

Abstract
The voltage-gated K+ channels Kv7.2 and Kv7.3 are located at the axon initial segment (AIS) and exert strong control over action potential generation. Therefore, changes in their localization or cell surface numbers are likely to influence neuronal signaling. However, nothing is known about the cell surface dynamics of Kv7.2/7.3 at steady state or during short-term neuronal stimulation. This is primarily attributable to their membrane topology, which hampers extracellular epitope tagging. Here we circumvent this limitation by fusing an extra phluorin-tagged helix to the N terminus of human Kv7.3. This seven transmembrane chimera, named super ecliptic phluorin (SEP)–TAC-7.3, functions and traffics as a wild-type (WT) channel. We expressed SEP–TAC-7.3 in dissociated rat hippocampal neurons to examine the lateral mobility, surface numbers, and localization of AIS Kv7.2/7.3 heteromers using live imaging. We discovered that they are extraordinarily stable and exhibit a very low surface mobility both during steady state and neuronal stimulation. In the latter case, we also found that neither localization nor cell surface numbers were changed. However, at high glutamate loads, we observed a rapid irreversible endocytosis of Kv7.2/7.3, which required the activation of NR2B-containing NMDA receptors, Ca2+ influx, and calpain activation. This excitotoxic mechanism may be specific to ankyrin G-bound AIS proteins because Nav1.2 channels, but not AIS GABAA receptors, were also endocytosed. In conclusion, we have, for the first time, characterized the cell surface dynamics of a full-length Kv7 channel using a novel chimeric strategy. This approach is likely also applicable to other Kv channels and thus of value for the additional characterization of this ion channel subfamily.
SIGNIFICANCE STATEMENT The voltage-gated K+ channels Kv7.2 and Kv7.3 exert strong control over action potential generation, but little is known about their cell surface dynamics. Using a novel phluorin-based approach, we here show that these channels are highly stable at steady state and different types of neuronal stimulation. However, at high glutamate loads, they undergo a rapid calpain-dependent endocytosis that likely represents an early response during excitotoxic states.
Keywords: excitotoxicity, FRAP, glutamate, Kv7 channels, live imaging, phluorin
Introduction
The voltage-gated potassium channels Kv7.2 and Kv7.3 are expressed widely in the brain. As heteromers, they constitute the molecular correlate of the axonal M-current: a slow non-inactivating potassium current (Schroeder et al., 1998). In vivo, they are located specifically at the axon initial segment (AIS), nodes of Ranvier, and presynaptic boutons (Pan et al., 2006; Rasmussen et al., 2007). At the AIS, Kv7.2/7.3 regulate the resting membrane potential and action potential threshold (Shah et al., 2008) and therefore exert strong control over neuronal firing. Thus, modulating the electrophysiological properties of Kv7.2./7.3 is one way of fine-tuning neuronal signaling (Hoshi et al., 2003). However, neurons are also able to regulate the number of channels at a given site in an activity-dependent manner. Such a change, either through exocytosis/endocytosis or lateral movement, is paralleled by a proportional change in conductance. This phenomenon is well characterized for glutamate receptors (Malenka, 2003) but also applies for other ionotropic receptors (Muir et al., 2010) and dendritic Kv channels (Misonou et al., 2004; Kim et al., 2007). Whether this could apply for the physiologically important Kv7.2/7.3 heteromers at the AIS remains to be determined. However, measurement of Kv7.2/7.3 cell surface dynamics requires the ability to image these channels in living cells. This has been hampered by the lack of suitable antibodies but also by their membrane topology: both termini are intracellular, and the extracellular loops very short. Consequently, insertion of large extracellular epitope tags is not possible.
Therefore, our aim was to develop a tool that bypasses this limitation and use it to study the dynamics of cell surface Kv7.2/7.3 heteromers. To do so, we added an extra phluorin-tagged transmembrane helix to Kv7.3, thereby generating a seven-transmembrane chimera. Using this construct, we show that the localization, lateral mobility, and cell surface numbers of Kv7.2/7.3 heteromers are highly stable, both during steady state and mild neuronal stimulation. However, at sustained NMDA receptor (NMDAR) activation, Kv7.2/7.3 are endocytosed rapidly in a Ca2+- and calpain-dependent manner.
Materials and Methods
DNA constructs.
We used a modified pcDNA3.1+ vector (M1 pcDNA3.1+) containing a cDNA sequence encoding a signal peptide fused to the M1 FLAG tag. The TAC vector was generated inserting a truncated version of the human TAC antigen (NM_000417) into M1 pcDNA3.1+. The super ecliptic phluorin (SEP)–TAC vector was generated inserting SEP upstream of TAC. Human Kv7.3 (NM_004319) were inserted downstream of TAC, as were the rat Nav1.2 II–III linker (NM_012647.1). SEP-tagged α2-GABAAR has been described previously (Muir et al., 2010).
Neuronal culture and transfections.
Neuronal hippocampal cultures from rat embryos of either sex were prepared as described previously (Rasmussen et al., 2007). Cells were transfected at 7–8 d in vitro (DIV) with a total of 0.9 μg of DNA using Lipofectamine2000 (Life Technologies) and imaged at 10–13 or 18 DIV. SEP–TAC-7.3 and Kv7.2 were transfected at equimolar ratios.
Live imaging of SEP–TAC channels.
Fluorescence was captured using a Zeiss LSM780 inverted laser-scanning confocal microscope equipped with an incubator and a 63×, 1.4 numerical aperture oil-immersion objective. Coverslips were mounted in an imaging chamber (Warner Instruments) and imaged at 37°C in artificial CSF (aCSF; in mm: 125 NaCl, 5 KCl, 1 MgCl2, 2 CaCl2, 10 d-glucose, and 10 HEPES, pH 7.4). SEP fluorescence was collected with a fully open pinhole, because the fluorescence in confocal mode was too low. In stimulation experiments, images were collected once before and once after treatment, thereby eliminating photobleaching. The first image was acquired in pure aCSF. The second image was acquired 10 min after addition of an equivalent volume of aCSF containing drug(s) at 2× final concentration. Cells were preincubated with APV (100 μm), nisoldipine (100 μm), ifenprodil (50 μm), Brilliant Blue G (100 nm), or Ni2+ (50 μm) for 10 min or with dynasore (80 μm), cyclosporin A (CsA; 1 μm), chlorpromazine (100 μm), KN93 (2-[N-(2-hydroxyethyl)]-N-(4-methoxybenzenesulfonyl)amino-N-(4-chlorocinnamyl)-N-methylbenzylamine; 10 μm), okadaic acid (50 nm), contotoxin MVIIC (100 nm), or MDL-28170 (N-[N-[(phenylmethoxy)carbonyl]-l-valyl]-phenylalaninal; 100 μm) for 30–60 min. In time-lapse stimulation experiments, cells were imaged at 0.2 Hz for 10 min and cells stimulated after the second frame. In pH time-lapse experiments, cells were imaged a 1 Hz for 1 min. An equivalent volume of aCSF containing 10 mm HCl was added after the fifth frame to decrease extracellular pH to 5.6. All drugs used were from Sigma and did not alter the pH of aCSF when added during experiments.
Fluorescence recovery after photobleaching (FRAP).
SEP–TAC-7.3-expressing neurons were imaged at 0.5 Hz for 2 min in aCSF. After five frames, a circular spot (4 μm diameter) in the AIS was bleached with a 488 nm laser (five iterations at 100% laser power giving 70–80% bleaching efficiency). Data were normalized to the initial fluorescence intensity (1) and that after bleaching (0) after background subtraction.
Antibodies.
The primary antibodies included the following: anti-M1 FLAG mouse Ig2b (1:500 dilution; Sigma), anti-MAP2 rabbit IgG (H-300, 1:100 dilution; Santa Cruz Biotechnology), anti-myc mouse IgG1 (9E10, 1:50 dilution; Developmental Studies Hybridoma Bank), anti-pan-Nav mouse IgG1 (K58/35, 1:500 dilution; Sigma), anti-pan-Neurofascin (external) mouse IgG2a (A12/18, 1:100 dilution; Neuromab), and anti-pan-Neurofascin mouse IgG1 (L11A/41, 1:1 dilution; Neuromab). The secondary antibodies included the following: Alexa Fluor 568-conjugated goat anti-mouse IgG2b or IgG2a, Alexa Fluor 488-conjugated goat anti-mouse IgG1, and Alexa Fluor 647-conjugated donkey anti-rabbit IgG (1:1000 dilution; Invitrogen).
AIS live labeling.
Live labeling of the AIS and propidium iodide (PI) staining was performed as described previously (Schafer et al., 2009). Imaging of SEP-tagged GABAAR was performed as for SEP–TAC-7.3 but in confocal mode.
Image analysis.
SEP fluorescence analysis was performed in Fiji (Fiji_Is_Just_ImageJ). A ROI was drawn around the AIS covering a distance of ∼30 μm starting from its base. After background subtraction, the intensity after treatment was normalized to the intensity before treatment. In time-lapse experiments, the StackReg macro was used to correct for drift in the x–y plane. For each frame, the ROI fluorescence intensity was normalized to that of the first frame. Kymographs were generated using the MultipleKymograph macro.
Electrophysiology.
Visually guided patch-clamp recordings were performed in whole-cell configuration with a Multiclamp 700B amplifier (Molecular Devices) in voltage-clamp mode. The pipette solution contained the following: 122 mm K-gluconate, 2.5 mm MgCl2, 5.6 mm Mg-gluconate, 5 mm K-HEPES, 5 mm H-HEPES, 5 mm Na2ATP, 1 mm EGTA, 2.5 mm biocytine, HCl to adjust pH to 7.4, and 250 μm Alexa Fluor 488 (Sigma-Aldrich). Cells was perfused with Ringer's solution (in mm: 120 NaCl, 5 KCl, 15 NaHCO3, 20 glucose, 2 MgCl2, and 3 CaCl2, pH 7.6) saturated with 98% O2 and 2% CO2. Electrodes had an input resistance of 2–5 MΩ. Data were sampled with a 16-bit analog-to-digital converter (DIGIDATA 1440; Molecular Devices) at 100 kHz and filtered offline with low-pass Bessel filter (2 kHz cutoff). Data are presented without liquid-junction potential correction.
Statistics.
All experiments were performed on neurons from at least three hippocampal cultures. Statistical analyses were one-sample Student's t test (μ0 = 1) or ANOVA with Tukey's post hoc test. Data are presented as box-and-whisker plots. F/F0 values are given as mean ± SEM.
Results
To enable the separate detection of cell surface and intracellular Kv7.3 populations, we added an extra FLAG-tagged α helix to the N terminus of human Kv7.3 and a myc tag to its C terminus. Thus, in this seven transmembrane chimera (named TAC-7.3), the FLAG epitope is located at the extracellular side and the myc epitope intracellularly (Fig. 1A). First, we tested whether TAC-7.3 was functional. Indeed, patch-clamp experiments in CHO cells revealed that its electrophysiological properties were similar to a WT channel (Fig. 1B). To test whether the trafficking and subcellular localization of TAC-7.3 were altered, we coexpressed it with Kv7.2 in dissociated hippocampal neurons that were stained at 10 DIV with anti-FLAG antibody before and anti-myc antibody after permeabilization to detect cell surface and total expression, respectively. Remarkably, whereas total expression was uniformly distributed (myc), the cell surface expressed population of TAC-7.3 (FLAG) was restricted to the axon (as indicated by absence of MAP2) and enriched at the AIS (Fig. 1C), demonstrating that its cell surface expression is tightly controlled. Furthermore, like endogenous Kv7.3 (Pan et al., 2006), TAC-7.3 expression began more distal to the soma than AIS Nav channels (Fig. 1D). Thus, the TAC-7.3 channel functions as a WT channel and enables the specific detection of cell surface Kv7.3 channels.
Figure 1.
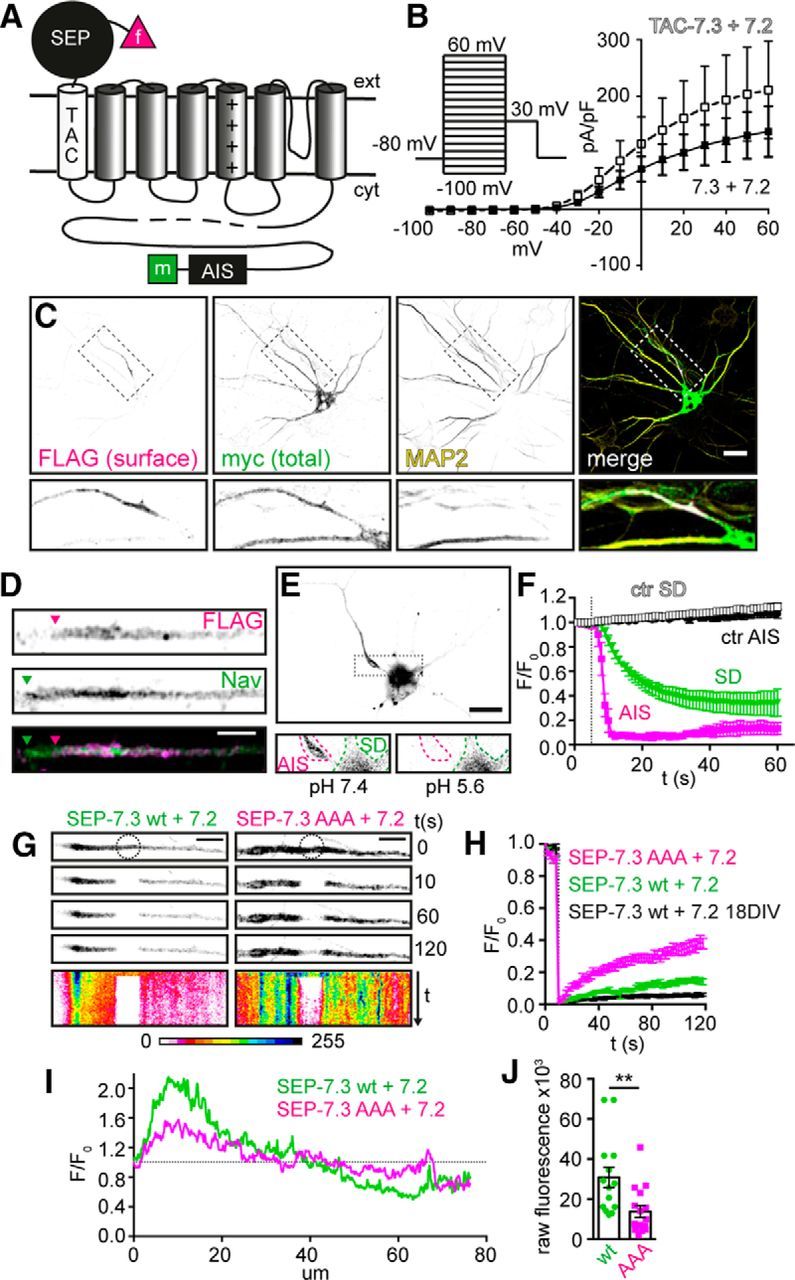
Adding an extra phluorin-tagged helix to Kv7.3 enables live-cell imaging of Kv7.2/7.3 at the AIS. A, Illustration of SEP–TAC-7.3 showing its extra helix (TAC; white) and extracellular SEP (black). f, FLAG tag; m, myc tag. B, I–V plot from CHO cells transfected with TAC-7.3 and Kv7.2 (white squares) or Kv7.3 and Kv7.2 (black squares). Presented as mean ± SEM, n = 7–9. C, 10 DIV hippocampal neuron transfected with TAC-7.3 and Kv7.2. Surface TAC-7.3 was stained with FLAG antibody before permeabilization (FLAG) and total TAC-7.3 with myc antibody after (total). Dendrites were stained with MAP2 antibody. D, Cell surface TAC-7.3 (FLAG) and endogenous Nav AIS expression in a 10 DIV neuron. Arrowheads indicate start positions. E, Fluorescence from a neuron expressing SEP–TAC-7.3 and Kv7.2 one frame before (left) and right after (right) a pH decrease from 7.4 to 5.6. SD, Somatodendritic compartment. F, Time course of SEP fluorescence at the AIS and SD during pH decrease or control (ctr) experiments. Cells were imaged at 1 Hz for 1 min and pH was decreased to 5.6 at 5 s. Presented as mean ± SEM, n = 10–15 cells. G, Representative FRAP experiments at the AIS of 10 DIV hippocampal neurons transfected with Kv7.2 and SEP–TAC-7.3 (left) or SEP–TAC-7.3 AAA (an ankG-binding domain mutant; right). Cells were imaged at 0.5 Hz for 2 min and bleached (circle) after five images. Kymographs (line scans through the AIS, projected over time) are shown at the bottom in 16-bit color look-up table. H, Quantification of FRAP experiments. SEP fluorescence in the bleach region was normalized to that of the first frame and is presented as mean ± SEM, n = 17. I, Mean intensity of SEP–TAC-7.3 WT (n = 9) and AAA (n = 11) axonal line scans. Normalized to the fluorescence at the AIS start. J, Raw AIS fluorescence of SEP–TAC-7.3 WT- and AAA-transfected neurons. Individual values and mean ± SEM are shown. **p < 0.01. Scale bars: C, E, 20 μm. D, G, 5 μm.
To be able to study the surface dynamics of Kv7.3, we fused a SEP to the N terminus of TAC-7.3 (Fig. 1A). This pH-sensitive GFP-variant fluoresces at neutral pH but is quenched at acidic pH. Thus, in our construct (SEP–TAC-7.3), SEP fluorescence originates from cell surface channels as the acidic environment of the endosomal system quenches that of intracellular channels. Importantly, changes in SEP fluorescence therefore reflect changes in the number of SEP–TAC-7.3 channels at the cell surface.
To test SEP–TAC-7.3, we coexpressed it with Kv7.2 and live-imaged transfected neurons using wide-field fluorescence microscopy. At neutral pH, SEP fluorescence was restricted predominantly to a single neurite and displayed a clear AIS-enriched profile similar to that of TAC-7.3 in fixed cells (Fig. 1E). Lowering the pH to 5.6 confirmed that the AIS fluorescence originated from cell surface-expressed channels only as it was instantly and completely quenched (Fig. 1E,F). In some cells, we also detected fluorescence in the somatodendritic compartment. However, this fluorescence disappeared slowly after decreasing the pH, indicating that it likely arises from accumulated intracellular channels (Fig. 1E,F).
We first used SEP–TAC-7.3 to examine the lateral mobility of AIS Kv7.2/7.3 heteromers using FRAP. Intriguingly, FRAP in cells expressing SEP–TAC-7.3 and Kv7.2 was very low within the recovery period in both young (10 DIV) and mature (18 DIV) neurons (Fig. 1G,H). This is likely attributable to the strong binding to ankyrin G (ankG) because mutation of the C-terminal ankG-binding domain (AAA) markedly increased lateral mobility (Fig. 1G,H). This was also reflected in AIS enrichment and cell surface levels, which were significantly lower for the mutant (Fig. 1I,J). That the mutant is enriched at all may reflect presence of endogenous Kv7.3 in the heteromers or contribution of ankG binding by Kv7.2. Thus, at steady state, AIS Kv7.2/7.3 WT heteromers are exceptionally stable and AIS enriched via binding to ankG.
We next asked whether short-term changes in neuronal activity modulate the number, lateral mobility, or localization of SEP–TAC-7.3 as observed for other CNS channels. To do so, we exposed neurons to depolarizing conditions (55 mm K+), 5 μm glutamate, or 100 μm GABA for 10 min and recorded images before and after treatment. In all conditions, the localization of SEP–TAC-7.3 did not change (Fig. 2A). Moreover, we only observed small or no variations in the fluorescence intensity (Fig. 2A,B), indicating that the number of channels essentially did not change during this period. This was also the case after 25 min (data not shown), although we observed a decrease in fluorescence in some glutamate-exposed neurons (see below). Finally, we found that the lateral mobility of SEP–TAC-7.3 in treated cells did not differ from untreated controls (Fig. 2C). Collectively, these data indicate that Kv7.2/7.3 heteromers at the AIS are exceptionally stable during short but relatively large changes in neuronal activity.
Figure 2.

Kv7.2/7.3 are highly stable at steady state and during neuronal stimulation but endocytose at high glutamate loads. A, Representative AIS images from neurons expressing SEP–TAC-7.3 and Kv7.2 before and after 10 min stimulation with 55 mm KCl, 5 μm Glu, or 100 μm GABA. B, Quantification of stimulation experiments. The number of cells included is indicated. *p < 0.05. C, Quantification of FRAP experiments performed in control (ctr), Glu, or GABA stimulated neurons. Presented as mean ± SEM, n = 17. D, Representative experiment (left) and quantification (right) of AIS SEP fluorescence in SEP–TAC-7.3- and Kv7.2- expressing 10 and 18 DIV neurons during 10 min treatment with 30 μm glutamate and 1 μm glycine (Glu/Gly). ***p < 0.0001. E, Illustration of SEP–TAC (left). Neurons expressing SEP–TAC (green) and mTagBFP2 (cell fill dye, white) were imaged at different pH (top; ensuring that SEP–TAC is cell surface expressed) and before/after Glu/Gly treatment (bottom). Quantification of Glu/Gly experiments (right). F, Representative images (left) and quantification (right) of SEP–TAC-7.3 fluorescence in Glu/Gly stimulated neurons pretreated with DMSO, 80 μm dynasore (dyna), or 100 μm chlorpromazine (cpz). ***p < 0.0001. G, Time course of SEP–TAC-7.3 fluorescence in representative control and Glu/Gly exposed neurons. Cells were imaged at 0.2 Hz for 10 min. The magneta- and blue-stapled lines indicate the rate shift. H, Left, Electrophysiological measurements of Kv7.2/7.3 currents in neurons expressing SEP–TAC-7.3 and Kv7.2 before (black trace) and after (magneta) Glu/Gly treatment. Middle, Top, Time course of tail current amplitude measured with 20 s trial intervals. Black and magneta arrows indicate traces shown to the left. Middle, Bottom, Changes in membrane conductance calculated with a 5 mV voltage-depolarizing step in the beginning of the voltage protocol. Right, Variations of average tail current amplitude before and after Glu/Gly exposure (n = 20 trial for each condition for each cell). I, PI staining of 10 DIV neurons 24 h after Glu/Gly (bottom) or sham (top) treatment. Differential interference contrast (DIC) images are shown left. Scale bars: A, D, F, 5 μm; E, 20 μm; I, 50 μm.
As mentioned, we occasionally observed that longer exposures to glutamate resulted in SEP fluorescence decreases, suggesting that, during sustained and high glutamate loads, Kv7.2/7.3 might be downregulated actively. To test this, we treated neurons with 30 μm glutamate and 1 μm glycine (Glu/Gly), a previously used combination in trafficking experiments (Muir et al., 2010). Indeed, 10 min after exposure, the SEP–TAC-7.3 fluorescence was decreased markedly in both 10 and 18 DIV neurons (Fig. 2D; F/F0, 0.49 ± 0.03 and 0.57 ± 0.05, respectively). This was not attributable to a decrease in extracellular pH, because the fluorescence in neurons expressing cell surface SEP–TAC (i.e., a single transmembrane helix coupled to an extracellular SEP) was not reduced after the same treatment (Fig. 2E, bottom). To verify that the fluorescence decrease was attributable to channel endocytosis, we pretreated neurons with dynasore (80 μm) or chlorpromazine (100 μm) before Glu/Gly treatment to inhibit dynamin- or clathrin-dependent endocytosis, respectively. Indeed, both pretreatments significantly reduced the decrease in SEP fluorescence on Glu/Gly exposure (Fig. 2F; dynasore F/F0, 0.84 ± 0.04; chlorpromazine F/F0, 0.64 ± 0.03), confirming that, on sustained glutamate exposure, Kv7.2/7.3 channels are endocytosed, which requires dynamin and clathrin-coated pits. We next examined the time course of Kv7.2/7.3 endocytosis using time-lapse imaging of SEP–TAC-7.3-expressing neurons. In most cells, the decrease in SEP fluorescence was biphasic in that it was relatively slow within the first 4–5 min after Glu/Gly addition (Fig. 2G, magneta line) and subsequently accelerated (Fig. 2G, blue line).
To test whether the endocytosis also resulted in a concomitant decrease in Kv7.2/7.3 current, we performed whole-cell patch clamp in neurons expressing SEP–TAC-7.3 and Kv7.2. Transfected neurons, identified by SEP fluorescence, were depolarized from −70 to −20 and then −40 mV (Fig. 2H, left). This evoked a stable slowly decaying outward tail current characteristic for Kv7-mediated M currents (Fig. 2H, middle). In agreement with our imaging data, a 3–5 min exposure to Glu/Gly induced fluctuations in the membrane conductance (Fig. 2H, middle) concomitant with a significant decrease of the tail current (Fig. 2H, right; n = 4; p < 0.0001 for each cell considered individually, Wilcoxon's signed-rank test).
The input conductance was stable >30 min after treatment, ensuring that the neurons were healthy during and a period after treatment. However, we found that the endocytosis was irreversible (data not shown), indicating that it is an excitotoxic event. To test this, we stained neurons 24 h after treatment with PI. Indeed, treated neurons were stained by PI and dissolved, whereas sham treated were not (Fig. 2I).
We next used a pharmacological approach to examine the molecular mechanism that regulates the Kv7.2/7.3 endocytosis. Interestingly, inhibiting NMDARs with 100 μm APV before Glu/Gly treatment completely blocked endocytosis (Fig. 3A). This was also the case for the NR2B-specific blocker ifenprodil (50 μm; Fig. 3A), indicating that the endocytosis is initiated by sustained activation of NR2B-containing NMDARs. Whereas Glu/Gly treatment in Ca2+-free medium also failed to initiate endocytosis, demonstrating that it requires influx of extracellular Ca2+, blocking P2X7 receptors with Brilliant Blue G (100 nm) did not (Fig. 3A). L- and T-type voltage-gated calcium channels (VGCCs) have been shown to mediate the slow depolarization-induced movement of AIS (Grubb and Burrone, 2010). However, in our case, inhibition of L-type (100 μm nisoldipine), R/T-type (50 μm Ni2+), or N/P/Q-type (50 nm conotoxin MVIIC) VGCCs all failed to inhibit the endocytosis (Fig. 3B). We next sought to determine which calcium-activated effectors are involved in the glutamate-induced endocytosis. Calcineurin/PP2B has been shown to drive the distal movement of AIS during long-term depolarization (Evans et al., 2013). To examine whether it contributes to the endocytosis of Kv7.2/7.3, we pretreated SEP–TAC-7.3-expressing neurons with 1 μm CsA before Glu/Gly exposure. However, inhibiting calcineurin did not block the endocytosis (Fig. 3C). This was also the case if we inhibited the phosphatases PP1 and PP2A (50 nm okadaic acid) or CaMKII kinase (10 μm KN-93; Fig. 3C). The protease calpain is responsible for proteolysis of the AIS cytoskeleton during ischemic insults (Schafer et al., 2009). Intriguingly, blocking this protease (100 μm MDL-28170) significantly reduced the endocytosis on glutamate exposure (Fig. 3C; F/F0, 0.80 ± 0.03). Thus, this suggests that activation of calpain by NMDAR-mediated calcium influx contributes to the rapid endocytosis of Kv7.2/7.3 from the AIS. Whereas Glu/Gly exposure did not change the position of AIS proteins within the treatment period (Fig. 3E), we asked whether the endocytosis of Kv7.2/7.3 is specific or part of a bulk mechanism. To answer this, we measured the fate of another ankG-bound and a non-ankG-bound AIS protein during treatment. In the first case, we developed a Nav reporter by fusing the rNav1.2 II–III linker to SEP–TAC. This construct trafficked to the AIS and, intriguingly, also endocytosed on Glu/Gly treatment (Fig. 3D). In the latter, we used an SEP-tagged GABAAR α2-subunit and imaged GABAAR clusters in dendrites and at the AIS, the latter of which was identified by live labeling with a Neurofascin antibody (Fig. 3F). GABAARs at dendritic synapses undergo declustering during NMDAR activation (Muir et al., 2010), and we also observed a pronounced dynasore-resistant decrease in SEP fluorescence during Glu/Gly treatment (Fig. 3F). Interestingly, the decrease was much less pronounced at the AIS compared with dendrites and, most importantly, was not affected by pretreatment with dynasore (Fig. 3F). This shows that, as opposed to SEP–TAC-7.3 and SEP–TAC–Nav1.2, AIS GABAARs are not endocytosed during Glu/Gly exposure but decluster instead.
Figure 3.

Kv7.2/7.3 endocytosis requires activation of NR2B-containing NMDARs, Ca2+ influx, and calpain activity. A, Representative images (left) and quantification (right) of SEP–TAC-7.3 AIS fluorescence in Glu/Gly stimulated neurons pretreated with 100 μm APV, 50 μm ifenprodil (Ifp), 100 nm Brilliant Blue G (BBG), or imaged in Ca2+-deficient aCSF (no Ca2+). ***p < 0.0001. B, Quantification of SEP–TAC-7.3 AIS fluorescence in Glu/Gly stimulated neurons pretreated with DMSO, 100 μm nisoldipine (nisol; L-type VGCC blocker), 50 μm Ni2+ (R-/T-type VGCC blocker), or 100 nm conotoxin MVIIC (ctx MVIIC; N/P/Q-type VGCC blocker). C, Quantification of SEP–TAC-7.3 AIS fluorescence in Glu/Gly stimulated neurons pretreated with DMSO, 100 μm CsA (calcineurin inhibitor), 50 nm okadaic acid (PP1/PP2A inhibitor), 10 μm KN93 (CaMKII kinase inhibitor), or 100 μm MDL-28170 (MDL; calpain inhibitor). **p < 0.001. D, Representative images (middle) and quantification (right) of SEP–TAC-Nav1.2 II–III AIS fluorescence in Glu/Gly stimulated neurons. E, Representative AIS images of ankG-, Neurofascin- (NF), or Nav-stained control or Glu/Gly-treated neurons. F, Left, 10 DIV hippocampal neuron expressing SEP-tagged GABAAR α2 subunit. Soma and neurites are outlined based on a DIC image. Middle, GABAAR clusters at the AIS and a dendrite before and after Glu/Gly exposure. The AIS was identified by Neurofascin live labeling (NF, bottom). Right, Quantification of AIS and dendritic GABAAR declustering in neurons pretreated with DMSO or 80 μm dynasore (dyna). Presented as mean ± SEM. ***p < 0.0001. Scale bars, 5 μm.
Discussion
Short-term activity-dependent changes of ion channel localization or cell surface numbers are emerging as an important mechanism to modulate neuronal signaling. Mechanistically, this depends on changes in lateral mobility or exocytosis/endocytosis. It has been reported for several ligand-gated channels, as well as Kv channels (Kim et al., 2007). However, this does not seem to apply to Kv7.2/7.3. First, we observed a very low rate of lateral diffusion at steady state as a result of the strong binding to ankG (Fig. 1H). Second, stimulating neurons with Glu or GABA did not change the diffusion rate of Kv7.2/7.3 (Fig. 2C). Collectively, this indicates that the ankG binding of Kv7.2/7.3, and hence the lateral mobility, is not dynamically altered on the short term. In line with this, we did not observe any change in localization or cell surface numbers of Kv7.2/7.3 on different short-term stimuli (Fig. 2B). This is opposed to long-term (i.e., days) depolarization in which the AIS start site and/or length are altered in a cell type-dependent manner (Grubb and Burrone, 2010).
We did observe a change during sustained activation of NMDARs in the form of a rapid endocytosis of Kv7.2/7.3 (Fig. 2D). This is an excitotoxic response, because it is irreversible and all neurons were dead 24 h after Glu/Gly treatment (Fig. 2I). Furthermore, the molecular mechanism shares significant features with AIS degradation in models of ischemia, including Ca2+-mediated calpain activation (Schafer et al., 2009). Of note, the endocytosis is selective, because AIS GABAARs were not endocytosed but rather declustered as opposed to Kv7.2/7.3 and Nav1.2, which did as indicated by our reporter constructs (Figs. 2, 3). Because it was shown previously that ankG-anchored Neurofascin also is endocytosed after large Ca2+ influxes (Schafer et al., 2009), it is conceivable that only ankG-bound proteins are endocytosed rapidly during excitotoxicity. Because these include the core machinery necessary for action potential generation, this may serve as a mechanism to prevent additional glutamate release.
In conclusion, our work has shed new light on the dynamics of Kv7.2/7.3 heteromers at the AIS. We anticipate that our approach is applicable to other Kv channel subfamilies and of value there.
Footnotes
This study was funded by grants from the Danish Council for Independent Research (T.B.-J.), the Lundbeck Foundation (T.B.-J., S.P.O., H.B.R., J.-F.P.), and the Danish Arrythmia Research Center (S.P.O., H.B.R., J.-F.P.). Dr. Joseph Kittler (University College London, London, UK) is thanked for providing SEP-tagged α2–GABAAR. Dr. Kenneth Lindegaard Madsen (University of Copenhagen, Copenhagen, Denmark) is thanked for providing the TAC segment. We acknowledge the Core Facility for Integrated Microscopy, Faculty of Health and Medical Sciences, University of Copenhagen.
The authors declare no competing financial interests.
References
- Evans MD, Sammons RP, Lebron S, Dumitrescu AS, Watkins TB, Uebele VN, Renger JJ, Grubb MS. Calcineurin signaling mediates activity-dependent relocation of the axon initial segment. J Neurosci. 2013;33:6950–6963. doi: 10.1523/JNEUROSCI.0277-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grubb MS, Burrone J. Activity-dependent relocation of the axon initial segment fine-tunes neuronal excitability. Nature. 2010;465:1070–1074. doi: 10.1038/nature09160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshi N, Zhang JS, Omaki M, Takeuchi T, Yokoyama S, Wanaverbecq N, Langeberg LK, Yoneda Y, Scott JD, Brown DA, Higashida H. AKAP150 signaling complex promotes suppression of the M-current by muscarinic agonists. Nat Neurosci. 2003;6:564–571. doi: 10.1038/nn1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Jung SC, Clemens AM, Petralia RS, Hoffman DA. Regulation of dendritic excitability by activity-dependent trafficking of the A-type K+ channel subunit Kv4.2 in hippocampal neurons. Neuron. 2007;54:933–947. doi: 10.1016/j.neuron.2007.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malenka RC. Synaptic plasticity and AMPA receptor trafficking. Ann N Y Acad Sci. 2003;1003:1–11. doi: 10.1196/annals.1300.001. [DOI] [PubMed] [Google Scholar]
- Misonou H, Mohapatra DP, Park EW, Leung V, Zhen D, Misonou K, Anderson AE, Trimmer JS. Regulation of ion channel localization and phosphorylation by neuronal activity. Nat Neurosci. 2004;7:711–718. doi: 10.1038/nn1260. [DOI] [PubMed] [Google Scholar]
- Muir J, Arancibia-Carcamo IL, MacAskill AF, Smith KR, Griffin LD, Kittler JT. NMDA receptors regulate GABAA receptor lateral mobility and clustering at inhibitory synapses through serine 327 on the γ2 subunit. Proc Natl Acad Sci U S A. 2010;107:16679–16684. doi: 10.1073/pnas.1000589107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan Z, Kao T, Horvath Z, Lemos J, Sul JY, Cranstoun SD, Bennett V, Scherer SS, Cooper EC. A common ankyrin-G-based mechanism retains KCNQ and NaV channels at electrically active domains of the axon. J Neurosci. 2006;26:2599–2613. doi: 10.1523/JNEUROSCI.4314-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen HB, Frøkjaer-Jensen C, Jensen CS, Jensen HS, Jørgensen NK, Misonou H, Trimmer JS, Olesen SP, Schmitt N. Requirement of subunit co-assembly and ankyrin-G for M-channel localization at the axon initial segment. J Cell Sci. 2007;120:953–963. doi: 10.1242/jcs.03396. [DOI] [PubMed] [Google Scholar]
- Schafer DP, Jha S, Liu F, Akella T, McCullough LD, Rasband MN. Disruption of the axon initial segment cytoskeleton is a new mechanism for neuronal injury. J Neurosci. 2009;29:13242–13254. doi: 10.1523/JNEUROSCI.3376-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder BC, Kubisch C, Stein V, Jentsch TJ. Moderate loss of function of cyclic-AMP-modulated KCNQ2/KCNQ3 K+ channels causes epilepsy. Nature. 1998;396:687–690. doi: 10.1038/25367. [DOI] [PubMed] [Google Scholar]
- Shah MM, Migliore M, Valencia I, Cooper EC, Brown DA. Functional significance of axonal Kv7 channels in hippocampal pyramidal neurons. Proc Natl Acad Sci U S A. 2008;105:7869–7874. doi: 10.1073/pnas.0802805105. [DOI] [PMC free article] [PubMed] [Google Scholar]