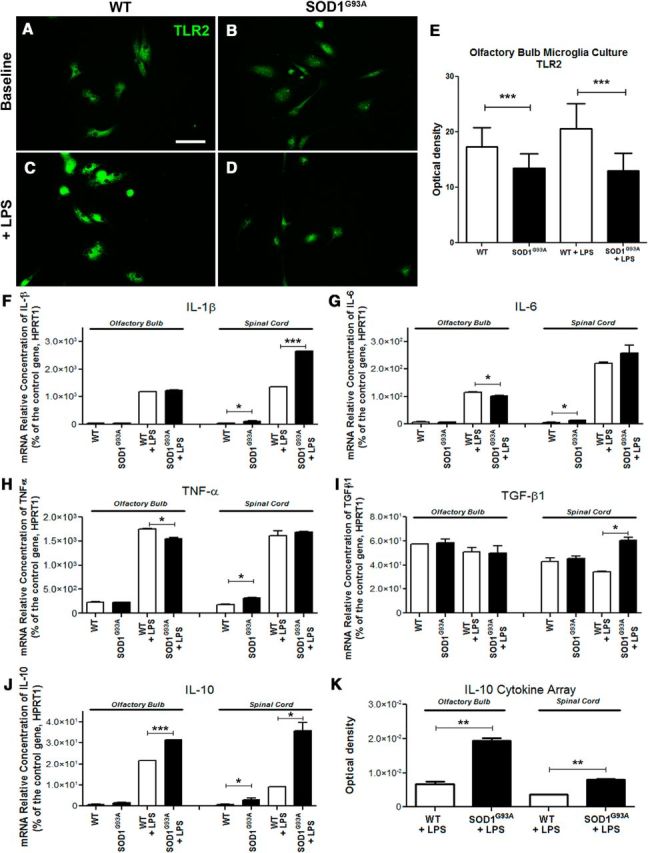
IL-10 overexpression in in vitro adult microglia primary cultures. A–D, Baseline expression of TLR2 detected by immunofluorescent staining in adult primary cultures derived from the olfactory bulb of 60-d-old WT controls (A) and SOD1G93A mice (B) and TLR2 upregulation following in vitro LPS stimulation (C, D), Scale bar, 50 μm. E, Optical density quantification of TLR2 immunofluorescent stained cultures (WT, n = 6; SOD1G93A, n = 6; p < 0.0001; WT + LPS, n = 6; SOD1G93A + LPS, n = 6; p < 0.0001). F–J, RT-PCR analysis of the adult microglia primary cultures from olfactory bulb and spinal cord of WT and SOD1G93A mice, before and after LPS stimulation, looking at relative mRNA concentrations of IL-1β (spinal cord, WT vs SOD1G93A, p = 0.0319; WT + LPS vs SOD1G93A + LPS, p < 0.0001; F), IL-6 (olfactory bulb, WT + LPS vs SOD1G93A + LPS, p = 0.0388; spinal cord, WT vs SOD1G93A, p = 0.0346; G), TNFα (olfactory bulb, WT + LPS vs SOD1G93A + LPS, p = 0.0170; spinal cord, WT vs SOD1G93A, p = 0.0105; H), TGF-β1 (spinal cord, WT + LPS vs SOD1G93A + LPS, p = 0.0103; I), and IL-10 (olfactory bulb, WT + LPS vs SOD1G93A + LPS, p = 0.0002; spinal cord, WT vs SOD1G93A, p = 0.0479; WT + LPS vs SOD1G93A + LPS, p = 0.0206; J). All mRNA relative concentrations were normalized on the control gene HPRT1, and n = 2 for all RT-PRC analyses. K, Quantification of the IL-10 cytokine array from in vivo olfactory bulb 24 h after LPS stimulation (WT + LPS, n = 6; SOD1G93A + LPS, n = 6; p = 0.0054) and from in vivo lumbar spinal cord after LPS stimulation (WT + LPS, n = 6; SOD1G93A + LPS, n = 6; p = 0.0014). Error bars indicate SEM.
Therefore, to determine whether observed overexpression of IL-10 is restricted to CNS and resident microglial cells and/or Tregs are also involved in this early immunosuppression, we performed a flow cytometry analysis of the blood of 50- to 60-d-old SOD1G93A mice and the age-matched littermates in control conditions and 24 h after LPS challenge. We quantified different immune cell type subpopulations and measured the mean IL-10 levels in Tregs. Because Beers et al. (2011) and Henkel et al. (2013) demonstrated previously that in the early symptomatic phase of disease Tregs can modulate inflammatory response via anti-inflammatory cytokine IL-4, we also measured levels of IL-4. As shown in Figure 4A–G, in baseline conditions, quantitative analysis of the numbers of CD4-, CD8-, and CD11b-positive cells revealed no difference between SOD1G93A mutant mice and WT littermates. As expected, the LPS challenge markedly increased the number of monocytes/macrophages in the blood; however, we did not observe any difference in the numbers of CD11b-positive cells between two experimental groups. Finally, we identified the CD25+/FoxP3+ subpopulation of cells (specific markers for Tregs; Fig. 4H,J). Quantitative analysis revealed no significant difference in the numbers of Tregs in control condition and following LPS challenge. As revealed in Figure 4, K and L, we did not observed statistically significant difference in the mean cellular levels of IL-10 and IL-4 in CD25+FoxP3+ cells. Together, our results suggest that, contrary to marked induction of IL-10 in microglial cells found in presymptomatic SOD1-mediated disease, no changes were observed in the profiles of peripheral immune cells and/or IL-10 levels in baseline conditions and in response to systemic LPS challenge. Hence, in presymptomatic SOD1G93A mice, early changes in cytokine profiles and deregulation of the innate immune responses were restricted to a population of the resident microglial cells.
Figure 4.
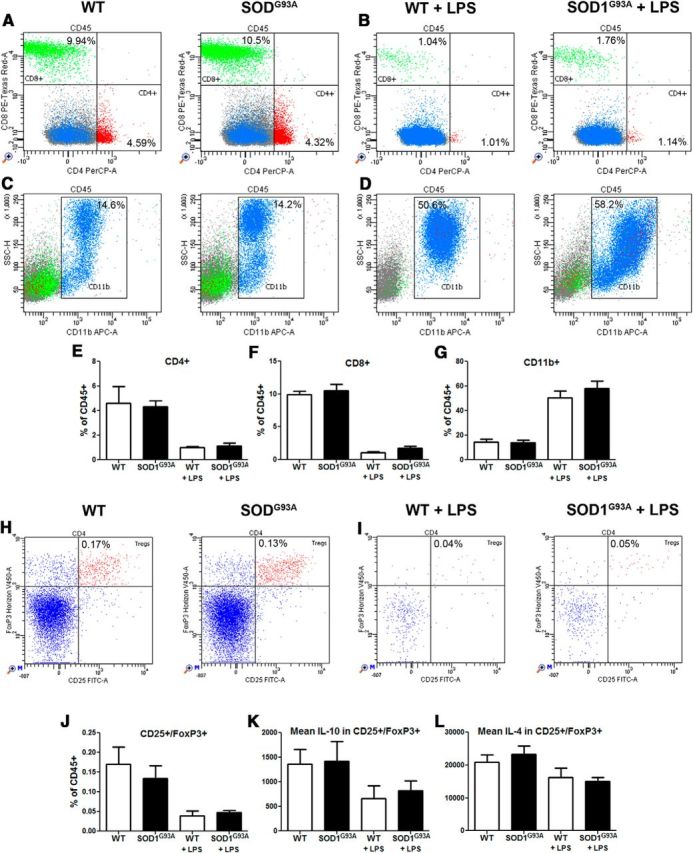
Presymptomatic microglial phenotype is not mediated by peripheral immune cells. Blood samples from presymptomatic (60 d) and 24 h LPS stimulated WT and SOD1G93A mice were collected to assess the potential of immune cells in the periphery to modulate the repressed microglial response observed in the CNS of presymptomatic SOD1G93A mice. The blood samples were stained for CD45, CD4, CD8, and CD11b or for CD45, CD4, CD25, FoxP3, IL-4, and IL-10 and analyzed in flow cytometry. A–D, In 60-d-old mice, the CD4+ and CD8+ lymphocyte proportions were unchanged between WT and SOD1G93A (A, B), as were the monocyte/macrophage CD11b+ populations (C, D). CD4+, CD8+ lymphocytes and CD11b+ monocytes/macrophages of WT and SOD1G93A mice all showed equivalent response to LPS stimulation. E–G, Overall FACS counts suggest no deregulation in the CD4+, CD8+, or CD11b+ population in presymptomatic 60-d-old SOD1G93A mice. CD25+/FoxP3+ Tregs were also assessed for their production of IL-4 and IL-10. H, I, Both at baseline (H) and after LPS stimulation (I), WT and SOD1G93A mice had similar Tregs population ratios. J, Tregs counts suggest no deregulation between presymptomatic WT and SOD1G93A mice at 60 d. K, L, Levels of IL-10 (K) and IL-4 (L) were estimated as mean levels of fluorescence intensity among the Tregs cells. Again, no differences were observed between WT and SOD1G93A presymptomatic 60 d Tregs (n = 4–6 in each groups). Error bars indicate SEM.
Deregulation of TLR2 is IL-10 dependent
One of the most intriguing findings in presymptomatic SOD1 mutant mice is a marked deregulation of the TLR2 biophotonic/bioluminescence signal observed in in vivo imaging. The above described primary culture results suggest a role of IL-10; however, the in vitro model may not recapitulate all aspects of the presymptomatic disease. Therefore, to determine the relevance of this finding and to gain additional insights into microglial activation and TLR2 regulation in early ALS, we took advantage of our established in vivo model system, a double-transgenic SOD1/TLR2 reporter mouse model. We next asked whether above described overexpression of IL-10 may be an underlying mechanism involved in downregulation of TLR2 response. To answer this question, and to block effects of IL-10, we used a specific neutralizing antibody raised against IL-10R1, the functional IL-10 receptor. This approach and antibody have been successfully tested in vivo in the model of murine West Nile virus infection (Bai et al., 2009). Mice were injected with high dose of the mIL-10R1 antibody (200 μg, i.p.) 24 h before LPS injection. As expected, the bioluminescence imaging of the OB/brain region of interest revealed that the systemic LPS induced a robust induction of the TLR2 signal in 60-d-old control mice, and the response was attenuated in presymptomatic SOD1G93A mice (Figure 5A,B). However, as shown in Figure 5, C and D, induction of the TLR2 signal was completely restored in mice pretreated with IL-10R neutralizing antibody (WT, 19.9 × 104 ± 1.6 × 104, n = 19; SOD1G93A, 15.0 × 104 ± 0.7 × 104, n = 24; p = 0.0047; WT + LPS; 3.66 × 106 ± 0.2 × 106, n = 24; SOD1G93A + LPS, 2.79 × 106 ± 0.3 × 106, n = 20; p = 0.0273; SOD1G93A + αIL-10R + LPS, 4.62 × 106 ± 0.8 × 106, n = 9; p = 0.0126). The same pattern of the TLR2 signal recovery was observed in the lumbar spinal cord. As revealed in Figure 5E–H, pretreatment with IL-10R antibody completely restored the TLR2 signal induction, following LPS challenge, in the spinal cord region of presymptomatic SOD1G93A mice (WT, 4.45 × 104 ± 0.3 × 104, n = 19; SOD1G93A, 3.60 × 104 ± 0.2 × 104, n = 22; p = 0.0119; WT + LPS, 3.91 × 105 ± 0.3 × 105, n = 16; SOD1G93A + LPS, 2.98 × 105 ± 0.2 × 105, n = 13; p = 0.0351; SOD1G93A + αIL-10R + LPS, 5.91 × 105 ± 0.8 × 105, n = 8; p = 0.0005). These data clearly suggest that IL-10 has a role in modulation of early TLR2 responses in presymptomatic ALS mice.
Figure 5.
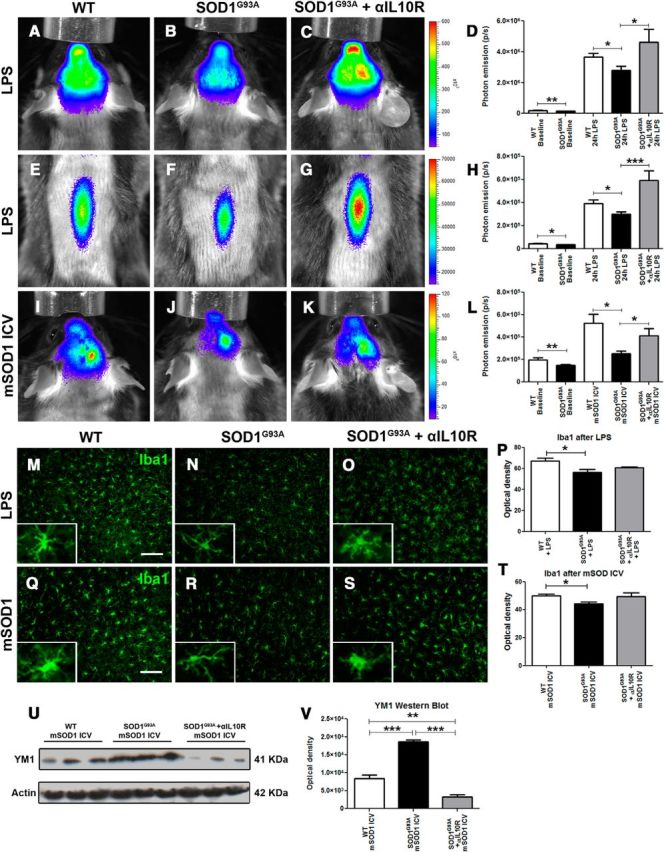
Modulating the microglial response by blocking the IL-10 pathway. A–C, E–G, Real-time imaging of TLR2 induction in the olfactory bulb and brain, 24 h after LPS stimulation, in WT controls (A), SOD1G93A mice (B), and SOD1G93A mice treated with αIL-10R antibodies (C) and the respective real-time imaging of the spinal cord (E–G) show a significant LPS-induced microglial activation increase in αIL-10R-treated SOD1G93A compared to untreated SOD1G93A mice. D, Quantitative analysis of the photonic emissions in the head (WT, n = 19; SOD1G93A, n = 24; p = 0.0047; WT + LPS, n = 24; SOD1G93A + LPS, n = 20; p = 0.0273; SOD1G93A + αIL-10R + LPS, n = 9; p = 0.0126). H, Quantitative analysis of the photonic emissions in the spinal cord (WT, n = 19; SOD1G93A, n = 22; p = 0.0119; WT + LPS, n = 16; SOD1G93A + LPS, n = 13; p = 0.0351; SOD1G93A + αIL-10R + LPS, n = 8; p = 0.0005). I–K, Real-time imaging of TLR2 induction in the olfactory bulb and brain, 48 h following intracerebroventricular injection of mSOD1, in WT controls (I), SOD1G93A mice (J), and SOD1G93A mice treated with αIL-10R antibodies (K) shows a significant mSOD1-induced microglial activation increase in the SOD1G93A αIL-10R-treated mice. L, Quantitative analysis of the photonic emissions in the head (WT, n = 19; SOD1G93A, n = 24; p = 0.0047; WT + LPS, n = 10; SOD1G93A + LPS, n = 6; p = 0.0221; SOD1G93A + αIL-10R + LPS, n = 7; p = 0.0490). M–P, Immunostaining of Iba1 after LPS stimulation in WT, SOD1G93A, and SODG93A +αIL-10R mice (M–O) and associated optical density quantification (P; WT + LPS, n = 5; SOD1G93A + LPS, n = 5; p = 0.0258; SOD1G93A + αIL-10R + LPS, n = 5). Q–T, Immunostaining of Iba1 after mSOD1 intracerebroventricular stimulation in WT, SOD1G93A, and SODG93A + αIL-10R mice (Q–S) and associated optical density quantification (T; WT + mSOD1, n = 5; SOD1G93A + mSOD1, n = 5; p = 0.0161; SOD1G93A + αIL-10R + mSOD1, n = 5). Scale bars: 100 μm. U, V, Western blots of Ym1 in WT, SOD1G93A, and SOD1G93A + αIL-10R brain extracts following mSOD1 intracerebroventricular stimulation (U) and optical density quantification (V) show a significant increase of Ym1 in the SOD1G93A + mSOD1 samples, but also a significant decrease of Ym1 levels in the SOD1G93A + αIL-10R + mSOD1 samples (WT + mSOD1, n = 3; SOD1G93A + mSOD1, n = 3; p = 0.0009; SOD1G93A + αIL-10R + mSOD1, n = 3; p < 0.0126). Error bars indicate SEM.
The next question is, what is the pathophysiological relevance and its role in early disease pathogenesis? IL-10 is a major anti-inflammatory cytokine involved in suppression of production of several proinflammatory molecules. Interestingly, work by Yanagawa and Onoé (2007) suggests that enhanced IL-10 levels and consequent downregulation of TLR responses may be a result of cellular priming with TLR4 and TLR2 ligands. It is noteworthy that mutated SOD1 protein early in the disease forms misfolded species that, as shown by Urushitani et al. (2006), can be secreted from affected neurons. Once secreted, the misfolded SOD1 species may act as danger-associated molecular patterns (DAMPs) and act as a ligand to TLR2 and/or TLR4 receptors. Previous work by Zhao et al. (2010) nicely demonstrated that misfolded SOD1 protein may indeed act as a TLR2 ligand and modify properties of microglial cells in vitro. Therefore, we hypothesized that early in disease, small amounts of the SOD1 misfolded protein species are secreted from neurons and, once extracellular, may act as TLR2 ligands. Preexposure to small amount of DAMPs (in this case, misfolded SOD1 species) would reduce sensitivity of microglial cells and the innate immune system to the second LPS or DAMP challenge. Thus, if our hypothesis is correct, the second exposure to known misfolded protein would result (as in case of the LPS challenge) in enhanced IL-10 production and attenuated microglial activation/TLR response to misfolded protein in early presymptomatic SOD1G93A mice, whereas the response would be very robust in WT age-matched mice. As shown in Figure 5H–L, intracerebroventricular injection of recombinant misfolded SOD1 protein induced a robust induction of the TLR2 signals in the WT mice, whereas the signal was attenuated in the presymptomatic SOD1 mutant mice (WT, 19.9 × 104 ± 1.6 × 104, n = 19; SOD1G93A, 15.0 × 104 ± 0.7 × 104, n = 24; p = 0.0047; WT + LPS, 5.24 × 105 ± 0.8 × 105, n = 10; SOD1G93A + LPS, 2.51 × 105 ± 0.2 × 105, n = 6; p = 0.0221). Importantly, pretreatment with IL-10R blocking antibody restored the microglial activation/TLR2 response in SOD1G93A mice, suggesting IL-10-mediated downregulation of TLR2 response (SOD1G93A + αIL-10R + LPS, 4.14 × 105 ± 0.6 × 105, n = 7; p = 0.0490).
Next, to investigate whether deregulation of the TLR2 signals observed by imaging in presymptomatic microglial cells is associated with the development of distinct polarization phenotypes, we analyzed by immunofluorescence brain sections of the imaged animals. Indeed, as shown in Figure 5M–T, microglial cells from the SOD1G93A mice following LPS or mSOD1 challenge acquire less activated phenotypes reflected by more ramified morphology and by a significant decrease in Iba1 immunoreactivity, as revealed by optical density measurement. Moreover, Western blot analysis of the brain homogenates from WT and SOD1 mutant mice exposed to mSOD1 protein revealed a twofold increase in Ym1 levels in SOD1 mutant mice. This increase was abolished by IL-10R blocking antibody (Fig. 5U,V). Ym1 is well established marker of microglia/macrophage alternative activation; therefore, its selective upregulation in presymptomatic SOD1G93A mice further supports a view of early immunosuppression in these mice.
Chronic infusion with IL-10R blocking antibody precipitates disease onset in the SOD1G93A mouse model
To determine whether attenuation of TLR2 response and overexpression of IL-10 in resident microglial cells is an adaptive and/or maladaptive cellular phenotype in presymptomatic disease, we initiated long-term infusion with IL-10R blocking antibodies. The treatment was initiated at 60 d of age (n = 16 per group) using an infusion cannula connected to osmotic minipumps (Alzet model 2006; pumping rate, 0.15 μl/h; duration of treatment, 42 d). The sterile brain infusion cannula was implanted into the right ventricle and was fixed with dental cement, and was removed at day 105. The control mice received the infusion of sterile saline. Because treatment was initiated before the clinical disease onset, the mice were closely monitored by a blinded investigator. As described previously (Urushitani et al., 2006; Keller et al., 2009), the clinical onset of disease was measured and scored as a loss of extension reflex and confirmed by a parallel loss of body weight, and body weight was measured on a daily basis (Fig. 6C). As shown in Figure 6, A and B, treatment with IL-10R antibody significantly shifted the clinical onset of disease by 7 d (74 vs 81 d for saline-treated animals). Whereas body weight measurements revealed that both experimental groups followed a classical bell-like shape of the curve (initial incremental increase of body weight followed by a rapid decline after clinical onset of disease; Fig. 6C), analysis of the values between the two groups revealed that treatment with IL-10R blocking antibody, consistent with the shift in disease onset, prevented early weight gain and precipitated weight loss at the time of the onset. The treatment ended at day 105, and animals were kept under observation for survival analysis. As shown in Figure 6D, the delivery of IL-10R blocking antibody from days 60 to 105 did not significantly affect the survival of SOD1G93A mice. Here it is noteworthy that the Alzet pumps stopped perfusion of the antibody at day 105; thus, we may not exclude the possibility that longer treatment may have some effects on survival. We also investigated whether treatment with IL-10R blocking antibody affects immune cell profiles at the periphery. Surprisingly, we did not observed any changes in the immune cell profiles and/or levels of IL-10 and IL-4 (data not shown). Because infusion of IL-10R blocking antibody precipitated disease in SOD1G93A mice, we hypothesized that overexpression of IL-10 in microglial cells can be used to therapeutically modulate microglial phenotypes in early ALS and temporally extend their adaptive/neuroprotective state, which may significantly decrease the initial burden of disease and delay neuronal dysfunction. To therapeutically target microglia, and to overexpress IL-10 selectively in microglial cells, a 1.3 kb IL-10 cDNA was expressed under human CD11b promoter and inserted into AAV2/9 viral vector (Fig. 6D). As described previously (Patel et al., 2014), the construct was injected intrathecally with a single injection of 3 × 109 viral particles in a 10 μl volume. The recipient SOD1G93A mice were 30- to 40-d-old, and an empty viral plasmid was used as control. As shown in Figure 6, E and F, treatment with AAV-encoded IL-10 significantly delayed the clinical onset of disease and prevented weight loss. In addition, AAV delivery of IL-10 significantly increased survival in SOD1G93A mice. As further shown in Figure 6, H and I, the efficiency and selectivity of the AAV infection was confirmed by immunodetection of the myc epitope. The double immunofluorescence of the lumbar spinal cord sections of the SOD1G93A mice that received AAV-CD11b-cMyc-IL-10 injection, at the end of the protocol, showed almost perfect colocalization of Iba1 microglail markers with the cMyc epitope, suggesting a selective and targeted overexpression of IL-10 in microglial cells. Collectively, our results provide in vivo evidence that IL-10 signaling in microglial cells may serve as an endogenous nonneuronal determinant of disease onset and may affect disease progression.
Figure 6.
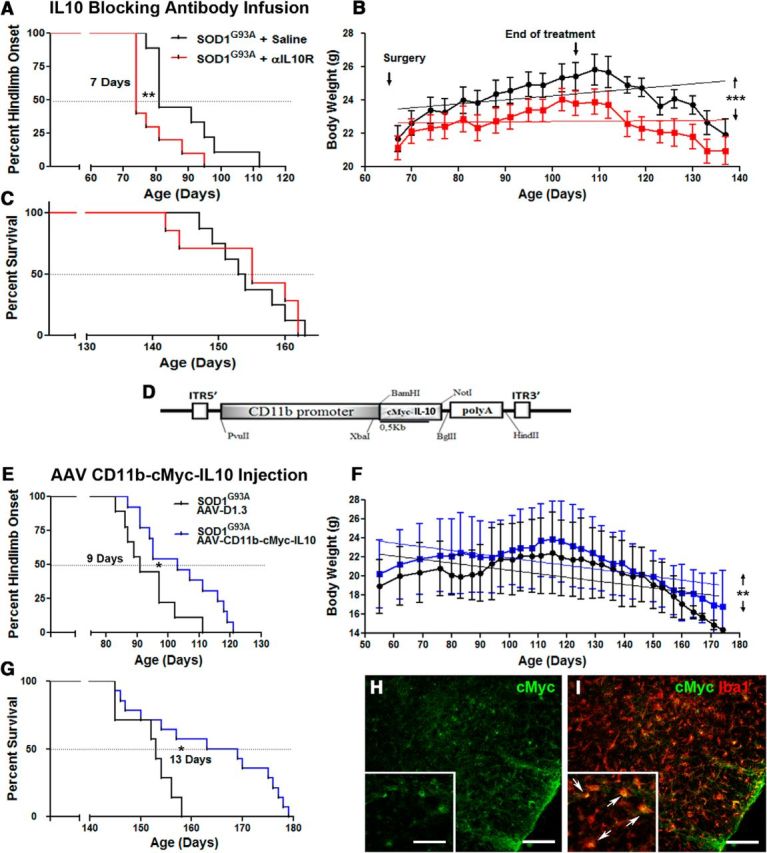
Treatment with blocking IL-10R antibodies accelerates disease onset in SOD1G93A mice, whereas AAV-CD11b-cMyc-IL10 virus intrathecal injection delays onset and extends survival. Longitudinal analysis of SOD1G93A mice subjected to a 42 d intracerebroventricular infusion of saline (black curves) or αIL-10R antibodies (red curves) started at presymptomatic 60 d of age. A, Kaplan–Meier analysis of the extension reflex revealed a significant median onset 7 d earlier in the αIL-10R infused group (saline treated mean onset, 81 d; n = 9; αIL-10R treated mean onset, 74 d; n = 10; p = 0.0101). B, Animals receiving the αIL-10R antibodies failed to gain weight like the saline-treated controls and maintained an overall significant lower weight through their lifespan (linear regression analysis shows that the elevation between the two curves is significant; p < 0.0001). C, The presymptomatic αIL-10R treatment did not however affect the survival rate between the two groups. D, Diagram of the viral AAV construct expressing cMyc-IL-10 under the control of the human CD11b gene promoter. Longitudinal analysis of SOD1G93A mice subjected to an intrathecal AAV virus injection, with an empty plasmid control virus (black curves) or AAA-CD11b-cMyc-IL10 virus (blue curves) injected at presymptomatic 30–35 d of age. E, Kaplan–Meier analysis of the extension reflex revealed a significant 9 d delayed median onset in the AAV-CD11b-cMyc-IL10-infected group (control AAV mean onset, 90 d; n = 8; AAV-Cd11b-cMyc-IL10 mean onset, 99 d; n = 14; p = 0.0157). F, Animals receiving the AAV-CD11b-cMyc-IL10 virus maintained an overall significantly higher weight through their lifespan (linear regression analysis shows that the elevation between the two curves is significant; p = 0.0039). G, The presymptomatic AAV-CD11b-cMYC-IL10 intrathecal injection also significantly increased survival by 13 d (control AAV mean survival, 153 d; n = 7; AAV-Cd11b-cMyc-IL10 mean survival, 166 d; n = 14; p = 0.0166). H, I, Immunostaining of cMyc in the spinal cord of AAV-CD11b-cMyc-IL10-injected mice shows colocalization with Iba1, confirming a microglial expression of the viral construct. Scale bars: 100 μm; insets, 50 μm. Error bars indicate SEM.
Finally we investigated how changes in microglial IL-10 signaling may affect microglial polarization phenotypes. We first investigated effects of IL-10R antibody treatment. As shown in Figure 7A–F, analysis of the different microglia activation markers revealed a significant increase of markers of microglial activation. When compared to controls, we observed increased immunoreactivity for Iba1 (saline, 1.05 × 104 ± 0.5 × 104, n = 5; αIL-10R, 1.31 × 104 ± 0.7 × 104, n = 8; p = 0.0298) and CD68 (saline, 13.63 ± 1.0, n = 6; αIl-10R, 16.87 ± 0.3, n = 6; p = 0.0128) in the lumbar spinal cord sections of the treated mice. Contrary to the proinflammatory effects of IL-10R blocking antibody, enhanced IL-10 levels have been associated with anti-inflammatory and neuroprotective microglial phenotypes. As shown in Figure 7, G and T, addition of recombinant IL-10 into mSOD1 exposed microglia significantly decreased expression levels of the proinflammatory cytokine TNFα and increased expression of an alternative microglia/macrophage activation marker, Ym1. Together, our results suggest that enhanced IL-10 levels together with downregulation of the TLR2 response represent an adaptive resident microglia-cell-specific mechanism that controls early disease propagation and onset. Inversely, and in keeping with this hypothesis, blocking the IL-10 signaling with a neutralizing antibody against its functional receptor IL-10R1 precipitated disease onset in SOD1G93A mice and shifted the microglia activation profiles toward the proinflammatory phenotype. Importantly, and in keeping with our hypothesis, a targeted overexpression of IL-10 in microglial cells significantly delayed the clinical onset of disease and increased survival in SOD1G93A mice. Hence, IL-10 may serve as an endogenous microglial mechanism involved in the early (presymptomatic) disease mechanisms and the control of disease onset. Moreover, targeted overexpression of IL-10 in microglial cells may have therapeutic potential in ALS.
Figure 7.
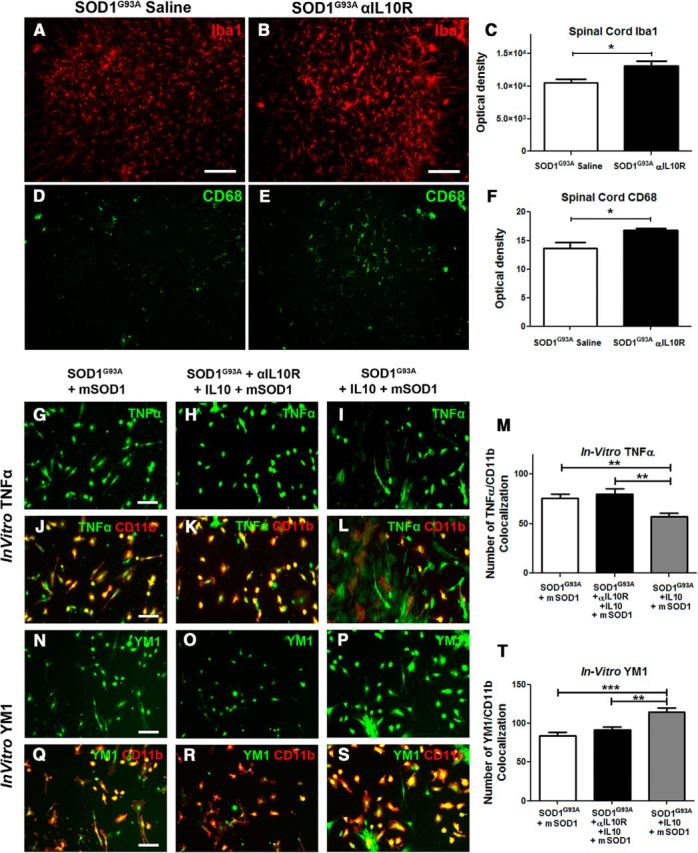
Intracerebroventricular infusion of blocking IL-10R antibodies increases inflammation in the lumbar spinal cord, whereas IL-10 increases anti-inflammatory markers. A–C, Immunostaining of Iba1 in the lumbar spinal cord ventral horn of mice following 42 d of saline (A) or αIL-10R (B) intracerebroventricular infusion in SOD1G93A mice reveals, by optical density quantification of the fluorescent signal, an increased Iba1 immunoreactivity in the αIL-10R-treated mice (C; saline, n = 5; αIL-10R, n = 8; p = 0.0298). D–F, Immunostaining for Cd68, a marker of microglial activation (D, E), similarly shows, by optical density quantification, an increased CD68 expression in αIL-10R-infused SOD1G93A mice (F; saline, n = 6; αIl-10R, n = 6; p = 0.0128). G–M, TNFα immunostaining following in vitro mSOD1 stimulation on SOD1G93A, SOD1G93A + αIL10R + IL10, and SOD1G93A + IL10 primary cultures and relative numbers of TNFα/CD11b colocalization (M) show a significant decrease of TNFα cells in the IL10-treated group (SOD1G93A + mSOD1, n = 16; SOD1G93A + αIL-10R + IL10 + mSOD1, n = 16; SOD1G93A + IL10 + mSOD1, n = 16; p = 0.0041). N–T, Ym1 immunostaining following in vitro mSOD1 stimulation on SOD1G93A, SOD1G93A + αIL-10R + IL10, and SOD1G93A + IL10 cultures and relative numbers of YM1/CD11b colocalization (T) show a significant increase of YM1 positive cells following IL10 treatment (SOD1G93A +mSOD1, n = 16; SOD1G93A +αIL-10R +Il10 +mSOD1, n = 16; SOD1G93A + IL10 +mSOD1, n = 16, p = 0.0002). Scale bars: 50 μm. Error bars indicate SEM.
Discussion
To date, it has been widely established that microglial cells actively contribute to disease progression and motor neuron degeneration in ALS (Robberecht and Philips, 2013). Yet, the role of microglial cells in early disease pathogenesis (before appearance of clinical symptoms) and in events initiating and/or precipitating disease onset is largely unknown. The work presented here provides important in vivo evidence of a distinct adaptive shift in functional microglial phenotypes in preclinical stages of SOD1-mutant-mediated disease. Using a mouse model for live imaging of microglial activation (Lalancette-Hébert et al., 2009) crossed with SOD1G93A mice, we discovered that, contrary to our expectations, the preonset phase of SOD1-mediated disease is characterized by development of distinct anti-inflammatory microglia profiles and attenuated innate immune/TLR2 responses to LPS and misfolded protein challenge. Remarkably, the same anti-inflammatory microglia profiles in the preonset stage of disease were observed in two different models of SOD1-mutant-mediated disease, the fast-progressing SOD1G93A and the slow-progressing SOD1G37R mouse models. Here we report (1) a 16-fold overexpression of anti-inflammatory cytokine IL-10 (SOD1G93A mutant microglia derived from 60-d-old mice) in baseline conditions followed by a 4.5-fold increase following LPS challenge, (2) that the shift in phenotypes and associated IL-10 overexpression was restricted to resident microglia and was not observed in peripheral immune cells, (3) that acute treatment with IL-10R blocking antibody restored the TLR2 response in vivo, (4) that intracerebroventricular long-term infusion of the IL-10R blocking antibody initiated at day 60 precipitated disease onset by 7 d and caused significant increase in markers of microglial activation, and (5) that selective overexpression of IL-10 in microglial cells using the AAV gene delivery system expressed under the human CD11b promoter significantly delayed the clinical onset of disease and increased survival of SOD1G93A mice. Based on these exciting findings, we propose that IL-10, a master immunoregulatory cytokine, may serve as an endogenous modifier of microglia (neuroprotective) phenotypes in preonset ALS and act as a nonneuronal determinant of clinical onset of the disease.
The direct role of microglial cells in SOD1-mutant-mediated disease pathogenesis was initially described in two landmark studies (Beers et al., 2006; Boillée et al., 2006). In the first study, using a mouse models with deletable transgenes, Boillée et al. (2006) demonstrated that the deletion of mutant SOD1 transgene within Cd11b-positive cells increased the life span of SOD1G37R mice by ∼100 d. Similar findings were described in a parallel paper (Beers et al., 2006) demonstrating that wild-type microglia extended survival in PU.1 knock-out mice bred with SOD1G93A mutants. Although there is an agreement on the neuroprotective role of wild-type microglia, the microglial cells carrying SOD1 mutations are markedly different relative to WT microglial cells. For example, in vitro studies revealed that microglial cells carrying the SOD1 mutation are more neurotoxic (Xiao et al., 2007). In addition, a previous study by Frakes et al. (2014) clearly demonstrated (in vivo and in vitro) NF-κB-driven activation and conversion of microglia phenotypes to neurotoxic inflammatory cells in SOD1G93A mice. Together, there is abundant evidence that after disease onset, microglial cells develop inflammatory and/or toxic phenotypes that may accelerate disease progression (Henkel et al., 2009). On the other hand, the contribution of microglial cells in the early disease pathogenesis remains unclear. In vivo preclinical studies using anti-inflammatory approaches clearly suggest a major shift in microglia profiles over the course of the disease. While treatments with anti-inflammatory compounds like minocycline or NF-κB inhibitor Withaferin A attenuated inflammation and conferred neuroprotection when initiated early, initiation of the treatment at later stages of disease increased inflammatory markers, suggesting a shift in cellular responses (Keller et al., 2011; Patel et al., 2014). Interestingly, results of our previous study (Patel et al., 2015) revealed that one of the parameters linked with a positive therapeutic outcome was indeed an increased level of IL-10. IL-10 is a key anti-inflammatory cytokine involved in regulation of innate and adaptive immune response (Couper et al., 2008; Saraiva and O'Garra, 2010). Induction of IL-10 represents a homeostatic mechanism that mediates a feedback inhibition loop and limits excessive production of proinflammatory cytokines, including TNFα, IL-1β, and IL-6 (Howard and O'Garra, 1992; Moore et al., 2001). Furthermore, evidence suggests that IL-10 signaling, through its functional IL-10R1, exerts its anti-inflammatory effects in part by selective induction/formation of the p50/p50 homodimers, known to suppress transcriptional NF-κB activity (Driessler et al., 2004). Although in infections an overexpression of IL-10 may induce so-called immune escape and correlate with poor pathogen control (Nylén and Sacks, 2007; Couper et al., 2008; Bai et al., 2009; Richter et al., 2013), the immune-regulatory properties of IL-10 conferred protection in brain injuries including ischemia (Pérez-de Puig et al., 2013), 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MTPT)-mediated striatal toxicity (Joniec-Maciejak et al., 2014), as well as in neurodegenerative disorders such as ALS. Our results clearly demonstrate that in SOD1-mutant-mediated ALS, experimental inhibition of IL-10 signaling markedly increases inflammatory markers in microglial cells and precipitates disease onset in SOD1G93A mutant mice (Figs. 6, 7), whereas targeted overexpression of IL-10 in microglial cells by AAV-mediated gene delivery confers neuroprotection; namely, a single intrathecal injection of viral vector encoding IL-10 under transcriptional control of the CD11b promoter in adult 30- to 40-d-old SOD1G93A mice delayed disease onset by 9 d and increased survival by 13 d. Our data are in accordance with the previous work of Ayers et al. (2015), who demonstrated that neonatal AAV-mediated delivery of IL-10 into the spinal axis increased the survival of SOD1G93A mice. Moreover, clinical evidence suggests that higher levels of IL-10 protein predict longer disease duration in ALS patients (Su et al., 2013). Intriguingly, the role of IL-10 signaling seems to be more complex and/or controversial in Alzheimer's disease. Previous studies reported that the presence of IL-10 promoter polymorphism associated with low production of IL-10 may be considered as an additive genetic risk factor for sporadic AD (Lio et al., 2003). However, previous work using an APP/PS1 mouse model revealed a negative role of IL-10 on Aβ accumulation and synaptic integrity (Chakrabarty et al., 2015; Guillot-Sestier et al., 2015). Thus, more studies are needed to clarify the role of IL-10 and adaptive immune homeostasis in different neurodegenerative disorders.
The questions to be addressed here are, what are the underlying mechanisms involved in 16-fold overexpression of IL-10 observed in presymptomatic microglia, and what is the functional relevance of IL-10-mediated downregulation of TLR2 response (Figs. 3, 4)? Work by Zhao et al. (2010) clearly demonstrated that extracellular toxicity of mSOD1 was mediated by microglia. Indeed, the mSOD1 extracellular protein was not directly toxic to motoneurons; it required microglial activation using TLR and CD14 pathways, thus suggesting that misfolded SOD1 acts as a DAMP and binds to TLRs expressed on microglial cells (Zhao et al., 2010). If misfolded SOD1 acts as a DAMP, the repetitive exposure to small amounts of secreted misfolded SOD1 in early stages of disease would initiate TLR2- and TLR4-mediated microglial priming and trigger enhanced production of IL-10. Overexpression of IL-10 in TLR2–TLR4 primed cells, as reported previously in dendritic cells (Yanagawa and Onoé, 2007; Chang et al., 2009), would then induce an attenuated innate immune response to pathogen-associated molecular patterns (LPS) and DAMP (misfolded mutant SOD1) challenges (Fig. 4). It has been established that misfolded SOD1 can be secreted in extracellular space (Urushitani et al., 2006); thus, a small amount of the extracellular protein, in early disease, can bind to microglial TLRs and prime the cells. Our results are in accordance with this view; namely, SOD1G93A mutant mice, when compared to age-matched WT littermates showed attenuated sensitivity to injected misfolded SOD1 species (Fig. 4). Furthermore, the presence of small amounts of misfolded protein may explain why changes in immune cell phenotypes and relative overexpression of IL-10 were restricted to the CNS (Figs. 5, 6E–J). It is noteworthy that misfolded SOD1 species have not been detected and/or secreted at the periphery; therefore, the TLR-mediated priming effect in presymptomatic disease is absent (Fig. 4).
In conclusion, our results provide important in vivo evidence of a distinct adaptive shift in functional microglial phenotypes in preclinical stages of SOD1-mutant-mediated disease. Based on our results, we propose a distinct role for IL-10 in early disease mechanisms. Here it is noteworthy that IL-10 production becomes deregulated with aging (Ciaramella et al., 2011). This may suggest that in ALS, the capacity of microglial cells to adequately produce IL-10 in response to different challenges may be compromised by aging, resulting in reactive microgliosis and neuronal stress (Figs. 6, 7), leading to a clinical stage of disease. Finally, our results clearly demonstrate that, early in disease, IL-10 is instrumental in the maintenance of immune homeostasis of microglial cells. Moreover, based on our results, we propose that targeted overexpression of IL-10 in microglia may have therapeutic potential in ALS; namely, IL-10 modulates microglial immune phenotypes and temporally extends their adaptive/neuroprotective state, which may significantly delay early neuronal dysfunction and decrease the initial burden of the disease.
Footnotes
This work was supported by the Muscular Dystrophy Association, Canadian Institutes of Health Research, and the Amyotrophic Lateral Sclerosis Society of Canada. J.K. holds a Senior Scholarship Award from Fonds de recherche du Québec en Santé. We thank Christine Bareil, Geneviève Roussin, and Sophie Vachon for their technical help.
The authors declare no competing financial interests.
References
- Ayers JI, Fromholt S, Sinyavskaya O, Siemienski Z, Rosario AM, Li A, Crosby KW, Cruz PE, DiNunno NM, Janus C, Ceballos-Diaz C, Borchelt DR, Golde TE, Chakrabarty P, Levites Y. Widespread and efficient transduction of spinal cord and brain following neonatal AAV injection and potential disease modifying effect in ALS mice. Mol Ther. 2015;23:53–62. doi: 10.1038/mt.2014.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai F, Town T, Qian F, Wang P, Kamanaka M, Connolly TM, Gate D, Montgomery RR, Flavell RA, Fikrig E. IL-10 signaling blockade controls murine West Nile virus infection. PLoS Pathog. 2009;5:e1000610. doi: 10.1371/journal.ppat.1000610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beach TG, White CL, 3rd, Hladik CL, Sabbagh MN, Connor DJ, Shill HA, Sue LI, Sasse J, Bachalakuri J, Henry-Watson J, Akiyama H, Adler CH, Arizona Parkinson's Disease C. Olfactory bulb alpha-synucleinopathy has high specificity and sensitivity for Lewy body disorders. Acta Neuropathol. 2009;117:169–174. doi: 10.1007/s00401-008-0450-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beers DR, Henkel JS, Xiao Q, Zhao W, Wang J, Yen AA, Siklos L, McKercher SR, Appel SH. Wild-type microglia extend survival in PU.1 knockout mice with familial amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A. 2006;103:16021–16026. doi: 10.1073/pnas.0607423103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beers DR, Henkel JS, Zhao W, Wang J, Huang A, Wen S, Liao B, Appel SH. Endogenous regulatory T lymphocytes ameliorate amyotrophic lateral sclerosis in mice and correlate with disease progression in patients with amyotrophic lateral sclerosis. Brain. 2011;134:1293–1314. doi: 10.1093/brain/awr074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boillée S, Yamanaka K, Lobsiger CS, Copeland NG, Jenkins NA, Kassiotis G, Kollias G, Cleveland DW. Onset and progression in inherited ALS determined by motor neurons and microglia. Science. 2006;312:1389–1392. doi: 10.1126/science.1123511. [DOI] [PubMed] [Google Scholar]
- Bosco DA, Morfini G, Karabacak NM, Song Y, Gros-Louis F, Pasinelli P, Goolsby H, Fontaine BA, Lemay N, McKenna-Yasek D, Frosch MP, Agar JN, Julien JP, Brady ST, Brown RH., Jr Wild-type and mutant SOD1 share an aberrant conformation and a common pathogenic pathway in ALS. Nat Neurosci. 2010;13:1396–1403. doi: 10.1038/nn.2660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakrabarty P, Li A, Ceballos-Diaz C, Eddy JA, Funk CC, Moore B, DiNunno N, Rosario AM, Cruz PE, Verbeeck C, Sacino A, Nix S, Janus C, Price ND, Das P, Golde TE. IL-10 alters immunoproteostasis in APP mice, increasing plaque burden and worsening cognitive behavior. Neuron. 2015;85:519–533. doi: 10.1016/j.neuron.2014.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang YC, Kao WC, Wang WY, Wang WY, Yang RB, Peck K. Identification and characterization of oligonucleotides that inhibit Toll-like receptor 2-associated immune responses. FASEB J. 2009;23:3078–3088. doi: 10.1096/fj.09-129312. [DOI] [PubMed] [Google Scholar]
- Ciaramella A, Spalletta G, Bizzoni F, Salani F, Caltagirone C, Bossù P. Effect of age on surface molecules and cytokine expression in human dendritic cells. Cell Immunol. 2011;269:82–89. doi: 10.1016/j.cellimm.2011.04.010. [DOI] [PubMed] [Google Scholar]
- Cordeau P, Jr, Lalancette-Hébert M, Weng YC, Kriz J. Live imaging of neuroinflammation reveals sex and estrogen effects on astrocyte response to ischemic injury. Stroke. 2008;39:935–942. doi: 10.1161/STROKEAHA.107.501460. [DOI] [PubMed] [Google Scholar]
- Couper KN, Blount DG, Riley EM. IL-10: the master regulator of immunity to infection. J Immunol. 2008;180:5771–5777. doi: 10.4049/jimmunol.180.9.5771. [DOI] [PubMed] [Google Scholar]
- Driessler F, Venstrom K, Sabat R, Asadullah K, Schottelius AJ. Molecular mechanisms of interleukin-10-mediated inhibition of NF-kappaB activity: a role for p50. Clin Exp Immunol. 2004;135:64–73. doi: 10.1111/j.1365-2249.2004.02342.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frakes AE, Ferraiuolo L, Haidet-Phillips AM, Schmelzer L, Braun L, Miranda CJ, Ladner KJ, Bevan AK, Foust KD, Godbout JP, Popovich PG, Guttridge DC, Kaspar BK. Microglia induce motor neuron death via the classical NF-kappaB pathway in amyotrophic lateral sclerosis. Neuron. 2014;81:1009–1023. doi: 10.1016/j.neuron.2014.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gowing G, Dequen F, Soucy G, Julien JP. Absence of tumor necrosis factor-alpha does not affect motor neuron disease caused by superoxide dismutase 1 mutations. J Neurosci. 2006;26:11397–11402. doi: 10.1523/JNEUROSCI.0602-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gowing G, Lalancette-Hébert M, Audet JN, Dequen F, Julien JP. Macrophage colony stimulating factor (M-CSF) exacerbates ALS disease in a mouse model through altered responses of microglia expressing mutant superoxide dismutase. Exp Neurol. 2009;220:267–275. doi: 10.1016/j.expneurol.2009.08.021. [DOI] [PubMed] [Google Scholar]
- Grad LI, Yerbury JJ, Turner BJ, Guest WC, Pokrishevsky E, O'Neill MA, Yanai A, Silverman JM, Zeineddine R, Corcoran L, Kumita JR, Luheshi LM, Yousefi M, Coleman BM, Hill AF, Plotkin SS, Mackenzie IR, Cashman NR. Intercellular propagated misfolding of wild-type Cu/Zn superoxide dismutase occurs via exosome-dependent and -independent mechanisms. Proc Natl Acad Sci U S A. 2014;111:3620–3625. doi: 10.1073/pnas.1312245111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillot-Sestier MV, Doty KR, Gate D, Rodriguez J, Jr, Leung BP, Rezai-Zadeh K, Town T. Il10 deficiency rebalances innate immunity to mitigate Alzheimer-like pathology. Neuron. 2015;85:534–548. doi: 10.1016/j.neuron.2014.12.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurney ME. Transgenic-mouse model of amyotrophic lateral sclerosis. N Engl J Med. 1994;331:1721–1722. doi: 10.1056/NEJM199412223312516. [DOI] [PubMed] [Google Scholar]
- Henkel JS, Beers DR, Zhao W, Appel SH. Microglia in ALS: the good, the bad, and the resting. J Neuroimmune Pharmacol. 2009;4:389–398. doi: 10.1007/s11481-009-9171-5. [DOI] [PubMed] [Google Scholar]
- Henkel JS, Beers DR, Wen S, Rivera AL, Toennis KM, Appel JE, Zhao W, Moore DH, Powell SZ, Appel SH. Regulatory T-lymphocytes mediate amyotrophic lateral sclerosis progression and survival. EMBO Mol Med. 2013;5:64–79. doi: 10.1002/emmm.201201544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard M, O'Garra A. Biological properties of interleukin 10. Immunol Today. 1992;13:198–200. doi: 10.1016/0167-5699(92)90153-X. [DOI] [PubMed] [Google Scholar]
- Joniec-Maciejak I, Ciesielska A, Wawer A, Sznejder-Pachołek A, Schwenkgrub J, Cudna A, Hadaczek P, Bankiewicz KS, Członkowska A, Członkowski A. The influence of AAV2-mediated gene transfer of human IL-10 on neurodegeneration and immune response in a murine model of Parkinson's disease. Pharmacol Rep. 2014;66:660–669. doi: 10.1016/j.pharep.2014.03.008. [DOI] [PubMed] [Google Scholar]
- Keller AF, Gravel M, Kriz J. Live imaging of amyotrophic lateral sclerosis pathogenesis: disease onset is characterized by marked induction of GFAP in Schwann cells. Glia. 2009;57:1130–1142. doi: 10.1002/glia.20836. [DOI] [PubMed] [Google Scholar]
- Keller AF, Gravel M, Kriz J. Treatment with minocycline after disease onset alters astrocyte reactivity and increases microgliosis in SOD1 mutant mice. Exp Neurol. 2011;228:69–79. doi: 10.1016/j.expneurol.2010.12.010. [DOI] [PubMed] [Google Scholar]
- Kleinewietfeld M, Hafler DA. Regulatory T cells in autoimmune neuroinflammation. Immunol Rev. 2014;259:231–244. doi: 10.1111/imr.12169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalancette-Hébert M, Gowing G, Simard A, Weng YC, Kriz J. Selective ablation of proliferating microglial cells exacerbates ischemic injury in the brain. J Neurosci. 2007;27:2596–2605. doi: 10.1523/JNEUROSCI.5360-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalancette-Hébert M, Phaneuf D, Soucy G, Weng YC, Kriz J. Live imaging of Toll-like receptor 2 response in cerebral ischaemia reveals a role of olfactory bulb microglia as modulators of inflammation. Brain. 2009;132:940–954. doi: 10.1093/brain/awn345. [DOI] [PubMed] [Google Scholar]
- Lalancette-Hébert M, Moquin A, Choi AO, Kriz J, Maysinger D. Lipopolysaccharide-QD micelles induce marked induction of TLR2 and lipid droplet accumulation in olfactory bulb microglia. Mol Pharm. 2010;7:1183–1194. doi: 10.1021/mp1000372. [DOI] [PubMed] [Google Scholar]
- Lalancette-Hébert M, Swarup V, Beaulieu JM, Bohacek I, Abdelhamid E, Weng YC, Sato S, Kriz J. Galectin-3 is required for resident microglia activation and proliferation in response to ischemic injury. J Neurosci. 2012;32:10383–10395. doi: 10.1523/JNEUROSCI.1498-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerner A, Bagic A. Olfactory pathogenesis of idiopathic Parkinson disease revisited. Mov Disord. 2008;23:1076–1084. doi: 10.1002/mds.22066. [DOI] [PubMed] [Google Scholar]
- Lio D, Licastro F, Scola L, Chiappelli M, Grimaldi LM, Crivello A, Colonna-Romano G, Candore G, Franceschi C, Caruso C. Interleukin-10 promoter polymorphism in sporadic Alzheimer's disease. Genes Immun. 2003;4:234–238. doi: 10.1038/sj.gene.6363964. [DOI] [PubMed] [Google Scholar]
- Liu Y, Hao W, Dawson A, Liu S, Fassbender K. Expression of amyotrophic lateral sclerosis-linked SOD1 mutant increases the neurotoxic potential of microglia via TLR2. J Biol Chem. 2009;284:3691–3699. doi: 10.1074/jbc.M804446200. [DOI] [PubMed] [Google Scholar]
- Maysinger D, Behrendt M, Lalancette-Hébert M, Kriz J. Real-time imaging of astrocyte response to quantum dots: in vivo screening model system for biocompatibility of nanoparticles. Nano Lett. 2007;7:2513–2520. doi: 10.1021/nl071611t. [DOI] [PubMed] [Google Scholar]
- Medzhitov R, Horng T. Transcriptional control of the inflammatory response. Nat Rev Immunol. 2009;9:692–703. doi: 10.1038/nri2634. [DOI] [PubMed] [Google Scholar]
- Moore KW, de Waal Malefyt R, Coffman RL, O'Garra A. Interleukin-10 and the interleukin-10 receptor. Annu Rev Immunol. 2001;19:683–765. doi: 10.1146/annurev.immunol.19.1.683. [DOI] [PubMed] [Google Scholar]
- Nguyen MD, Julien JP, Rivest S. Induction of proinflammatory molecules in mice with amyotrophic lateral sclerosis: no requirement for proapoptotic interleukin-1beta in neurodegeneration. Ann Neurol. 2001;50:630–639. doi: 10.1002/ana.1256. [DOI] [PubMed] [Google Scholar]
- Nylén S, Sacks D. Interleukin-10 and the pathogenesis of human visceral leishmaniasis. Trends Immunol. 2007;28:378–384. doi: 10.1016/j.it.2007.07.004. [DOI] [PubMed] [Google Scholar]
- Patel P, Kriz J, Gravel M, Soucy G, Bareil C, Gravel C, Julien JP. Adeno-associated virus-mediated delivery of a recombinant single-chain antibody against misfolded superoxide dismutase for treatment of amyotrophic lateral sclerosis. Mol Ther. 2014;22:498–510. doi: 10.1038/mt.2013.239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel P, Julien JP, Kriz J. Early-stage treatment with withaferin A reduces levels of misfolded superoxide dismutase 1 and extends lifespan in a mouse model of amyotrophic lateral sclerosis. Neurotherapeutics. 2015;12:217–233. doi: 10.1007/s13311-014-0311-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez-de Puig I, Miró F, Salas-Perdomo A, Bonfill-Teixidor E, Ferrer-Ferrer M, Márquez-Kisinousky L, Planas AM. IL-10 deficiency exacerbates the brain inflammatory response to permanent ischemia without preventing resolution of the lesion. J Cereb Blood Flow Metab. 2013;33:1955–1966. doi: 10.1038/jcbfm.2013.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter K, Perriard G, Behrendt R, Schwendener RA, Sexl V, Dunn R, Kamanaka M, Flavell RA, Roers A, Oxenius A. Macrophage and T cell produced IL-10 promotes viral chronicity. PLoS Pathog. 2013;9:e1003735. doi: 10.1371/journal.ppat.1003735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robberecht W, Philips T. The changing scene of amyotrophic lateral sclerosis. Nat Rev Neurosci. 2013;14:248–264. doi: 10.1038/nrn3430. [DOI] [PubMed] [Google Scholar]
- Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, Donaldson D, Goto J, O'Regan JP, Deng HX, Rahmani Z, Krizus A, McKenna-Yasek D, Cayabyab A, Gaston SM, Berger R, Tanzi RE, Halperin JJ, Herzfeldt B, Van den Bergh R, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362:59–62. doi: 10.1038/362059a0. [DOI] [PubMed] [Google Scholar]
- Saraiva M, O'Garra A. The regulation of IL-10 production by immune cells. Nat Rev Immunol. 2010;10:170–181. doi: 10.1038/nri2711. [DOI] [PubMed] [Google Scholar]
- Saxena S, Roselli F, Singh K, Leptien K, Julien JP, Gros-Louis F, Caroni P. Neuroprotection through excitability and mTOR required in ALS motoneurons to delay disease and extend survival. Neuron. 2013;80:80–96. doi: 10.1016/j.neuron.2013.07.027. [DOI] [PubMed] [Google Scholar]
- Su XW, Simmons Z, Mitchell RM, Kong L, Stephens HE, Connor JR. Biomarker-based predictive models for prognosis in amyotrophic lateral sclerosis. JAMA Neurol. 2013;70:1505–1511. doi: 10.1001/jamaneurol.2013.4646. [DOI] [PubMed] [Google Scholar]
- Synofzik M, Ronchi D, Keskin I, Basak AN, Wilhelm C, Gobbi C, Birve A, Biskup S, Zecca C, Fernández-Santiago R, Kaugesaar T, Schöls L, Marklund SL, Andersen PM. Mutant superoxide dismutase-1 indistinguishable from wild-type causes ALS. Hum Mol Genet. 2012;21:3568–3574. doi: 10.1093/hmg/dds188. [DOI] [PubMed] [Google Scholar]
- Urushitani M, Sik A, Sakurai T, Nukina N, Takahashi R, Julien JP. Chromogranin-mediated secretion of mutant superoxide dismutase proteins linked to amyotrophic lateral sclerosis. Nat Neurosci. 2006;9:108–118. doi: 10.1038/nn1603. [DOI] [PubMed] [Google Scholar]
- Wesson DW, Levy E, Nixon RA, Wilson DA. Olfactory dysfunction correlates with amyloid-beta burden in an Alzheimer's disease mouse model. J Neurosci. 2010;30:505–514. doi: 10.1523/JNEUROSCI.4622-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Q, Zhao W, Beers DR, Yen AA, Xie W, Henkel JS, Appel SH. Mutant SOD1(G93A) microglia are more neurotoxic relative to wild-type microglia. J Neurochem. 2007;102:2008–2019. doi: 10.1111/j.1471-4159.2007.04677.x. [DOI] [PubMed] [Google Scholar]
- Yanagawa Y, Onoé K. Enhanced IL-10 production by TLR4- and TLR2-primed dendritic cells upon TLR restimulation. J Immunol. 2007;178:6173–6180. doi: 10.4049/jimmunol.178.10.6173. [DOI] [PubMed] [Google Scholar]
- Zetterström P, Andersen PM, Brännström T, Marklund SL. Misfolded superoxide dismutase-1 in CSF from amyotrophic lateral sclerosis patients. J Neurochem. 2011;117:91–99. doi: 10.1111/j.1471-4159.2011.07177.x. [DOI] [PubMed] [Google Scholar]
- Zhang R, Hadlock KG, Do H, Yu S, Honrada R, Champion S, Forshew D, Madison C, Katz J, Miller RG, McGrath MS. Gene expression profiling in peripheral blood mononuclear cells from patients with sporadic amyotrophic lateral sclerosis (sALS) J Neuroimmunol. 2011;230:114–123. doi: 10.1016/j.jneuroim.2010.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao W, Beers DR, Henkel JS, Zhang W, Urushitani M, Julien JP, Appel SH. Extracellular mutant SOD1 induces microglial-mediated motoneuron injury. Glia. 2010;58:231–243. doi: 10.1002/glia.20919. [DOI] [PMC free article] [PubMed] [Google Scholar]