

Abstract
Basolateral amygdala (BLA) is critical for fear learning, and its heightened activation is widely thought to underpin a variety of anxiety disorders. Here we used chemogenetic techniques in rats to study the consequences of heightened BLA activation for fear learning and memory, and to specifically identify a mechanism linking increased activity of BLA glutamatergic neurons to aberrant fear. We expressed the excitatory hM3Dq DREADD in rat BLA glutamatergic neurons and showed that CNO acted selectively to increase their activity, depolarizing these neurons and increasing their firing rates. This chemogenetic excitation of BLA glutamatergic neurons had no effect on the acquisition of simple fear learning, regardless of whether this learning led to a weak or strong fear memory. However, in an associative blocking task, chemogenetic excitation of BLA glutamatergic neurons yielded significant learning to a blocked conditioned stimulus, which otherwise should not have been learned about. Moreover, in an overexpectation task, chemogenetic manipulation of BLA glutamatergic neurons prevented use of negative prediction error to reduce fear learning, leading to significant impairments in fear inhibition. These effects were not attributable to the chemogenetic manipulation enhancing arousal, increasing asymptotic levels of fear learning or fear memory consolidation. Instead, chemogenetic excitation of BLA glutamatergic neurons disrupted use of prediction error to regulate fear learning.
SIGNIFICANCE STATEMENT Several neuropsychiatric disorders are characterized by heightened activation of the amygdala. This heightened activation has been hypothesized to underlie increased emotional reactivity, fear over generalization, and deficits in fear inhibition. Yet the mechanisms linking heightened amygdala activation to heightened emotional learning are elusive. Here we combined chemogenetic excitation of rat basolateral amygdala glutamatergic neurons with a variety of behavioral approaches to show that, although simple fear learning is unaffected, the use of prediction error to regulate this learning is profoundly disrupted, leading to formation of inappropriate fear associations and impaired fear inhibition.
Keywords: amygdala, anxiety, DREADD, extinction, fear conditioning, prediction error
Introduction
Pavlovian fear conditioning enables learning about, and adaptive responding to, sources of danger in the world. The amygdala is critical for the formation, consolidation, and retrieval of fear memories (Davis, 1992; Schafe et al., 2001; Maren and Quirk, 2004; Paré et al., 2004; Lüthi and Lüscher, 2014). Principal cells of the basolateral amygdala (BLA) receive glutamatergic inputs from thalamus and cortex conveying information about the conditioned stimulus (CS) and aversive footshock unconditioned stimulus (US) (Farb and Ledoux, 1999; Shi and Davis, 1999; Sah et al., 2003; Lanuza et al., 2008), and their activity is sufficient for fear learning (Johansen et al., 2010a). These glutamatergic neurons are subject to complex regulation by multiple families of GABAergic interneurons (Ehrlich et al., 2009; Wolff et al., 2014; Tovote et al., 2015), show synaptic plasticity during fear conditioning, and form fear memories in an NMDA receptor-dependent manner (McKernan and Shinnick-Gallagher, 1997; Maren and Quirk, 2004; Marek et al., 2013).
The amygdala has also long been implicated in normal and clinical human anxiety and fear. Several neuropsychiatric disorders are characterized by heightened activation of the amygdale; and in the case of anxiety, such heightened activation is widely thought to underpin clinical aberrations in fear (Rauch et al., 2006; Jovanovic and Ressler, 2010; Shin and Liberzon, 2010). For example, in human neuroimaging studies, sufferers of clinical anxiety, including simple phobia (Goossens et al., 2007), post-traumatic stress (Shin et al., 1997, 2005), and social anxiety (Tillfors et al., 2001, 2002), show heightened amygdala activation during fear provocation or during failures of fear inhibition (Rauch et al., 2006; Jovanovic and Ressler, 2010). Such heightened amygdala activation has also been linked to augmented fear responding in nonhuman animal studies (Desmedt et al., 2015).
However, the mechanisms linking excessive amygdala activation to excessive fear learning and fear responding remain elusive. Here we used chemogenetic excitation of rat BLA glutamatergic neurons (Armbruster et al., 2007; Alexander et al., 2009; Urban and Roth, 2013; Sternson and Roth, 2014) to identify the consequences of heightened amygdala glutamatergic neuronal activity for BLA neurons, fear learning, and fear memory formation. We show that, although simple fear learning is unaffected by chemogenetic excitation of BLA glutamatergic neurons, the use of prediction error to regulate this fear learning is profoundly disrupted, leading to formation of inappropriate fear associations and impaired learning of fear inhibition.
Materials and Methods
Subjects
A total of 153 experimentally naive male Sprague Dawley rats (260–350 g) were obtained from Animal Resources Centre (Murdoch, Western Australia). Animals were housed in groups of four maximum in ventilated racks in a climate-controlled colony room. The colony room was maintained on a 12 h light/dark cycle (lights on at 7:00 A.M.). Three days before commencement of experimental procedures, rats were food restricted to 85%–90% of their free feeding weight. Otherwise, rats had free access to food and water. The University of New South Wales Animal Care and Ethics Committee and the University of Sydney Animal Ethics Committee approved the procedures.
Apparatus
All behavioral procedures were conducted in eight identical operant chambers with dimensions 24 (length) × 30 (width) × 21 cm (height). The top, rear wall and hinged door of the chambers were constructed of Perspex. The sidewalls of the chambers were constructed of stainless steel panels. All chambers had a grid floor constructed of stainless steel rods, 4 mm in diameter spaced 15 mm apart. The grid floor was connected to a constant current generator. A magazine (entry space 5 × 5 cm) was built in to the left side panel and was attached to a pellet delivery system that delivered 45 g grain pellets (Able Scientific Biotechnology). A lever was mounted 4 cm to the right of the magazine hopper. Each chamber was placed in a larger sound-attenuating box, dimensions 83 (length) × 59 (width) × 59 cm (height). A fan was attached to the right side wall to provide sufficient ventilation during behavioral testing. Two CS were used. The auditory CS was a 60 s 80 dB clicker delivered through a speaker attached to the right side of the rear wall of the operant chamber and the visual CS was a 60 s flashing LED (8 [length] × 5 [width] × 3 cm [height]) mounted on the ceiling of the sound attenuating chamber. The US was a scrambled footshock delivered to the grid floor and was 0.5 s in duration. The intensity of the US depended on the behavioral procedure used. All behavioral protocols were controlled through Med-PC software (Med Associates).
Viral vectors
Adeno-associated viral (AAV) vectors encoding the hM3Dq DREADD or eYFP were obtained from the University of North Carolina Vector Core (Chapel Hill, NC). The vectors used in these experiments were AAV5-CaMKII α-HA-hM3Dq-IRES-mCitrine (3 × 1012 vp/ml) and AAV5-CaMKII α-eYFP (4 × 1012 vp/ml).
Surgery
Before surgery, rats received an intraperitoneal injection of a mixture of 100 mg/ml ketamine (Ketapex, Apex Laboratories) and 0.3 ml/kg xylazine (Rompun; Bayer). Once anesthetized, rats were placed in a stereotaxic apparatus (model 942, Kopf) and shaved to expose the skin surface of the head. Before incision, rats received a subcutaneous injection of carprofen (5 mg/kg) and an injection of 0.5% bupivacaine (Cenvet) just under the surface of the incision site. Following incision, a hand drill was used to make two craniotomies above the BLA and a 5 μl, 30-gauge conical tipped microinfusion syringe (Hamilton) was used to infuse 0.75 μl of AAV vectors into BLA (anteroposterior −3.00; mediolateral ±5.00; dorsoventral −8.60 in mm from bregma) (Paxinos and Watson, 2007) over a 3 min period at a rate of 0.25 μl/min (UMP3 with SYS4 Microcontroller; World Precision Instruments). The syringe was left in place for 5–7 min to permit diffusion of the injected vectors. Bone wax (Coherent Scientific) was used to seal the opening of the skull. After surgery, rats were injected intraperitoneally with 0.3 ml of 300 mg/ml solution of procaine penicillin (Benicillin; Illium) and subcutaneously with 0.3 ml of a 100 mg/ml solution of cephazolin (Hospira). Daily postoperative and recovery procedures, including weight and infection management, were conducted for the remainder of the experiment. All behavioral procedures commenced a minimum of 3 weeks after surgery.
Experiment 1: chemogenetic activation of BLA glutamatergic neurons
We validated hM3Dq expression in BLA CaMKIIα-positive cells and confirmed BLA cellular response to clozapine-n-oxide (CNO; RTI International). After a minimum of 3 weeks after surgery, rats were killed for immunohistochemical detection of mCitrine and CaMKIIα (n = 4). For c-Fos and mCitrine immunohistochemistry, all rats had been infused unilaterally into the right BLA with AAV5-CaMKII α-HA-hM3Dq-IRES-mCitrine so that the nontransduced hemisphere could serve as a within-subjects control. These rats received an intraperitoneal injection of 3 mg/kg CNO (diluted in 5% DMSO and saline; n = 6) or vehicle (n = 6) 3 weeks following surgery and then killed with an intraperitoneal injection of sodium pentobarbital 150 min later.
Acute slices were prepared from both unilaterally and bilaterally injected animals at least 6 weeks following viral injections (rats aged 15–17 weeks). Rats were anesthetized with isoflurane, decapitated, and their brains were quickly removed and chilled in ice-cold cutting solution (in mm) as follows: 125 NaCl, 25 NaHCO3, 11 d-glucose, 2.5 KCl, 1.25 NaH2PO4.2H2O, 2.5 MgCl2, and 0.5 CaCl2, saturated with carbogen (95% O2/5% CO2). Coronal slices (280 μm) containing the amygdala were cut using a V1200S vibratome (Leica). Slices were hemisected and initially incubated in an NMDG-HEPES recovery solution (in mm) as follows: 93 NMDG chloride, 2.5 KCl, 1.2 NaH2PO4, 30 NaHCO3, 20 HEPES, 25 d-glucose, 5 sodium ascorbate, 2 thiourea, 3 sodium pyruvate, 10 MgCl2, 0.5 CaCl2, pH 7.3, 300–310 mOsm/L, heated to 34°C and saturated with carbogen for 10 min (Zhao et al., 2011). Slices were then returned to carbogenated cutting solution and incubated for a further 20 min at 34°C, then allowed to equilibrate to room temperature for at least 30 min.
For recording, slices were transferred to the recording chamber and continually perfused (∼2 ml/min) with aCSF containing (in mm) the following: 125 NaCl, 25 NaHCO3, 11 d- glucose, 2.5 KCl, 1.25 NaH2PO4.2H2O, 1 MgCl2, and 2 CaCl2, saturated with carbogen and heated to 32°C–34°C. In all cases, NBQX (5 μm, Abcam Biochemicals) and picrotoxin (100 μm, Sigma) were included in the circulating aCSF to block fast excitatory and inhibitory transmission respectively. hM3Dq-transfected BLA neurons were readily identified using an Olympus BX51 microscope equipped with 40× water-immersion objective, Dodt gradient contrast optics, and epifluorescence illumination. Whole-cell current-clamp recordings were made from both mCitrine+ and mCitrine− BLA neurons using patch pipettes (3–5 mΩ) containing the following (in mm): 135 potassium gluconate, 8 NaCl, 0.5 EGTA, 10 HEPES, 2 Mg-ATP, 0.3 Na-GTP, and 0.1% biocytin, pH 7.3, 280–285 mOsm/L. Because of high levels of hM3Dq expression in the BLA that received AAV injections, recordings from mCitrine− BLA neurons were performed on slices from noninjected hemispheres of unilaterally injected animals. To monitor changes in membrane voltage, continuous chart recordings were made from cells held in current-clamp mode with zero current injected. To elicit minimal firing, a 1 s depolarizing current pulse (typically 100–300 pA) was applied every 10 s. Following CNO application, mCitrine+ cells were held up to 1 h to monitor drug washout, whereas mCitrine− cells were allowed 10 min washout before the application of the broad-spectrum muscarinic agonist carbachol (10 μm for 5 min; Sigma). Recordings were performed using a MultiClamp 700B amplifier (Molecular Devices), filtered at 5 kHz, and sampled at 10 kHz. Data were acquired and analyzed using Axograph software (Molecular Devices). Series resistance was monitored throughout each experiment, and data were discarded if this fluctuated >20% or if series resistance was >20 mΩ.
Experiment 2: chemogenetic activation of BLA glutamatergic neurons and fear learning
In these experiments, as per our previous work (Arico and McNally, 2014; Yau and McNally, 2015), we used conditioned suppression of lever pressing for food to assess Pavlovian-conditioned fear. Conditioned suppression as a measure of fear has several advantages for the study of fear prediction errors. It has a nonzero baseline because rats lever press for a pellet reward at a constant rate and so can reveal decreases and increases in fear; there are relatively high levels of baseline activity during training and testing sessions; it is equally sensitive to visual and auditory CS despite these CS eliciting different amounts of freezing; each of the seminal behavioral preparations identifying the actions of prediction error on fear association formation was established using conditioned suppression; and, finally, assessment of conditioned suppression is completely automated.
Baseline lever pressing.
Rats were trained to lever press to establish a stable baseline lever pressing response. On days 1 and 2, rats received magazine training in which every lever press was rewarded with the delivery of a pellet. In addition, rats also received free pellet deliveries on a fixed interval 300 s schedule, in which a pellet was delivered on average every 300 s. Magazine training sessions were terminated after 60 min or if the rat reached 100 lever presses. If rats failed to reach 100 lever presses, they were shaped to lever press by the experimenter. On day 3, rats underwent a 60 min session of lever press training under a variable interval (VI) 30 s schedule. From day 4 until the end of the experiment, rats were maintained on a VI 120 s schedule of lever pressing. These sessions lasted 120 min unless otherwise noted. On days 9 and 10, rats received preexposure to a visual CS (flashing LED). This CS was presented four times for 60 s, inter-trial interval (ITI) ranging from 1200 to 1800 s.
Acquisition.
Rats were randomly allocated to a weak US group or a strong US group. All rats underwent fear conditioning on days 11 and 12. Rats received four 60 s presentations of the visual CS with a randomized ITI of 1200–1800 s while being maintained on a VI 120 s. These presentations coterminated with a 0.5 s, 0.6 mA US for the strong US groups or a 0.5 s, 0.3 mA US for the weak US groups. These sessions lasted 120 min. Thirty minutes before these sessions, rats were injected intraperitoneally with CNO (3 mg/kg) or vehicle.
Extinction test.
Rats were tested across days 13–16. Rats received four 60 s presentations of CSA alone with an ITI of 900 s while being maintained on a VI 120 s schedule. Each test session lasted 70 min.
Experiment 3: chemogenetic activation of BLA glutamatergic neurons prevents use of prediction error to regulate fear learning
Baseline lever pressing.
The procedures used for baseline lever press training were as described previously. On days 9 and 10, rats received preexposure to CSA (flashing LED) and CSB (clicker). CS were presented for 60 s, four times each with ITIs ranging from 1200 to 1800 s.
Stage I.
Rats were randomly allocated to groups Block and Control. On days 11–13, groups Block received four 60 s presentations of CSA with a randomized ITI of 1200–1800 s while being maintained on a VI 120 s lever pressing schedule. Each CSA presentation coterminated with a 0.5 s footshock US (0.8 mA). Each Stage I session lasted for 120 min. Rats in groups Control received further VI 120 training.
Stage II.
On days 14 and 15, all rats received four 60 s presentations of a compound comprising CSA and CSB (CSAB) with a randomized ITI of 490–1200 s while being maintained on a VI 120 s lever pressing schedule. The compound CSAB coterminated with a 0.5 s US (0.8 mA). These sessions lasted 70 min each. Thirty minutes before the commencement of each session rats received intraperitoneal injection of either CNO (3 mg/kg) or vehicle.
Test.
All rats were tested on day 16. Rats received four 60 s presentations of CSB with an ITI of 900 s while being maintained on a VI 120 s lever pressing schedule. This session lasted 70 min.
Experiment 4: chemogenetic activation of BLA glutamatergic neurons prevents use of negative prediction error to reduce fear
Baseline lever pressing.
The procedures used for baseline lever press training and CS preexposure (days 1–10) were the same as described previously with one exception. All procedures in this experiment were conducted with the house light on.
Stage I.
Rats underwent Stage I training on days 11–13. They received two 60 s presentations of CSA and two 60 s presentations of CSB with a randomized ITI of 1200–1800 s while being maintained on a VI 120 s schedule. Each stimulus was reinforced with a 0.5 s, 0.8 mA US. The order of CS presentations was counterbalanced. Each Stage I session lasted 120 min.
Stage II.
Rats underwent Stage II training on days 14–16. Rats were randomly allocated to groups Over and groups Control. Rats in groups Over received four 60 s presentations of a CSAB compound with a randomized ITI of 490–1200 s while being maintained on a VI 120 s schedule. Each compound presentation coterminated with a 0.5 s, 0.8 mA US. Each Stage II session lasted 70 min. Rats in groups Control received further VI 120 s lever press training without any CS or US presentations. Thirty minutes before each Stage II session, rats received an intraperitoneal injection of CNO (3 mg/kg) or vehicle.
Test.
All rats were tested for both CSA and CSB in two separate test sessions on days 17 and 18. These sessions were counterbalanced for CS identity. Rats received four 60 s presentations of CSA and CSB alone with an ITI of 900 s between presentations. Each session lasted for 70 min.
Immunohistochemistry
Following transcardial perfusion with 0.9% saline, 1% sodium nitrite solution, and 360 μl/L heparin, then 4% buffered PFA, brains were postfixed and then placed in 20% hypertonic sucrose solution for 24–48 h. A cryostat (Leica Microsystems) was used to collect 40 μm coronal sections, preserved in phosphate buffered azide (0.1% sodium azide) at 4°C before immunohistochemistry.
Immunoreactivity for mCitrine (AAV5-CaMKII α-HA-hM3Dq-IRES-mCitrine) and eYFP (AAV5- CaMKIIα-eYFP) was performed (chicken anti-eGFP; Invitrogen; A10262). CaMKIIα and eGFP immunoreactivity (IR) were processed using two-color immunofluorescence. Free-floating sections were washed for 20 min in 0.1 m phosphate buffer (PB; pH 7.4), followed by 5 min in PB Triton X (PBTx; 0.2%; pH 7.4), then treated with sodium citrate buffer, pH 6.0, at 75°C for 15 min and 10 min off heat. Sections were washed with PBTx (10 min), blocked (2 h, 5% NGS in PBTx), and then placed in 1:500 rabbit anti-CaMKIIα (Abcam; ab92332) and 1:750 chicken anti-GFP (Invitrogen; A10262, diluted in a solution of 0.2% PBTx and 2% NGS) at 4°C for 48 h. Sections were washed in PB for 30 min and then incubated in 1:500 Alexa-555 goat anti-rabbit (Invitrogen; A21429) and 1:750 Alexa-488 goat anti-chicken (Invitrogen; A11039), diluted in PBTx (0.2%) and 2% NGS at room temperature for 3 h. Sections were washed for 30 min in PB, mounted, and coverslipped (Permfluor, Thermo Scientific). CaMKIIα-IR and eGFP-IR used a confocal microscope (Olympus BX61) and Fluoview FV1200 software.
c-Fos-IR and eGFP IR were processed using two-color peroxidase immunohistochemistry. Free-floating sections were washed for 30 min in 0.1 m phosphate buffer (PB; pH 7.4), followed by 50% ethanol (EtOH) for 30 min and then 3% hydrogen peroxide diluted in 50% EtOH for 30 min. Sections were blocked (30 min with 5% NHS diluted in PB), then placed in 1:1000 rabbit anti-c-fos (Santa Cruz Biotechnology; sc-52) and 1:2000 chicken anti-GFP (Invitrogen; A10262) diluted in 0.3% Triton-X, 2% NHS, and 0.1 m PB, pH 7.4, and incubated at 4°C for 48 h. Sections were washed three times for 20 min each (PB, pH 7.4), incubated in 1:3000 biotinylated donkey anti-rabbit (Jackson ImmunoResearch Laboratories; 711, 065, 152) (diluted in a solution of 0.3% Triton-X, 2% NHS, and 0.1 m PB, pH 7.4), overnight at room temperature. They were then washed in PB, pH 7.4, and incubated in Avidin-Biotin (ABC reagent; Vector Elite kit 6 μl/ml avidin and 6 μl/ml biotin) diluted with PB containing 0.2% Triton, pH 7.4. c-Fos IR was identified using a nickel intensified DAB (D5637–56, Sigma) reaction. Immediately before this, sections were washed twice in PB, pH 7.4, and once in 0.1 m acetate buffer, pH 6.0. Sections were incubated in DAB solution (0.025% DAB, 0.04% ammonium chloride, and 0.2% d-glucose in 0.1 m acetate buffer, pH 6.0). The peroxidase reaction was catalyzed by 0.2 μl/ml glucose oxidase and then rinsed with 0.1 m acetate buffer. Sections were then washed with PB and incubated in biotinylated donkey anti-chicken (1:3000; Jackson ImmunoResearch Laboratories; 703, 065, 155) overnight before a second DAB reaction, without nickel intensification, to identify brown eGFP-IR cells. Sections were then washed thoroughly with PB and mounted, dehydrated, cleared, and coverslipped (Entellan, ProSciTech). cFos-IR and eGFP-IR were imaged at 20× using a transmitted light microscope (Olympus BX51) and counted using Photoshop (Adobe).
Data analysis
Suppression ratios were calculated as SR = a/(a + b) (Annau and Kamin, 1961), where a represents the number of lever presses during the CS period and b represents the number of lever presses recorded 60 s pre CS. A SR of 0.5 indicates low suppression or low fear, whereas an SR of 0 indicates complete suppression or high fear. For the acquisition experiment, data from each trial of acquisition and the first trial on each day of test were analyzed. For the blocking experiment, the first trial each day was reported and analyzed. For overexpectation, the first trial for each day of Stages I and II and trials across both test days (CSA and CSB) were analyzed. Behavioral data and c-Fos-IR neuronal counts were analyzed via planned contrasts (Harris, 2004). The Hay's Decision-wise error rate (α) was controlled at 0.05 for each contrast.
Results
Experiment 1: chemogenetic activation of BLA glutamatergic neurons
First, we used AAV5-CaMKII α-HA-hM3D(Gq)-IRES-mCitrine (hereafter referred to as hM3Dq) to express the designer muscarinic receptor bilaterally in rat BLA glutamatergic neurons (n = 4), and we quantified immunoreactivity (IR) for CaMKIIα and mCitrine/eGFP in BLA to confirm expression in this cell type (Fig. 1A). There were significantly more dual CaMKIIα-IR/eGFP-IR cells than single eGFP-IR cells (F(1,3) = 19.35, p < 0.05), and almost all eGFP-IR cells were localized to CaMKIIα-IR cells (Fig. 1B).
Figure 1.
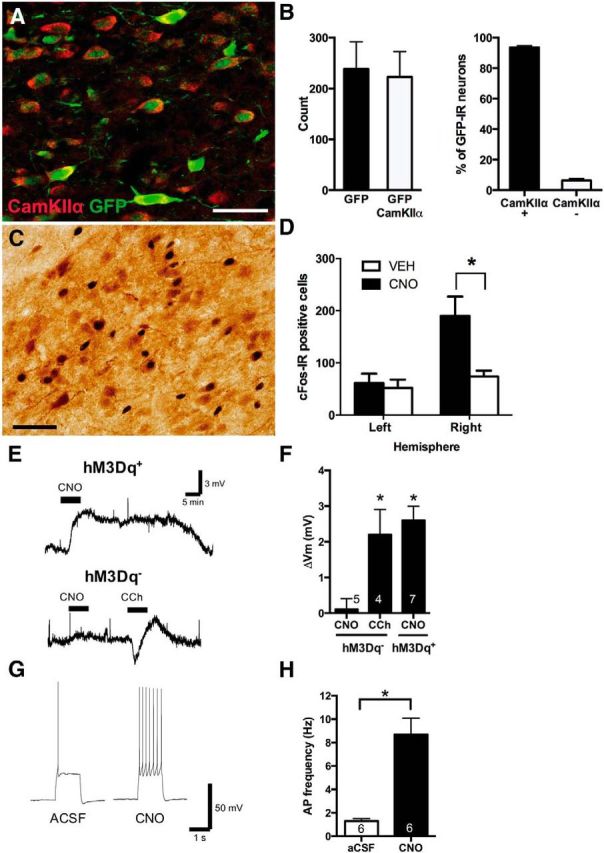
Chemogenetic activation of BLA glutamatergic neurons. A, Colocalization of CaMKIIα-IR and DREADD+ (eGFP-IR) BLA cells (red represents CaMKIIα-IR; green represents eGFP-IR). Scale bar, 25 μm. B, Total eGFP-IR and eGFP-IR/CaMKII α-IR dual-labeled cells in rat BLA and percentage of CaMKIIα+ and CaMKIIα− eGFP-IR cells. *p < 0.05. Group size: CaMKIIα- hM3Dq, n = 4. C, Representative c-Fos-IR (black) and eGFP-IR (brown) cells in an hM3Dq animal injected with CNO (3 mg/kg, i.p.) Scale bar, 50 μm. D, Total c-Fos-IR in transduced (right) and nontransduced (left) BLA. *p < 0.05 (hemisphere × drug interaction). Group size: CaMKIIα- hM3Dq-CNO, n = 6; CaMKIIα- hM3Dq-Veh, n = 6. E, Representative voltage traces from hM3Dq+ and hM3Dq− BLA neurons. F, CNO (1–5 μm) depolarized hM3Dq+/mCitrine+ neurons but had no effect on hM3Dq−/mCitrine− neurons, which remained responsive to carbachol. *p < 0.05. G, Representative voltage response from hM3Dq+/mCitrine+ neurons to minimal depolarizing current injection (100–300 pA, 1 s) before (aCSF) and during CNO (5 μm) application. H, CNO significantly increased action potential (AP) frequency in hM3Dq+/mCitrine+ neurons. *p < 0.05. Data are mean ± SEM; the number of neurons per experiment is indicated at the corresponding bars. Group size: CaMKIIα- hM3Dq, n = 7.
Then we used detection of the activity marker c-Fos to determine the effects of chemogenetic excitation of BLA glutamatergic neurons. Rats (n = 12) with expression of hM3Dq in the right BLA received intraperitoneal injection of either CNO (3 mg/kg) or vehicle before examination of c-Fos expression in the right (transduced) and left (nontransduced) BLA. CNO (n = 6) caused significant recruitment of BLA neurons (Fig. 1C) relative to injection of vehicle (n = 6) (main effect: F(1,10) = 11.34, p < 0.05), and this increase in c-Fos expression was selective to the right BLA (interaction: F(1,10) = 6.18, p < 0.05) (Fig. 1D).
Next, we made whole-cell current-clamp recordings from hM3Dq+ BLA neurons to determine the effects of hM3Dq receptor activation on neuronal activity. Bath application of CNO (1–5 μm) for 5 min significantly depolarized hM3Dq+ BLA neurons by 2.6 ± 0.3 mV compared with baseline membrane potentials (paired t test, p < 0.05; n = 7; Fig. 1E,F). This depolarization was sufficient to markedly increase firing rate (Fig. 1G) compared with baseline firing evoked by a 1 s depolarizing current injection (100–300 pA) that elicited minimal firing (paired t test, p < 0.05, n = 6; Fig. 1H). In contrast, no significant change in membrane potential was observed for hM3Dq− BLA neurons from the nontransduced hemisphere (paired t test, p > 0.05; n = 5; Fig. 1E,F). Importantly, we found no deficit in endogenous muscarinic receptor activity in these hM3Dq− cells because the broad-spectrum muscarinic agonist carbachol (10 μm) induced an initial hyperpolarization followed by a marked depolarization in hM3Dq− BLA neurons (paired t test, p < 0.01; n = 4, Fig. 1E,F). This biphasic response is consistent with changes in conductance of various ion channels following broad-spectrum muscarinic activation (McQuiston and Madison, 1999; Bell et al., 2013). Hence, CNO selectively depolarizes and increases the firing rate of hM3Dq expressing BLA neurons while not affecting nonexpressing neurons.
Experiment 2: chemogenetic activation of BLA glutamatergic neurons and fear learning
We then asked whether this chemogenetic excitation of BLA glutamatergic neurons yields heightened fear learning and memory formation. Rats with bilateral BLA expression of hM3Dq (n = 22) or AAV5-CaMKII α-eYFP (hereafter referred to as eYFP) (n = 18) (Fig. 2A) were trained to fear a visual CS via pairings with a 0.3 mA (weak) or 0.6 mA (strong) footshock to establish weak or strong fear memories, respectively (Fig. 2B). They were injected with CNO 30 min before these training sessions. Rats were later tested for their fear responses (conditioned suppression) to the visual CS in the absence of CNO.
Figure 2.
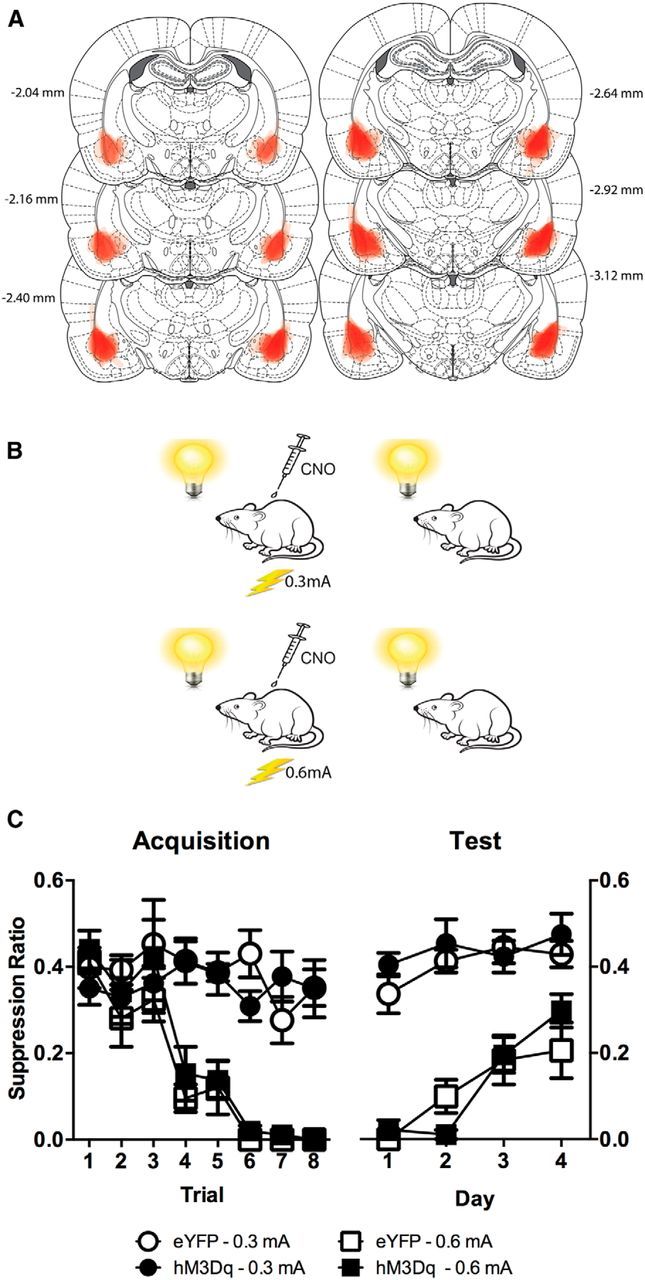
Chemogenetic activation of BLA glutamatergic neurons and fear learning. A, Extent of eGFP-IR for each rat included in the analysis represented at 10% opacity. B, Behavioral procedure. Groups 0.3 mA received visual CS-0.3 mA shock training and the Groups 0.6 mA received visual CS-0.6 mA training during acquisition. CNO (3 mg/kg, i.p.) was injected 30 min before acquisition training. C, Mean ± SEM suppression ratios are shown for acquisition and test. A ratio of 0.5 = no suppression of lever pressing during the CS (no fear), and 0 = complete suppression of lever pressing during the CS (high fear). The 0.6 mA footshock conditioned more fear than 0.3 mA (p < 0.05), but there was no effect of chemogenetic excitation (p > 0.05). Group sizes: CaMKIIα-hM3Dq-0.6 mA, n = 5; CaMKIIα-eYFP-0.6 mA, n = 7; CaMKIIα-hM3Dq-0.3 mA, n = 7; CaMKIIα-eYFP-0.3 mA, n = 9.
Histology
Figure 2A shows the extent of eGFP-IR across all rats included in the analyses with each rat represented at 10% opacity. Twelve rats were excluded due to expression of the eYFP or hM3Dq being located predominantly outside the BLA.
Behavior
The 0.6 mA footshock supported more fear learning than the 0.3 mA footshock across training (Fig. 2C) (F(1,24) = 48.15, p < 0.05) and test (F(1,24) = 149.88, p < 0.05), confirming that the different shock intensities used during training established weak and strong fear memories. CNO was injected before acquisition training only. However, chemogenetic excitation of BLA glutamatergic neurons had no effect on this fear learning and memory formation, assessed across the course of CS–US pairings or during the subsequent extinction tests (all F(1,24) < 1, p > 0.05), regardless of whether such memories were weak or strong. So, chemogenetic excitation was not able to convert weak fear learning into stronger learning, was unable to shift asymptotic levels of fear learning, and did not augment fear memory consolidation.
Experiment 3: chemogenetic activation of BLA glutamatergic neurons prevents use of prediction error to regulate fear learning
The failure of chemogenetic excitation to affect simple fear learning and memory formation shows that the consequences of heightened BLA glutamatergic neuronal activation for fear learning and memory are subtle. A key feature of normal fear learning and amygdala synaptic plasticity is that they are tightly regulated by prediction error (McNally and Westbrook, 2006; McNally et al., 2011). In this way, only mismatches between predicted and actual danger (i.e., prediction errors) trigger fear learning (Rescorla and Wagner, 1972).
To examine the impact of chemogenetic excitation of BLA glutamatergic neurons on use of fear prediction errors, rats expressing hM3Dq (n = 28) or eYFP (n = 28) bilaterally in BLA (Fig. 3A) were assessed in an associative blocking procedure (Kamin, 1968) (Fig. 3B) with injections of CNO 30 min before Stage II. In this task, rats were first trained that CSA (visual CS) predicts footshock. Subsequently, CSA was presented in compound with a neutral CSB (auditory CS) and followed by the same footshock. The prior fear learning to CSA blocks learned fear to CSB. This occurs because the animal's expectations are matched to the danger posed, generating minimal prediction error and minimal fear learning. A control group, on the other hand, receiving just Stage II training of a neutral CSA presented in compound with a neutral CSB and followed by footshock, does learn fear to CSB. This occurs due to positive prediction error. If chemogenetic excitation of BLA glutamatergic neurons disrupts this regulation of fear learning by prediction error, then it will cause inappropriate fear learning to the blocked CSB.
Figure 3.
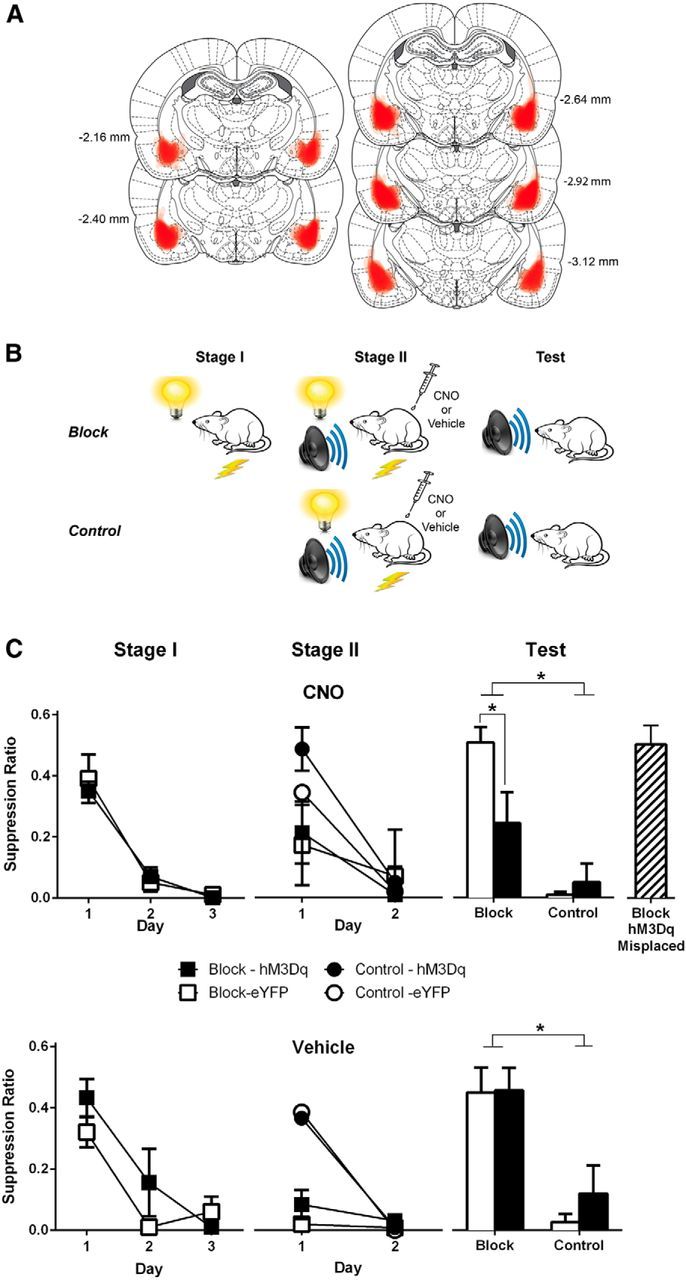
Chemogenetic activation of BLA glutamatergic neurons prevents use of prediction error to regulate fear learning. A, Extent of eGFP-IR for each rat included in the analysis represented at 10% opacity. B, Behavioral procedure. Groups Block received Stage I CSA-shock training. In Stage II, all groups received CSAB-shock pairings. CNO (3 mg/kg, i.p.) or Vehicle (i.p.) was injected 30 min before Stage II sessions. All rats were then tested for fear to CSB. C, Mean ± SEM suppression ratios during Stage I, Stage II, and test for CNO (top) or vehicle (bottom). There was evidence for blocking in CaMKIIα-eYFP-Veh and CaMKII-hM3Dq-Veh and CaMKIIα-hM3Dq-CNO-Misplaced groups, versus CaMKII-hM3Dq-Veh. Blocking was attenuated in CaMKIIα-hM3Dq-CNO versus CaMKIIα-eYFP-CNO. *p < 0.05. Group sizes: CaMKIIα-hM3Dq-Block-CNO, n = 5; CaMKIIα-hM3Dq-Control-CNO, n = 6; CaMKIIα-eYFP-Block-CNO, n = 7; CaMKIIα-eYFP-Control-CNO, n = 5; CaMKIIα-hM3Dq-Block-Vehicle, n = 5; CaMKIIα-hM3Dq-Control-Vehicle, n = 6; CaMKIIα-eYFP-Block-Vehicle, n = 7; CaMKIIα-eYFP-Control-Vehicle, n = 7; CaMKIIα-hM3Dq-CNO-Misplaced, n = 6.
Histology
Figure 3A shows the extent of eGFP-IR across all rats included in the analyses with each rat represented at 10% opacity. Eight animals were excluded due to misplaced hM3Dq or eYFP expression, primarily in the surrounding CeA or endopiriform cortex or unilaterally in BLA.
Behavior
All block groups acquired fear across Stage I (main effect of day: F(1,20) = 80.11, p < 0.05) (Fig. 3C), and there were no differences between groups (hM3Dq vs eYFP; F(1,20) < 1, p > 0.05; CNO vs Veh; F(1,20) < 1, p > 0.05; interactions: F(1,20) < 1, p > 0.05).
During Stage II, the block groups expressed fear to the compound CSAB (main effect Block vs Control: F(1,36) = 12.27, p < 0.05), whereas the control groups acquired fear (main effect of day: F(1,36) = 71.92, p < 0.05; group × day interaction: F(1,36) = 25.53, p < 0.05). There was no effect of DREADD (main effect: F(1,36) < 1, p > 0.05), CNO injection (F(1,36) = 2.9, p > 0.05), or interaction (group × drug × DREADD interaction: F(1,36) < 1, p > 0.05). Further simple effect analyses showed no differences between the DREADD and eYFP groups during Stage II. So, there was no effect of the DREADD on fear expression during Stage II (all p < 0.05).
The results from test revealed normal use of prediction error to regulate learning among the vehicle groups. Animals that received vehicle injections during Stage II demonstrated associative blocking (i.e., less fear to CSB in Block than Control groups, main effect: F(1,21) = 23.25, p < 0.05) regardless of the DREADD (main effect: F(1,21) < 1, p > 0.05; group × DREADD interaction: F(1,21) < 1, p > 0.05). So, there was fear learning to the CS signaling an unexpected footshock (positive prediction error) and no fear learning to the CS signaling an expected footshock (no prediction error).
In contrast, chemogenetic activation of BLA glutamatergic neurons prevented this use of prediction error to guide fear learning (main effect of DREADD: F(1,19) = 5.22, p < 0.05; group × DREADD interaction F(1,19) = 4.43, p < 0.05). Associative blocking was attenuated in hM3Dq animals (CNO-hM3Dq-Block vs CNO-eYFP-Block: F(1,19) = 9.96, p < 0.05), causing inappropriate fear learning to the blocked CS in hM3Dq animals. This inappropriate fear learning was selective to the associative blocking manipulation because chemogenetic excitation had no effect on fear learning in the Control groups (F(1,19) < 1, p > 0.05).
To verify that this DREADD-driven disruption of prediction error was anatomically specific to BLA, we examined animals treated with CNO that had robust but misplaced expression of hM3Dq. This misplaced expression was typically in central nucleus of the amygdala or piriform cortex (Fig. 3C; CNO-hM3Dq-Block-Misplaced, n = 6). In contrast to animals with BLA hM3Dq expression, these animals with misplaced hM3Dq expression showed normal use of prediction error. Associative blocking was intact yielding no fear learning to expected danger (vs CNO-Block-eYFP group: F(1,15) < 1, p > 0.05; vs CNO-hM3Dq-Block group: F(1,15) = 5.29, p < 0.05). This confirms the anatomical selectivity of disrupted prediction error by BLA chemogenetic manipulation.
Experiment 4: chemogenetic activation of BLA glutamatergic neurons prevents use of negative prediction error to inhibit fear
Finally, we asked whether this failure of prediction error to regulate learning extends to the inhibition of fear. Fear inhibition is profoundly disrupted in anxiety disorders, and such disruption is associated with hyperactivity of the amygdala (Rachman, 1994; Guthrie and Bryant, 2006; Michael et al., 2007; Jovanovic et al., 2010, 2012; Dunsmoor and Paz, 2015). In rodents, such fear inhibition is often studied via extinction. However, deficits in extinction learning have numerous causes that do not necessarily reflect deficits in use of negative prediction error (Storsve et al., 2010, 2012).
We used an overexpectation procedure (Rescorla, 1970; Lattal and Nakajima, 1998; McNally et al., 2004; Rescorla, 2007) to specifically isolate negative prediction error causing fear inhibition. Rats expressing hM3Dq (n = 19) or eYFP bilaterally in BLA (n = 15) (Fig. 4A) were trained in an overexpectation procedure (Fig. 4B) so that in Stage I two CS, CSA (visual) and CSB (auditory), separately predicted footshock. In Stage II, the two cues were presented in compound and followed by the same footshock for the overexpectation group. This causes a reduction in learned fear to both CS. This reduction occurs, despite the footshock US being present, because the animal's expectations of danger have been increased: it sums predictions from both CS to predict two footshocks, but it receives only one. This generates a negative prediction error causing fear inhibition. If chemogenetic activation of BLA glutamatergic neurons disrupts use of negative prediction error to inhibit fear, then CNO injections before Stage II training should prevent overexpectation of fear.
Figure 4.
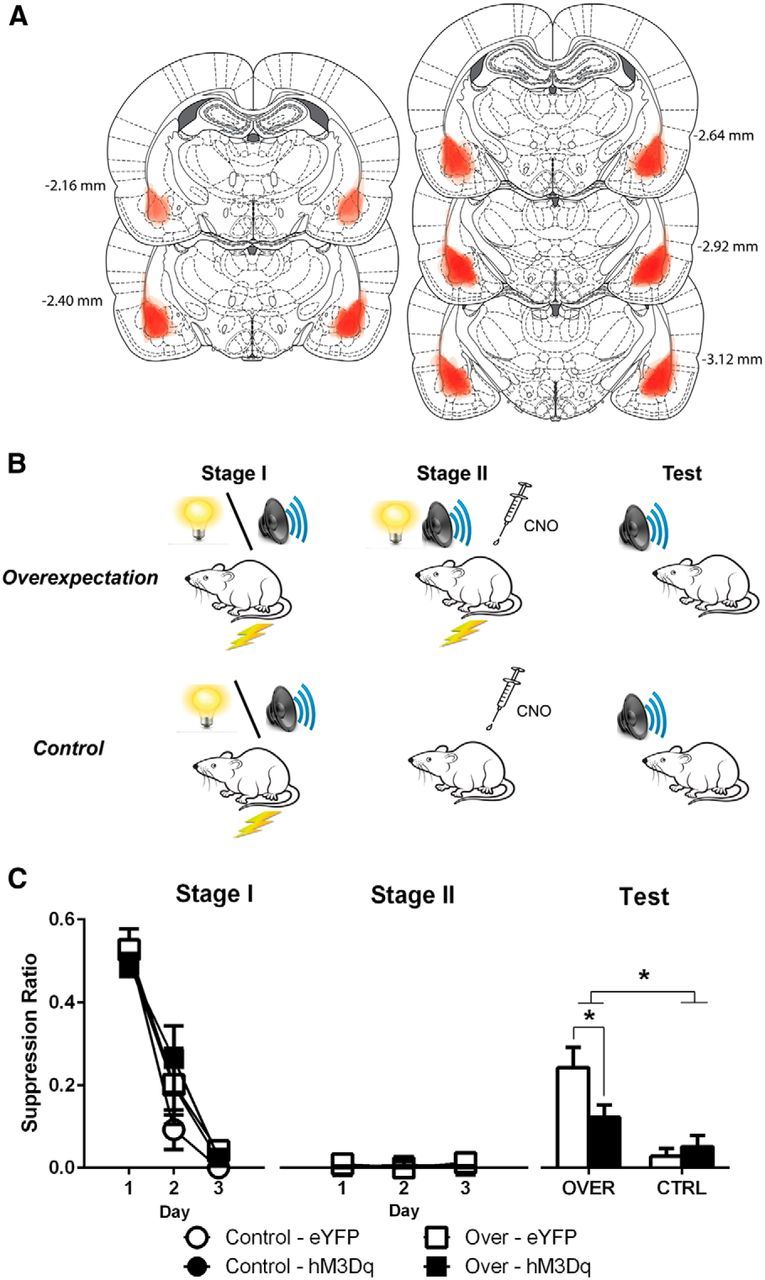
Chemogenetic activation of BLA glutamatergic neurons prevents use of negative prediction error to inhibit fear. A, Extent of eGFP-IR for each rat included in the analysis represented at 10% opacity. B, Behavioral procedure. All groups received CSA-shock and CSB-shock pairings in Stage I. In Stage II, Groups Over received CSA-shock presentations. Groups Control received further VI 120 training. CNO (3 mg/kg, i.p.) was injected 30 min before Stage II training. All groups were then tested for fear to CSA and CSB. C, Mean ± SEM suppression ratios during Stage I, Stage II, and test. Overexpectation was attenuated in CaMKIIα-hM3Dq-Over versus CaMKIIα-eYFP-Over. *p < 0.05. Group sizes: CaMKIIα-hM3Dq-Over, n = 9; CaMKIIα-eYFP-Over, n = 6; CaMKIIα-hM3Dq-Control, n = 6; CaMKIIα-eYFP-Control, n = 6.
Histology
Figure 4A shows the extent of eGFP-IR across all rats included in the analyses with each rat represented at 10% opacity. Seven rats were excluded as per exclusion criteria outlined previously. Two of these animals were in the hM3Dq-Over group.
Behavior
All groups learned to fear CSA and CSB across Stage I (main effect of day: F(1,23) = 743.51, p < 0.05; no main effect of group: F(1,23) = 1.45, p > 0.05; no interaction: F(1,23) < 1, p > 0.05) (Fig. 4C). During Stage II, both the eYFP and hM3Dq groups were identical: expressing high levels of fear (no main effect of DREADD: F(1,13) < 1, p > 0.05; no main effect of day: F(1,13) < 1, p > 0.05; no day × group interaction: F(1,13) < 1, p > 0.05).
Overexpectation caused inhibitory learning and loss of fear (Fig. 4C) (main effect of group: F(1,23) = 17.71, p < 0.05; no main effect of DREADD: F(1,23) = 2.05, p > 0.05). Importantly, chemogenetic activation of BLA glutamatergic neurons prevented this use of negative prediction error to reduce fear, yielding a significant impairment in learned fear inhibition (group × DREADD interaction: F(1,23) = 4.50, p < 0.05; hM3Dq-Over vs eYFP-Over: F(1,23) = 6.95, p < 0.05; hM3Dq-Control vs eYFP-Control: F(1,23) < 1, p > 0.05).
Discussion
We used chemogenetic excitation of BLA glutamatergic neurons to identify the mechanisms linking heightened amygdala activation to excessive fear. First, we validated our DREADD approach via immunohistochemistry to show that the CaMKIIα-hM3Dq DREADD predominantly colocalized to BLA CaMKIIα-IR cells. We then demonstrated that injections of CNO caused c-Fos expression in the transduced but not nontransduced BLA. Finally, we showed that CNO significantly depolarized hM3Dq+ BLA neurons and markedly increased firing rates evoked by current injection. Next, we used this chemogenetic approach to determine the effects of BLA activation on the acquisition of fear. Chemogenetic excitation of BLA glutamatergic neurons did not augment simple fear learning regardless of whether this learning led to a weak or strong fear memory. Instead, chemogenetic excitation of these neurons acted selectively to prevent the associative blocking of fear learning and also acted to prevent inhibitory fear learning in an overexpectation task.
Methodological considerations
There was no evidence here that these effects of chemogenetic manipulation on fear learning were due to AAV-mediated expression of the hM3Dq DREADD disrupting general BLA function and/or behavior. In slices, the hM3Dq DREADD caused expected membrane depolarization and increased firing rates. As in our previous work (Yau and McNally, 2015), hM3Dq and eYFP animals acquired the baseline lever pressing task at the same rate and to the same level; they also acquired, retained, and expressed fear at the same levels as the eYFP groups. The hM3Dq groups were largely indistinguishable from eYFP controls. These findings argue strongly against a nonselective effect of BLA hM3Dq expression on learning or memory.
There was also no evidence that CNO had nonspecific effects that confound interpretation (Rogan and Roth, 2011), at least in the measures we assessed. In BLA slices, there was no detectable effect of CNO on membrane potentials in hM3Dq− neurons, despite these neurons showing intact sensitivity to the broad-spectrum muscarinic agonist carbachol. At the behavioral level, there was no effect of CNO injection on simple fear learning or memory; and in the associative blocking as well as overexpectation experiments, there was no difference between groups until the test phases. In addition, in the associative blocking experiment, the CNO-injected hM3Dq misplaced group showed normal behavioral function and learning.
Together, these findings argue strongly against nonselective or nonspecific effects of the hM3Dq or CNO manipulations on BLA function and fear learning. Instead, our findings indicate that chemogenetic excitation of BLA glutamatergic neurons has subtle and selective effects on fear learning.
Heightened amygdala activation and disrupted prediction error
Our data show that chemogenetic excitation of BLA glutamatergic neurons does not alter asymptotic levels of fear, increase the perceived intensity of the footshock, or augment fear memory consolidation because there were no within session or between session differences between hM3Dq and eYFP in acquisition or expression of simple forms of fear learning. This is consistent with observations that such simple fear learning is often unaltered in clinically anxious populations (Lissek et al., 2005) (but see below).
Rather, we detected a specific deficit in the use of prediction error to regulate fear learning. Prediction error regulates fear learning by matching increments and decrements in fear precisely to the danger posed. This error instructs fear learning to unexpected events (positive prediction error), prevents fear learning to expected events (no prediction error), and instructs inhibitory learning to omission of expected events (negative prediction error). hM3Dq excitation of BLA glutamatergic neurons disrupted use of this error to regulate fear learning. Specifically, this excitation yielded two consequences. First, it prevented associative blocking, causing inappropriate fear memory formation: animals learned to fear a CS that was not a valid predictor of danger in the associative blocking task. Animals in the Block-hM3Dq-CNO group showed significantly more fear to the blocked CS than the Block-hM3Dq-Vehicle and Block-eYFP-CNO controls. Second, chemogenetic excitation impaired fear inhibition. Animals were unable to use negative prediction error to reduce fear in the overexpectation task. Animals in the Over-hM3Dq group showed impaired overexpectation compared with the Over-eYFP group. The finding of impaired inhibition of fear in overexpectation is important because, unlike fear extinction, which has multiple causes for fear loss (Rescorla, 2001; Myers and Davis, 2002), overexpectation selectively reveals actions of negative prediction error in causing fear loss.
Fear prediction error acts by regulating the activity of BLA neurons. Expected aversive events yield less recruitment of BLA and less fear learning than unexpected ones. This diminution of BLA activity during fear learning has been reported in rodents (Furlong et al., 2010; Johansen et al., 2010b) and humans (Dunsmoor et al., 2008; Eippert et al., 2012), including in human associative blocking tasks. It is due to periaqueductal gray-based circuits causing reductions in transmission of aversive US information to the BLA and reducing fear learning commensurately (Fanselow, 1998; McNally and Westbrook, 2006; McNally et al., 2011; Herry and Johansen, 2014). Chemogenetic excitation of BLA glutamatergic neurons may bypass circuit level feedback control over the activity of BLA glutamatergic neurons during fear learning. The hM3Dq excitation effectively circumvents control over BLA glutamatergic neuron activity by fear prediction error, enabling learning to a CS that is not a valid predictor of danger when it would not otherwise occur (blocking) and preventing fear inhibition (overexpectation). This insensitivity to prediction error during heightened BLA glutamatergic neuronal activation may provide a common mechanism for overgeneralization of fear and failures of fear inhibition characteristic of pathological anxiety (Jovanovic and Ressler, 2010; Dunsmoor and Paz, 2015).
Amygdala and prediction error
There are three additional features of these results that deserve comment. First, the effects of chemogenetic excitation in the overexpectation design could suggest that BLA glutamatergic neurons show bidirectional sensitivity to prediction error: positive error and increments in fear are coded by increases in activity, whereas negative error and decrements in fear are coded by decreases in activity. However, it is equally plausible that such errors are coded by excitation in distinct populations of BLA glutamatergic neurons (e.g., fear “on” and “off” cells) (Herry et al., 2008; Senn et al., 2014). The chemogenetic BLA manipulation does not allow easy distinction between these possibilities. Second, BLA lesions do not affect prediction error driven upregulation of attention to CS in Pavlovian appetitive conditioning (Holland et al., 2001). This suggests that BLA contributions to the effects of prediction error may differ in appetitive and fear conditioning, a suggestion supported by the role of nucleus accumbens in attentional selection of danger signals (Li and McNally, 2015). Finally, the deficit in use of prediction error was restricted to the blocking and overexpectation designs. There was no effect on simple fear acquisition. BLA glutamatergic neurons may be especially sensitive to fear learning based on pooled or summed prediction errors (e.g., during blocking and overexpectation) and less sensitive to learning based on unique or individual errors (e.g., during single cue conditioning) (Le Pelley, 2004). However, given that manipulations of BLA glutamatergic neurons affect learning to individual cues (Wolff et al., 2014), this is an interesting but unlikely possibility. Alternatively, despite sensitivity to differences in asymptotic levels of fear learning, our measure may have been less sensitive to differences in rate of learning. This could obscure any effect on the DREADD manipulation on fear learning to the single cues. Indeed, DREADDs modulate fear acquisition in mice using immobility as the measure of learned fear (Yiu et al., 2014).
In conclusion, here we combined chemogenetic techniques with behavioral approaches isolating the actions of fear prediction error to show that heightened activation of BLA glutamatergic neurons acts selectively to disrupt use of prediction error in regulating fear learning. Animals were less able to use their past experience with danger to regulate their future learning about this danger, as shown by impaired associative blocking, and were less able to reduce fear when fear expectations exceeded the danger posed, as shown by impaired overexpectation. Chemogenetic excitation of BLA glutamatergic neurons could therefore serve as a useful model for identifying the cellular and circuit level consequences of heightened amygdala activation as well as for understanding fear prediction error signals in pathological anxiety.
Footnotes
A.S. was supported by an Australian Postgraduate Award. G.P.M. was supported by a Future Fellowship from Australian Research Council FT120100250. This work was supported by Australian Research Council Grant FT120100250 to G.P.M. and National Health and Medical Research Council Grants 1077806 to G.P.M. and E.E.B. and 1047372 to E.E.B. and G.P.M. We thank Pascal Carrive and Adam Hamlin for advice on CaMKIIα immunohistochemistry; and Nathan Marchant, Rick Richardson, Fred Westbrook, and Nathan Holmes for discussions of these experiments.
The authors declare no competing financial interests.
References
- Alexander GM, Rogan SC, Abbas AI, Armbruster BN, Pei Y, Allen JA, Nonneman RJ, Hartmann J, Moy SS, Nicolelis MA, McNamara JO, Roth BL. Remote control of neuronal activity in transgenic mice expressing evolved G-protein coupled receptors. Neuron. 2009;63:27–39. doi: 10.1016/j.neuron.2009.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Annau Z, Kamin LJ. The conditioned emotional response as a function of the intensity of the US. J Comp Physiol Psychol. 1961;54:428–432. doi: 10.1037/h0042199. [DOI] [PubMed] [Google Scholar]
- Arico C, McNally GP. Opioid receptors regulate blocking and overexpectation of fear learning in conditioned suppression. Behav Neurosci. 2014;128:199–206. doi: 10.1037/a0036133. [DOI] [PubMed] [Google Scholar]
- Armbruster BN, Li X, Pausch MH, Herlitze S, Roth BL. Evolving the lock to fit the key to create a family of G protein-coupled receptors potently activated by an inert ligand. Proc Natl Acad Sci U S A. 2007;104:5163–5168. doi: 10.1073/pnas.0700293104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell LA, Bell KA, McQuiston AR. Synaptic muscarinic response types in hippocampal CA1 interneurons depend on different levels of presynaptic activity and different muscarinic receptor subtypes. Neuropharmacology. 2013;73:160–173. doi: 10.1016/j.neuropharm.2013.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis M. The role of the amygdala in fear and anxiety. Annu Rev Neurosci. 1992;15:353–375. doi: 10.1146/annurev.ne.15.030192.002033. [DOI] [PubMed] [Google Scholar]
- Desmedt A, Marighetto A, Piazza PV. Abnormal fear memory as a model for posttraumatic stress disorder. Biol Psychiatry. 2015;78:290–297. doi: 10.1016/j.biopsych.2015.06.017. [DOI] [PubMed] [Google Scholar]
- Dunsmoor JE, Paz R. Fear generalization and anxiety: behavioral and neural mechanisms. Biol Psychiatry. 2015;78:336–343. doi: 10.1016/j.biopsych.2015.04.010. [DOI] [PubMed] [Google Scholar]
- Dunsmoor JE, Bandettini PA, Knight DC. Neural correlates of unconditioned response diminution during Pavlovian conditioning. Neuroimage. 2008;40:811–817. doi: 10.1016/j.neuroimage.2007.11.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrlich I, Humeau Y, Grenier F, Ciocchi S, Herry C, Lüthi A. Amygdala inhibitory circuits and the control of fear memory. Neuron. 2009;62:757–771. doi: 10.1016/j.neuron.2009.05.026. [DOI] [PubMed] [Google Scholar]
- Eippert F, Gamer M, Büchel C. Neurobiological mechanisms underlying the blocking effect in aversive learning. J Neurosci. 2012;32:13164–13176. doi: 10.1523/JNEUROSCI.1210-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fanselow MS. Pavlovian conditioning, negative feedback, and blocking: mechanisms that regulate association formation. Neuron. 1998;20:625–627. doi: 10.1016/S0896-6273(00)81002-8. [DOI] [PubMed] [Google Scholar]
- Farb CR, Ledoux JE. Afferents from rat temporal cortex synapse on lateral amygdala neurons that express NMDA and AMPA receptors. Synapse. 1999;33:218–229. doi: 10.1002/(SICI)1098-2396(19990901)33:3<218::AID-SYN6>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- Furlong TM, Cole S, Hamlin AS, McNally GP. The role of prefrontal cortex in predictive fear learning. Behav Neurosci. 2010;124:574–586. doi: 10.1037/a0020739. [DOI] [PubMed] [Google Scholar]
- Goossens L, Sunaert S, Peeters R, Griez EJ, Schruers KR. Amygdala hyperfunction in phobic fear normalizes after exposure. Biol Psychiatry. 2007;62:1119–1125. doi: 10.1016/j.biopsych.2007.04.024. [DOI] [PubMed] [Google Scholar]
- Guthrie RM, Bryant RA. Extinction learning before trauma and subsequent posttraumatic stress. Psychosom Med. 2006;68:307–311. doi: 10.1097/01.psy.0000208629.67653.cc. [DOI] [PubMed] [Google Scholar]
- Harris RJ. ANOVA: an analysis of variance primer. Itasca, IL: Peacock; 2004. [Google Scholar]
- Herry C, Johansen JP. Encoding of fear learning and memory in distributed neuronal circuits. Nat Neurosci. 2014;17:1644–1654. doi: 10.1038/nn.3869. [DOI] [PubMed] [Google Scholar]
- Herry C, Ciocchi S, Senn V, Demmou L, Müller C, Lüthi A. Switching on and off fear by distinct neuronal circuits. Nature. 2008;454:600–606. doi: 10.1038/nature07166. [DOI] [PubMed] [Google Scholar]
- Holland PC, Hatfield T, Gallagher M. Rats with basolateral amygdala lesions show normal increases in conditioned stimulus processing but reduced conditioned potentiation of eating. Behav Neurosci. 2001;115:945–950. doi: 10.1037/0735-7044.115.4.945. [DOI] [PubMed] [Google Scholar]
- Johansen JP, Hamanaka H, Monfils MH, Behnia R, Deisseroth K, Blair HT, LeDoux JE. Optical activation of lateral amygdala pyramidal cells instructs associative fear learning. Proc Natl Acad Sci U S A. 2010a;107:12692–12967. doi: 10.1073/pnas.1002418107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansen JP, Tarpley JW, LeDoux JE, Blair HT. Neural substrates for expectation-modulated fear learning in the amygdala and periaqueductal gray. Nat Neurosci. 2010b;13:979–986. doi: 10.1038/nn.2594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jovanovic T, Ressler KJ. How the neurocircuitry and genetics of fear inhibition may inform our understanding of PTSD. Am J Psychiatry. 2010;167:648–662. doi: 10.1176/appi.ajp.2009.09071074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jovanovic T, Norrholm SD, Blanding NQ, Davis M, Duncan E, Bradley B, Ressler KJ. Impaired fear inhibition is a biomarker of PTSD but not depression. Depress Anxiety. 2010;27:244–251. doi: 10.1002/da.20663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jovanovic T, Kazama A, Bachevalier J, Davis M. Impaired safety signal learning may be a biomarker of PTSD. Neuropharmacology. 2012;62:695–704. doi: 10.1016/j.neuropharm.2011.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamin LJ. Attention-like processes in classical conditioning. In: Campbell BJ, Church RM, editors. Miami symposium on the prediction of behavior: aversive stimulation. Miami: University of Miami; 1968. pp. 9–33. [Google Scholar]
- Lanuza E, Moncho-Bogani J, Ledoux JE. Unconditioned stimulus pathways to the amygdala: effects of lesions of the posterior intralaminar thalamus on foot-shock-induced c-Fos expression in the subdivisions of the lateral amygdala. Neuroscience. 2008;155:959–968. doi: 10.1016/j.neuroscience.2008.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lattal KM, Nakajima S. Overexpectation in appetitive Pavlovian and instrumental conditioning. Anim Learn Behav. 1998;26:351–360. doi: 10.3758/BF03199227. [DOI] [Google Scholar]
- Le Pelley ME. The role of associative history in models of associative learning: a selective review and a hybrid model. Q J Exp Psychol. 2004;57:193–243. doi: 10.1080/02724990344000141. [DOI] [PubMed] [Google Scholar]
- Li SS, McNally GP. Selecting danger signals: dissociable roles of nucleus accumbens shell and core glutamate in predictive fear learning. Eur J Neurosci. 2015;41:1515–1523. doi: 10.1111/ejn.12892. [DOI] [PubMed] [Google Scholar]
- Lissek S, Powers AS, McClure EB, Phelps EA, Woldehawariat G, Grillon C, Pine DS. Classical fear conditioning in the anxiety disorders: a meta-analysis. Behav Res Ther. 2005;43:1391–1424. doi: 10.1016/j.brat.2004.10.007. [DOI] [PubMed] [Google Scholar]
- Lüthi A, Lüscher C. Pathological circuit function underlying addiction and anxiety disorders. Nat Neurosci. 2014;17:1635–1643. doi: 10.1038/nn.3849. [DOI] [PubMed] [Google Scholar]
- Marek R, Strobel C, Bredy TW, Sah P. The amygdala and medial prefrontal cortex: partners in the fear circuit. J Physiol. 2013;591:2381–2391. doi: 10.1113/jphysiol.2012.248575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maren S, Quirk GJ. Neuronal signalling of fear memory. Nat Rev Neurosci. 2004;5:844–852. doi: 10.1038/nrn1535. [DOI] [PubMed] [Google Scholar]
- McKernan MG, Shinnick-Gallagher P. Fear conditioning induces a lasting potentiation of synaptic currents in vitro. Nature. 1997;390:607–611. doi: 10.1038/37605. [DOI] [PubMed] [Google Scholar]
- McNally GP, Westbrook RF. Predicting danger: the nature, consequences, and neural mechanisms of predictive fear learning. Learn Mem. 2006;13:245–253. doi: 10.1101/lm.196606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNally GP, Pigg M, Weidemann G. Blocking, unblocking, and overexpectation of fear: a role for opioid receptors in the regulation of Pavlovian association formation. Behav Neurosci. 2004;118:111–120. doi: 10.1037/0735-7044.118.1.111. [DOI] [PubMed] [Google Scholar]
- McNally GP, Johansen JP, Blair HT. Placing prediction into the fear circuit. Trends Neurosci. 2011;34:283–292. doi: 10.1016/j.tins.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McQuiston AR, Madison DV. Muscarinic receptor activity has multiple effects on the resting membrane potentials of CA1 hippocampal interneurons. J Neurosci. 1999;19:5693–5702. doi: 10.1523/JNEUROSCI.19-14-05693.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michael T, Blechert J, Vriends N, Margraf J, Wilhelm FH. Fear conditioning in panic disorder: enhanced resistance to extinction. J Abnorm Psychol. 2007;116:612–617. doi: 10.1037/0021-843X.116.3.612. [DOI] [PubMed] [Google Scholar]
- Myers KM, Davis M. Behavioral and neural analysis of extinction. Neuron. 2002;36:567–584. doi: 10.1016/S0896-6273(02)01064-4. [DOI] [PubMed] [Google Scholar]
- Paré D, Quirk GJ, Ledoux JE. New vistas on amygdala networks in conditioned fear. J Neurophysiol. 2004;92:1–9. doi: 10.1152/jn.00153.2004. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The rat brain in stereotaxic coordinates. Ed 6. Amsterdam: Elsevier; 2007. [DOI] [PubMed] [Google Scholar]
- Rachman S. The overprediction of fear: a review. Behav Res Ther. 1994;32:683–690. doi: 10.1016/0005-7967(94)90025-6. [DOI] [PubMed] [Google Scholar]
- Rauch SL, Shin LM, Phelps EA. Neurocircuitry models of posttraumatic stress disorder and extinction: human neuroimaging research—past, present, and future. Biol Psychiatry. 2006;60:376–382. doi: 10.1016/j.biopsych.2006.06.004. [DOI] [PubMed] [Google Scholar]
- Rescorla RA. Reduction in the effectiveness of reinforcement after prior excitatory conditioning. Learn Motivation. 1970;1:372–381. doi: 10.1016/0023-9690(70)90101-3. [DOI] [Google Scholar]
- Rescorla RA. Experimental extinction. In: Klein SB, Mowrer, editors. Handbook of contemporary learning theories. Hillsdale, NJ: Erlbaum; 2001. pp. 119–154. [Google Scholar]
- Rescorla RA. Renewal after overexpectation. Learn Behav. 2007;35:19–26. doi: 10.3758/BF03196070. [DOI] [PubMed] [Google Scholar]
- Rescorla RA, Wagner AR. A theory of Pavlovian conditioning: variations in the effectiveness of reinforcement and nonreinforcement. In: Black AH, Prokasy WF, editors. Classical conditioning II: current research and theory. New York: Appleton-Century Crofts; 1972. pp. 64–99. [Google Scholar]
- Rogan SC, Roth BL. Remote control of neuronal signaling. Pharmacol Rev. 2011;63:291–315. doi: 10.1124/pr.110.003020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sah P, Faber ES, Lopez De Armentia M, Power J. The amygdaloid complex: anatomy and physiology. Physiol Rev. 2003;83:803–834. doi: 10.1152/physrev.00002.2003. [DOI] [PubMed] [Google Scholar]
- Schafe GE, Nader K, Blair HT, LeDoux JE. Memory consolidation of Pavlovian fear conditioning: a cellular and molecular perspective. Trends Neurosci. 2001;24:540–546. doi: 10.1016/S0166-2236(00)01969-X. [DOI] [PubMed] [Google Scholar]
- Senn V, Wolff SB, Herry C, Grenier F, Ehrlich I, Gründemann J, Fadok JP, Müller C, Letzkus JJ, Lüthi A. Long-range connectivity defines behavioral specificity of amygdala neurons. Neuron. 2014;81:428–437. doi: 10.1016/j.neuron.2013.11.006. [DOI] [PubMed] [Google Scholar]
- Shi C, Davis M. Pain pathways involved in fear conditioning measured with fear-potentiated startle: lesion studies. J Neurosci. 1999;19:420–430. doi: 10.1523/JNEUROSCI.19-01-00420.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin LM, Liberzon I. The neurocircuitry of fear, stress, and anxiety disorders. Neuropsychopharmacology. 2010;35:169–191. doi: 10.1038/npp.2009.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin LM, Kosslyn SM, McNally RJ, Alpert NM, Thompson WL, Rauch SL, Macklin ML, Pitman RK. Visual imagery and perception in posttraumatic stress disorder. Arch Gen Psychiatry. 1997;54:233–241. doi: 10.1001/archpsyc.1997.01830150057010. [DOI] [PubMed] [Google Scholar]
- Shin LM, Wright CI, Cannistraro PA, Wedig MM, McMullin K, Martis B, Macklin ML, Lasko NB, Cavanagh SR, Krangel TS, Orr SP, Pitman RK, Whalen PJ, Rauch SL. A functional magnetic resonance imaging study of amygdala and medial prefrontal cortex responses to overtly presented fearful faces in posttraumatic stress disorder. Arch Gen Psychiatry. 2005;62:273–281. doi: 10.1001/archpsyc.62.3.273. [DOI] [PubMed] [Google Scholar]
- Sternson SM, Roth BL. Chemogenetic tools to interrogate brain functions. Annu Rev Neurosci. 2014;37:387–407. doi: 10.1146/annurev-neuro-071013-014048. [DOI] [PubMed] [Google Scholar]
- Storsve AB, McNally GP, Richardson R. US habituation, like CS extinction, produces a decrement in conditioned fear responding that is NMDA dependent and subject to renewal and reinstatement. Neurobiol Learn Mem. 2010;93:463–471. doi: 10.1016/j.nlm.2009.12.011. [DOI] [PubMed] [Google Scholar]
- Storsve AB, McNally GP, Richardson R. Renewal and reinstatement of the conditioned but not the unconditioned response following habituation of the unconditioned stimulus. Behav Processes. 2012;90:58–65. doi: 10.1016/j.beproc.2012.03.007. [DOI] [PubMed] [Google Scholar]
- Tillfors M, Furmark T, Marteinsdottir I, Fischer H, Pissiota A, Långström B, Fredrikson M. Cerebral blood flow in subjects with social phobia during stressful speaking tasks: a PET study. Am J Psychiatry. 2001;158:1220–1226. doi: 10.1176/appi.ajp.158.8.1220. [DOI] [PubMed] [Google Scholar]
- Tillfors M, Furmark T, Marteinsdottir I, Fredrikson M. Cerebral blood flow during anticipation of public speaking in social phobia: a PET study. Biol Psychiatry. 2002;52:1113–1119. doi: 10.1016/S0006-3223(02)01396-3. [DOI] [PubMed] [Google Scholar]
- Tovote P, Fadok JP, Lüthi A. Neuronal circuits for fear and anxiety. Nat Rev Neurosci. 2015;16:317–331. doi: 10.1038/nrn3945. [DOI] [PubMed] [Google Scholar]
- Urban DJ, Roth BL. DREADDs (designer receptors exclusively activated by designer drugs): chemogenetic tools with therapeutic utility. Annu Rev Pharmacol Toxicol. 2013;55:399–417. doi: 10.1146/annurev-pharmtox-010814-124803. [DOI] [PubMed] [Google Scholar]
- Wolff SB, Gründemann J, Tovote P, Krabbe S, Jacobson GA, Müller C, Herry C, Ehrlich I, Friedrich RW, Letzkus JJ, Lüthi A. Amygdala interneuron subtypes control fear learning through disinhibition. Nature. 2014;509:453–458. doi: 10.1038/nature13258. [DOI] [PubMed] [Google Scholar]
- Yau JO, McNally GP. Pharmacogenetic excitation of dorsomedial prefrontal cortex restores fear prediction error. J Neurosci. 2015;35:74–83. doi: 10.1523/JNEUROSCI.3777-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yiu AP, Mercaldo V, Yan C, Richards B, Rashid AJ, Hsiang HL, Pressey J, Mahadevan V, Tran MM, Kushner SA, Woodin MA, Frankland PW, Josselyn SA. Neurons are recruited to a memory trace based on relative neuronal excitability immediately before training. Neuron. 2014;83:722–735. doi: 10.1016/j.neuron.2014.07.017. [DOI] [PubMed] [Google Scholar]
- Zhao S, Ting JT, Atallah HE, Qiu L, Tan J, Gloss B, Augustine GJ, Deisseroth K, Luo M, Graybiel AM, Feng G. Cell type-specific channelrhodopsin-2 transgenic mice for optogenetic dissection of neural circuitry function. Nat Methods. 2011;8:745–752. doi: 10.1038/nmeth.1668. [DOI] [PMC free article] [PubMed] [Google Scholar]