

Abstract
Copper is an essential element in biological systems. Its potent redox activity renders it necessary for life, but at the same time, misregulation of its cellular pools can lead to oxidative stress implicated in aging and various disease states. Copper is commonly thought of as a static cofactor buried in protein active sites; however, evidence of a more loosely bound, labile pool of copper has emerged. To help identify and understand new roles for dynamic copper pools in biology, we have developed selective molecular imaging agents for this metal, drawing inspiration from both biological binding motifs and synthetic model complexes that reveal thioether coordination as a general design strategy for selective and sensitive copper recognition. In this review, we summarize some contributions, primarily from our own laboratory, on fluorescence- and magnetic resonance-based molecular-imaging probes for studying copper in living systems using thioether coordination chemistry.
Keywords: copper, fluorescent probes, molecular imaging, MR probes, thioether coordination
1. Introduction
Copper is an essential element for life.[1] Owing to its potent redox activity, the participation of this metal as a catalytic and structural cofactor in enzymes that function in biological processes spanning energy generation, oxygen transport, cellular metabolism, and signal transduction renders it vital for the life of eukaryotic organisms.[2] However, this same redox promiscuity, when mis-regulated, can also lead to aberrant generation of reactive oxygen species (ROS) that have been linked to aging and different disease states, including genetic disorders like Menkes[3–5] and Wilson’s[6–8] diseases, neurodegenerative diseases like Alzheimer’s,[9–12] Parkinson’s,[13] and Huntington’s[14] diseases, and metabolic disorders such as diabetes and obesity.[15–17] For this reason, copper and other redox-active metals have been canonically thought of as buried cofactors within enzyme active sites and part of a tightly bound metal pool. However, a more loosely bound pool, termed the labile pool, in which copper and other metals can dynamically exchange with ligands on timescales commensurate with signaling, has been observed. This metal pool, in which rapid ligand exchange and changes in metal concentration gradients can mediate signaling processes, has been extensively studied in regard to redox-inactive alkali, alkaline earth and transition metals, such as calcium, sodium, potassium, and zinc. Nonetheless, recent work from our lab and others has revealed that despite its high reactivity, dynamic copper fluxes are observed in important physiological processes, such as neuronal calcium signaling pathways,[18] spontaneous activity in neural circuits,[19] metabolic processes such as lipolysis,[20] and the activation of the mitogen-activated protein (MAP) kinase pathway relevant in normal physiology and in oncogenic serine/threonine-protein kinase B-Raf (BRAF) signaling and tumorogenesis.[21,22]
Against this backdrop, molecular imaging provides a versatile approach that can be used to monitor labile metal fluxes in real time with spatial and temporal resolution.[23–25] In combination with complementary biochemical and genetic methods, as well as direct readouts of total metal content, small-molecule probes with different readout signals, including fluorescence, colorimetric, and magnetic resonance (MR) relaxivity modalities, can be used effectively as pilot screening tools for quickly assessing metal status in different physiological states. As such, the development of metal-selective small-molecule probes provides a potentially powerful toolbox that allows for the mapping of labile metal pools and the study of the roles of these dynamic fluxes. In this review, we focus on the use of thioether coordination chemistry as a general design strategy for achieving copper-selective probes for molecular imaging – optical and magnetic resonance imaging, as an illustrative example of opportunities at the interface between inorganic chemistry and biology.
2. Bioinspired Strategies for Selective Copper Recognition
Owing to environmental heterogeneities in biological systems, the development of small-molecule probes for biological use is inherently challenging. Because transition metals such as copper are far less abundant than their biologically relevant alkali/alkaline earth counterparts, selectivity poses one of the main challenges in probe design. Additionally for copper, redox specificity is required, as it can exist in two major oxidation states, Cu(I) and Cu(II). In the cellular setting, most of the copper that is undergoing dynamic exchange is in the Cu(I) state, owing to the reducing intracellular environment (average potential of – 0.25 V)[26] buffered in part by the thiol-containing low-molecular weight peptide glutathione (GSH). Preceding import across the plasma membrane, extracellular Cu(II) is reduced to the Cu(I) state by an insufficiently understood mechanism that presumably involves membrane reductases,[27] after which intracellular accumulation of the metal ion is achieved primarily by the high-affinity Cu(I)-specific channel copper transporter 1 (CTR1). The prevalence of the intracellular Cu(I) state is further evidenced by the identification of Cu(I)-specific metallochaperones such as Atox-1,[28] which delivers Cu(I) to the p-type ATPases ATP7A/B, the copper chaperone for superoxide dismutase (CCS),[29,30] and the various Sco[31] and Cox[32] copper chaperones for mitochondrial copper pools to metallate enzymes such as cytochrome c oxidase (CcO). The specificity of this emerging class of proteins for copper binding is crucial for maintaining cellular homeostasis of this metal.
2.1. Metal and Ligand Considerations for Copper Coordination
Inspection of biological copper sites can give us insight into its preferred modes of binding. Coordination in protein copper-binding sites is typically dominated by histidine, cysteine, and methionine residues. The makeup of each ligand set depends on the specific role, copper transport or enzymatic activity, of the binding site. However, the general use of these amino-acid side chains can be viewed as also governed by the hard and soft acids and bases (HSAB) principle.[33,34] In line with this theory, soft acid Cu(I) and borderline Cu(II) will preferentially bind ligands with borderline to soft basicities, respectively, leading to a favored use of amino-acid residues with nitrogen and sulfur donor atoms. Additionally, the number of donors, as well as the coordination geometry, can also impart a degree of selectivity between the possible metal oxidation states for these binding sites. Even though Cu(I) does not have a geometric preference based on a lack of ligand field stabilization energy (LFSE), it is often found in low-coordinate systems with 2, 3, or 4 ligands in linear, trigonal planar, or tetrahedral geometries. Conversely, LFSE for Cu(II) results in higher coordinate (4, 5, or 6 ligands) systems with square planar and square pyramidal geometries.
Moreover, the chemical identity of the ligating groups can also lead to favored oxidation states for copper in the binding site. Coordination environments including histidine and cysteine residues are pH-dependent, as deprotonation of the imidazole (pKa~6 and 14) and thiol (pKa~8.5) side chains lead to anionic donors. While the chemical identity of the coordinating groups and their arrangement in a particular three-dimensional geometry confer a degree of specificity for metal binding, it is important to remember that proteins also control selectivity of their binding sites by additional secondary sphere coordination effects that can further alter the local protein environment upon metal binding. For example, in the electron-transfer enzyme Cu,Zn-superoxide dismutase (SOD1), the bridging histidine residue between the Cu and Zn sites has an altered pKa, causing the imidazolate to form at physiological pH. (Type II center, Figure 1).
Figure 1.

Examples of copper-containing sites in Nature.
Furthermore, the ability of cysteines to form the thiolate renders them prone to oxidation, which can be correlated to a prevalence of cysteine-rich copper sites in reducing cellular compartments in comparison with more oxidizing intracellular locales. In contrast, the thioether side chain in methionine provides a neutral donor with pH-independent coordination, as well as less susceptibility to oxidation. This inability to be ionized, in combination with a longer side chain, also gives methionine a slight hydrophobic nature.
2.2. Biological Systems Reveal Methionine as an Ideal Ligand for Cu(I) Binding
The use of sulfur-based ligands enhances the selectivity for the cuprous over the cupric state, as shown by higher Cu(II) affinity when including nitrogen donors in synthetic copper ligands.[35] However, the use of the sulfur donor cysteine over methionine (or vice versa) in cuproprotein binding sites can be associated with that specific protein’s chemical role and surrounding biological environment. Cysteine copper binding is pH-dependent and susceptible to redox activity, but these same residues achieve tighter binding to the metal than methionine, owing to their anionic charge. In contrast, methionine binds with weaker affinities than the cysteine thiolate, but at the same time, varying the number of methionine ligands can tune the affinity of the binding site. Additionally, methionine-rich sites provide a unique environment that stabilizes Cu(I) coordination, as well as excellent selectivity over other biologically-relevant cations like Zn(II) and Fe(II).[36,37]
As a result, methionine has emerged as an important ligand for copper in biological systems. Copper centers involving methionine residues are prevalent in systems ranging from electron-transfer proteins to copper trafficking proteins. Even though most S-donors in electron-transfer protein copper sites are thiolate-based, there are a few examples of methionine copper interactions. Cupredoxins, such as plastocyanin and azurin, show the typical 2-histidine 1-cysteine copper coordination in a trigonal planar geometry. Additionally, these blue copper proteins possess a weakly bound methionine thioether sulfur in the axial position[38,39] (Figure 1). Other electron-transfer proteins with methionine binding within their active sites include Cytochrome c oxidase (CcO), which has a methio-nine ligand in its mixed valence CuA center.[40] As with electron-transfer proteins, the copper trafficking counterparts also show prevalent cysteine-thiolate coordination to copper instead of the thioether moiety. Bis-cysteinate Cu(I) coordination is a hallmark of cytosolic copper ligation, with many transport proteins exhibiting a CXXC binding motif (Figure 1). Among the copper sensing and trafficking proteins that exhibit this motif are the eukaryotic copper chaperones CCS[29,30,41,42] and Atox1,[43–47] as well as the Cu(I)-binding P-type ATPases ATP7A/B.[48–50] In comparison with cytosolic Cu(I) binding, which occurs in a very reducing environment, transport of extracellular copper into the cell is highly associated with methionine thioether binding. The homotrimeric copper transporter CTR1 contains a cell surface-located N-terminal region with histidine and methionine-rich domains (Mets motif) that are implicated in copper transport across the membrane[51–53] (Figure 1). Methionine motifs (MX3M) in the transmembrane region are also important in shuttling copper down the pore of the protein.[51,54]
Perhaps the best-characterized methionine-copper coordination chemistry is that of protein networks involved in copper resistance, predominantly found in less redox-balanced spaces of prokaryotic organisms. To relieve the cytosol of excess copper, bacteria export the metal using the ATPase CopA, which binds copper in a similar fashion as ATP7A/B.[55] For gram-negative bacteria, this detoxifying process is not sufficient, as there is still a need to manage the copper that accumulates in the periplasm. A canonical example of periplasmic, copper-handling machinery is the Cus pathway, an export system that is composed of three proteins, CusA, CusB, and CusC.[56,57] The CusA and CusB proteins possess three-coordinate methionine-only binding sites[57,58] (Figure 1), whereas CusC does not appear to have metal-binding features as observed by crystallographic experiments.[59] In contrast, CusF, a copper chaperone for CusB,[60] binds copper in a 2-methionine, 1-histidine binding site with a tryptophan residue that provides a cation-π interaction with the metal[61,62] (Figure 1). In the relatively less studied Pco and Cop copper resistance pathways, where proteins are encoded by plasmid DNA in comparison with chromosomal DNA, methionine coordination is observed in the Cu(I) binding sites of PcoC and CopC, but not in their separate Cu(II) sites that mainly offer N and O donors as ligands.[63–67] Copper-handling systems like the Cus pathway, as well as the Pco/Cop copper resistance pathways, are characterized by higher-coordinate Cu(I) complexes (three or four ligands), when compared with the cysteine-dominated environments of the cytosolic copper proteins. This situation is in part due to the need to compensate for the weaker Cu(I)-binding affinity of thioethers, in comparison with thiolates.
2.3. Synthetic Compounds have Exploited Thioether Donors for Copper Coordination
The ability of thioethers to coordinate to a variety of metals, and the effect of their moderate π-acidity[68] on the stabilization of low metal oxidation states have been historically important in motivating the study of thioether-metal complexes. The need for homoleptic thioether complexes, in order to evaluate the effect of thioether ligation on the electronic structure of metal ions, was satisfied by the development of crown thioethers. In seminal work, Cooper and coworkers studied the synthesis, conformational analysis and metal coordination of macrocycles including the 9S3, 12S3, 18S6, and 24S6 variations[69] (Figure 2). Extensive efforts on the investigation of different metal complexes of crown thioethers (including nickel, cobalt, copper, ruthenium, rhodium, and silver) indicate that thioethers present a marked preference for the lower, “softer” metal oxidation states. This behavior manifests itself in the redox properties of the complexes, as well as in their magnetic and EPR features.[70]
Figure 2.

Metal-binding (a) crown thioethers; and (b) mixed-donor macrocyclic ligands, with NxS4−x and NxS5−x donor sets.
Another important avenue for motivation of the study of thioether-metal complexation arose from the previous speculation (since confirmed)[71,72] of methionine copper coordination in blue copper proteins. In efforts to elucidate the origin of the unexpected redox, optical, and EPR properties of these copper centers, Rorabacher and coworkers carried out an impressive set of studies on the copper complexes of macrocyclic tetra- and pentadentate thioethers, in which they showed that coordination to thioethers shifts the Cu(II/I) potential to more positive values, enhances the rate of Cu(II/I) redox self-exchange kinetics, and generates unusually intense optical bands.[73,74] Furthermore, studies on mixed-donor macro-cyclic ligands with NxS4−x and NxS5−x donor sets[35,75] (Figure 2) established important trends in copper coordination: 1) stability constants for Cu(II) complexes increase by 5–6 orders of magnitude for each replacement of a thioether sulfur donor by an amine nitrogen donor atom; and 2) the more positive Cu(II/I) redox potential of macrocyclic donors with sulfur donors, compared with aliphatic nitrogen atoms, is credited to a destabilization of the Cu(II) oxidation state, rather than a stabilization of the Cu(I) state. Additional studies of tripodal ligands with mixed nitrogen and sulfur donors to investigate the effect of binding geometry on electron-transfer kinetics of Cu(II/I) systems[76,77] provided further insight on copper- thioether coordination, demonstrating a narrow distribution of the stability constants of Cu(I) complexes, in comparison with the Cu(II) counterparts, and a noticeable effect of the geometry on the Cu(II) complexes. Interestingly, the compatibility of these ligands for copper sensing has been established with regard to their better stability towards oxidation, in comparison with thiolate ligands, and in that the reported Kd values of these complexes fall within the window of 10−11 M (GSH) to 10−18 M (SOD1) for biological Cu(I) ligands.
3. Fluorescence-based Copper(I) Sensing Probes
Inspired by Nature’s use of thioether ligation, as well as the aforementioned work on copper coordination chemistry, we and others have extensively employed thioether receptor motifs to develop a series of recognition-based fluorescent probes for the detection of Cu(I).[78–80] Using this strategy, a fluorophore reporter is coupled to a chelator (receptor) that is specific for the metal of interest. Once bound to the analyte, a change in the optical properties of the sensor is observed, which can be reversed upon analyte dissociation. For a more in-depth discussion on the mechanisms and modes of small-molecule fluorescence sensing of metal cations, we refer to the following review articles.[81,82]
Selectivity of the receptor for the metal of interest can be achieved by careful design of the ligand using fundamental principles of coordination chemistry. Considering some of the basics of copper homeostasis (e.g., sulfur coordination, low coordination number), one can envision a few possible strategies for building receptors that can chelate Cu(I) with good selectivity and competitive binding constants. The common use of a photoinduced electron transfer (PeT) quenching mechanism as a turn-on/off switch for metal sensing sets up a particular stipulation for the receptor moiety; a substituted amine is customarily used as the PeT switch due to its lone-pair interaction with the cationic analyte.[82–84] In PeT-based fluorescent sensors, two important entities, the electron donor (the substituted amine) and the electron acceptor (the excited-state fluorophore), interact to produce an optical response upon analyte binding. In the unbound state, photoexcitation of the system leads to a charge-separated state in which the highest occupied molecular orbital (HOMO) of the donor lies in an energetically favorable state to transfer an electron to the excited-state fluorophore and hence quench the fluorescence relaxation pathway of this state. Upon analyte binding, the energy level of the HOMO of the electron-rich donor is lowered, causing electron transfer to be less thermodynamically favored, alleviating PeT, and thus restoring fluorescence in this state.
Combining what has been learned from the studies of macrocyclic and aliphatic NxS4−x and NxS5−x copper complexes, as well as the characterization of numerous copper-binding sites involved in copper homeostasis, we reason that thioether-rich ligands can satisfy the requirement for amine coordination needed for PeT modulation by metal binding while maintaining selectivity for Cu(I), and as a result, can be used as the receptors of choice for synthetic copper sensors.
3.1. Use of a Thioether-rich Receptor (NS4) for Cu+ Sensing: Design of a First-generation Cu+ Sensor
Use of a boron dipyrromethene (BODIPY) fluorophore coupled to an azatetrathia receptor (termed NS4’) gave rise to Coppersensor 1 (CS1),[85] the first Cu+-responsive probe with visible excitation and emission profiles (Figure 3). The NS4’ receptor, an acyclic analog to the Cu(I)-binding tetrathiaza crown ether used in the important first small-molecule fluorescent sensor for Cu(I), CTAP-1,[86] binds the metal ion to produce a selective response. CS1 features a 10-fold increase in probe fluorescence upon metal binding with Kd=4×10−12 M. (Figure 4) Additionally, the dye responds to exogenous CuCl2 addition to HEK 293T cells in confocal imaging experiments, where the fluorescence enhancement can be reversed by the addition of a membrane-permeable Cu(I) chelator. The ability of CS1 to reversibly detect labile copper in this system was validated by an independent study;[87] however, its use to detect copper changes upon supplementation with CuCl2 or Cu(gtsm) (Cu(II)-glyoxalbis(N4-methyl-3-thiosemicarbazonato)) in other mammalian cell lines, including CHO, M17, U87Mg, and SHSY5Y, proved unsuccessful. These observations point to the fact that there is no one-size-fits-all chemical probe for every biological model, and therefore characterization of the sensor in its intended biological or environmental system of use is necessary. Indeed, when used in combination with complementary techniques, such as direct metal detection via inductively coupled plasma mass spectrometry (ICP-MS) and biochemical assays like DNA microarrays and protein profiling, CS1 has been applied to study copper dynamics in a wide range of bacterial,[88] plant,[89] and yeast[90–92] models of copper accumulation and misregulation.
Figure 3.

Cu(I)-selective fluorescent probes bearing thioether receptors.
Figure 4.

Selective Cu(I) recognition by CS1 and turn-on response in probe fluorescence.
3.2. Design of More Sensitive Next-generation Sensors for Detection of Endogenous Levels of Cu+ in Living Cells and Tissue
Application of the NS4’ receptor to different fluorophore platforms has yielded next-generation probes that allow the detection of endogenous copper levels in different biological models. For example, replacement of the fluorine atoms in the BODIPY core with the more electron-rich methoxy substituents afforded Coppersensor 3 (CS3)[18] (Figure 3). Increasing the electron density at the fluorophore (PeT acceptor component) resulted in a turnon response enhancement (75-fold for CS3 vs. 10-fold for CS1), as well as higher quantum yield (Φ=0.40 for CS3 vs. Φ =0.13 for CS1). These improvements, in combination with a tighter binding affinity for Cu(I) (Kd=9× 10−14 M), allowed for the visualization of endogenous levels of labile copper in HEK 293T that could be depleted by treatment with the cell-impermeable chelator bathocuproine disulfonate (BCS). Additionally, CS3 was able to report on neuronal copper translocation upon depolarization with KCl. The biological interpretation suggested by these pilot experiments was confirmed by X-ray fluorescence microscopy (XFM) of analogous fixed samples, which directly showed that copper moves from somatic cell bodies to peripheral dendritic processes upon stimulation. (Figure 5A).
Figure 5.
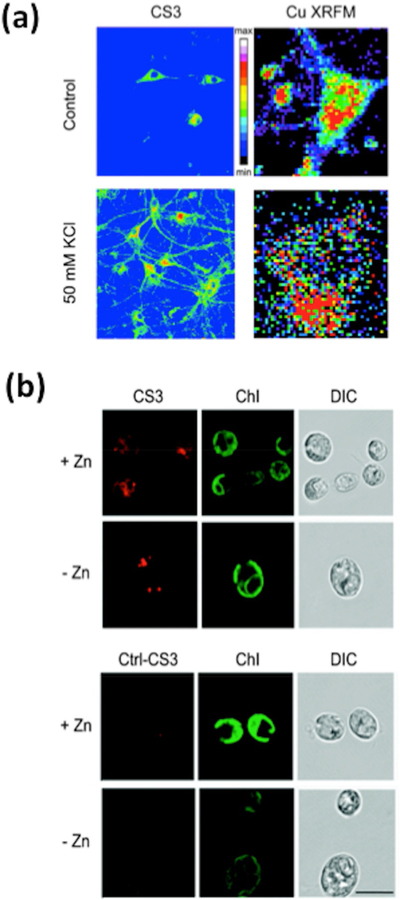
(a) Labile copper pool detection in rat hippocampal neuron cells with CS3 and XFM. (b) Monitoring copper accumulation in zinc-limited and zinc-replete wild-type Chlamydomonas rein-hardtii (CC-4532) cells by using CS3 and Ctrl-CS3.
Additionally, work from others has exploited the use of CS3 as a complementary tool to study copper in other biological models. Among these studies are included the evaluation of CTR2 functions,[93,94] the use of the probe to track the efficiency of copper depletion or supplementation treatments,[95] and the implementation of the dye in screening assays to study the endogenous ATP7A-transport of copper into lysosomes as a response to elevation in copper levels.[96] More recently, a comprehensive suite of techniques, which were pursued after observations with the CS3 probe, revealed a new organelle termed the “cuprosome” that acts as storage for reversible sequestration of copper in response to zinc deficiency in the green algae Chlamydomonas reinhardtii.[97] The development and further use of Ctrl-CS3, a matched control dye to CS3 that is unable to bind copper due to the replacement of the thioether motifs with methylene units (Figure 3), allowed for the assessment of fluorescent hot spots that were observed with CS3 (but not with the control probe) in higher frequency in zinc-deficient cells than in control cells (Figure 5B). In addition, CS3 (but not Ctrl-CS3) fluorescence was diminished upon copper chelation. Direct metal-analysis techniques like Nano-SIMS and X-ray absorption spectroscopy, in combination with supplementary biochemical assays, were used to confirm the existence of these copper traps and suggest that the apparent reversible sequestration of copper in these compartments may play a role in minimizing protein mismetallation during zinc deficiency while maintaining copper stores for future use.
Despite the utility of the BODIPY-based probes in various biological models, the use of this fluorophore scaffold has been limited in other applications. Its relatively high hydrophobicity has led to restricted implementation of the probes to study more complex systems, such as tissue and cell types with differential hydrophilicities due to uneven distribution and staining, as well as limited photostability, particularly in the methoxy-substituted BODIPY forms, has hindered their use for prolonged imaging experiments. In efforts to improve the properties of these reagents, we recently developed a series of Cu(I)-responsive probes based on the rhodol scaffold. This hybrid fluorescein-rhodamine fluorophore shows high tunability in visible excitation/emission profiles and signal-to-noise responses, and high optical brightness, as well as improved hydrophilicity and photostability.[98] Indeed, the use of different amine substituents on the xanthone core gave rise to the Copper Rhodol (CR) family.[19] Of the five initial sensors, Copper Rhodol 3 (CR3) was the best performing, with a 13-fold turn-on response to Cu(I) in vitro (Figure 3). Further modification to the pendant aryl ring by substitution of the ortho methyl, with a bulkier trifluoromethyl analog, afforded Copper Fluor 3 (CF3, Figure 3). This CH3 to CF3 modification introduces two advantages: 1) non-radiative decay pathways by rotations about the aryl-aryl bond that decrease fluorescence quantum yield are diminished; and 2) reduced electron density in the aryl ring (PeT donor component) favors PeT quenching of the unbound probe. Consequently, CF3 exhibits an improved 40-fold fluorescence enhancement upon Cu(I) binding with a dissociation constant Kd=3×10−13 M. The more hydrophilic nature of CR3 and CF3, compared with CS3, was confirmed by measurements of apparent octanol/water distribution coefficients (logD values of 0.96, 1.15, and 3.46, respectively, where larger values indicate greater hydrophobicity).
The improved properties of the CF3 probe allowed its use for assessing labile Cu(I) dynamics in dissociated hippocampal neuronal cultures and retinal tissue by one- and two-photon microscopy. Molecular imaging of CF3 and the non-Cu(I)-responsive Ctrl-CF3 analog on dissociated hippocampal neurons and mouse retinal tissue that had been acutely treated with the copper chelator BCS showed decreased fluorescence for CF3, but an unchanged signal for the control dye (Figure 6). After observing the presence of a labile copper pool in both systems, we sought to probe the effects of pharmacological (chelation with BCS) and genetic (CTR1 knockout) alteration of these copper pools on spontaneous activity, a fundamental property of developing neural circuits. In both cases, disruption of copper homeostasis resulted in increases in event frequency and the fraction of cells involved in spontaneous correlated activity, confirmed by calcium imaging. Taken together, the results identify a fundamental physiological role for copper in neural function. We have recently expanded this work to red-shifted versions that enable real-time monitoring of copper fluxes in other cell types, such as adipocytes, and these chemical probes have helped identify an essential role for copper in lipolysis, a major metabolic process in the body for burning fat.[20]
Figure 6.

Two-photon imaging of: (a) CF3; and (b) Ctrl-CF3 in hippocampal neurons in the absence and presence of a copper chelator BCS.
Finally, an added challenge for fluorescent probe design lies in the development of sensors with specific localization to assess exclusive pools of metal in subcellular compartments. To address this issue, further modifications to the BODIPY core afforded Mito-CS1,[99] a mitochondrially-targetable probe that makes use of a triphenyl phosphonium targeting group for specific subcellular localization (Figure 3).[100] Mito-CS1 features a 10-fold turn-on response upon Cu(I) ligation with Kd=7×10−12 M, localizes to the mitochondria in HEK 293T cells and patient fibroblasts, and responds well in cellulo in copper supplementation and chelation experiments. In combination with genetic models of copper misregulation and direct bulk metal measurements, the use of Mito-CS1 helped establish the concept of a prioritization of mitochondrial copper homeostasis over other cellular compartments, even in situations of systemic copper deficiency and mitochondrial metallochaperone malfunction. Specifically, molecular-imaging experiments with patient-derived fibroblasts with mutations in the copper exporter ATP7A showed a relative increase in probe fluorescence that was confirmed by inductively coupled plasma optical emission spectroscopy (ICP-OES). Patient fibroblasts with mutations in mitochondrial copper metallochaper-ones SCO1 and SCO2 showed no appreciable difference, compared with control fibroblasts in Mito-CS1 signal or ICP-OES measurements on isolated mitochondria, but showed decreased total copper levels vs. wild-type measured by ICP-OES (Figure 7). Combining these complementary techniques, Mito-CS1 imaging and ICP-OES results indicate that mitochondrial copper homeostasis is prioritized over other compartments, even in the course of overall copper deficiency, presumably to preserve CcO and SOD activity.
Figure 7.

Live-cell molecular imaging of mitochondrial copper homeostasis in: (a) control; (b) ATP7a; (c) SCO1; and (d) SCO2 patient fibro-blasts with Mito-CS1. (e) Mean fluorescence intensities of (a)–(d). Measured total copper level in: (f) patient fibroblasts; and (g) mitochondria.
3.3. Use of an NS3 Receptor for Sensing Labile Copper Pools in Living Animals
Owing to the poor tissue-penetrating ability of visible light and the desire to apply Cu(I)-responsive probes to study thicker specimens like tissue and whole animals, efforts to develop sensors with longer-wavelength/near-infrared (NIR) excitation profiles have been applied. We combined an NS3 modified thioether receptor platform first reported by Fahrni[101] to the NIR fluorophore scaffold cyanine 7 to produce Coppersensor 790 (CS790)[102] (Figure 3). CS790 has a selective 15-fold turn-on response to Cu(I) with 760 nm excitation and a 790 nm emission profile. Capping of the carboxylates with acetoxymethyl esters to aid in cell permeability and retention afforded CS790AM, which showed increases in fluorescence signal in HEK 293T cells that were pretreated with CuCl2, compared with the control, as well as a reversal of this fluorescence enhancement when the copper chelator tris[2-(ethylthio)ethyl]amine (NS3) was added. The probe was further capable of reporting changes in live hairless SKH-1 mice upon copper supplementation with CuCl2 and chelation with ATN-224, the choline salt of tetrathiomolyb-date currently in development as a treatment for Wilson’s disease,[103] marking the first example of live-mouse copper imaging with a fluorescent sensor (Figure 8). Furthermore, CS790AM was used to monitor copper levels in Atp7b−/− mice, a murine model that is metabolically and phenotypically similar to Wilson’s disease due to the inactivation of the ATP7B gene and subsequent anomalous accumulation of copper in several tissues.[6] Live-animal imaging showed increased fluorescence in the livers of mutant mice relative to those from wild-type mice, which could be reversed with copper chelator treatment. The collective results point to a path forward for the application of fluorescent copper probes to study physiology, disease diagnosis, and monitor treatment of copper imbalance in whole animals.
Figure 8.

(a) Live SKH-1 mice imaging with fluorescent CS790AM probe. 1: vehicle only; 2: vehicle and CS790AM; 3: vehicle, CuCl2, and CS790AM; 4: CuCl2, ATN-224, and CS790AM; and 5: vehicle, ATN-224, and CS790AM. (b) Calculated total photon flux from each mouse, 5 minutes after the injection of probe.
4. Magnetic Resonance-based Copper-sensing Probes
Whereas metal-responsive fluorescent sensors provide a great tool to assess relative metal levels in living biological systems from the cell to the tissue to the organism level, the translational application to medicine can be limited by the intrinsic properties of the use of visible to near-infrared light as a readout for metal activity. In this regard, magnetic resonance imaging (MRI) is a powerful, clinically-used molecular-imaging technique that allows for the capture of three-dimensional images of organisms with up to cellular resolution.[104] Even though the most common contrast observed in MR images results from water molecules in different environments, the use of contrast agents with paramagnetic metals, such as Mn2+, Mn3+, Fe3+, Cu2+, and Gd3+, can be implemented to enhance image contrast. Of these paramagnetic metals, high spin Gd3+ is particularly well suited for this application and is indeed used in 40–50% of all clinical MRI applications.[105] The image enhancing abilities of coordination complexes of these metals relies on the ability to efficiently relax nearby nuclei and increase the relaxation rates of water protons.[106] Modulation of the relaxivity, the efficiency of a contrast agent to enhance the relaxation rate, by a specific analyte can therefore be applied to a sensing strategy.
The design of metal-responsive MR probes can implement the idea of relaxivity modulation to afford a toolbox that can be useful in studying complex contributions of metals to physiology and disease. There are a number of ways to affect the degree of relaxivity in a contrast agent sensor, including the number of inner-sphere water molecules (q-modulation) as well as rotational tumbling time (τR-modulation). For a more in-depth discussion on the development and criteria for responsive MRI contrast agents as chemical sensors, we refer the reader to the following review articles.[107–109]
4.1. Development of a MR-contrast Agent for Selective Copper(II) Sensing
Inspired by the seminal work of Meade and coworkers in the development of EGad, a “smart” contrast agent that reports on β-galactosidase activity,[110,111] our laboratory reported the first copper-responsive MR-sensor Copper-Gad 1 (CG1)[112] (Figure 9). CG1 features a Gd3+ contrast agent platform coupled to an iminodiacetate ligand for Cu2+ binding, the prevalent oxidation state in extracellular fluids. In the absence of Cu2+, the anionic carboxylate donors of the receptor hinder inner-sphere water access to the Gd3+ thereby minimizing relaxivity. Upon binding to Cu2+, the Gd3+ is less sterically hindered, leading to increased inner-sphere water access and proton relaxivity affording a turn-on response. CG1 presents a 41 % increase in relaxivity upon selective binding of Cu2+ with Kd=167±48 μM. We note that while binding was mostly selective to Cu2+, the turn-on response was partially muted in the presence of 10-fold excess Zn2+. Nevertheless, the response to Cu2+ was stronger, owing to its higher affinity on the Irving-Williams series, and provided a prototype candidate for prospective MR-based copper imaging in living systems.
Figure 9.

Copper selective MR-contrast agents with thioether receptors.
4.2. Design of MR-contrast Agents with Thioether-rich Receptors for Copper (I) Sensing
Following the initial results from the CG1 probe, we sought to improve upon the relatively modest sensitivity and change in relaxivity, as well as selectivity, versus Zn2+. Additionally, we designed a new family of copper-responsive MR probes to be activated by Cu+ and/or Cu2+ to potentially track copper in its two biologically relevant oxidation states. To achieve this goal, we introduced various thioether-rich receptors to the Gd3+ contrast agent scaffold through a 2,6-dimethylpyridine linker to afford Copper-Gad probes 2–6 (CG2–CG6, Figure 9).[113] The pyridyl linker acts as a coordination switch, where in the absence of analyte, the apo-receptor caps the Gd3+ center to minimize the interaction with inner-sphere water molecules, thus lowering the proton relaxivity. Upon analyte binding, the linker switches to a conformation that lowers the steric bulk around the Gd3+, allowing for inner-sphere access of water and producing higher relaxivity. Introduction of thioether donors shifted the selectivity towards Cu+ binding in CG2–CG5, with relaxivity increases ranging from 92% to 360%, where higher turn-on responses correlate to higher S/N donor ratios. CG6 had a unique and equal response to Cu+ and Cu2+ due to a quick redox equilibration that favors the higher copper oxidation state, possibly due to the N/O/S donor set provided by the receptor. The composition and number of potential donors in the ligand set influences the binding affinities as well, where compounds with higher thioether coordination CG2 and CG3 presented higher Cu+ binding affinities, Kd=3.7×10−14 M and 2.6×10−13 M, respectively. In comparison, sensors with only three potential donors (CG4 and CG5) showed lower affinities to Cu+ (Kd=1.4×10−11 M and 3.2× 10−11 M, respectively). The tetradentate N/S/O donor system in CG6 afforded a higher affinity for Cu2+ (Kd=9.9×10−16 M) in comparison with the first-generation probe CG1. Additionally, to validate the potential use of these sensors for molecular-imaging applications, we established that CG complexes are capable of visualizing changes in biologically relevant copper levels in T1 phantom images at clinical field strengths.
We next evaluated the effects of anions on relaxivity changes for the CG probes. Relaxivity increases upon copper binding seen in the presence of phosphate anions are unaffected; however, carboxylate-type anions, including citrate, lactate, and carbonate, presumably bind to the Gd3+ contrast agent core and yield significantly lower increases in relaxivity upon analyte binding. To minimize anion sensitivity, we designed Copper-Gad 7 (CG7),[114] an analog to CG2 where the acetate groups on the Gd3+ contrast agent core are substituted by hexanedioate derivatives to shield the paramagnetic metal from biologically abundant anions by steric and electrostatic action (Figure 9). This modification led to a comparable relaxivity turn-on ratio to CG2 (340% for CG7 vs. 360% for CG2), as well as an unperturbed selective binding of Cu+ over other biologically relevant cations in the N2S2 receptor. However, the Cu+ response for CG7 is not significantly affected by carboxylate or phosphate anions, revealing that the installation of peripheral carboxylate functionalities improves anion compatibility for MR-based copper sensing.
4.3. Design of a Copper-responsive MR Agent for Biological Imaging
As the CG series of MR-based probes features excellent in vitro properties, we were eager to explore their use in living systems, namely in in cellulo applications. However, due to the large size of contrast agent probes (like other typical Gd3+-containing MR contrast agents such as Gd-DTPA and Gd-DOTA), we reasoned and confirmed that the Cu+-responsive CG probes have poor cell membrane permeability and are largely confined to the extracellular space. To circumvent this issue, we introduced a polyargi-nine tag on the CG2 scaffold to improve cellular uptake of the probe. Synthesis of Arg8CG2 resulted in a cell-permeable copper-responsive MR-based probe with comparable spectroscopic properties and response to Cu+ that allowed for the visualization of biological perturbations of copper levels in a murine Menkes disease model cell line.[115] Indeed, Arg8CG2 showed greater cellular uptake compared with CG2 in HEK 293T cells as confirmed by ICP-MS studies of the lysed cells. Superior in cellulo relaxivity of Arg8CG2 over CG2 was also established by copper supplementation and chelation experiments where HEK 293T cells treated with either CuCl2 or the copper chelator BCS and later treated with probe (Arg8CG2 or CG2) showed more pronounced differences in relaxivity (increases or decreases in the case of supplementation or chelation respectively) for Arg8CG2. Relaxivity measurements of the Menkes model fibroblast line WG1005, which bears a mutation in the Atp7a gene, compared with control fibroblasts MCH58, showed clear differences between the two cell lines, as reported by Arg8CG2. Furthermore, phantom imaging experiments of the two cell lines treated with copper or copper chelator resulted in clear contrast variations, pointing to the applicability of the MR-based probe for in vivo imaging (Figure 10) and representing a unique application for copper-responsive MRI.
Figure 10.

(a) Structure of the Arg8CG2 construct. (b) Cellular uptake of CG2 (black) and Arg8CG2 (gray) in HEK 293T cells at different dosing concentrations. Cells were lysed with RIPA buffer after probe incubation and analyzed by ICP-MS. (c) Relaxivity measurements of CG2 and Arg8CG2 in HEK 293T cells. Bars represent the incubation of cells with control vehicle (black), copper (light gray), or BCS (dark grey). (d) T1-weighed phantom images of MCH58 and WG1005 cells incubated with CG2 or Arg8CG2.
5. Concluding Remarks
The use of thioether coordination chemistry for the selective ligation of copper has been exploited by biology and synthetic chemists alike. Structural and spectroscopic characterization of cuproprotein binding sites, in combination with the study of synthetic small-molecule model complexes, has revealed the privileged nature of the thioether moiety, via methionine coordination, as an important strategy for pH-independent and oxidation-resistant binding of the cuprous ion. Consequently, this type of binding has been extensively implemented in the development of copper-specific recognition moieties for molecular-imaging applications. Indeed, the diverse array of small-molecule fluorescent copper sensors with varying properties (visible to NIR-excitation profiles, signal-to-noise contrast, hydrophilicity, etc.) has led to the identification of novel biological roles of copper as a signaling entity, in addition to its canonical roles as a static cofactor within protein active sites. Furthermore, the use of thioether coordination of copper in different imaging modalities with more translatable potential, such as MRI, has pointed the way towards possible applications for disease diagnosis and treatment monitoring.
Despite the success of thioether coordination of copper as a strategy for selective metal sensing, the structural variety of the receptor moieties has been rather limited to the use of macrocyclic or aliphatic amines containing thioether donors. As such, apparent binding affinities of the resulting sensors, which can be narrowly altered by inclusion or modification of different reporter moieties (i.e., fluorophores), leave room for future innovation. Indeed, development of new ligands to explore the effects of binding geometry and ligand topology/architecture on the affinity and selective binding of copper in biological systems is a worthwhile goal. The resulting expansion of the chemical toolbox of molecular probes will undoubtedly lead to new opportunities to discover and study new biology of copper and other metals.
Acknowledgements
We thank the National Institutes of Health (GM 79465) for supporting our work on metal imaging probes. K. M. R.-T. was partially supported by a Chemical Biology Training Grant from the NIH (T32 GM066698). S. K. thanks TUBITAK for postdoctoral support. C. J. C. is an Investigator of the Howard Hughes Medical Institute.
Biographies

Karla Ramos-Torres graduated from the University of Puerto Rico, Río Piedras in 2011 with a B.S. in Chemistry. She is currently a Ph.D. candidate at UC Berkeley, where she works in the laboratory of Prof. Chris Chang. Her current research interests include the development of molecular probes for the detection of biological copper. Outside of research, Karla enjoys playing volleyball, hiking, and dancing.

Safacan Kolemen is a postdoctoral researcher in the laboratory of Prof. Chris Chang at UC Berkeley. His work focuses on development of pH-sensitive and copper-selective molecular probes. He received his Ph.D. in 2014 from Bilkent University, Turkey, where he worked under the supervision of Prof. Engin U. Akkaya on designing photosensitizers for photodynamic therapy applications.

Chris Chang is the Class of 1942 Chair Professor in the Departments of Chemistry and Molecular and Cell Biology at UC Berkeley, Howard Hughes Medical Institute Investigator, and Faculty Scientist in the Chemical Sciences Division of Lawrence Berkeley National Laboratory. He is a Senior Editor of ACS Central Science. Chris received his B.S. and M.S. from Caltech in 1997, working with Harry Gray, studied as a Fulbright scholar in Strasbourg, France with Jean-Pierre Sauvage, and received his Ph.D. from MIT in 2002 with Dan Nocera. After postdoctoral studies with Steve Lippard, Chris joined the UC Berkeley Faculty in 2004. His laboratory focuses on chemical biology and inorganic chemistry with particular interests in molecular imaging and catalysis applied to neuroscience and sustainable energy. His group’s work has been recognized by honors from the Dreyfus, Beckman, Sloan, and Packard Foundations, Amgen, Astra Zeneca, and Novartis, Technology Review, ACS (Cope Scholar, Eli Lilly, Nobel Laureate Signature, Baekeland), RSC (Transition Metal Chemistry), SBIC, and the Blavatnik Foundation.
Footnotes
We dedicate this submission to Harry Gray, who has been a teacher, mentor, friend, and inspiration to us all.
References
- [1].Lippard SJ, Berg JM, Principles of Bioinorganic Chemistry, University Science Books, Mill Valley, CA, 1994. [Google Scholar]
- [2].Tapiero H, Townsend DM, Tew KD, Biomed. Pharmac-other. 2003, 57, 386–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Vulpe C, Levinson B, Whitney S, Packman S, Gitschier J, Nat. Genet 1993, 3, 7–13. [DOI] [PubMed] [Google Scholar]
- [4].Kaler SG, Nat. Rev. Neurol 2011, 7, 15–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].La Fontaine S, Mercer JF, Arch. Biochem. Biophys 2007, 463, 149–167. [DOI] [PubMed] [Google Scholar]
- [6].Lutsenko S, Biochem. Soc. Trans 2008, 36, 1233–1238. [DOI] [PubMed] [Google Scholar]
- [7].Huster D, Hoppert M, Lutsenko S, Zinke J, Lehmann C, Mossner J, Berr F, Caca K, Gastroenterology 2003, 124, 335–345. [DOI] [PubMed] [Google Scholar]
- [8].Huster D, Ann. N. Y. Acad. Sci 2014, 1314, 37–44. [DOI] [PubMed] [Google Scholar]
- [9].Ayton S, Lei P, Bush AI, Free Radical Biol. Med 2013, 62, 76–89. [DOI] [PubMed] [Google Scholar]
- [10].Barnham KJ, Masters CL, Bush AI, Nat. Rev. Drug Discovery 2004, 3, 205–214. [DOI] [PubMed] [Google Scholar]
- [11].Savelieff MG, Lee S, Liu Y, Lim MH, ACS Chem. Biol 2013, 8, 856–865. [DOI] [PubMed] [Google Scholar]
- [12].Matlack KE, Tardiff DF, Narayan P, Hamamichi S, Caldwell KA, Caldwell GA, Lindquist S, Proc. Natl. Acad. Sci. U.S.A 2014, 111, 4013–4018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Vonk WI, Kakkar V, Bartuzi P, Jaarsma D, Berger R, Hofker MH, Klomp LW, Wijmenga C, Kampinga HH, van de Sluis B, PLoS One 2014, 9, e92408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Xiao G, Fan Q, Wang X, Zhou B, Proc. Natl. Acad. Sci. U.S.A. 2013, 110, 14995–15000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Burkhead JL, Lutsenko S, in Lipid Metabolism (Ed.: Baez RV), InTech, 2013, DOI: 10.5772/51819. [DOI] [Google Scholar]
- [16].Huster D, Purnat TD, Burkhead JL, Ralle M, Fiehn O, Stuckert F, Olson NE, Teupser D, Lutsenko S, J. Biol. Chem 2007, 282, 8343–8355. [DOI] [PubMed] [Google Scholar]
- [17].Huster D, Lutsenko S, Mol. BioSyst 2007, 3, 816–824. [DOI] [PubMed] [Google Scholar]
- [18].Dodani SC, Domaille DW, Nam CI, Miller EW, Finney LA, Vogt S, Chang CJ, Proc. Natl. Acad. Sci. U.S.A 2011, 108, 5980–5985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Dodani SC, Firl A, Chan J, Nam CI, Aron AT, Onak CS, Ramos-Torres KM, Paek J, Webster CM, Feller MB, Chang CJ, Proc. Natl. Acad. Sci. U.S.A 2014, 111, 16280–16285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Krishnamoorthy L, Cotruvo JAJ, Chan J, Kaluarachchi H, Muchenditsi A, Pendyala VS, Jia S, Aron AT, Vander Wal MN, Guan T, Smaga LP, Farhi SS, New EJ, Lutsenko S, Chang CJ, Nat. Chem. Biol 2016, DOI: 10.1038/nchembio.2098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Turski ML, Brady DC, Kim HJ, Kim BE, Nose Y, Counter CM, Winge DR, Thiele DJ, Mol. Cell. Biol 2012, 32, 1284–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Brady DC, Crowe MS, Turski ML, Hobbs GA, Yao X, Chaikuad A, Knapp S, Xiao K, Campbell SL, Thiele DJ, Counter CM, Nature 2014, 509, 492–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Que EL, Domaille DW, Chang CJ, Chem. Rev 2008, 108, 1517–1549. [DOI] [PubMed] [Google Scholar]
- [24].Domaille DW, Que EL, Chang CJ, Nat. Chem. Biol 2008, 4, 168–175. [DOI] [PubMed] [Google Scholar]
- [25].Chan J, Dodani SC, Chang CJ, Nat. Chem 2012, 4, 973–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Go YM, Jones DP, Biochim. Biophys. Acta 2008, 1780, 1273–1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Lutsenko S, Curr. Opin. Chem. Biol 2010, 14, 211–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Hatori Y, Lutsenko S, Antioxid. Redox Signaling 2013, 19, 945–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Culotta VC, Klomp LWJ, Strain J, Casareno RLB, Krems B, Gitlin JD, J. Biol. Chem 1997, 272, 23469–23472. [DOI] [PubMed] [Google Scholar]
- [30].Rae TD, Schmidt PJ, Pufahl RA, Culotta VC, O’Halloran TV, Science 1999, 284, 805–808. [DOI] [PubMed] [Google Scholar]
- [31].Leary SC, Cobine PA, Kaufman BA, Guercin GH, Mattman A, Palaty J, Lockitch G, Winge DR, Rustin P, Horvath R, Shoubridge EA, Cell Metab. 2007, 5, 9–20. [DOI] [PubMed] [Google Scholar]
- [32].Punter FA, Adams DL, Glerum DM, Hum. Genet 2000, 107, 69–74. [DOI] [PubMed] [Google Scholar]
- [33].Pearson RG, J. Am. Chem. Soc 1963, 85, 3533–3539. [Google Scholar]
- [34].Ahrland S, Chatt J, Davies NR, Q. Rev. Chem. Soc 1958, 12, 265–276. [Google Scholar]
- [35].Westerby BC, Juntunen KL, Leggett GH, Pett VB, Koenigbauer MJ, Purgett MD, Taschner MJ, Ochrymowycz LA, Rorabacher DB, Inorg. Chem 1991, 30, 2109–2120. [Google Scholar]
- [36].Reedijk J, J. Inorg. Biochem 2012, 115, 182–185. [DOI] [PubMed] [Google Scholar]
- [37].Davis AV, O’Halloran TV, Nat. Chem. Biol 2008, 4, 148–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Colman PM, Freeman HC, Guss JM, Murata M, Norris VA, Ramshaw JAM, Venkatappa MP, Nature 1978, 272, 319–324. [Google Scholar]
- [39].Nar H, Messerschmidt A, Huber R, Vandekamp M, Canters GW, J. Mol. Biol 1991, 221, 765–772. [DOI] [PubMed] [Google Scholar]
- [40].Tsukihara T, Aoyama H, Yamashita E, Tomizaki T, Yamaguchi H, Shinzawaitoh K, Nakashima R, Yaono R, Yoshikawa S, Science 1995, 269, 1069–1074. [DOI] [PubMed] [Google Scholar]
- [41].Lamb AL, Wernimont AK, Pufahl RA, O’Halloran TV, Rosenzweig AC, Biochemistry 2000, 39, 1589–1595. [DOI] [PubMed] [Google Scholar]
- [42].Lamb AL, Wernimont AK, Pufahl RA, Culotta VC, O’Halloran TV, Rosenzweig AC, Nat. Struct. Biol 1999, 6, 724–729. [DOI] [PubMed] [Google Scholar]
- [43].Wernimont AK, Huffman DL, Lamb AL, O’Halloran TV, Rosenzweig AC, Nat. Struct. Biol 2000, 7, 766–771. [DOI] [PubMed] [Google Scholar]
- [44].Anastassopoulou I, Banci L, Bertini I, Cantini F, Kat-sari E, Rosato A, Biochemistry 2004, 43, 13046–13053. [DOI] [PubMed] [Google Scholar]
- [45].Hamza I, Schaefer M, Klomp LWJ, Gitlin JD, Proc. Natl. Acad. Sci. U.S.A 1999, 96, 13363–13368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Larin D, Mekios C, Das K, Ross B, Yang AS, Gilliam TC, J. Biol. Chem 1999, 274, 28497–28504. [DOI] [PubMed] [Google Scholar]
- [47].Walker JM, Tsivkovskii R, Lutsenko S, J. Biol. Chem 2002, 277, 27953–27959. [DOI] [PubMed] [Google Scholar]
- [48].Banci L, Bertini I, Ciofi-Baffoni S, Huffman DL, O’Halloran TV, J. Biol. Chem 2001, 276, 8415–8426. [DOI] [PubMed] [Google Scholar]
- [49].Banci L, Bertini I, Del Conte R, D’Onofrio M, Rosato A, Biochemistry 2004, 43, 3396–3403. [DOI] [PubMed] [Google Scholar]
- [50].DeSilva TM, Veglia G, Opella SJ, Proteins Struct. Funct. Bioinf 2005, 61, 1038–1049. [DOI] [PubMed] [Google Scholar]
- [51].Puig S, Lee J, Lau M, Thiele DJ, J. Biol. Chem 2002, 277, 26021–26030. [DOI] [PubMed] [Google Scholar]
- [52].Rubino JT, Riggs-Gelasco P, Franz KJ, J. Biol. Inorg. Chem 2010, 15, 1033–1049. [DOI] [PubMed] [Google Scholar]
- [53].Pope CR, Flores AG, Kaplan JH, Unger VM, Curr. Top. Membr 2012, 69, 97–112. [DOI] [PubMed] [Google Scholar]
- [54].De Feo CJ, Aller SG, Siluvai GS, Blackburn NJ, Unger VM, Proc. Natl. Acad. Sci. U.S.A 2009, 106, 4237–4242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Rensing C, Fan B, Sharma R, Mitra B, Rosen BP, Proc. Natl. Acad. Sci. U.S.A 2000, 97, 652–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Paulsen IT, Park JH, Choi PS, Saier MH Jr., FEMS Microbiol. Lett 1997, 156, 1–8. [DOI] [PubMed] [Google Scholar]
- [57].Long F, Su CC, Zimmermann MT, Boyken SE, Rajashankar KR, Jernigan RL, Yu EW, Nature 2010, 467, 484–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Bagai I, Liu W, Rensing C, Blackburn NJ, McEvoy MM, J. Biol. Chem 2007, 282, 35695–35702. [DOI] [PubMed] [Google Scholar]
- [59].Kulathila R, Kulathila R, Indic M, van den Berg B, Plos One 2011, 6, e15610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Franke S, Grass G, Rensing C, Nies DH, J. Bacteriol. 2003, 185, 3804–3812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Loftin IR, Franke S, Blackburn NJ, McEvoy MM, Protein Sci 2007, 16, 2287–2293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Xue Y, Davis AV, Balakrishnan G, Stasser JP, Staehlin BM, Focia P, Spiro TG, Penner-Hahn JE, O’Halloran TV, Nat. Chem. Biol 2008, 4, 107–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Peariso K, Huffman DL, Penner-Hahn JE, O’Halloran TV, J. Am. Chem. Soc 2003, 125, 342–343. [DOI] [PubMed] [Google Scholar]
- [64].Arnesano F, Banci L, Bertini I, Mangani S, Thompsett AR, Proc. Natl. Acad. Sci. U.S.A 2003, 100, 3814–3819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Arnesano F, Banci L, Bertini I, Thompsett AR, Structure 2002, 10, 1337–1347. [DOI] [PubMed] [Google Scholar]
- [66].Arnesano F, Banci L, Bertini I, Felli IC, Luchinat C, Thompsett AR, J. Am. Chem. Soc 2003, 125, 7200–7208. [DOI] [PubMed] [Google Scholar]
- [67].Zhang L, Koay M, Maher MJ, Xiao Z, Wedd AG, J. Am. Chem. Soc 2006, 128, 5834–5850. [DOI] [PubMed] [Google Scholar]
- [68].Murray SG, Hartley FR, Chem. Rev 1981, 81, 365–414. [Google Scholar]
- [69].Cooper SR, Acc. Chem. Res 1988, 21, 141–146. [Google Scholar]
- [70].Cooper SR, Rawle SC, Struct. Bonding 1990, 72, 1–72. [Google Scholar]
- [71].Guss JM, Freeman HC, J. Mol. Biol 1983, 169, 521–563. [DOI] [PubMed] [Google Scholar]
- [72].Adman ET, Stenkamp RE, Sieker LC, Jensen LH, J. Mol. Biol 1978, 123, 35–47. [DOI] [PubMed] [Google Scholar]
- [73].Rorabacher DB, Martin MJ, Koenigbauer MJ, Malik M, Schroeder RR, Endicott JF, Ochrymowycz LA, in Copper Coordination Chemistry: Biochemical and Inorganic Perspectives (Eds.: Karlin KD, Zubieta J), Adenine Press, Guilderland, NY, 1983, p. 167. [Google Scholar]
- [74].Diaddario LL, Dockal ER, Glick MD, Ochrymowycz LA, Rorabacher DB, Inorg. Chem 1985, 24, 356–363. [Google Scholar]
- [75].Bernardo MM, Heeg MJ, Schroeder RR, Ochrymowycz LA, Rorabacher DB, Inorg. Chem 1992, 31, 191–198. [Google Scholar]
- [76].Ambundo EA, Ochrymowycz LA, Rorabacher DB, Inorg. Chem 2001, 40, 5133–5138. [DOI] [PubMed] [Google Scholar]
- [77].Ambundo EA, Yu QY, Ochrymowycz LA, Rorabacher DB, Inorg. Chem 2003, 42, 5267–5273. [DOI] [PubMed] [Google Scholar]
- [78].Cotruvo JA, Aron AT, Ramos-Torres KM, Chang CJ, Chem. Soc. Rev 2015, 44, 4400–4414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Aron AT, Ramos-Torres KM, Cotruvo JA, Chang CJ, Acc. Chem. Res 2015, 48, 2434–2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Fahrni CJ, Curr. Opin. Chem. Biol 2013, 17, 656–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].de Silva AP, Gunaratne HQN, Gunnlaugsson T, Huxley AJM, McCoy CP, Rademacher JT, Rice TE, Chem. Rev 1997, 97, 1515–1566. [DOI] [PubMed] [Google Scholar]
- [82].de Silva AP, Moody TS, Wright GD, Analyst 2009, 134, 2385–2393. [DOI] [PubMed] [Google Scholar]
- [83].Valeur B, Leray I, Coord. Chem. Rev 2000, 205, 3–40. [Google Scholar]
- [84].Chandross EA, Thomas HT, Chem. Phys. Lett 1971, 9, 393–396. [Google Scholar]
- [85].Zeng L, Miller EW, Pralle A, Isacoff EY, Chang CJ, J. Am. Chem. Soc 2006, 128, 10–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Yang L, McRae R, Henary MM, Patel R, Lai B, Vogt S, Fahrni CJ, Proc. Natl. Acad. Sci. U.S.A 2005, 102, 11179–11184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Price KA, Hickey JL, Xiao Z, Wedd AG, James SA, Liddell JR, Crouch PJ, White AR, Donnelly PS, Chem. Sci 2012, 3, 2748–2759. [Google Scholar]
- [88].Espirito Santo C, Lam EW, Elowsky CG, Quaranta D, Domaille DW, Chang CJ, Grass G, Appl. Environ. Microbiol 2011, 77, 794–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Bernal M, Casero D, Singh V, Wilson GT, Grande A, Yang H, Dodani SC, Pellegrini M, Huijser P, Connolly EL, Merchant SS, Kramer U, Plant Cell 2012, 24, 738–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Beaudoin J, Ioannoni R, López-Maury L, Bähler J, Ait-Mohand S, Guérin B, Dodani SC, Chang CJ, Labbé S, J. Biol. Chem 2011, 286, 34356–34372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Cusick KD, Minkin SC, Dodani SC, Chang CJ, Wilhelm SW, Sayler GS, Environ. Sci. Technol 2012, 46, 2959–2966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Quaranta D, Krans T, Espirito Santo C, Elowsky CG, Domaille DW, Chang CJ, Grass G, Appl. Environ. Microbiol 2011, 77, 416–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Ohrvik H, Nose Y, Wood LK, Kim BE, Gleber SC, Ralle M, Thiele DJ, Proc. Natl. Acad. Sci. U.S.A 2013, 110, E4279–E4288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Huang CP, Fofana M, Chan J, Chang CJ, Howell SB, Metallomics 2014, 6, 654–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Ding X, Xie H, Kang YJ, J. Nutr. Biochem 2011, 22, 301–310. [DOI] [PubMed] [Google Scholar]
- [96].Polishchuk EV, Concilli M, Iacobacci S, Chesi G, Pastore N, Piccolo P, Paladino S, Baldantoni D, van Ijzendoorn SC, Chan J, Chang CJ, Amoresano A, Pane F, Pucci P, Tarallo A, Parenti G, Brunetti-Pierri N, Settembre C, Ballabio A, Polishchuk RS, Dev. Cell 2014, 29, 686–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Hong-Hermesdorf A, Miethke M, Gallaher SD, Kropat J, Dodani SC, Chan J, Barupala D, Domaille DW, Shirasaki DI, Loo JA, Weber PK, Pett-Ridge J, Stemmler TL, Chang CJ, Merchant SS, Nat. Chem. Biol 2014, 10, 1034–1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Whitaker JE, Haugland RP, Ryan D, Hewitt PC, Haugland RP, Prendergast FG, Anal. Biochem 1992, 207, 267–279. [DOI] [PubMed] [Google Scholar]
- [99].Dodani SC, Leary SC, Cobine PA, Winge DR, Chang CJ, J. Am. Chem. Soc 2011, 133, 8606–8616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Murphy MP, Smith RA, Annu. Rev. Pharmacol. Toxicol 2007, 47, 629–656. [DOI] [PubMed] [Google Scholar]
- [101].Chaudhry AF, Verma M, Morgan MT, Henary MM, Siegel N, Hales JM, Perry JW, Fahrni CJ, J. Am. Chem. Soc 2010, 132, 737–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Hirayama T, Van de Bittner GC, Gray LW, Lutsenko S, Chang CJ, Proc. Natl. Acad. Sci. U.S.A 2012, 109, 2228–2233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Brewer GJ, Metallomics 2009, 1, 199–206. [DOI] [PubMed] [Google Scholar]
- [104].Kuhn W, Angew. Chem. Int. Ed. Engl 1990, 29, 1–19. [Google Scholar]
- [105].Raymond KN, Pierre VC, Bioconjugate Chem 2005, 16,3–8. [DOI] [PubMed] [Google Scholar]
- [106].Caravan P, Ellison JJ, McMurry TJ, Lauffer RB, Chem. Rev 1999, 99, 2293–2352. [DOI] [PubMed] [Google Scholar]
- [107].Yoo B, Pagel MD, Front. Biosci 2008, 13, 1733–1752. [DOI] [PubMed] [Google Scholar]
- [108].Major JL, Meade TJ, Acc. Chem. Res 2009, 42, 893–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Que EL, Chang CJ, Chem. Soc. Rev 2010, 39, 51–60. [DOI] [PubMed] [Google Scholar]
- [110].Moats RA, Fraser SE, Meade TJ, Angew. Chem. Int. Ed. Engl 1997, 36, 726–728. [Google Scholar]
- [111].Louie AY, Huber MM, Ahrens ET, Rothbacher U, Moats R, Jacobs RE, Fraser SE, Meade TJ, Nat. Biotechnol 2000, 18, 321–325. [DOI] [PubMed] [Google Scholar]
- [112].Que EL, Chang CJ, J. Am. Chem. Soc 2006, 128, 15942–15943. [DOI] [PubMed] [Google Scholar]
- [113].Que EL, Gianolio E, Baker SL, Wong AP, Aime S, Chang CJ, J. Am. Chem. Soc 2009, 131, 8527–8536. [DOI] [PubMed] [Google Scholar]
- [114].Que EL, Gianolio E, Baker SL, Aime S, Chang CJ, Dalton Trans 2010, 39, 469–476. [DOI] [PubMed] [Google Scholar]
- [115].Que EL, New EJ, Chang CJ, Chem. Sci 2012, 3, 1829–1834. [DOI] [PMC free article] [PubMed] [Google Scholar]