

Abstract
Growth-associated protein 43 (GAP43), a protein kinase C (PKC)-activated phosphoprotein, is often implicated in axonal plasticity and regeneration. In this study, we found that GAP43 can be induced by the endotoxin lipopolysaccharide (LPS) in rat brain astrocytes both in vivo and in vitro. The LPS-induced astrocytic GAP43 expression was mediated by Toll-like receptor 4 and nuclear factor-κB (NF-κB)- and interleukin-6/signal transducer and activator of transcription 3 (STAT3)-dependent transcriptional activation. The overexpression of the PKC phosphorylation-mimicking GAP43S41D (constitutive active GAP43) in astrocytes mimicked LPS-induced process arborization and elongation, while application of a NF-κB inhibitory peptide TAT-NBD or GAP43S41A (dominant-negative GAP43) or knockdown of GAP43 all inhibited astrogliosis responses. Moreover, GAP43 knockdown aggravated astrogliosis-induced microglial activation and expression of proinflammatory cytokines. We also show that astrogliosis-conditioned medium from GAP43 knock-down astrocytes inhibited GAP43 phosphorylation and axonal growth, and increased neuronal damage in cultured rat cortical neurons. These proneurotoxic effects of astrocytic GAP43 knockdown were accompanied by attenuated glutamate uptake and expression of the glutamate transporter excitatory amino acid transporter 2 (EAAT2) in LPS-treated astrocytes. The regulation of EAAT2 expression involves actin polymerization-dependent activation of the transcriptional coactivator megakaryoblastic leukemia 1 (MKL1), which targets the serum response elements in the promoter of rat Slc1a2 gene encoding EAAT2. In sum, the present study suggests that astrocytic GAP43 mediates glial plasticity during astrogliosis, and provides beneficial effects for neuronal plasticity and survival and attenuation of microglial activation.
SIGNIFICANCE STATEMENT Astrogliosis is a complex state in which injury-stimulated astrocytes exert both protective and harmful effects on neuronal survival and plasticity. In this study, we demonstrated for the first time that growth-associated protein 43 (GAP43), a well known growth cone protein that promotes axonal regeneration, can be induced in rat brain astrocytes by the proinflammatory endotoxin lipopolysaccharide via both nuclear factor-κB and signal transducer and activator of transcription 3-mediated transcriptional activation. Importantly, LPS-induced GAP43 mediates plastic changes of astrocytes while attenuating astrogliosis-induced microglial activation and neurotoxicity. Hence, astrocytic GAP43 upregulation may serve to indicate beneficial astrogliosis after CNS injury.
Keywords: astrogliosis, EAAT2, GAP43, microglial activation, MKL1, neurotoxicity
Introduction
Astrogliosis occurs in response to CNS insults, such as inflammation, ischemia, and excitotoxicity, to maintain the homeostasis and provide trophic support in the injured brain (Ridet et al., 1997; Sofroniew and Vinters, 2010). Astrogliosis induced by moderate insult involves morphological hypertrophy with actin-dependent process, arborization, and several beneficial features including upregulation of excitatory amino acid transporter 2 (EAAT2), trophic factors, and aquaporin 4 (AQP4) to attenuate excessive glutamate-induced neurotoxicity, provide survival signaling, and maintain water/osmolarity homeostasis, respectively (Desilva et al., 2008; Sofroniew, 2009). Whereas severe astrogliosis occurs when being aggravated by harmful insults, leading to detrimental effects such as a robust proinflammatory cytokine release and glial scar formation to trigger microglia-mediated neuroinflammation and inhibit axonal regeneration, respectively. (Zhang et al., 2010; Iseki et al., 2012). Yet, specific mediators that can promote beneficial effects and attenuate detrimental effects of astrogliosis to facilitate postinjury repair remain unknown.
Actin reorganization during astrogliosis determines astrocytic plasticity by increasing the astrocytic processes and endfeet for the glia–neuron interaction (Baorto et al., 1992; Molotkov et al., 2013). These cytoskeletal dynamics also mediate the increased expression and surface localization of EAAT2 and AQP4 (Nicchia et al., 2008; Lau et al., 2011). Growth-associated protein 43 (GAP43) is an activity-dependent plasticity protein enriched in axons to promote actin polymerization for filopodia formation and axon regeneration (Nguyen et al., 2009). GAP43 is activated by protein kinase C-mediated phosphorylation at serine 41 (S41), which promotes the polymerization and stabilization of filamentous actin (F-actin), and is inactivated by calcineurin to give the S41-dephosphorylated GAP43 form that blocks actin polymerization (He et al., 1997; Lautermilch and Spitzer, 2000). In astrocytes, the basal level of GAP43 expression is low, although it is essential for astrocyte differentiation (Deloulme et al., 1993; Shen et al., 2004). Interestingly, a recent genomic analysis revealed that Gap43 mRNA was upregulated in reactive astrocytes in both cerebral ischemia and an intracerebroventricular endotoxin lipopolysaccharide (LPS) treatment (Zamanian et al., 2012). However, the functional characterization of astrocytic GAP43 in the injured CNS has not been determined.
Brain inflammation is one of the major triggers of astrogliosis (Fitch and Silver, 2008), and attenuation of the proinflammatory response is known to be beneficial for functional recovery after CNS injuries (Guo et al., 2012). The gene expression associated with astrogliosis is mostly triggered by the activation of the following two transcription factors: nuclear factor-κB (NF-κB) and signal transducer and activator of transcription 3 (STAT3; Sofroniew, 2009). NF-κB is activated by the proinflammatory cytokine receptors and Toll-like receptor 4 (TLR4; Gorina et al., 2011), and then targets proinflammatory genes, such as tumor necrosis factor-α (TNF-α) and interleukin-6 (IL-6), that further activate microglia to amplify neuroinflammation. STAT3 activation during astrocyte reactivation is often triggered by the NF-κB-induced IL-6 release that activate the IL-6 receptor (Harré et al., 2003; Lin et al., 2008), and astrocyte-specific STAT3 knockout impaired the injury-induced astrogliosis in neurodegenerative diseases and neurotrauma animal models (Herrmann et al., 2008; Ben Haim et al., 2015). The best known downstream target of STAT3 involved in astrogliosis is glial fibrillary acidic protein (GFAP; Kahn et al., 1997; Yeo et al., 2013), the astrogliosis marker that is used most often. However, GFAP is not associated with the functional outcome of astrogliosis (Hol and Pekny, 2015). Notably, STAT3 is a transcription factor for Gap43 gene expression in CNS injury (Qiu et al., 2005; Tsai et al., 2007). Thus, astrocytic GAP43 induction observed in the injured brain might involve the astrogliosis-associated STAT3 activation.
In this study, we show that GAP43 expression in primary cultured rat astrocytes as induced by LPS plays beneficial roles in immune modulation and neuronal survival and plasticity during astrogliosis. Both NF-κB and STAT3 were found to be critical for the transcriptional activation of astrocytic GAP43. Furthermore, glutamate uptake activity of the LPS-reactivated astrocytes was found to be dependent upon the astrocytic GAP43 induction, and the mechanism involved the actin-dependent transcriptional regulation of EAAT2.
Materials and Methods
Animals.
Sprague Dawley rats were obtained from BioLASCO Taiwan Co. and National Yang-Ming University (Taipei, Taiwan). The neonatal rat pups at postnatal days 1–2 were used for the astrocyte preparation, and pregnant female rats were used to harvest embryonic rats for the primary culture of cortical neurons. Animal experimentation procedures were approved by the Experimental Animal Review Committee at National Yang-Ming University and were conducted in keeping with the U.S. National Institutes of Health Guide for the Care and Use of Laboratory Animals and the U.S. National Institutes of Health guidelines for the care and use of animals for experimental procedures.
Drug treatment.
Lipopolysaccharide, methylprednisolone (MP), and glutamate were obtained from Sigma-Aldrich. LPS Rhodobacter sphaeroides (LPS-RS) and polyinosinic:polycytidylic acid [Poly(I:C)] were obtained from InvivoGen. AG490 and parthenolide (PTN) were obtained from Calbiochem. For the NF-κB pathway inhibitor peptide (NBD), TAT-NBD (ygrkkrrqrr–TALDWSWLQTE; Nijboer et al., 2008) and its mutant peptide TAT-NBDmut (ygrkkrrqrr–TALDASALQTE) were synthesized by Kelowna International Scientific.
Primary cultures of rat cortical astrocytes, microglia, neurons, and SM826 microglia cell line.
Primary cultured rat astrocytes and microglia were prepared from Sprague Dawley rat pups at postnatal days 1–2, as previously described (Sun et al., 2013; Lee et al., 2015). In brief, the dissected cerebral cortices were mechanically triturated in glia culture medium [GCM; DMEM/F12 (Invitrogen) with 10% FBS, 100 U/ml penicillin, and 100 μg/ml streptomycin], filtered through a 40 μm cell strainer, and the filtered cells were plated in 75 mm flasks coated with poly-l-ornithine and grown in GCM for 12–14 d followed by orbital shaking at 100 rpm in 37°C for 6 h to obtain microglia, which were cultured in GCM. After removing the residual microglia and oligodendrocyte precursor cells by repeating the shaking twice, the final attached cells, mostly astrocytes, were subcultured in the following astrocyte culture medium containing heparin-binding EGF-like growth factor (HBEGF, abbreviated as HB) to obtain stellate-shape HB astrocytes (Foo, 2013): 50% DMEM/50% Neural Basal medium (Invitrogen) containing 292 μg/ml l-glutamine, 100 μg/ml bovine serum albumin, 100 μg/ml transferrin, 16 μg/ml putrescine dihydrochloride, 60 ng/ml progesterone, 40 ng/ml sodium selenite, 5 μg/ml N-acetyl-l-cysteine, 5 ng/ml HBEGF (Sigma-Aldrich), 1 mm sodium pyruvate, 100 U/ml penicillin, and 100 μg/ml streptomycin. The cultured HB astrocytes were used for experiments at day 3 after final seeding, and the purity was ∼98%, as determined by GFAP immunostaining. Primary rat microglia and mouse SM826 microglia cell line were cultured as previous description (Lee et al., 2015). Primary cultured rat cortical neurons were prepared from Sprague Dawley rat embryos harvested from pregnant female Sprague Dawley rats at gestation day 17 (Wang et al., 2015). The culture yielded >85% of neuronal population, as characterized by NeuN immunostaining, and 10 d in vitro (DIV) cultures were used.
Intracerebroventricular injection of LPS and immunohistochemistry.
Adult male Sprague Dawley rats (8–9 weeks of age) were anesthetized and placed in a stereotactic device (David Kopf Instruments). LPS at 20 or 100 μg/5 μl (Sigma-Aldrich) or vehicle (saline) was injected into the right lateral ventricle. After the injection, the rat recovered for 24 h, and then was anesthetized and killed by intracardial perfusion with 4% paraformaldehyde to harvest the brain for cryosection (30 μm thickness). The brain sections were permeabilized with Tris-buffered saline (TBS) containing 0.05% Tween-20 and then immunostained with rabbit anti-GFAP (1:100; Millipore) and mouse anti-GAP43 (1:50; Millipore) diluted in TBS with 0.1% Tween-20, followed by secondary antibody incubation [Alexa Fluor 488- or Alexa Fluor 594-conjugated goat anti-mouse or rabbit IgG (1:400; Invitrogen)], and nuclear staining with 4′,6′-diamidino-2-phenylindole dihydrochloride (DAPI; Abcam). The fluorescent images were acquired using an FV1000 (Olympus) confocal microscope, and the colocalization signals and area of GAP43 and GFAP were quantified using MetaMorph version 7.7.0.0 software (Molecular Devices). The “Color Separate” tool of the MetaMorph Display mode was used to separate the GAP43 and GFAP signals. The thresholded images of GFAP and GAP43 staining were used to select the region in the corpus callosum and cingulum, and then the area of the total GFAP+ with or without GAP43-overlapped signals was obtained by using the “Measure Colocalization” tool of the MetaMorph Measurement mode. The percentage of GAP43+ and GAP43− area in total GFAP+ area were also calculated.
Western blot analysis and ELISA.
For Western blotting, total proteins of cultured cells were extracted by cold lysis buffer [50 mm Tris, 150 mm NaCl, 1% Triton-X, 0.5% SDS, 1 mm Na3VO4, phosphatase inhibitor cocktail I (Sigma-Aldrich), and protease inhibitor cocktail (GE Healthcare), pH 7.4]. The proteins were electrophoresed, electrotransferred to PVDF membrane, and then probed with primary antibodies including rabbit anti-GAP43 (1:1000; Millipore), anti-S41-phosphorylated GAP43 (1:300; Millipore), anti-GFAP (1:2000; Millipore) diluted in TBS with 0.05% Tween-20 (TBST), anti-lamin A (1:1000; Santa Cruz Biotechnology), anti-STAT3 (1:1000; Cell Signaling Technology), and anti-cyclooxygenase 2 (COX-2; 1:2000; Abcam) antibodies diluted in PBS with 0.05% Tween-20 (PBST); mouse anti-inducible nitric oxide synthase (iNOS; 1:1000; BD Transduction Laboratories), anti-β-actin (1:10,000; Abcam), anti-microtubule-associated protein 2 (MAP2; 1:1000; Millipore), anti-postsynaptic density-95 (PSD-95; 1:1000; NeuroMab) protein, and anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH; 1:1000; Biogenesis) antibodies diluted in PBST. The immune complex was further probed with horseradish peroxidase-conjugated secondary antibodies (Jackson ImmunoResearch Laboratories), visualized using enhanced chemiluminescence reagents (Western Lightning Plus-ECL, PerkinElmer), and detected by ImageQuant LAS 4000 (GE Healthcare). The band intensity was analyzed by GelAnalyzer software. ELISA was performed to measure the protein levels of IL-6 and TNF-α in cultured medium derived from the astrocyte cultures using the commercial Quantikine rat IL-6 and TNF-α ELISA kits (R&D Systems) according to manufacturer instructions.
Immunofluorescence confocal imaging of primary cultured rat brain cells.
Primary cultured astrocytes, microglia, and neurons were fixed with 4% paraformaldehyde, permeabilized, and incubated with PBST-diluted antibodies, as follows: (1) rabbit anti-GFAP and mouse anti-GAP43 antibodies in primary rat astrocytes; (2) rabbit anti-GAP43 and mouse anti-NeuN (Millipore) antibodies in primary rat cortical neurons; and (3) mouse anti-OX42 (CD11b; Serotec) and rabbit anti-Iba1 (Abcam) antibodies in primary rat microglia. DAPI and the rhodamin-conjugated phalloidin were used for labeling the cell nucleus and actin filament, respectively. The fluorescent images were acquired using an FV1000 Olympus confocal microscope, and the colocalization signals of GAP43 and GFAP were quantified using MetaMorph version 7.7.0.0 software. The average number and length of the astrocytic process per cell were quantified using ImageJ [National Institutes of Health (http://rsb.info.nih.gov/ij/)]. For the quantification of the cell number and immunoreactivity area of the OX42+ microglia, the “Arithmetic” tool of the MetaMorph Process mode was used to transfer the confocal images to 16 bit. The 16 bit images of OX42 and DAPI staining were used to measure the double-positive cell number by using the “Multi Wavelength Cell Scoring” tool of the MetaMorph Apps mode, in which both the percentage of OX42+ cells in the total DAPI number and the area of OX42+ signal were calculated. For the quantification of GAP43+ neurite length in primary cortical neurons, the total length of GAP43-labeled neurites was obtained from five to six microscopic fields, and then divided by the number of NeuN-positive neurons in the respective microscopic field.
Flow cytometry.
The flow cytometry that was performed was modified from a previously described procedure (Liang et al., 2010) to detect the population of GAP43+ astrocytes in the primary rat astrocyte culture with or without LPS treatment. In brief, primary rat astrocytes were trypsinized, fixed by 4% paraformaldehyde/PBS, and permeabilized by cold methanol. The permeabilized cells were incubated with both mouse anti-GAP43 (1:50; Millipore) and rabbit anti-AQP4 (1:50; Millipore) antibodies and then with goat anti-mouse IgG-Alexa Fluor 488, anti-rabbit IgG-Alexa Fluor 633 (1:300; Invitrogen), and DAPI. Immunolabeled cells were then washed and resuspended in PBS for the cell population determination using FACSAria (BD Biosciences), and the data were analyzed using FlowJo software (Tree Star Inc.). The analysis included a gating for cell size, and the cell population was presented as a dot plot divided into four quadrants to show the percentage of AQP4−/GAP43−, AQP4−/GAP43+, AQP4+/GAP43−, and AQP4+/GAP43+ in total gated cells.
Quantitative RT-PCR.
Total RNA was extracted with TRIzol reagent (Invitrogen), and 1.5 μg of total RNA was used to synthesize cDNA using a reverse transcriptase kit (Applied Biosystems). The quantitative RT-PCR was performed using ABI StepOne sequence detector system (Applied Biosystems) by SYBR Green methodology and was normalized to the expression of a housekeeping gene (18S), as previously described (Lee et al., 2015). Primer sets and the product size of each cDNA of interest were as follows: rat Gap43 (5′-CCTGCTGCTGTCACTGATGCTG-3′ and 5′-CTCATCCTGTCGGGCACTTTCC-3′, 159 bp); Stat3 (5′-AGCTCTTAGG GCCTGGTGTGAACTACT-3′ and 5′-GGATGGCCCTCTCCCGCTCCTTGCTGA-3′, 201 bp); Slc1a2 (5′-GGTGGCACCTCCATCCGAGGA-3′ and 5′-CAGCT TGGCCTGCTCACCCA-3′, 205 bp); Mkl1 (5′-AGCAGCAGGAAAATGGCTCC TCCAG-3′ and 5′-GTTCACTCAAAGGCGAGGGTTGTGG-3′, 180 bp); iNOS (5′-ACAAGCTGCATGTGACTCCATC-3′ and 5′-GTCCTCTGGTCAAACTCTT GGAG-3′, 171 bp); and 18S (5′-GAGGTGAAATTCTTGGACCGG-3′ and 5′-CGAACCTCCGACTTTCGT TCT-3′, 93 bp).
Chromatin immunoprecipitation.
A chromatin immunoprecipitation (ChIP) assay was performed as described previously (Lin et al., 2009). In brief, the chromatin isolated from primary rat astrocytes was immunoprecipitated with rabbit anti-STAT3 (Cell Signaling Technology), anti-P65/RelA (Cell Signaling Technology), or anti-MRTF-A [megakaryoblastic leukemia 1 (MKL1); Santa Cruz Biotechnology] antibodies at 4°C for 16 h, followed by incubation with a salmon sperm DNA/protein agarose slurry (Millipore) to immobilize the DNA–protein–antibody complex. The protein-bound DNA fragments were purified using a PCR purification kit (Qiagen) for PCR amplification. For the determination of STAT3 and P65/RelA bound on the rat Gap43 gene promoter, PCR primers were designed to amplify the fragments around the rGap43 promoter region containing STATx- and NF-κB-predicted binding sites. The primer sequences were as follows: forward, 5′-TCCATCCAACCTCTCCAACTGTCTG-3′; and reverse, 5′-TTCTCAGAA TTGTTCACTAGCTTCC-3′. For the determination of MKL1 binding on the putative serum response element (SRE)-containing rat Slc1a2 gene promoter fragment, the primer sets were as follows: forward, 5′-TGTAGGTGTCTGCTGTGTCCCAGTCCC-3′; and reverse, 5′-GCTGGAGACGTATCTCTTCCACCAACCG-3′. Semi-quantitative PCR were performed to examine the ChIP results. The band intensity was quantified, and the enrichment of protein–DNA binding in each group was expressed as the relative band intensity to the respective control group.
Plasmid construction and transfection.
A luciferase reporter plasmid driven by rat GAP43 (rGAP43) promoter (approximately −864 to −402, AC_000079.1) was prepared, as described previously with modification (Takahashi et al., 2006). The genomic DNA was extracted from a Sprague Dawley rat brain, the fragment of Gap43 promoter was obtained by PCR following gel extraction, and the nucleotide sequences of this PCR product were verified by sequencing. The DNA fragment was cloned and subsequently constructed into KpnI and XbaI sites of pGL3-basic vector (Promega) to obtain the pGL3-rGap43 promoter firefly luciferase construct. To examine the Gap43 promoter activity in astrocytes, the cultured astrocytes in a 24-well plate were cotransfected with 0.5 μg/well pGL3-rGap43 promoter construct and 0.05 μg/well pRL-TK Renilla luciferase normalization construct (Promega) using Lipofectamine 3000 (Invitrogen) according to manufacturer instructions. The transfected astrocytes were then subjected to LPS treatment for 24 h, observed to measure the luciferase activity using the Dual-Luciferase Assay System (Promega) and to detect the luminescence using a luminometer (Berthold). The relative luciferase activity driven by the rGap43 promoter was calculated by normalizing to Renilla signals and ratio to the control group. For the GAP43 overexpression experiment, the EGFP-tagged rat GAP43 wild-type (pEGFPN1-rGAP43-WT) and S41 mutants that mimic phosphorylated S41 (pEGFPN1-rGAP43-S41D) and dephosphorylated GAP43 (pEGFPN1-rGAP43-S41A) were established as described previously. The constructs were transfected into primary astrocytes using Lipofectamine 3000 for 24 h followed by the LPS treatment, and the empty vector transfectant was used as the control. The GFP-positive cells were examined for image analysis.
RNA interference.
Silencing of the gene expression of interest was performed by transfecting small interfering RNAs (siRNAs) specifically targeting mRNAs of rat Gap43, Stat3, Mkl1, or scramble siRNA (Silencer Select Predesigned siRNA, Ambion) into cultured astrocytes using Lipofectamine 3000 according to manufacturer instructions. The siRNA sequences are as following: GAP43 (rat Gap43; NM_017195), 5′-AGAAUGAUGAGGACCAAAAtt-3′ (exon 2) and 5′-ACAGGAUGAGGGU AAAGAAtt-3′ (exon 3); STAT3 (rat stat3; NM_012747) 5′-GAGUUGAAUUAUCA GCUUAtt-3′ (exon 11) and 5′-GCAGAGUUCAAGCACCUGAtt-3′ (exon 13); and MKL1 (rat Mkl1; XM_003750385), 5′-AGGACUAUUUGAAACGGAAtt-3′ (exon 4) and 5′-GCACAUGGAUGAUCUGUUUtt-3′ (exon 12). The cultured astrocytes were incubated in opti-MEM containing siRNAs for 5 h, and then the medium was replaced with astrocyte medium and incubated for another 43 h, followed by the designated treatments. The knock-down efficiencies of these genes were tested by quantitative real-time PCR or Western blotting at 48 h after the siRNA transfection.
Astrocyte-conditioned medium.
Astrocyte media derived from primary rat astrocytes with designated treatments were prepared as previously described with minor modification (Lin et al., 2011). Cultured astrocytes were transfected with Gap43 siRNA or scrambled RNA in opti-MEM for 5 h, and then the medium was renewed to remove siRNAs and was incubated for another 43 h to make the total 48 h transfection. The transfected astrocytes were then treated with or without 1 μg/ml LPS for 1 h, and then the medium was renewed to remove LPS and incubated for another 23 h to harvest the astrocyte-conditioned medium (ACM). The ACM obtained did not contain siRNA or LPS, and were used to treat primary cultured rat cortical neurons and microglia. In parallel, the concentrations of IL-6 and TNF-α in ACM were determined by ELISA.
Cell damage and survival assessment.
Astrocytic damage and survival were examined by lactate dehydrogenase (LDH) release assay (Lin et al., 2009) and water-soluble tetrazolium salt-1 (WST-1) assay, respectively. For LDH assay, the cultured media from the treated astrocytes or neurons were harvested to measure the released LDH activity. For astrocytes, the percentage of LDH release was calculated by the ratio to the respective control. For neurons, the intraneuronal LDH activities were also measured, and the percentage of LDH release in each well was obtained by dividing the medium LDH by the total LDH level (medium plus intracellular). For the WST-1 assay, cells were incubated with diluted WST-1 reagent (Roche) for 2 h in an incubator at 37°C. After shaking for 1 min, the cell suspension was subjected to an ELISA reader, and absorbance at A450 with reference at A600 was measured.
Glutamate uptake assay.
The glutamate uptake activity of astrocytes was assessed by measuring the glutamate clearance capacity, as modified from a previous report (Roybon et al., 2013). Briefly, the cultured rat astrocytes were equilibrated in Na+ buffer (5 mm Tris-HCl, pH 7.2, 10 mm HEPES, 140 mm NaCl, 2.5 mm KCl, 1.2 mm CaCl2, 1.2 mm MgCl2, 1.2 mm K2HPO4, and 10 mm d-glucose) for 10 min at room temperature, and then shifted to Na+ buffer containing 2 mm glutamate (Sigma) for another 10 min at 37°C, followed by two rapid washes to remove extracellular glutamate with ice-cold Na+-free buffer (5 mm Tris-HCl, pH 7.2, 10 mm HEPES, 140 mm choline-Cl, 2.5 mm KCl, 1.2 mm CaCl2, 1.2 mm MgCl2, 1.2 mm K2HPO4 and 10 mm d-glucose). The cells were then lysed to determine the intracellular glutamate content using Glutamate Colorimetric Assay kit (BioVision) according to the manufacturer protocol. Absorbance was measured at 450 nm using a microplate reader. The concentration of intracellular glutamate was calculated from the standard curve and normalized to the total protein amount per sample.
Statistic analysis.
Statistical analyses were performed using GraphPad Prism software version 5.0. Data were expressed as the mean ± SEM. Statistical analysis was performed using the unpaired t test. p < 0.05 was considered significant. The p values were as follows: *p < 0.05, **p < 0 0.01, and ***p < 0.001 versus the respective control group; #p < 0.05, ##p < 0 0.01, and ###p < 0.001 versus the LPS group, or the designated pair of groups unless stated otherwise.
Results
Proinflammatory LPS significantly increased GAP43 expression via TLR4 in primary cultured astrocytes
We examined whether the proinflammatory LPS treatment, which induces astrogliosis, would affect the GAP43 expression in primary cultured rat astrocytes. Rat astrocytes were cultured in medium containing HBEGF (for details, see Materials and Methods), so-called HB astrocytes, with a stellate-shaped phenotype, which is similar to their morphology in the brain. Western blot analysis showed that LPS concentrations at 2.5, 50, and 1000 ng/ml elevated iNOS, a proinflammatory mediator that indicates astrocyte reactivation, in a dose-dependent manner. These concentrations of LPS treatments also significantly increased the total GAP43 (2.5 ng/ml, 1.7-fold; 50 ng/ml, 1.7-fold; 1000 ng/ml, 1.9-fold) and S41-phosphorylated GAP43 (2.5 ng/ml, 1.4-fold; 50 ng/ml, 1.6-fold; 1000 ng/ml, 1.8-fold; Fig. 1A,B). However, these LPS treatments did not affect the expression of the astrocyte marker GFAP. Notably, the phosphorylation of GAP43 at S41, which indicates its capacity to promote actin polymerization, was not enhanced in all of the LPS concentration treatments, as indicated by the ratio of S41-phosphorylated GAP43 to total GAP43. Thus, GAP43 and its active form can be upregulated in parallel in reactivated astrocytes induced by proinflammatory LPS treatment. LPS at 50 ng/ml was thus chosen for subsequent in vitro studies given its submaximal effect on the GAP43 induction.
Figure 1.

LPS-induced GAP43 upregulation in primary rat astrocytes. A, Western blot analysis of GAP43, S41-phosphorylated GAP43 (pGAP43), GFAP, and proinflammatory iNOS proteins in primary rat astrocytes cultured with HBEGF-containing medium (HB astrocytes) treated with LPS for 24 h at indicated concentrations. The intensity of the bands of each target protein was quantified and normalized by Lamin A. B, Quantitative results of iNOS, GAP43, pGAP43, pGAP43/GAP43 ratio, and GFAP in A. n = 3–5. C, Immunofluorescent confocal images of GAP43 and GFAP in primary cultured rat HB astrocytes exposed to 50 ng/ml LPS with or without 1 h pretreatment with a TLR4 antagonist, RS (500 ng/ml), or a TLR3 agonist, Poly(I:C) (10 μg/ml) for 24 h. D, Quantification of the percentage of GAP43-GFAP colocalization area in C. The number of microscopic fields was counted as indicated. E, Immunofluorescent confocal images of GAP43, GFAP, and F-actin (blue) in primary cultured rat MD astrocytes treated with LPS (50 ng/ml) or its vehicle (CTL; equal volume of H2O) for 24 h. F, Flow cytometry analysis of GAP43-labeled and astrocyte marker AQP4-labeled cells in HB astrocytes treated with 50 or 1000 ng/ml LPS for 24 h. The two-parameter histogram represented AQP4-Alexa Fluor 633 staining (APC) on the x-axis and GAP43-Alexa Fluor 488 staining (GFP) on the y-axis. The numbers indicated the percentage of AQP4+ astrocytes below or above the horizontal line that indicated the threshold signal of GAP43.
Immunofluorescent staining of GAP43 and GFAP in the HB astrocytes showed that the basal level of GAP43 immunoreactivity was much lower than the GFAP signals in the untreated astrocytes, and the former was markedly elevated by the LPS treatment (Fig. 1C). Notably, LPS induced GFAP–GAP43 colocalization in the astrocytic processes, as shown by confocal microscopy. LPS is an agonist of TLR4, and its GAP43-inducing effect was blocked by TLR4 antagonist (LPS-RS), whereas the TLR3 agonist Poly(I:C) did not induce such an effect (Fig. 1C). Quantitative results indicated that LPS significantly increased the colocalization signals from 17.7% to 33.9% (p = 0.0022), and this LPS effect was blocked to 17.8% by LPS-RS. The colocalization signal in the Poly(I:C) group was 23.4%, with no significant difference from the control (Fig. 1D). Notably, when the same LPS treatment was performed in McCarthy-de Vellis (MD) astrocytes, which were cultured in the absence of HBEGF supplement (McCarthy and de Vellis, 1980), the astrocytes showed a flat-shaped phenotype without stellation in both the control and LPS-treated groups (Fig. 1E). GAP43 in the LPS-activated MD astrocytes was found to be redistributed, not increased, from the perinuclear cytoplasm to the inner surface membrane, and the actin filaments were also rearranged to the inner surface membrane and highly colocalized with GAP43.
GAP43 upregulation by LPS may increase the population of GAP43+ astrocytes. We examined this assumption by using flow cytometry to double label primary HB astrocytes with GAP43 and AQP4, an astrocyte-specific water channel. The flow cytometry data showed that, indeed, LPS increased the population of GAP43+/AQP4+ cells from 46.17% (control) to 67.8% (50 ng/ml LPS) and 69.12% (1000 ng/ml LPS). In contrast, the percentage of GAP43−/AQP4+ astrocytes was decreased from 17.17% (control) to 4.71% (50 ng/ml LPS) and 3.17% (1000 ng/ml LPS; Fig. 1F).
In sum, these results suggest that proinflammatory LPS stimulation can upregulate GAP43 in stellate-shaped astrocytes via TLR4, and thereby can increase the population of GAP43+ astrocytes. Flat-shaped MD astrocytes were less responsive to this GAP43-elevating effect induced by LPS.
LPS stimulation induced astrocytic GAP43 expression in rat brain
The astrocytic GAP43 induction by the LPS stimulation was further validated in vivo by using intracerebroventricular injection of LPS at 20 or 100 μg into the rat brain, in which a dose close to the former was reported to induce secretion of S100B from astrocytes in vivo (Guerra et al., 2011), whereas the latter was presumed to induce severe neuroinflammation. Confocal microscopy showed that both 20 and 100 μg LPS treatments significantly increased the total levels and colocalization of the GFAP-GAP43 signals in the corpus callosum and cingulum (Fig. 2A), whereas the GAP43 signals were mostly separated from the GFAP signals in the saline-injected brain. Quantitative data further indicated that the percentage of GAP43+-GFAP+ area in total GFAP+ area was increased from 7% (saline, i.c.v.) to 69% (20 μg LPS, p < 0.001) and 56% (100 μg LPS, p < 0.001) in the corpus callosum (Fig. 2B, left), and from 16% to 65% (20 μg LPS, p < 0.001) and 60% (100 μg LPS, p < 0.001) in the cingulum (Fig. 2B, right). Notably, we found that the proportion of the GAP43-GFAP colocalization in the total GFAP+ area seems to be lower in the 100 μg LPS group compared with the 20 μg LPS group in both brain regions, with their difference in the corpus callosum reached statistical significance (Fig. 2B, left; p = 0.0021). Thus, inflammatory LPS stimulation can also increase GAP43 in reactive astrocytes in vivo, and this effect seems to be more prominent under moderate rather than severe insult.
Figure 2.
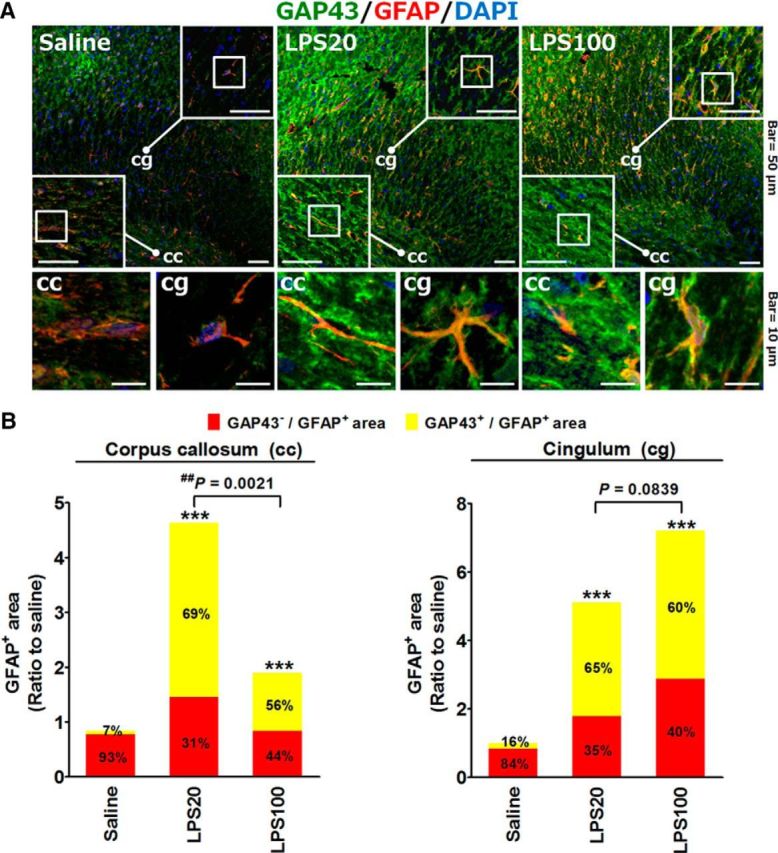
LPS-induced GAP43 expression in GFAP-positive astrocytes in vivo. A, Immunohistochemistry and confocal microscopy of GAP43 and GFAP in the corpus callosum (cc) and cingulum (cg) sections of rats treated with unilateral injection of LPS (20 or 100 μg, i.c.v., in 5 μl of saline solution) or an equal volume of saline solution for 24 h. Representative confocal images of corpus callosum and cingulum regions in the square frames were enlarged to indicate the GAP43+/GFAP+ immunoreactivities. DAPI (blue) indicated the cell nucleus. B, Quantitative graph of the total GFAP+ area in cc (left) and cg (right), and the proportion of GAP43+/GFAP+ vs GAP43−/GFAP+ signals in the total GFAP+ area calculated from two individual rat brains with two sections (bregma, −2.56 mm) per rat in each experimental group in A. ***p < 0.001 compared with the saline group by Fisher's exact test for the proportion of astrocytic GAP43+ signal.
NF-κB and STAT3 mediate the transcriptional activation of GAP43 in LPS-activated astrocytes
To investigate the mechanism of astrocytic GAP43 induction by LPS/TLR4, a time-dependent experiment was performed, and the result indicated that the induction was not apparent until 16 h after the LPS treatment (Fig. 3A). We then examined the following signaling mediators: (1) an anti-inflammatory glucocorticoid known to attenuate LPS-induced proinflammatory responses and nitric oxide production in astrocytes (Kozuka et al., 2007); (2) NF-κB, a TLR4 downstream signaling that mediates astrogliosis-induced proinflammatory responses; (3) STAT3, the astrogliosis-associated transcription factor that can be activated by proinflammatory IL-6, one of the proinflammatory NF-κB target genes (Harré et al., 2002; Rego et al., 2011), released from reactivated astrocytes. Using pharmacological approaches, we found that LPS-induced astrocytic Gap43 mRNA expression can be reduced by a clinically used synthetic glucocorticoid MP (1 μm) from 1.5-fold to 0.5-fold (MP plus LPS; p = 0.0004; Fig. 3B). An NF-κB inhibitor, PTN, and a pan JAK/STAT signaling inhibitor, AG490, also blocked LPS-enhanced Gap43 mRNA expression (p = 0.0026 in LPS vs PTN plus LPS; p = 0.009 in LPS vs AG490 plus LPS; Fig. 3B). Thus, astrocytic GAP43 induction is mediated by the LPS-induced signaling mechanisms involved in proinflammatory astrogliosis, and is regulated by anti-inflammatory glucocorticoids.
Figure 3.

LPS-induced astrocytic GAP43 expression was mediated by both NF-κB and STAT3. A, Quantitative RT-PCR analysis showed Gap43 mRNA expression at different time points in LPS (50 ng/ml)-treated primary rat HB astrocytes. B, The increase of Gap43 mRNA expression after LPS treatment (50 ng/ml, 16 h) was inhibited by the anti-inflammatory glucocorticoid MP (1 μm), NF-κB inhibitor PTN (10 μm), and JAK inhibitor AG490 (10 μm), respectively). n = 3. Note that the control group (CTL) was treated with 0.1% DMSO, the vehicle for MP, PTN, and AG490. C, Schematic representation of rat Gap43 (rGAP43) promoter fragment. The predicted NF-κB and STATx binding sites locate in promoter region at a nearby TATA box. The binding activities of NF-κB and STAT3 in this promoter region were examined by ChIP assay with the indicated primers. A luciferase reporter DNA construct of rat Gap43 promoter fragment (rGap43 promoter-Luc) was prepared for promoter activity determination. D, ChIP assay of NF-κB binding on rGap43 promoter by using p65/RelA antibody to precipitate the protein–DNA complex in HB astrocytes treated with 60 min 0.1% DMSO (CTL) or LPS (50 ng/ml) with or without 90 min pretreatment with PTN (10 μm). E, ChIP assay of STAT3 and p65/RelA binding on rGap43 promoter in HB astrocytes treated with LPS at 0, 30, and 60 min. The PCR primers (−633 to −414 bp; C) were used for the DNA fragment amplification by semi-quantitative PCR. The band intensity of the gel images in D and E was quantified and normalized to the input control to obtain the enrichment of PCR products ratio to the CTL or the 0 min values (bottom panel in D and E). n = 3. F, Luciferase reporter assay of LPS (50 ng/ml)-treated HB astrocytes transfected with rGap43 promoter-Luc. The increased luciferase activity after 24 h of LPS treatment was completely blocked by the AG490 (10 μm) pretreatment. n = 6. G, The effects of siRNA-mediated STAT3 knockdown (siSTAT3) on LPS-induced GAP43 expression were determined by Western blotting. n = 3. H, ELISA of IL-6 release at different time point of LPS (50 ng/ml)-treated cultured rat astrocytes. The concentrations of IL-6 released in the culture medium were reported in picograms per milliliter. n = 3. I, The effects of siRNA-mediated IL-6 knockdown (si-IL6) on IL6 and Gap43 mRNA expression at 16 h after the LPS treatment were determined by qRT-PCR. n = 3.
To further dissect the transcriptional regulation of LPS-induced GAP43 expression, we searched for the putative binding sites for NF-κB p65/RelA and STAT3 on the Gap43 gene promoter using the TRANSFAC database version 8.3 (Farré et al., 2003). The Gap43 promoter has been identified upstream of the start cordon and surrounding TATA box, in which the STATs and NF-κB p65/RelA binding sites were identified (Fig. 3C). We then used a ChIP assay to examine the binding of NF-κB p65/RelA and STAT3 on the rat Gap43 gene promoter. The genomic DNA fragment in the Gap43 promoter spanning from −633 to −414 was immunoprecipitated by anti-p65/RelA or anti-STAT3 antibodies followed by PCR amplification. Our data showed that the binding of NF-κB P65/RelA, the direct downstream signaling of TLR4, onto the Gap43 promoter had a 3.4-fold increase compared with the control at 60 min after the LPS treatment in HB astrocytes, and the effect was completely blocked by a NF-κB pathway inhibitor PTN (Fig. 3D). We further examined whether NF-κB and STAT3 bind to the Gap43 promoter sequentially. The ChIP assay data show that NF-κB p65/RelA but not STAT3 bound to the Gap43 promoter at 30 min after the LPS treatment, and then at the 60 min the p65/RelA binding slightly declined, whereas the STAT3 binding became significant (Fig. 3E). These results suggested that the binding of NF-κB and STAT3 to the Gap43 promoter may occur sequentially after the LPS stimulation. We then established an rGap43 promoter–luciferase construct (rGap43 promoter–Luc), in which the DNA fragment spanning approximately −864 to −402 of the rat Gap43 gene promoter was cloned and subsequently constructed into the pGL3-basic vector, followed by transfection into primary rat astrocytes to examine whether the Gap43 promoter activity was responsive to the LPS stimulation. The data showed that LPS can induce Gap43 promoter activity by twofold, and the effect was abolished by AG490 (p = 0.0007 in LPS vs AG490 plus LPS; Fig. 3F). Since AG490 is a pan JAK inhibitor and not specific for STAT3, we further validated the essential role of STAT3 in astrocytic GAP43 expression by using siRNA to knock down STAT3 expression, The data showed that the Stat3 siRNA (siSTAT3) effectively reduced STAT3 as well as completely abolished the LPS-induced astrocytic GAP43 compared with the scrambled (Scr) RNA-transfected cells (p = 0.003 in Scr-LPS vs siSTAT3-LPS; Fig. 3G).
STAT3 activation during inflammation-induced astrogliosis is known to be mediated by the IL-6 receptor activated by the glia-derived IL6. We examined the temporal profile of the LPS-induced IL-6 release, found that the IL-6 level in the astrocyte culture medium started to ascend as early as 2 h and then exponentially elevated over time after the LPS treatment (Fig. 3H). The timing of the IL-6 induction was much earlier than the LPS-induced GAP43 expression (Fig. 3A), although later than the STAT3 binding to the Gap43 promoter. We further examined whether IL-6 knockdown would attenuate LPS-induced GAP43 expression. Transfection of siRNA specific for IL-6 showed a 58% knockdown efficiency of the robust induction of IL6 mRNA in LPS-treated primary rat astrocytes (Fig. 3I, top), and this condition blocked the LPS-induced Gap43 mRNA expression (Fig. 3I, bottom). Together, these data suggest that the LPS-induced astrocytic GAP43 gene expression is mediated by NF-κB and STAT3, and that the latter seems to be contributed, at least in part, by LPS-induced IL-6 release.
NF-κB blocking peptide reduced LPS-induced GAP43 in the astrocytic processes
Pharmacological agents that inhibit NF-κB, such as PTN, often have other targets (Gopal et al., 2007). To further confirm the NF-κB-dependent effect, the specific NF-κB blocking peptide approach was used to examine the expression and localization of GAP43 induction in LPS-stimulated HB astrocytes. NF-κB was inhibited by a cell-permeable peptide, TAT-NBD, a 22 aa peptide containing the NF-κB essential modulator (NEMO)/inhibitor of κB kinase γ-binding domain coupled to the transduction sequence of the HIV-TAT protein to block the interaction of NEMO with IκB kinase complex and exert a anti-inflammatory effect (Yang et al., 2013), in the LPS-treated primary astrocytes. Western blot results indicated that TAT-NBD, not the mutant TAT-NBD, can block the LPS-increased astrocytic GAP43 (Fig. 4A). Immunofluorescent staining indicated that LPS-induced GAP43 upregulation in the astrocytic process as well as the process arborization were both diminished by the TAT-NBD treatment (Fig. 4B). Quantitative results showed that the average process number per cell in the mutant NBD- and NBD-treated LPS-reactivated HB astrocytes were 4.8 and 2.7, respectively (p = 0.0042; Fig. 4C); the colocalization of GAP43 and GFAP in the astrocytic process were 38.5% and 16.3%, respectively (p < 0.0001; Fig. 4D). In addition, we noted that the morphology of NBD-treated astrocytes seems unhealthy, although the assessment of cell viability (WST-1 assay) and damage (LDH release assay) indicated that neither TAT-NBD nor TAT-NBDmut caused appreciable damage to astrocytes under the LPS treatment (Fig. 4E,F). In sum, the NF-κB signaling pathway that mediates the LPS-induced process arborization may also contribute to the induction and process localization of GAP43 in astrocytes.
Figure 4.

NF-κB mediated LPS-induced GAP43 expression in the astrocytic processes. A, Western blotting of GAP43 in primary HB astrocytes treated with vehicle of TAT-peptide (CTL; 0.1% DMSO) or LPS (50 ng/ml) for 24 h with the pretreatment of 20 μm TAT-NF-κB inhibitory peptide (TAT-NBD) or its mutant control, TAT-NBDmut, for 2 h. Lamin A was used as the internal control. The bar graphs showed the quantification of band intensity normalized by the respective internal control. n = 3 in each group. B, Immunofluorescent double staining of GAP43 and GFAP in LPS-treated HB astrocytes with TAT-NBDmut or TAT-NBD pretreatments under the same condition as in A. C, D, Imaging analysis and quantification of the average number of astrocytic processes per cell (C) and the percentage of GAP43 signals in the GFAP-positive astrocytic process [GAP43 intensity/(GAP43+GFAP) intensity × 100%] (D) obtained from B. The number of microscopic images counted in each group was as indicated. E, F, Cell viability (WST-1 assay; E) and damage (LDH release assay; F) of the LPS-treated astrocytes with or without TAT-NBDmut or TAT-NBD pretreatments. n = 3.
S41-phosphorylated GAP43 mediates astrocytic plasticity
The GAP43 upregulation in the astrocytic process implies its contribution to the morphological plasticity in reactivated astrocytes (Strittmatter et al., 1994; Wolff et al., 1998; Murk et al., 2013). Since Figure 1, A and B, has shown that the S41-phosphorylated GAP43 that promotes actin polymerization was increased proportionally with the total GAP43 in the LPS-reactivated astrocytes, we further used the previously established EGFP-tagged wild-type GAP43 (GAP43WT), GAP43S41D, and GAP43S41A mutants, which respectively mimic S41-phosphorylated and unphosphorylated GAP43 (Wang et al., 2015) in the primary HB rat astrocytes, to investigate the role of S41-phosphorylated GAP43 in astrocytic plasticity. Immunofluorescent double labeling of GFP and GFAP (Fig. 5A) for quantification of the number (Fig. 5B, left) and length (Fig. 5B, right) of processes in GFP-positive astrocytes was performed to examine the arborization and elongation of astrocytic processes, respectively. The data showed that both parameters were profoundly increased by GAP43S41D overexpression compared with the GAP43WT-expressing astrocytes (process number/cell in GAP43S41D-control, 6.5 ± 0.61 vs GAP43WT-control, 3 ± 0.26, p < 0.0001; process length/cell in GAP43S41D-control, 728.45 ± 32.67 μm vs GAP43WT- control, 264.48 ± 34.6 μm, p < 0.0001), and these two parameters were not further increased by the LPS treatment. In contrast, GAP43S41A overexpression did not affect the basal level, but significantly inhibited the LPS-induced increase in process number compared with the GAP43WT group. GAP43S41A did not affect the average process length of astrocytes compared with the GAP43WT groups (Fig. 5A,B). In contrast, when transfecting the GAP43 WT and mutant constructs into the flat-shaped MD astrocytes, we found that cells expressing GFP-WT or S41D mutant were hypertrophic in the cell body without forming processes (i.e., stellation of the flat astrocytes; Fig. 5C). Together, these data suggested that S41-phosphorylated GAP43 has differential effects on morphological plasticity in different astrocyte phenotypes (i.e., with the enhanced process arborization and elongation in the stellate astrocytes), whereas cell body hypertrophy occurs in the flat-shaped astrocytes.
Figure 5.

GAP43 mediated LPS-induced astrocyte process arborization. A, Immunofluorescent confocal images of GFP (green), GFAP (red), and DAPI (blue) in primary rat HB astrocytes transfected with GFP-tagged WT-GAP43, S41D, or S41A GAP43 mutants for 24 h followed by 24 h of LPS (50 ng/ml) treatment. B, Quantification of the process number (left) and length (right) per cell in A. n.s., No significant difference. The number of microscopic images counted in each group was as indicated in the bar graph. C, Immunofluorescent confocal images of GFP (green) and F-actin (red) in primary rat MD astrocytes transfected with GFP-tagged WT-GAP43, S41D, or S41A GAP43 mutants followed by LPS treatment of the same condition as in A. Note that cellular hypertrophy, not astrocytic process formation, was observed in the S41D-GAP43-transfected or LPS-treated WT-GAP43-transfected astrocytes. D, Western blotting and qRT-PCR (bottom) of GAP43 expression in HB astrocytes transfected with siGAP43 or scrambled RNA (Scr) followed by vehicle (CTL) or LPS (50 ng/ml) treatment for 24 h to indicate the efficiency of the knockdown of GAP43. n = 3. E, Confocal immunofluorescence of GAP43 and F-actin (phalloidin labeling in red) indicated that siGAP43 transfection attenuated the LPS-induced GAP43 and affected astrocyte process arborization. F, Quantification of average astrocyte process number (left) and length (right) per cell obtained from E. The number of microscopic images counted in each group was as indicated below the graph.
We then used Gap43 siRNA (siGAP43) transfection to examine the effect of GAP43 reduction on the LPS-induced astrocytic plasticity. The knock-down efficiency, as assessed by Western blotting (Fig. 5D, top) and quantitative RT-PCR (Fig. 5D, bottom) showed a >80% reduction of Gap43 mRNA in siGAP43-transfected primary rat astrocytes with or without the LPS treatment. Immunostaining of GAP43 and phalloidin labeling of F-actin further indicated that Gap43 siRNA transfection diminished the GAP43 immunoreactivities in both control and LPS-treated astrocytes (Fig. 5E). The LPS treatment increased the number of phalloidin-labeled astrocytic processes in Scr-RNA-transfected astrocytes, and this effect was diminished in siGAP43-transfected astrocytes (Scr/LPS, 6.28 ± 0.7 vs Scr/CTL, 3.83 ± 0.35, p = 0.0036; siGAP4/LPS, 4.38 ± 0.44 vs siGAP43/CTL, 3.47 ± 0.29, p = 0.0803; Fig. 5E,F, left), which is similar to the GAP43S41A effect shown in Figure 5B. The similarity of siGAP43 with the GAP43S41A overexpression also appeared in the lack of effects on the process number in the resting astrocytes (Fig. 5F, left) and the average length of astrocytic processes in both resting and LPS-treated astrocytes (Fig. 5F, right).
Together, these results suggest that, similar to its role in neuronal plasticity, astrocytic GAP43 induction and phosphorylation are critical for LPS-induced astrocytic plasticity.
Astrocytic GAP43 knockdown augmented astrogliosis-induced microgliosis and inhibited axonal plasticity
Next, we investigated the role of astrocytic GAP43 on the microgliosis and neuronal plasticity, as the actin reorganization is known to be involved in the reactive astrocyte-regulated immune response, synaptic plasticity, and neuronal survival (Tancredi et al., 2000; Haber et al., 2006; Burgos et al., 2007; Strehl et al., 2014). We applied ACM derived from LPS-treated primary astrocytes transfected with Scr or Gap43 siRNA onto primary rat microglia and cortical neurons (Fig. 6A). Of note, we used transient high-concentration LPS treatment (1 μg/ml) for 1 h followed by refreshing of the medium to remove LPS, and then incubated for 23 h to obtain the LPS-free ACM. This transient LPS treatment also induced astrocytic GAP43 expression (Fig. 6A, inset), with the fold increase that was similar to the sustained treatment with LPS at 50 ng/ml (Fig. 3A,B). We found that microglia treated with ACM from siGAP43-transfected LPS-treated (siGAP43/LPS) astrocytes led to the increased expression of proinflammatory mediator COX-2 than those treated with ACM from scrambled RNA-transfected vehicle-treated (Scr/CTL) astrocytes (Fig. 6B). The purity of the primary microglial culture used was confirmed by using Iba1 immunostaining to label all (both resting and activated) microglia and double labeled with OX42 for CD11b. The immunostaining images (Fig. 6C, top) showed that all OX42+ cells were Iba1+ microglia (indicated by white arrows), and quantitative data (Fig. 6D, left) indicated that >95% of the cells were Iba1+ (red) in both the control and LPS-treated groups; the OX42+/Iba1+ cells in the control and LPS groups were 23% and 56%, respectively. Astrocytic GAP43 knockdown markedly increased the population of LPS-ACM-induced reactivated microglia, as identified by the OX-42 immunostaining that labels the activated microglia marker CD11b (49.4 ± 2.19% in the Scr/LPS-ACM group vs 62.2 ± 2.48% in the siGAP43/LPS-ACM group, p = 0.005; Fig. 6C, bottom, D, middle). The area of the OX42+ immunoreactivities was also increased in the cultured microglia treated with siGAP43/LPS-ACM (96 ± 12 μm2/cell in the Scr/LPS-ACM group vs 142 ± 16 μm2/cell in the siGAP43/LPS-ACM group, p = 0.048; Fig. 6C,D, right). These results suggested that LPS-induced astrocytic GAP43 may attenuate reactive astrocyte-induced microglial activation and proinflammatory response.
Figure 6.
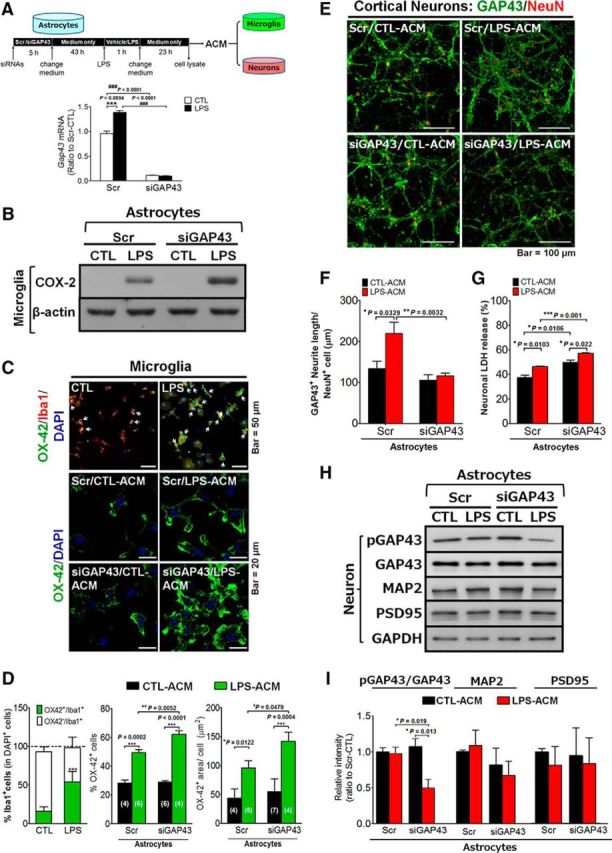
Effects of astrocytic GAP43 knockdown on ACM-induced microglial activation and neurotoxicity. A, A diagram indicates the timeline of ACM preparation from siGAP43-transfected, LPS-treated HB astrocytes. The obtained ACMs were used to treat cultured microglia or cortical neurons, and the cell lysates were subjected to qRT-PCR to determine the LPS-induced GAP43 expression and the siGAP43-mediated knockdown efficiency. B, Western blotting of proinflammatory protein COX-2 expression in ACM-treated SM826 microglia. C, Representative images of OX42 immunostaining of primary rat microglia treated with ACM. Top, The double staining of microglia marker Iba1 (red) and activated microglia expressing-OX42 (green) showed the microglia population and the increased CD11b/OX42 signals in the primary rat microglia treated with 10 ng/ml LPS for 24 h. White arrows indicated the OX42+/Iba1+ cells. Bottom, Immunofluorescent staining of OX42+-activated microglia after treatments with ACM from the indicated conditions. D, Quantification of the population of Iba1+ and OX42+ cells in C (top, left), and the population (top, middle), and the immunoreactivity area of OX42+ cells (top, right) normalized by the total cell number (DAPI+ nuclei) in each microscopic field in C (bottom). The number labeled in each bar indicated the number of microscopic images counted in each group. E, Confocal images of GAP43 (green) and NeuN (red) immunostaining in primary rat cortical neurons treated with ACM obtained from indicated conditions for 24 h. F, Quantification of GAP43-labeled neurite length per NeuN+ cell in E. Note the decreased GAP43-labeled neurite density in neurons treated with siGAP43/LPS-ACM. G, The neuronal damage of cortical neurons treated with ACMs for 24 h was measured by LDH release assay. H, Western blotting of neuroplasticity-related proteins, including pGAP43, GAP43, MAP2, and PSD-95 in cortical neurons treated with ACMs for 24 h. GAPDH serves as a loading control. I, Quantification results of pGAP43/GAP43 ratio, MAP2, and PSD-95 in H.
We further examined the effect of astrocytic GAP43 reduction on neuronal plasticity, with a special focus on the neuronal GAP43 that labels growing axons, in 10 DIV primary rat cortical neurons to mimic the effect of inflammatory stimuli to brain neurons in vivo. The immunostaining result of GAP43 in ACM-treated cortical neurons showed that Scr/LPS-ACM markedly increased the axonal plasticity, as indicated by the average length of GAP43+ neurites per NeuN-labeled neuron (133.4 ± 18.54 μm/neuron in Scr/CTL-ACM-treated group vs 219.1 ± 27.65 μm/neuron in Scr/LPS-ACM-treated group, p = 0.0329; Fig. 6E,F), whereas the siGAP43/LPS-ACM treatment failed to increase axon length (115.87 ± 6.54 μm/cell) compared with the Scr/LPS-ACM-treated group (p = 0.0032 vs Scr/LPS-ACM-treated neurons; Fig. 6E,F). Notably, the inhibitory effect of astrocytic GAP43 knockdown on axonal plasticity was accompanied by an elevation of ACM-induced neurotoxicity, as indicated by the LDH release in primary rat cortical neurons (Fig. 6G), in which Scr/CTL-ACM and Scr/LPS-ACM, respectively, induced 37.4% and 46.4%, whereas siGAP43/CTL-ACM and siGAP43/LPS-ACM, respectively, induced 49.8% in and 57%, of neuronal LDH release.
Because GAP43-mediated axon elongation requires S41-phosphorylated GAP43, we further examined neuronal GAP43 phosphorylation after the ACM treatments. LPS-ACM did not affect neuronal GAP43 phosphorylation compared with the CTL-ACM from the Scr-transfected astrocytes. However, ACM from siGAP43/LPS-treated astrocytes markedly reduced neuronal GAP43 phosphorylation, but not expression, compared with ACMs from siGAP43/CTL-, Scr/CTL-, and Scr/LPS-treated astrocytes (p = 0.019 in Scr/LPS-ACM vs siGAP43/LPS-ACM; Fig. 6H,I). This finding correlates well with the reduction of axon length by the siGAP43/LPS-ACM (Fig. 6F), suggesting that reactive astrocytes without GAP43 induction may inhibit axon growth by suppressing neuronal GAP43 phosphorylation. Notably, the levels of dendritic proteins (i.e., MAP2 and PSD-95) were not affected by ACMs obtained from LPS-activated astrocytes with either Scr or siGAP43 transfection (Fig. 6I).
In sum, these data suggested that the LPS-induced astrocytic GAP43 may function to attenuate the detrimental effects of reactivated astrocytes on microglial activation, neuronal survival, and axonal plasticity.
Astrocytic GAP43 dampened proinflammatory mediator induction and mediated glutamate transporter upregulation in LPS-treated astrocytes
We further searched for the astrocytic GAP43-regulated mediators that attenuate microgliosis and neurotoxicity in LPS-stimulated rat astrocytes. For the microgliosis-dampening effect, we examined the release of IL-6 and TNF-α and the expression of iNOS, and found that these proinflammatory mediators were all significantly enhanced by astrocytic GAP43 knockdown in LPS-treated astrocytes (Fig. 7A–C). This finding correlated well with the enhancement of microgliosis by siGAP43/LPS-ACM (Fig. 6B,C), suggesting that astrocytic GAP43 may mediate a dampening effect on the LPS-induced proinflammatory responses in reactivated astrocytes.
Figure 7.

GAP43 knockdown aggravated proinflammatory responses and reduced the expression and activity of glutamate transporter EAAT2 in LPS-treated astrocytes. A, B, ELISA of IL-6 and TNF-α release in ACMs collected from primary rat HB astrocytes treated with conditions as indicated in Figure 6A. The released cytokine levels were expressed in picograms per micrograms total protein. n = 3. C, Quantitative RT-PCR of iNOS mRNA in HB astrocytes treated with conditions, as indicated in Figure 6A. D–F, Western blot analysis of EAAT2 protein (D), qRT-PCR of the mRNA of rat EAAT2 gene (Slc1a2; E), and glutamate uptake activity assay (F) in primary astrocytes transfected with Scr or siGAP43 followed by the 50 ng/ml LPS treatment for 24 h. β-actin was used as an internal control in D.
We further examined the effect of GAP43 knockdown on the expression of astrocyte-specific EAAT2, which mediates the uptake of excessive extracellular glutamate and may contribute to the reactivated astrocyte-induced neurotoxicity. The data showed that LPS upregulated the EAAT2 protein 1.9-fold in rat astrocytes (p = 0.0017 vs control group; Fig. 7D). The rat Slc1a2 mRNA encoding EAAT2 was also increased by LPS treatment in the Scr-transfected astrocytes, and siGAP43 transfection significantly reduced Slc1a2 mRNA in both CTL and LPS groups compared with their respective Scr groups, although LPS increased Slc1a2 mRNA in siGAP43-transfected astrocytes (Fig. 7E). Importantly, the glutamate uptake activity of the Scr-transfected astrocytes was increased 1.4-fold by LPS treatment, and this effect was reversed in siGAP43-transfected astrocytes, in which LPS reduced glutamate uptake activity by 28% of the vehicle-treated group (Fig. 7F; p = 0.0259 vs siGAP43/control). These results correlated well with the neurotoxicity enhancement effects of siGAP43/LPS-ACM (Fig. 6E–I), suggesting that astrocytic GAP43 may contribute to the excessive glutamate removal to facilitate neuronal survival by mediating EAAT2 expression and its glutamate uptake activity.
GAP43 mediated EAAT2 expression through the activation of MKL1
GAP43 mediates actin polymerization, but how this mode of action can mediate EAAT2 expression is unknown. MKL1 is a transcriptional coactivator for serum response factor (SRF) and is activated when dissociated from G-actin during actin polymerization (Olson and Nordheim, 2010). In searching for the putative SRF binding element (i.e., SRE) we found a putative SRE spanning −725 to −714 on the rat Slc1a2 gene promoter using the TRANSFAC database version 8.3 (Farré et al., 2003). We thus assumed that MKL1 might be involved in the GAP43-mediated Slc1a2 gene expression. By knocking down MKL1 expression in primary astrocytes using siMKL1 (Fig. 8A), in which Mkl1 expression in both vehicle-treated (control) and LPS-treated astrocytes was reduced by 63% and 78%, respectively, we found that LPS-induced Slc1a2 mRNA expression was abolished, whereas the Gap43 expression was not affected (Fig. 8B,C). To demonstrate that MKL1 can be activated by LPS, which presumably induces actin polymerization, at least in part, via GAP43 induction and thereby causes the dissociation of MKL1 from G-actin and binding to the Slc1a2 promoter (Fig. 8D), we performed an anti-MKL1-based ChIP assay, in which we designed a set of PCR primers that flank the rat Slc1a2 promoter fragment containing an SRE (Fig. 8D; from −854 to −665). We found that LPS treatment significantly increased the MKL1 association with the SRE-containing Slc1a2 promoter fragment in scrambled RNA-transfected astrocytes, as indicated by semi-quantitative PCR (Fig. 8E), and this effect was completely abolished in the siGAP43-transfected astrocytes. Therefore, GAP43 that mediates actin polymerization may mediate the transcriptional activation of the glutamate transporter and other astrogliosis-associated genes via activation of MKL1.
Figure 8.

GAP43 mediated EAAT2 expression through the activation of MKL1 in LPS-stimulated astrocytes. A, The knock-down efficiency of Mkl1 siRNA in HB astrocytes was determined by qRT-PCR of Mkl1 mRNA. B, C, MKL1 knockdown reduced the LPS-induced Slc1a2 mRNA expression (B), but did not affect Gap43 mRNA expression (C). The qPCR results for each mRNA expression were normalized by the 18S mRNA. n = 3. D, A scheme indicates a hypothesis that GAP43 regulates the actin dynamic to affect MKL1 coactivator activity with SRF, and a predicted SRE binding site on the rat Slc1a2 promoter may mediate the actin-MKL1-SRF cascade to activate Slc1a2 gene transcription. E, Primary HB astrocytes transfected with Scr or Gap43 siRNA were incubated without (CTL) or with LPS (50 ng/ml) for 1 h, followed by MKL1-based ChIP assay on the SRE-containing Slc1a2 promoter with the fragment amplified by PCR using primers indicated in D. The enrichment of PCR products was expressed as the ratio of band intensity in each group to the Scr/CTL group.
Discussion
GAP43 mediates actin polymerization to promote neurite outgrowth, and its neuron-specific expression was mostly attributed to its basal level abundance (Benowitz and Routtenberg, 1997; He et al., 1997). In this study, we used the primary cultured rat brain astrocytes with stellate phenotype that had morphology similar to astrocytes in vivo to unravel the underlying mechanisms of the expression and function of GAP43 in reactive astrocytes, which might have been overlooked in the flat-shaped primary astrocytes and the neuronal GAP43-dominant immunohistochemistry of brain sections. Previous notions about the functions of GAP43 were dominated by its activity-dependent neuronal plasticity, and here we revealed for the first time that GAP43 plays important roles in mediating several important features of astrogliosis. Intriguingly, astrocytic GAP43 not only mediates morphological changes but also regulates proinflammatory responses and glutamate transporter expression in the reactivated astrocytes. In particular, astrocytic GAP43 deficiency leads to several detrimental outcomes, including aggravation of microglial activation as well as reduced axonal density and S41-phosphorylated GAP43 in cortical neurons. A summarized diagram indicating the upstream mediators and downstream effectors of astrocytic GAP43 elucidated in this study is illustrated in Fig. 9. Astrogliosis possesses both beneficial and deleterious effects that counterbalance each other, and beneficial astrogliosis is an important adaptation mechanism that provides trophic support and maintains homeostasis for neuronal survival under mild/moderate insults. Here the factors that are facilitated or attenuated by astrocytic GAP43 appear to converge toward a favorable microenvironment for neuronal survival and plasticity in the inflamed brain.
Figure 9.

A proposed model of astrocytic GAP43 functions in mediating astrocytic plasticity; and regulating microglial activation, axonal plasticity, and neuronal survival. In astrocytes, inflammatory stimuli that activate TLR4 trigger both NF-κB and STAT3 signaling pathways and lead to their binding onto the Gap43 gene promoter. NF-κB also mediates the expression and release of IL-6 to activate IL-6 receptor (IL-6R), which then activates a second-phase JAK/STAT3 signaling to give rise to the upregulation of astrocytic GAP43 expression. Astrocytic GAP43, especially its S41-phosphorylated form that promotes actin polymerization, functions to mediate morphological plasticity, including process arborization and elongation in stellate, not flat-shaped, astrocytes during astrogliosis. Furthermore, astrocytic GAP43 may attenuate the release of proinflammatory cytokines (IL-6 and TNF-α), thereby dampening the activation and proinflammatory response in microglia. Astrocytic GAP43 can also facilitate axonal plasticity by attenuating the inhibitory effect of reactivated astrocytes on axon growth and neuronal GAP43 phosphorylation possibly involved the regulation of astrocyte-derived inhibitory factors (In.F.). Finally, an actin-dependent MKL1 activation that contributes to the GAP43-mediated transcriptional activation of EAAT2 was revealed, which increases the glutamate uptake activity and may lead to reduction of excessive glutamate to attenuate the astrogliosis-induced neurotoxicity.
Inflammatory insult-induced astrogliosis is mainly initiated by activating NF-κB and STAT3, and the NF-κB-mediated IL-6 production is known to be the major upstream signal of the STAT3 activation. While STAT3 is a transcription factor for Gap43 gene activation (Qiu et al., 2005; Tsai et al., 2007), this study showed for the first time that NF-κB may be directly involved in the GAP43 gene transcription via binding to GAP43 gene promoter. Although it appears that both early (STAT3 binding to Gap43 promoter before IL-6 release) and late (IL-6-dependent STAT3 activation) responses occurred after the LPS/TLR4 activation, the latter seems more essential for the final output of astrocytic GAP43 induction as it was completely abolished by si-IL-6. Nonetheless, the early activation of NF-κB may contribute to the early production of IL-6, though its early binding to Gap43 promoter may not be strong enough to activate Gap43 gene transcription until other transcription factors, such as STAT3, are incorporated into the promoter activation machinery.
Reduction of GAP43 expression in astrocytes would aggravate LPS-induced proinflammatory IL-6 and TNF-α production (Fig. 7A,B), suggesting that GAP43 might mediate negative feedback regulation on the NF-κB signaling pathway to attenuate outburst of neuroinflammatory responses (i.e., the deleterious effect of astrogliosis). Because GAP43 mediates actin polymerization, how this biological activity intervenes in the NF-κB signaling remains to be resolved. Notably, it was reported that the disruption of actin filaments would prime LPS-induced p65/NF-κB signaling (Eswarappa et al., 2008; Kustermans et al., 2008), and LPS-induced actin polymerization was found essential for the NF-κB-driven proinflammatory responses in endothelial cells (Cuschleri et al., 2003). Whether GAP43-mediated actin filament stabilization can affect LPS-induced NF-κB activity requires further study.
Process arborization is a typical feature of reactive astrogliosis, and this dynamic event allows astrocytes to expand their surface area and territory to interact with neighboring neurons (Halassa et al., 2007; Perea et al., 2009; Ricci et al., 2009). In this study, we found that both the number and the length of processes were increased by overexpression of phosphorylation-mimicking GAP43S41D, whereas they were decreased by the dephosphorylation-mimicking GAP43S41A in HB astrocytes (Fig. 5B). These effects were in accordance with the role of GAP43 in promoting axonal/neurite outgrowth, suggesting that the phosphorylation status of GAP43 may be useful for indicating both axonal and astrocytic plasticity. Notably, the reduction of GAP43 by RNA knockdown reduced only arborization, not length (Fig. 5F), and the overexpression of WT did not affect these two morphological properties (Fig. 5B). In fact, we found that LPS treatment did not have a consistent effect on the length of astrocytic processes, probably because LPS did not increase the phosphorylation of S41-GAP43 (i.e., the pGAP43/GAP43 ratio; Fig. 1A,B). Thus, the total level and the phosphorylation status of astrocytic GAP43 might have different indications for astrocytic plasticity. In addition, it was reported that a GAP43-3 fragment derived from the m-calpain-mediated cleavage of S41-unphosphorylated GAP43 is a feedback inhibitor of m-calpain to prevent further activation of calpain-mediated cell apoptosis in neurons (Zakharov and Mosevitsky, 2007). Since LPS seems to increase both S41-phosphorylated and S41-unphosphorylated GAP43 in parallel (Fig. 1B), it is possible that the latter could be the substrate of calpain to generate GAP43-3 fragments in the reactivated astrocytes. It would be interesting to study whether the phosphorylation status of GAP43 is also involved in the survival of activated astrocytes via the GAP43–calpain feedback regulation.
In addition to GAP43, other actin-associated proteins, such as profilin and Rac1, were also reported to affect astrocytic process and stellation (Racchetti et al., 2012; Molotkov et al., 2013). However, most of the studies were performed in the flat-shaped astrocytes. Importantly, the inhibition of Rho kinase and Rac1, which would presumably inhibit actin polymerization and stabilization, can induce stellation of the flat-shaped astrocytes (Racchetti et al., 2012). However, GAP43 is less inducible in MD astrocytes (Fig. 1E), and neither GAP43S1D nor GAP43S41A overexpression could induce stellation of these flat-shaped astrocytes (Fig. 5C). Therefore, it is likely that the inducibility of GAP43 involves the basal activity of Rho kinase signaling, which presumably would be lower in stellate astrocytes. Because astrocytic GAP43 is beneficial for neuronal survival and plasticity, future study is warranted to understand how astrocyte phenotypes and the Rho kinase signaling determine its inducibility during astrogliosis.
One of the most intriguing findings in this study is that astrocytic GAP43 knockdown led to a decrease in the neuronal S41-phosphorylated GAP43 and neurite length (Fig. 6E–I). Because this finding was obtained using the ACM from siGAP43/LPS astrocytes to treat cortical neurons, it is possible that certain soluble factors derived from astrocytes were regulated by astrocytic GAP43 to mediate neuronal GAP43 phosphorylation. Notably, the reduction of neuronal GAP43 phosphorylation by ACM derived from siGAP43/LPS-activated, but not siGAP43/control-activated, astrocytes further suggested that the LPS-activated astrocytes without GAP43 might be more inhibitory to axon growth. Reactive astrocytes are known to inhibit axon regeneration in the injured CNS by expressing/releasing inhibitory factors, such as chondroitin sulfate proteoglycan (Liu et al., 2008). It is thus possible that astrocytic GAP43 might attenuate inhibitory factor induction in the reactive astrocytes. In addition, LPS-ACM from either Scr-GAP43-transfected or siGAP43-transfected astrocytes did not reduce dendritic proteins MAP2 and PSD-95 (Fig. 6H,I). Whether astrocytic GAP43 differentially affects axonal versus dendritic plasticity requires further investigation. Nevertheless, future work on the astrocytic GAP43-mediated disinhibition for axon regeneration would provide important insight into the therapeutics for neuroregeneration in CNS injury.
The characteristics of astrogliosis involve activation of protein mediators for cytoskeletal reorganization, which can change the subcellular localization of functional proteins, such as the surface expression of EAAT2 and AQP4 (Nicchia et al., 2008; Lau et al., 2011; Sheean et al., 2013). However, whether the cytoskeletal dynamics could change astrocytic gene expression was unknown. In this study, we revealed for the first time that an actin-dependent transcriptional regulation mechanism (i.e., actin polymerization-dependent activation of MKL1) is causally related to the GAP43-mediated Slc1a2 gene transcription. This finding is important, as the direct mediator of actin-dependent transcriptional regulation (i.e., MKL1/MRTF-A) is now proven to be critical for the astrocyte-mediated glutamate homeostasis, and its activation requires the induction of astrocytic GAP43. A growing body of evidence has shown that actin-mediated gene expression is not only restricted to proteins for cell morphology and migration, but also for secretary proteins such as growth factors and cytokines (Muehlich et al., 2007; Velasquez et al., 2013; Giehl et al., 2015). Moreover, a recent study (Flouriot et al., 2014) showed that the activation of MKL1 would lead to a large-scale chromatin reorganization and histone post-translational modification by generating a widely open chromatin state and a global increase in biosynthetic activity associated with cell growth. RhoA, another actin polymerization mediator that mediates LPS-induced astrocyte migration (Sato et al., 2011) and MKL1 activation in various cell types (Smith et al., 2013), was also found to affect Slc1a2 gene transcription in a transcriptome analysis of RhoA kinase inhibitor-treated astrocytes (Lau et al., 2012). On the contrary, the activation of astrocytic RhoA with injury or chemical insults has been shown to play an adverse role in suppressing astrocyte stellation (Burgos et al., 2007) and exacerbating the inflammatory response (Dusaban et al., 2013) in addition to increasing astrocyte apoptosis (Miñambres et al., 2006). In the present study, astrocytic GAP43 appears to modulate astrogliosis toward beneficial features in various aspects. The impact of the GAP43/F-actin/MKL1 axis on beneficial astrogliosis and its potential implication on neuroprotection and neuroregeneration would be a promising direction to pursue.
Footnotes
This study was financially supported by Grants NSC 99-2321-B-010-015 and NSC 100-2321-B-010-008 from the National Science Council, Grant MOST 104-2320-B-010-015-MY3 from the Ministry of Science and Technology, and Aim for the Top University Plan from the Ministry of Education, Taiwan.
The authors declare no competing financial interests.
References
- Baorto DM, Mellado W, Shelanski ML. Astrocyte process growth induction by actin breakdown. J Cell Biol. 1992;117:357–367. doi: 10.1083/jcb.117.2.357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben Haim L, Ceyzériat K, Carrillo-de Sauvage MA, Aubry F, Auregan G, Guillermier M, Ruiz M, Petit F, Houitte D, Faivre E, Vandesquille M, Aron-Badin R, Dhenain M, Déglon N, Hantraye P, Brouillet E, Bonvento G, Escartin C. The JAK/STAT3 pathway is a common inducer of astrocyte reactivity in Alzheimer's and Huntington's diseases. J Neurosci. 2015;35:2817–2829. doi: 10.1523/JNEUROSCI.3516-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benowitz LI, Routtenberg A. GAP-43: an intrinsic determinant of neuronal development and plasticity. Trends Neurosci. 1997;20:84–91. doi: 10.1016/S0166-2236(96)10072-2. [DOI] [PubMed] [Google Scholar]
- Burgos M, Calvo S, Molina F, Vaquero CF, Samarel A, Llopis J, Tranque P. PKCepsilon induces astrocyte stellation by modulating multiple cytoskeletal proteins and interacting with Rho A signalling pathways: implications for neuroinflammation. Eur J Neurosci. 2007;25:1069–1078. doi: 10.1111/j.1460-9568.2007.05364.x. [DOI] [PubMed] [Google Scholar]
- Cuschleri J, Gourlay D, Garcia I, Jelacic S, Maier RV. Endotoxin-induced endothelial cell proinflammatory phenotypic differentiation requires stress fiber polymerization. Shock. 2003;19:433–439. doi: 10.1097/01.shk.0000051762.08171.ea. [DOI] [PubMed] [Google Scholar]
- Deloulme JC, Laeng P, Janet T, Sensenbrenner M, Baudier J. Expression of neuromodulin (GAP-43) and its regulation by basic fibroblast growth factor during the differentiation of O-2A progenitor cells. J Neurosci Res. 1993;36:147–162. doi: 10.1002/jnr.490360205. [DOI] [PubMed] [Google Scholar]
- Desilva TM, Billiards SS, Borenstein NS, Trachtenberg FL, Volpe JJ, Kinney HC, Rosenberg PA. Glutamate transporter EAAT2 expression is up-regulated in reactive astrocytes in human periventricular leukomalacia. J Comp Neurol. 2008;508:238–248. doi: 10.1002/cne.21667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dusaban SS, Purcell NH, Rockenstein E, Masliah E, Cho MK, Smrcka AV, Brown JH. Phospholipase C epsilon links G protein-coupled receptor activation to inflammatory astrocytic responses. Proc Natl Acad Sci U S A. 2013;110:3609–3614. doi: 10.1073/pnas.1217355110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eswarappa SM, Pareek V, Chakravortty D. Role of actin cytoskeleton in LPS-induced NF-kappaB activation and nitric oxide production in murine macrophages. Innate Immun. 2008;14:309–318. doi: 10.1177/1753425908096856. [DOI] [PubMed] [Google Scholar]
- Farré D, Roset R, Huerta M, Adsuara JE, Roselló L, Albà MM, Messeguer X. Identification of patterns in biological sequences at the ALGGEN server: PROMO and MALGEN. Nucleic Acids Res. 2003;31:3651–3653. doi: 10.1093/nar/gkg605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitch MT, Silver J. CNS injury, glial scars, and inflammation: inhibitory extracellular matrices and regeneration failure. Exp Neurol. 2008;209:294–301. doi: 10.1016/j.expneurol.2007.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flouriot G, Huet G, Demay F, Pakdel F, Boujrad N, Michel D. The actin/MKL1 signalling pathway influences cell growth and gene expression through large-scale chromatin reorganization and histone post-translational modifications. Biochem J. 2014;461:257–268. doi: 10.1042/BJ20131240. [DOI] [PubMed] [Google Scholar]
- Foo LC. Purification of rat and mouse astrocytes by immunopanning. Cold Spring Harb Protoc. 2013;2013:421–432. doi: 10.1101/pdb.prot074211. [DOI] [PubMed] [Google Scholar]
- Giehl K, Keller C, Muehlich S, Goppelt-Struebe M. Actin-mediated gene expression depends on RhoA and Rac1 signaling in proximal tubular epithelial cells. PLoS One. 2015;10:e0121589. doi: 10.1371/journal.pone.0121589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gopal YN, Arora TS, Van Dyke MW. Parthenolide specifically depletes histone deacetylase 1 protein and induces cell death through ataxia telangiectasia mutated. Chem Biol. 2007;14:813–823. doi: 10.1016/j.chembiol.2007.06.007. [DOI] [PubMed] [Google Scholar]
- Gorina R, Font-Nieves M, Márquez-Kisinousky L, Santalucia T, Planas AM. Astrocyte TLR4 activation induces a proinflammatory environment through the interplay between MyD88-dependent NFkappaB signaling, MAPK, and Jak1/Stat1 pathways. Glia. 2011;59:242–255. doi: 10.1002/glia.21094. [DOI] [PubMed] [Google Scholar]
- Guerra MC, Tortorelli LS, Galland F, Da Ré C, Negri E, Engelke DS, Rodrigues L, Leite MC, Gonçalves CA. Lipopolysaccharide modulates astrocytic S100B secretion: a study in cerebrospinal fluid and astrocyte cultures from rats. J Neuroinflammation. 2011;8:128. doi: 10.1186/1742-2094-8-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo RB, Wang GF, Zhao AP, Gu J, Sun XL, Hu G. Paeoniflorin protects against ischemia-induced brain damages in rats via inhibiting MAPKs/NF-kappaB-mediated inflammatory responses. PLoS One. 2012;7:e49701. doi: 10.1371/journal.pone.0049701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haber M, Zhou L, Murai KK. Cooperative astrocyte and dendritic spine dynamics at hippocampal excitatory synapses. J Neurosci. 2006;26:8881–8891. doi: 10.1523/JNEUROSCI.1302-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halassa MM, Fellin T, Takano H, Dong JH, Haydon PG. Synaptic islands defined by the territory of a single astrocyte. J Neurosci. 2007;27:6473–6477. doi: 10.1523/JNEUROSCI.1419-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harre EM, Roth J, Pehl U, Kueth M, Gerstberger R, Hubschle T. Selected contribution: role of IL-6 in LPS-induced nuclear STAT3 translocation in sensory circumventricular organs during fever in rats. J Appl Physiol (1985) 2002;92:2657–2666. doi: 10.1152/japplphysiol.00822.2001. [DOI] [PubMed] [Google Scholar]
- Harré EM, Roth J, Gerstberger R, Hübschle T. Interleukin-6 mediates lipopolysaccharide-induced nuclear STAT3 translocation in astrocytes of rat sensory circumventricular organs. Brain Res. 2003;980:151–155. doi: 10.1016/S0006-8993(03)02923-8. [DOI] [PubMed] [Google Scholar]
- He Q, Dent EW, Meiri KF. Modulation of actin filament behavior by GAP-43 (neuromodulin) is dependent on the phosphorylation status of serine 41, the protein kinase C site. J Neurosci. 1997;17:3515–3524. doi: 10.1523/JNEUROSCI.17-10-03515.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrmann JE, Imura T, Song B, Qi J, Ao Y, Nguyen TK, Korsak RA, Takeda K, Akira S, Sofroniew MV. STAT3 is a critical regulator of astrogliosis and scar formation after spinal cord injury. J Neurosci. 2008;28:7231–7243. doi: 10.1523/JNEUROSCI.1709-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hol EM, Pekny M. Glial fibrillary acidic protein (GFAP) and the astrocyte intermediate filament system in diseases of the central nervous system. Curr Opin Cell Biol. 2015;32:121–130. doi: 10.1016/j.ceb.2015.02.004. [DOI] [PubMed] [Google Scholar]
- Iseki K, Hagino S, Nikaido T, Zhang Y, Mori T, Yokoya S, Hozumi Y, Goto K, Wanaka A, Tase C. Gliosis-specific transcription factor OASIS coincides with proteoglycan core protein genes in the glial scar and inhibits neurite outgrowth. Biomed Res. 2012;33:345–353. doi: 10.2220/biomedres.33.345. [DOI] [PubMed] [Google Scholar]
- Kahn MA, Huang CJ, Caruso A, Barresi V, Nazarian R, Condorelli DF, de Vellis J. Ciliary neurotrophic factor activates JAK/Stat signal transduction cascade and induces transcriptional expression of glial fibrillary acidic protein in glial cells. J Neurochem. 1997;68:1413–1423. doi: 10.1046/j.1471-4159.1997.68041413.x. [DOI] [PubMed] [Google Scholar]
- Kozuka N, Kudo Y, Morita M. Multiple inhibitory pathways for lipopolysaccharide- and pro-inflammatory cytokine-induced nitric oxide production in cultured astrocytes. Neuroscience. 2007;144:911–919. doi: 10.1016/j.neuroscience.2006.10.040. [DOI] [PubMed] [Google Scholar]
- Kustermans G, El Mjiyad N, Horion J, Jacobs N, Piette J, Legrand-Poels S. Actin cytoskeleton differentially modulates NF-kappaB-mediated IL-8 expression in myelomonocytic cells. Biochem Pharmacol. 2008;76:1214–1228. doi: 10.1016/j.bcp.2008.08.017. [DOI] [PubMed] [Google Scholar]
- Lau CL, O'Shea RD, Broberg BV, Bischof L, Beart PM. The Rho kinase inhibitor Fasudil up-regulates astrocytic glutamate transport subsequent to actin remodelling in murine cultured astrocytes. Br J Pharmacol. 2011;163:533–545. doi: 10.1111/j.1476-5381.2011.01259.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau CL, Perreau VM, Chen MJ, Cate HS, Merlo D, Cheung NS, O'Shea RD, Beart PM. Transcriptomic profiling of astrocytes treated with the Rho kinase inhibitor fasudil reveals cytoskeletal and pro-survival responses. J Cell Physiol. 2012;227:1199–1211. doi: 10.1002/jcp.22838. [DOI] [PubMed] [Google Scholar]
- Lautermilch NJ, Spitzer NC. Regulation of calcineurin by growth cone calcium waves controls neurite extension. J Neurosci. 2000;20:315–325. doi: 10.1523/JNEUROSCI.20-01-00315.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YH, Lin CH, Hsu PC, Sun YY, Huang YJ, Zhuo JH, Wang CY, Gan YL, Hung CC, Kuan CY, Shie FS. Aryl hydrocarbon receptor mediates both proinflammatory and anti-inflammatory effects in lipopolysaccharide-activated microglia. Glia. 2015;63:1138–1154. doi: 10.1002/glia.22805. [DOI] [PubMed] [Google Scholar]
- Liang YJ, Kuo HH, Lin CH, Chen YY, Yang BC, Cheng YY, Yu AL, Khoo KH, Yu J. Switching of the core structures of glycosphingolipids from globo- and lacto- to ganglio-series upon human embryonic stem cell differentiation. Proc Natl Acad Sci U S A. 2010;107:22564–22569. doi: 10.1073/pnas.1007290108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CH, Chen CC, Chou CM, Wang CY, Hung CC, Chen JY, Chang HW, Chen YC, Yeh GC, Lee YH. Knockdown of the aryl hydrocarbon receptor attenuates excitotoxicity and enhances NMDA-induced BDNF expression in cortical neurons. J Neurochem. 2009;111:777–789. doi: 10.1111/j.1471-4159.2009.06364.x. [DOI] [PubMed] [Google Scholar]
- Lin MS, Hung KS, Chiu WT, Sun YY, Tsai SH, Lin JW, Lee YH. Curcumin enhances neuronal survival in N-methyl-d-aspartic acid toxicity by inducing RANTES expression in astrocytes via PI-3K and MAPK signaling pathways. Prog Neuropsychopharmacol Biol Psychiatry. 2011;35:931–938. doi: 10.1016/j.pnpbp.2010.12.022. [DOI] [PubMed] [Google Scholar]
- Lin ST, Wang Y, Xue Y, Feng DC, Xu Y, Xu LY. Tetrandrine suppresses LPS-induced astrocyte activation via modulating IKKs-IkappaBalpha-NF-kappaB signaling pathway. Mol Cell Biochem. 2008;315:41–49. doi: 10.1007/s11010-008-9787-4. [DOI] [PubMed] [Google Scholar]
- Liu WL, Lee YH, Tsai SY, Hsu CY, Sun YY, Yang LY, Tsai SH, Yang WC. Methylprednisolone inhibits the expression of glial fibrillary acidic protein and chondroitin sulfate proteoglycans in reactivated astrocytes. Glia. 2008;56:1390–1400. doi: 10.1002/glia.20706. [DOI] [PubMed] [Google Scholar]
- McCarthy KD, de Vellis J. Preparation of separate astroglial and oligodendroglial cell cultures from rat cerebral tissue. J Cell Biol. 1980;85:890–902. doi: 10.1083/jcb.85.3.890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miñambres R, Guasch RM, Perez-Aragó A, Guerri C. The RhoA/ROCK-I/MLC pathway is involved in the ethanol-induced apoptosis by anoikis in astrocytes. J Cell Sci. 2006;119:271–282. doi: 10.1242/jcs.02723. [DOI] [PubMed] [Google Scholar]
- Molotkov D, Zobova S, Arcas JM, Khiroug L. Calcium-induced outgrowth of astrocytic peripheral processes requires actin binding by Profilin-1. Cell Calcium. 2013;53:338–348. doi: 10.1016/j.ceca.2013.03.001. [DOI] [PubMed] [Google Scholar]
- Muehlich S, Cicha I, Garlichs CD, Krueger B, Posern G, Goppelt-Struebe M. Actin-dependent regulation of connective tissue growth factor. Am J Physiol Cell Physiol. 2007;292:C1732–C1738. doi: 10.1152/ajpcell.00552.2006. [DOI] [PubMed] [Google Scholar]
- Murk K, Blanco Suarez EM, Cockbill LM, Banks P, Hanley JG. The antagonistic modulation of Arp2/3 activity by N-WASP, WAVE2 and PICK1 defines dynamic changes in astrocyte morphology. J Cell Sci. 2013;126:3873–3883. doi: 10.1242/jcs.125146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen L, He Q, Meiri KF. Regulation of GAP-43 at serine 41 acts as a switch to modulate both intrinsic and extrinsic behaviors of growing neurons, via altered membrane distribution. Mol Cell Neurosci. 2009;41:62–73. doi: 10.1016/j.mcn.2009.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicchia GP, Rossi A, Mola MG, Procino G, Frigeri A, Svelto M. Actin cytoskeleton remodeling governs aquaporin-4 localization in astrocytes. Glia. 2008;56:1755–1766. doi: 10.1002/glia.20724. [DOI] [PubMed] [Google Scholar]
- Nijboer CH, Heijnen CJ, Groenendaal F, May MJ, van Bel F, Kavelaars A. A dual role of the NF-kappaB pathway in neonatal hypoxic-ischemic brain damage. Stroke. 2008;39:2578–2586. doi: 10.1161/STROKEAHA.108.516401. [DOI] [PubMed] [Google Scholar]
- Olson EN, Nordheim A. Linking actin dynamics and gene transcription to drive cellular motile functions. Nat Rev Mol Cell Biol. 2010;11:353–365. doi: 10.1038/nrm2890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perea G, Navarrete M, Araque A. Tripartite synapses: astrocytes process and control synaptic information. Trends Neurosci. 2009;32:421–431. doi: 10.1016/j.tins.2009.05.001. [DOI] [PubMed] [Google Scholar]
- Qiu J, Cafferty WB, McMahon SB, Thompson SW. Conditioning injury-induced spinal axon regeneration requires signal transducer and activator of transcription 3 activation. J Neurosci. 2005;25:1645–1653. doi: 10.1523/JNEUROSCI.3269-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Racchetti G, D'Alessandro R, Meldolesi J. Astrocyte stellation, a process dependent on Rac1 is sustained by the regulated exocytosis of enlargeosomes. Glia. 2012;60:465–475. doi: 10.1002/glia.22280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rego D, Kumar A, Nilchi L, Wright K, Huang S, Kozlowski M. IL-6 production is positively regulated by two distinct Src homology domain 2-containing tyrosine phosphatase-1 (SHP-1)-dependent CCAAT/enhancer-binding protein beta and NF-kappaB pathways and an SHP-1-independent NF-kappaB pathway in lipopolysaccharide-stimulated bone marrow-derived macrophages. J Immunol. 2011;186:5443–5456. doi: 10.4049/jimmunol.1003551. [DOI] [PubMed] [Google Scholar]
- Ricci G, Volpi L, Pasquali L, Petrozzi L, Siciliano G. Astrocyte-neuron interactions in neurological disorders. J Biol Phys. 2009;35:317–336. doi: 10.1007/s10867-009-9157-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridet JL, Malhotra SK, Privat A, Gage FH. Reactive astrocytes: cellular and molecular cues to biological function. Trends Neurosci. 1997;20:570–577. doi: 10.1016/S0166-2236(97)01139-9. [DOI] [PubMed] [Google Scholar]
- Roybon L, Lamas NJ, Garcia-Diaz A, Yang EJ, Sattler R, Jackson-Lewis V, Kim YA, Kachel CA, Rothstein JD, Przedborski S, Wichterle H, Henderson CE. Human stem cell-derived spinal cord astrocytes with defined mature or reactive phenotypes. Cell Rep. 2013;4:1035–1048. doi: 10.1016/j.celrep.2013.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato K, Horiuchi Y, Jin Y, Malchinkhuu E, Komachi M, Kondo T, Okajima F. Unmasking of LPA1 receptor-mediated migration response to lysophosphatidic acid by interleukin-1beta-induced attenuation of Rho signaling pathways in rat astrocytes. J Neurochem. 2011;117:164–174. doi: 10.1111/j.1471-4159.2011.07188.x. [DOI] [PubMed] [Google Scholar]
- Sheean RK, Lau CL, Shin YS, O'Shea RD, Beart PM. Links between L-glutamate transporters, Na+/K+-ATPase and cytoskeleton in astrocytes: evidence following inhibition with rottlerin. Neuroscience. 2013;254:335–346. doi: 10.1016/j.neuroscience.2013.09.043. [DOI] [PubMed] [Google Scholar]
- Shen Y, Mani S, Meiri KF. Failure to express GAP-43 leads to disruption of a multipotent precursor and inhibits astrocyte differentiation. Mol Cell Neurosci. 2004;26:390–405. doi: 10.1016/j.mcn.2004.03.004. [DOI] [PubMed] [Google Scholar]
- Smith EC, Teixeira AM, Chen RC, Wang L, Gao Y, Hahn KL, Krause DS. Induction of megakaryocyte differentiation drives nuclear accumulation and transcriptional function of MKL1 via actin polymerization and RhoA activation. Blood. 2013;121:1094–1101. doi: 10.1182/blood-2012-05-429993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sofroniew MV. Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci. 2009;32:638–647. doi: 10.1016/j.tins.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sofroniew MV, Vinters HV. Astrocytes: biology and pathology. Acta Neuropathol. 2010;119:7–35. doi: 10.1007/s00401-009-0619-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strehl A, Lenz M, Itsekson-Hayosh Z, Becker D, Chapman J, Deller T, Maggio N, Vlachos A. Systemic inflammation is associated with a reduction in Synaptopodin expression in the mouse hippocampus. Exp Neurol. 2014;261:230–235. doi: 10.1016/j.expneurol.2014.04.033. [DOI] [PubMed] [Google Scholar]
- Strittmatter SM, Valenzuela D, Fishman MC. An amino-terminal domain of the growth-associated protein GAP-43 mediates its effects on filopodial formation and cell spreading. J Cell Sci. 1994;107:195–204. doi: 10.1242/jcs.107.1.195. [DOI] [PubMed] [Google Scholar]
- Sun YY, Lin SH, Lin HC, Hung CC, Wang CY, Lin YC, Hung KS, Lien CC, Kuan CY, Lee YH. Cell type-specific dependency on the PI3K/Akt signaling pathway for the endogenous Epo and VEGF induction by baicalein in neurons versus astrocytes. PLoS One. 2013;8:e69019. doi: 10.1371/journal.pone.0069019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi M, Sato Y, Nakagami Y, Miyake K, Iijima S. Identification of cis-acting regions that contribute to neuron-specific expression of the GAP-43 gene. Biosci Biotechnol Biochem. 2006;70:1492–1495. doi: 10.1271/bbb.50576. [DOI] [PubMed] [Google Scholar]
- Tancredi V, D'Antuono M, Cafè C, Giovedì S, Buè MC, D'Arcangelo G, Onofri F, Benfenati F. The inhibitory effects of interleukin-6 on synaptic plasticity in the rat hippocampus are associated with an inhibition of mitogen-activated protein kinase ERK. J Neurochem. 2000;75:634–643. doi: 10.1046/j.1471-4159.2000.0750634.x. [DOI] [PubMed] [Google Scholar]
- Tsai SY, Yang LY, Wu CH, Chang SF, Hsu CY, Wei CP, Leu SJ, Liaw J, Lee YH, Tsai MD. Injury-induced Janus kinase/protein kinase C-dependent phosphorylation of growth-associated protein 43 and signal transducer and activator of transcription 3 for neurite growth in dorsal root ganglion. J Neurosci Res. 2007;85:321–331. doi: 10.1002/jnr.21119. [DOI] [PubMed] [Google Scholar]
- Velasquez LS, Sutherland LB, Liu Z, Grinnell F, Kamm KE, Schneider JW, Olson EN, Small EM. Activation of MRTF-A-dependent gene expression with a small molecule promotes myofibroblast differentiation and wound healing. Proc Natl Acad Sci U S A. 2013;110:16850–16855. doi: 10.1073/pnas.1316764110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang CY, Lin HC, Song YP, Hsu YT, Lin SY, Hsu PC, Lin CH, Hung CC, Hsu MC, Kuo YM, Lee YJ, Hsu CY, Lee YH. Protein kinase C-dependent growth-associated protein 43 phosphorylation regulates gephyrin aggregation at developing GABAergic synapses. Mol Cell Biol. 2015;35:1712–1726. doi: 10.1128/MCB.01332-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolff JR, Stuke K, Missler M, Tytko H, Schwarz P, Rohlmann A, Chao TI. Autocellular coupling by gap junctions in cultured astrocytes: a new view on cellular autoregulation during process formation. Glia. 1998;24:121–140. doi: 10.1002/(SICI)1098-1136(199809)24:1<121::AID-GLIA12>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- Yang D, Sun YY, Lin X, Baumann JM, Dunn RS, Lindquist DM, Kuan CY. Intranasal delivery of cell-penetrating anti-NF-kappaB peptides (Tat-NBD) alleviates infection-sensitized hypoxic-ischemic brain injury. Exp Neurol. 2013;247:447–455. doi: 10.1016/j.expneurol.2013.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeo S, Bandyopadhyay S, Messing A, Brenner M. Transgenic analysis of GFAP promoter elements. Glia. 2013;61:1488–1499. doi: 10.1002/glia.22536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zakharov VV, Mosevitsky MI. M-calpain-mediated cleavage of GAP-43 near Ser41 is negatively regulated by protein kinase C, calmodulin and calpain-inhibiting fragment GAP-43–3. J Neurochem. 2007;101:1539–1551. doi: 10.1111/j.1471-4159.2007.04452.x. [DOI] [PubMed] [Google Scholar]
- Zamanian JL, Xu L, Foo LC, Nouri N, Zhou L, Giffard RG, Barres BA. Genomic analysis of reactive astrogliosis. J Neurosci. 2012;32:6391–6410. doi: 10.1523/JNEUROSCI.6221-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D, Hu X, Qian L, O'Callaghan JP, Hong JS. Astrogliosis in CNS pathologies: is there a role for microglia? Mol Neurobiol. 2010;41:232–241. doi: 10.1007/s12035-010-8098-4. [DOI] [PMC free article] [PubMed] [Google Scholar]