

Abstract
Metastasis, involving the spread of systemic cancer to the brain, results in neurologic disability and death. Current treatments are largely palliative in nature; improved therapeutic approaches represent an unmet clinical need. However, recent experimental and clinical advances challenge the bleak longterm outcome of this disease. Encompassing key recent findings in epidemiology, genetics, microenvironment, leptomeningeal disease, neurocognition, targeted therapy, immunotherapy, and prophylaxis, we review preclinical and clinical studies to provide a comprehensive picture of contemporary research and the management of secondary brain tumors.
Landmarks in an Emerging Landscape
The true prevalence of brain metastasis is not well known, but is probably higher than the available epidemiological estimates (8.5–9.6%) [1–3], and is certainly more frequent than any other primary tumor in the brain [1]. Upon diagnosis, cancer patients with disease disseminated to their brains continue to face a dismal prognosis, which includes higher morbidity, mortality [4], and a dramatic increase in the cost of their treatment [5]. Current therapies are largely palliative and fail to improve survival for the majority of patients. However, advances in several therapeutic modalities have effectively challenged the lethal status of brain metastasis for particular subsets of patients [6]. In parallel, mechanistic interrogation of experimental models of brain metastasis continues to increase our limited knowledge of the complex underlying biology. Translation of these observations has resulted in innovative management approaches.
Several targeted therapies given to subgroups of patients with brain metastasis harboring specific molecular alterations can control secondary brain tumors and improve their overall prognosis. However, most patients do not fall into these subgroups, and consequently alternative strategies are necessary to discover additional vulnerabilities of brain metastasis. An essential emerging concept proposes that brain metastases evolve in a distinct manner compared to extracranial disease. Cancer cell evolution within the microenvironment might be more accentuated in the brain than in other secondary organs because of the inhospitable nature of the nave organ for incoming cancer cells [7,8]. Identification of forces driving this evolution (i.e., components of the microenvironment) and consequences for cancer cells (i.e., genomic alterations that are selected in brain metastatic cells locally) might allow novel therapeutic approaches to be developed that could be applied to more patients. Those cancer cells able to adapt will grow and modify their surroundings, generating an environment favoring metastatic outgrowth. A brain damaged by established metastases is vulnerable to infiltration by extracranial cell types, including leucocytes. Tumor-infiltrating lymphocytes have been found to surround brain metastasis [9], and therapeutic approaches to activate the immune system against cancer cells in the brain are being used in patients with brain metastasis. If more novel and effective therapies continue to expand and apply to more patients, it is tempting to speculate that survival might be extended, as has been reported in melanoma patients with brain metastasis [10]. In this novel scenario, deterioration of cognition as a result of disease progression or unwanted effects of therapy would represent a growing concern.
In addition to considering metastases that are already established, targeting clinically silent infiltrating ‘seeds’ would prevent their occurrence. Preclinical models (Box 1) have proposed that key molecules and mechanisms are necessary to succeed during the initial steps of brain colonization. However, translation of experimental findings to the clinical arena constitutes a major bottleneck in preventing brain metastasis.
Box 1. Preclinical Models of Brain Metastasis.
Cell Une-Derived Xenotransp/ants (c/DX)
Cell lines are engineered with different reporters, including reporters that are compatible with non-invasive imaging (e.g., luciferase, Luc, for bioluminescence) and/or histology (e.g., GFP). Multiple models are available derived from the main cancer types that generate brain metastasis 0ung cancer, breast cancer, and melanoma). After intracardiac inoculation, brain metastases develop and mice reach the endpoint of the disease 0.e., extensive weight loss, neurological symptoms, other) 5–7 weeks later. The incidence of extracranial multiorgan metastasis is limited. Alternative inoculation sites include intracarotid and orthotopic; this latter must usually be combined with surgery to remove the tumor mass at the inoculation point to give enough time for brain metastasis to develop.
Cell Une-Derived Allotransplants
Syngeneic mouse cell lines tropic to the brain have been developed that represent the main cancer types. In contrast to clDX, these models reach the endpoint of the experiment 2–3 weeks after of intracardiac inoculation, and, even after multiple rounds of in vivo selection for increasing brain tropism, they tend to develop high extracranial multiorgan metastasis. In addition, these models can be implanted into GEMMs harboring alterations to study the contribution of different organs, cell types, pathways, or specific molecules to the process of brain metastasis and to study the interactions with host cells by the use of specific mouse reporter strains.
GEMMs that Develop Brain Metastasis
GEMMs in which brain metastases have been reported are a minority [43,173,174]. The time required to detect brain metastasis is very variable, usualy requiring many weeks and more frequently many months. None of the three models that develop brain metastasis have reporters in cancer cells, and thus they are not compatible with most advanced imaging approaches which might facilitate detection of brain seeding. This consideration might well be extended to many other models (particularly those that represent oncogenomic profiles of high incidence of brain metastasis; see Figure 7 in main text) in which brain metastases have neither been reported nor excluded.
The current landscape of brain metastasis combines clinical observations with the search for novel biology through the use of experimental models to radically expand treatment options. This multimodal, intellectually cooperative approach will result in concrete improvements in outcomes for patients with either parenchymal or leptomeningeal brain metastasis.
Epidemiology
According to population-based estimates, the incidence of brain metastases ranges from 8.3 to 14.3 per 100 000 [11]. However, many population-based studies of brain metastases predate modern imaging and treatments, and likely underestimate the true incidence. Because patients with cancer live longer due to earlier detection and/or better systemic therapies, the incidence of brain metastases is believed to be rising, although data supporting this assumption are sparse. One study based on patients in the Swedish National Cancer Registry found that patients diagnosed with breast cancer between 2004 and 2006 were at 44% increased risk of being admitted with brain metastasis compared to 1998–2000, suggesting that the incidence of brain metastasis among breast cancer patients has increased [12].
Incidence proportions of brain metastases vary by primary site, stage, and even subtype of cancer, among other factors [1,13]. One study of patients in the Detroit metropolitan area diagnosed with lung, melanoma, breast, renal, or colorectal cancer from 1973 to 2001 found that the incidence of brain metastases was highest for lung (19.9%), followed by melanoma (6.9%), renal (6.5%), breast (5.1%), and colorectal (1.8%) cancers [1]. Lung cancer patients are most likely to present with brain metastases at diagnosis: 15–26% of non-small cell lung cancer (NSCLC) patients and 24% of small-cell lung cancer (SCLC) patients presenting with metastatic disease will have brain metastases [14]. Within metastatic breast cancer patients, ~8% will have brain metastases at presentation, although the risk of brain metastases is highest in human epidermal growth factor receptor 2 (HER2)-positive and triple-negative metastatic breast cancers [15]. Indeed, up to half of HER2-positive metastatic breast cancer patients will develop brain metastases sometime in their disease course, and this can occur at any time, including many years after their initial diagnosis [16]. In melanoma, 20–30% of patients with metastatic melanoma present with brain metastases at diagnosis, and nearly 50% will develop brain metastases during their disease course [14,17]. Thus patients with HER2 breast cancer, triple-negative breast cancer, melanoma, SCLC, or non-squamous NSCLC have the highest risk of developing brain metastasis [18,19].
The prognosis of patients with brain metastases can also vary depending on the primary site and key prognostic factors. The graded prognostic assessment (GPA) is a prognostic index for brain metastasis patients refined with diagnosis-specific prognostic indices for breast carcinoma, SCLC, NSCLC, gastrointestinal cancers, melanoma, and renal cell carcinoma [20]. GPA takes into account patient age, the degree of functional impairment defined by the Karnofsky performance status (KPS). which groups patients into three groups from poor, <40, to good status, >80, co-occurrence of extracranial metastases, and number of brain metastases. Each parameter is scored with O (worst), 0.5, or 1 (best). What is notable is the wide array of outcomes for brain metastasis patients. For example, the median survival for a breast cancer patient with a low GPA score of O (KPS ≤50, estrogen receptor (ER)/progesterone receptor (PR)/human epidermal growth factor receptor 2 (HER2)-negative, multiple brain metastases, age ≥60 years) is approximately 3 months while that for a breast cancer patient with a high GPA of 4.0 (KPS 90–100, ER/PR/HER2-positive, single metastasis, age <60 years) is a little over 2 years.
Genomics
Comparative genomic characterization of primary tumors, extracranial metastasis, and brain metastasis has emphasized that brain metastases may require distinct targeted therapeutic approaches. Early research on brain metastasis experimental models noted elevated expression of genes specifically enriched in cell lines with increased brain tropism (Figure 1A,B) [21,22]. Some of these genes have been functionally validated and have been scored in human samples as providing an increased risk of brain relapse [8,21–28]. Although the molecular mechanisms leading to enriched brain tropism in experimental models have not been fully explained, the ability of the best fitted cancer cell clones to achieve adaptation to the brain microenvironment seems a plausible explanation [29] in the absence of recurrent mutations [30].
Figure 1. Preclinical Strategies To Study Brain Metastasis.
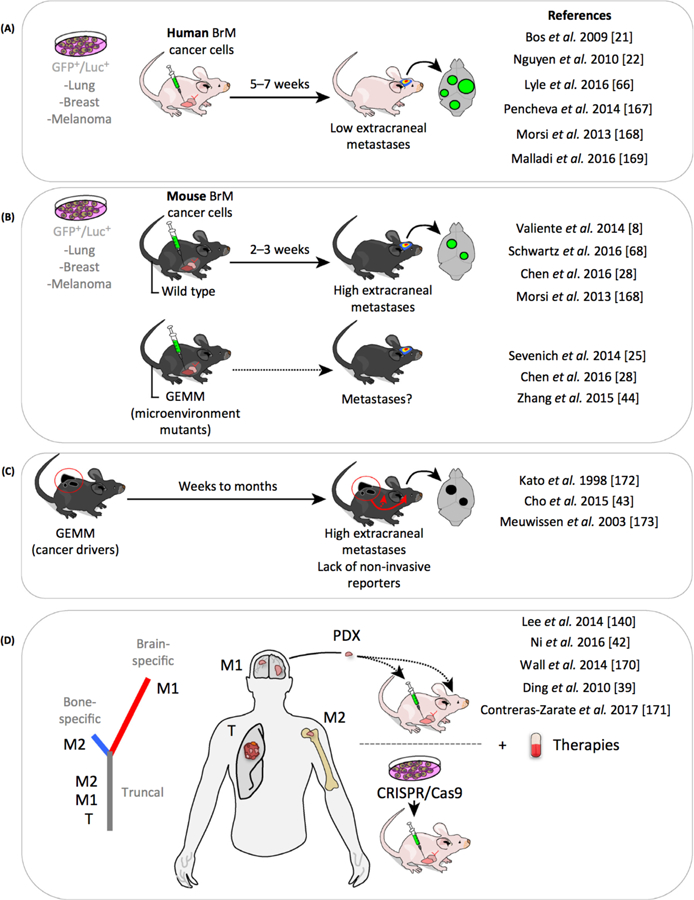
(A) Cell line-derived xenotransplants (clDX) (human cancer cell lines implanted in immunosuppressed animals) have been extensively used [21,22,66,168–170]. clDX correspond to organotropic cell lines that target preferentially the brain. (B) The use of syngeneic mouse cell lines tropic to the brain allows using immunocompetent mice and to test them in genetically engineered mouse models (GEMMs) with an altered brain microenvironment [25,28,44]. (C) Few of the many GEMM or chemically induced cancer models result in brain metastases; three such models have been reported to generate brain metastasis. (D) Patient-derived brain metastasis xenotransplants (POX) reproduce the main histological and genomic findings in humans [39,42,140,171,172]. and are potential resources for investigation of targeted therapies. Abbreviation: BrM, brain metastasis.
These early findings (molecular differences between brain-tropic and non-tropic cancer cells) have been recently confirmed in humans. Collected materials from neurosurgeries have been compared with matched and unmatched primary samples [31–39]. The identification of mutations that are only present in the brain suggests that cancer cells develop branched evolution [40], indicating that clones of cancer cells growing in the brain evolve following alternative routes to those followed in the primary tumor (Figure 1D) [31,39].
Beyond the question of the functional role for these mutations in adapting cancer cells to the brain, these data suggest a possible therapeutic intervention: the application of targeted therapies tailored to the genomics of brain metastasis (Figure 1D). Interestingly clinically actionable alterations were enriched in four main pathways, including CDK, Pl3K, EGFR, and MAPK [31]. Thus, the efficacy of targeted therapies in metastatic sites will not necessarily recapitulate the responses of the primary tumor. Consequently, profiling the metastatic compartment might be advantageous in planning therapeutic interventions, predicting responses, and discovering new targets that could be absent in the primary disease.
Among enriched pathways in brain metastasis, the Pl3K-AKT-mTOR pathway has been confirmed as a viable therapeutic target in various preclinical models of brain metastasis from different primary tumor types [41], including novel POX models (Figure 1D) [42] and genetically engineered mouse models (GEMM) (Figure 1C and Box 1) [43]. A mechanistic explanation was provided to illustrate how cancer cells could evolve in the brain to further activate this pathway, which included transfer of miRNA from reactive astrocytes to cancer cells targeting PTEN (Figure 2C) [44] as well as enrichment in activating mutations in PIK3CA or PIK3R1, homozygous deletions in PTEN, or amplifications in AKT2 or MET, as described in human samples [31].
Figure 2. The Complexity of the Brain Metastasis Microenvironment: Reactive Astrocytes.
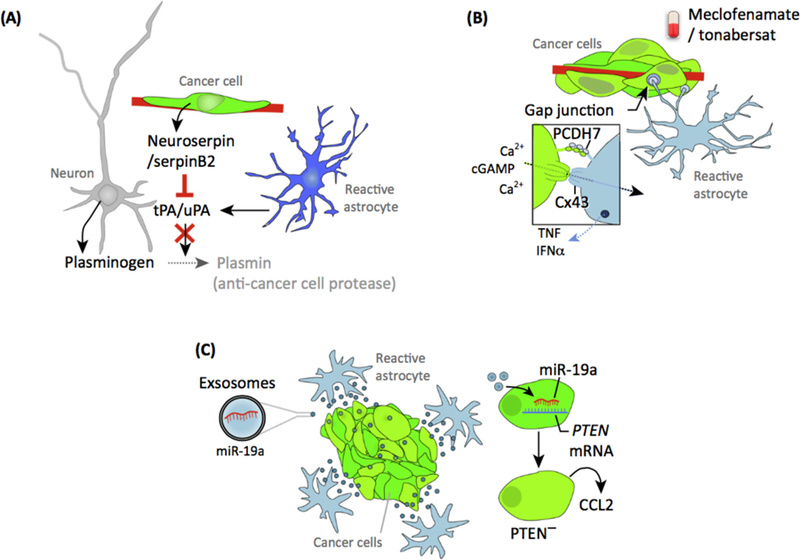
(A) Reactive astrocytes initiate a defensive program aimed to eliminate recently extravasated metastatic cells. Astrocytes produce the plasminogen activators (PA) IPA and uPA that activate plasminogen into plasmin. The active enzyme eliminates many cancer cells. A limited number of metastatic cells block this response through anti-PA serpins (neuroserpin, NS; and serpin 82, S82). Serpins inhibit plasmin generation and thus prevent its deleterious effects on cancer cells. (8) Surviving cancer cells continue to interact with reactive astrocytes during brain colonization, establishing gap junctions with reactive astrocytes. Metastatic cells employ these C43 gap junctions to send calcium and cGAMP to astrocytes. Within astrocytes, cGAMP activates a signaling pathway leading to secretion of TNF and IFN-? to induce cancer cell proliferation. (C) Astrocytes are known secretory cells and also produce extracellular vesicles, including exosomes. In the context of brain metastasis, astrocyte-derived exosomes contain miR-19a. Once internalized by metastatic cells, miR-19a targets PTEN. Loss of PTEN increases cancer cell prolliferation and induces secretion of CCL2. Secreted CCL2 attracts prometastatic myeloid cells to favor metastatic brain colonization.
There are also examples of the importance of brain metastasis genomics in the accurate classification of clinically relevant subtypes. For instance, it has been reported that positive ERα, PR, and HER2+ primary tumors could give rise to receptor-negative brain metastasis [45]; it is also possible for HER2-positive brain metastasis to arise from a HER2-negative primary tumor [45]. It remains unknown whether this is because pre-existing and low-abundance clones in the primary tumor eventually colonize the brain, or whether genetic alterations take place in those cancer cells that colonize the brain.
Microenvironment
Experimental models highlight the importance of molecular crosstalk between metastatic cells and the surrounding microenvironment. Excellent reviews have described interactions between primary brain tumors and the brain tumor microenvironment [46,47]. However, relatively limited information exists regarding the microenvironment of metastatic brain tumors. To date, these research efforts have been largely focused on reactive astrocytes. The wide variety of cancer cell-astrocyte interactions provides several novel therapeutic opportunities.
After extravasation, single cancer cells are immediately surrounded by reactive astrocytes [8,48] likely alerted by damage-associated molecular patterns (DAMPs) [49]. Astrocytes serve as an efficient first line of protection in the central nervous system (CNS) [8,50,51]. In the context of brain metastasis, they reduce the numbers of potential metastasis-initiating cells by activating plasmin (Figure 2A) [8]. This natural defense contributes, in part, to the high inefficiency of brain colonization by cancer cells [7]. Select cancer cells evade this response through the expression of serpins that prevent protease activation [8]. Fittingly, cancer cells may be rendered sensitive to brain defenses by targeting serpins (Figure 2A).
Surviving cancer cells remain located in the perivascular niche [7,8,52] alongside neural stem cells [53]. Potential benefits of this location for cancer cells include increased access to nutrients and oxygen, contact with the basal lamina of capillaries, and preferential access to angiocrine factors produced by endothelial cells [8,52–55]. The interaction with the pre-existing vasculature, independent of angiogenesis [8], is termed vascular cooption [56] and has been demonstrated experimentally and clinically in lung cancer, breast cancer, and melanoma [7,8,22,48,57,58]. Vascular cooption during brain metastasis initiation is dependent on β1-integrin and L1CAM [8,52]: L1CAM is cleaved and its function blocked by the action of plasmin (Figure 2A) [8]. Whether these cell adhesion molecules (L1CAM and β1-integrin) are molecularly linked and share downstream targets that may explain the link between vascular cooption and proliferation remains unknown.
Proliferation of metastasis-initiating cells establishes a variable number of micrometastases in experimental models, as determined by histology [8] and time-lapse video-microscopy studies [7]. However, only a fraction of these will reach a clinically detectable size [59]. Recent work suggests that the microenvironment participates in the regulation of this differential growth potential. Some micrometastases physically interact with reactive astrocytes [28,60]. This interaction depends on the presence of protocadherin 7 (PCDH7) which allows contact to be established between cancer cells and reactive astrocytes [28]. Homophilic PCDH7 interactions induce the generation of C×43-dependent gap junctions and the exchange of ions and secondary messengers [61–63]. These heterotypic cell-cell interactions increase cancer cell growth and resistance to chemotherapy-induced apoptosis by decreasing intracellular Ca2+ levels [61] and activating Stat1/p65 signaling in the cancer cells (Figure 2B) [28]. Thus, systemic therapies encounter multiple barriers to efficacy within the brain, beyond that of simple drug penetration [64,65], which involve reactive astrocytes and other cells from the microenvironment such as pericytes [66].
Further evidence for the importance of astrocytes in brain metastasis derives from their secretory capacity [49,67]. Both cytokines and extracellular vesicles have been shown to influence brain metastasis progression. Exosomes containing miR-19a secreted from reactive astrocytes reach cancer cells, where it induces downregulation of PTEN and increases the aggressiveness of cancer cells in the brain (Figure 2C) [44]. This finding illustrates how the microenvironment influences the evolution of cancer cells in situ. The secretory products of reactive astrocytes, which have been suggested to reach the systemic circulation [67], have also been linked to the ability to attract circulating tumor cells to the brain [68].
The complex behavior of reactive astrocytes, which may elicit both pro- and antitumorigenic effects, is reminiscent of patterns found in other components of the microenvironment, including microglia [69–74]. This might reflect the presence of an underlying heterogeneity within the microenvironment that is exploited by cancer cells. The presence of astrocytes and microglia subpopulations have been identified in other experimental models of CNS disorders [75–79]. Dissection of their specific contribution to the progression of disease is expected to provide a rationale for the design of more specific and effective drugs [80].
Leptomeningeal Metastasis
Although parenchymal metastases are the most frequently encountered, leptomeningeal metastasis (LM), namely the spread of tumor cells within the subarachnoid space, occurs in up to 10% of cancer patients, and may result from any malignancy [81–83].
The leptomeninges encase the brain and spinal cord, and contain the circulating cerebrospinal fluid (CSF). Cancer cells may access the leptomeningeal space through four main points of entry: from the arterial circulation through the choroid plexus [84,85], from the venous circulation through Bateson’s plexus [86], via direct invasion along spinal and cranial nerves [84], or by invasion from parenchymal disease through the glia limitans [87]. Once within the leptomeninges, cancer cells must adapt to a microenvironment completely different from that of the blood or the primary tumor and, as in the case of parenchymal metastasis, cancer cells will modify the surrounding environment to accommodate their own metabolic needs [85].
LM has been difficult to study given the lack of relevant models. However, a recent report on mouse models of LM demonstrated that these metastatic cells are distinct from parenchymal brain metastatic cells from the same primary tumor (Figure 3A,B) [85], suggesting that tailoring therapeutic approaches to this site of disease may be warranted.
Figure 3. Experimental Models of Leptomeningeal Disease.
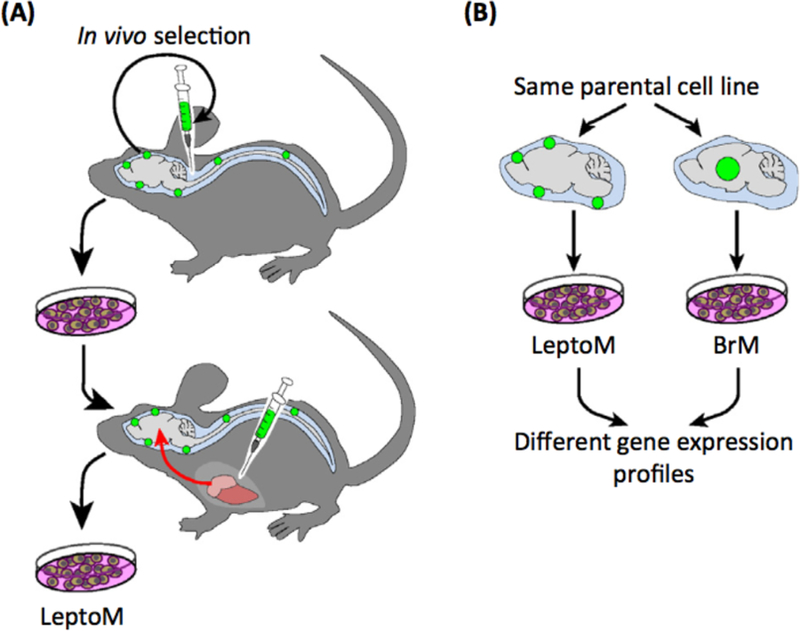
(A) Leptomeningeal disease has been established in immunocompetent and immunosuppressed mouse models. These models were generated by in vivo selection of cells proliferating in the cerebrospinal fluid (CSF) after inoculation into the cistema magna. After several rounds of in vivo selection, cell lines were inoculated intracardiacally. The subpopulation of cells targeting the leptomeninges after intracardiac inoculation was termed LeptoM. (B) Transcriptomic analysis of LeptoM and BrM (parenchymal brain metastasis) generated from the same parental cells revealed different profiles, indicating that leptomeningeal tropism constitutes a unique biological entity.
Diagnosis of LM is traditionally complex given the variability of clinical presentation [83]. However, the combination of neuroimaging and CSF cytology obtained from lumbar puncture yields specificity and sensitivity of up to 95% and 85%, respectively. Additional experimental diagnostic options include circulating tumor cells analysis [88] and cell-free DNA (cfDNA) [34], which potentially provide additional information regarding oncogenomic alterations.
Typically, diagnosis of leptomeningeal disease prompts a prognosis ranging from 8 weeks to 6 months, despite aggressive treatments [89–91]. Treatment options include radiation [92] and/ or intra-CSF or systemic chemotherapy [93,94] applied according to recent guidelines for tailoring treatment [95]. Recent clinical studies with blood-brain barrier (BBB)-permeable third-generation EGFR inhibitors in patients with EGFR-mutant lung cancer who have leptomeningeal disease have shown significant efficacy, improving mean overall survival as much as 63% [96]. In addition, preclinical studies have provided interesting hypothesis regarding treating leptomeningeal disease by impairing C3 signaling [85].
Neurocognition
Multiple brain metastases induce cognitive dysfunction in a large proportion of patients, and neurocognitive outcomes can be improved by whole-brain radiation therapy (WBR1) in responding patients [97]. However, given recent progress in systemic therapy of many cancers, an important issue is to protect neurocognition in patients with limited brain disease who have a life expectancy estimated to be more than several months. Very limited data are available regarding the impact of brain metastases on neural circuits despite preclinical findings indicating their negative influence on neuronal homeostasis [98,99]. However, radiation therapy-induced neurocognitive decline is well described.
Historically, radiation-induced dementia with ataxia and urinary incontinence was described in up to 30% of patients 1 year after receiving unconventionally large dosages of WBRT (6–8.5 Gy) [100]. These patients demonstrated a radiographic picture compatible with leukoencephalopathy and associated hydrocephalus, and such dosage schemes are no longer used. When using more conventional, modern dosages (of up to 3 or 4 Gy), recent clinical trials demonstrate that early cognitive decline may occur. The addition of WBRT to stereotactic radiosurgery (SRS) increases the risk of decline in learning and memory function at 4 months after treatment [101], which was also reproduced in a Phase Ill trial using a primary neurocognitive endpoint [102], including deterioration in immediate recall (31% vs 8%), delayed recall (51% vs 20%), and verbal fluency (19% vs 2%). The 22952–26001 trial of the European Organisation for Research and Treatment of Cancer (EORTC) [103] reported lower cognitive functioning accompanied by reduced physical functioning and increased fatigue within 1 year from treatment. It is still unknown whether the early decline is predictive of long-term or permanent decline. Re-evaluation of a Japanese trial suggested that >50% of long-term survivors developed some deterioration of neurocognitive functions up to 36 months after WBRT [104].
The pathogenesis of radiation-induced damage may be due to injury of the endothelium of small vessels, which leads to an accelerated atherosclerosis and ultimately to chronic ischemia [105]. or to injury of neuronal stem cells, specifically affecting those located in the hippocampus, an area of the brain that is involved in preserving memory (Figure 4) [106]. When radiation-induced neurocognitive dysfunction develops, few options with modest effect are available for treatment.
Figure 4. Pathophysiology of Radiation-Induced Damage.
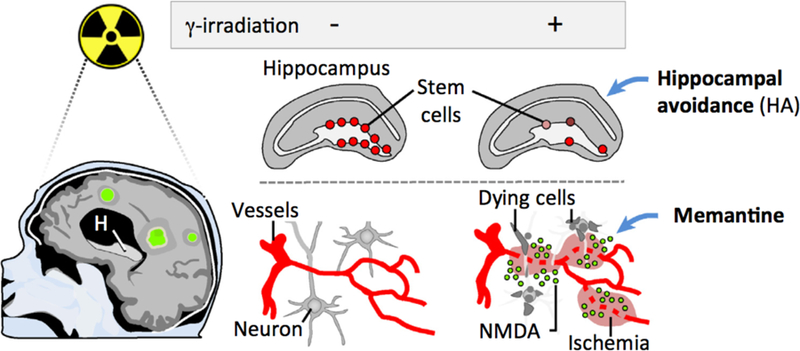
Whole-brain radiation therapy (WBRT) induces secondary neurocognitive effects through two proposed mechanisms: destruction of neural stem cells located in the hippocampus, and/or damage to brain capillaries generating localized ischemic areas. By either mechanism, death of neuronal cells increases extracellular NMDA levels, resulting in toxicity for other cells. Abbreviation: H, hippocampus.
Given that the vascular injury from radiation is very similar to that observed in vascular dementia, a line of clinical research hypothesized that the underlying pathophysiology could be extrapolated [107]. Memantine is a non-competitive, low-affinity antagonist of the N-methyl-D-aspartate (NMDA) receptor, which is one of the receptors activated by glutamate, the principal excitatory neurotransmitter. Memantine has the potential to block the excessive NMDA stimulation following ischemia which could lead to excitotoxic damage of the normal brain [108]. Memantine used in a Phase II trial during and after WBRT modestly delayed time to cognitive decline with a tendency to reduce the rates of decline in memory (Figure 4) [107].
Another avenue of research to preserve cognitive function during WBRT is to spare the hippocampus. The basis for this is studies that demonstrate that low-dose radiation in rodents results in blockade of hippocampal neurogenesis and damage to the neurogenic microenvironment, resulting in significant short-term memory impairment [106]. It has therefore been hypothesized that sparing the hippocampus during WBRT could prevent damage to neuronal progenitor cells and better preserv memory functions [109]. A Phase II trial proved that hippocampal avoidance (HA) during WBRT reduced memory decline in 23% of patients (Figure 4) [110]. An ongoing Phase III trial (NRGCC001) will test both approaches (memantine combined with HA) during WBRT for brain metastases. Additional strategies also include potentiation of cholinergic transmission in the brain by the use of donepezil, a reversible inhibitor of acetylcholinesterase [111].
Given the potential toxicity from WBRT, it is often avoided in favor of locally delivered high doses of radiation using SRS, or alternatively radiation is avoided altogether and replaced by available systemic therapies with efficacy in the CNS.
Targeted Therapies
The inability of many systemic chemotherapeutic agents to cross the BBB has limited their use in treatment of these patients [112]. The assumption that the BBB is disrupted in brain metastasis has proved to be incorrect because drug conjugates only reached therapeutic levels in 15% of lesions [65], and side-by-side comparison of a drug with and without chemical modifications conferring BBB permeability found that only the brain-permeable compound was able to impair brain metastasis [113]. Thus it remains to be determined whether the properties of the BBB are not altered or instead whether the barrier is modified into the so-called blood/ tumor-brain barrier (BTB). The BTB might conserve limited permeability via different regulatory mechanisms, such as altered pericyte subpopulations that might impose alternative constrains [66]. Because of these potential limitations, it seems reasonable to utilize drugs that are able to cross the normal BBB.
Several drugs that cross the BBB and target molecular alterations in key oncogenic drivers of the primary tumor have induced positive responses in secondary brain tumors (Figure 5A–C). Although this has established additional therapeutic options, patients with brain metastasis harboring these vulnerabilities in the primary tumor are a minority (Figure 5D). Approximately 2– 4% and 5% of lung cancer brain metastasis derive from EGFR mutant [114] or ALK-translocated primary tumors, respectively (Figure 5A) [115]; 25% of breast cancer brain metastases belong to HER2+ primary tumors (Figure 5B) [Surveillance, Epidemiology, and End Results Program (SEER) database; https://seer.cancer.gov/] and 50% of melanoma brain metastasis result from BRAF mutant primaries (Figure 5C) [116]. Together, these findings suggest that ~18% of the patients diagnosed with brain metastasis will be eligible for targeted therapies (Figure 5C). These patients could benefit significantly from these therapies and thus have a relatively better prognosis than patients without targetable driver mutations.
Figure 5. Patients with Brain Metastasis Eligible for Targeted Therapies.
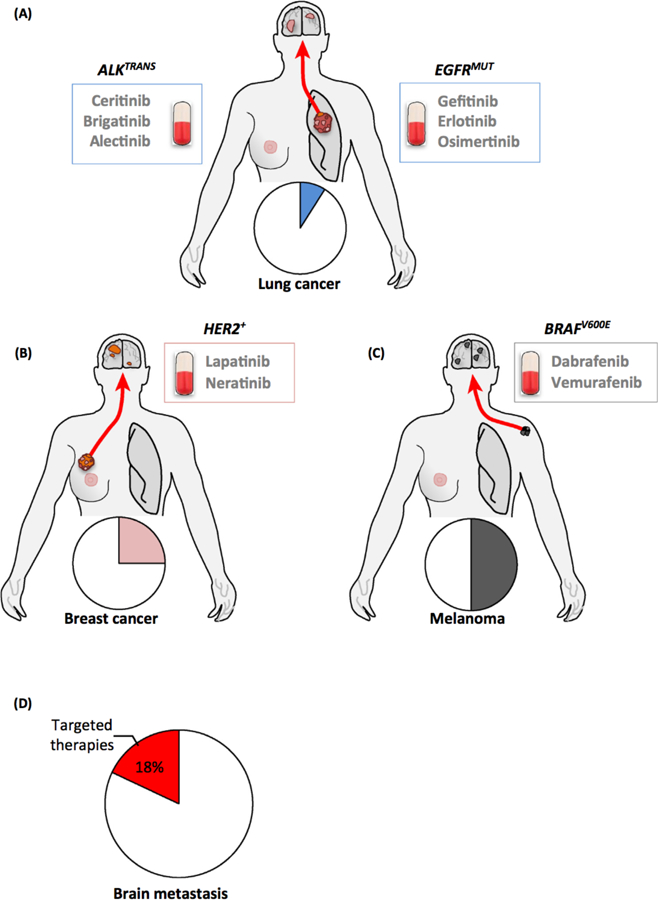
Patients with lung cancer (A), breast cancer (B), or melanoma (C) brain metastasis may benefrt from targeted therapies. The main oncogenomic alterations that qualify these patients for this advanced treatment are shown, as well as the corresponding drugs that have shown efficacy in the brain. The percentage of patients harboring brain metastases that are susceptible to targeted therapy is very low (D).
lntracranial activity of gefitinib and erlotinib, the two first-generation EGFR tyrosine kinase inhibitors (TKls) approved for management of advanced EGFR mutant NSCLC [117]. was initially seen in retrospective series [118,119], with additional evidence of brain responses (decrease of tumor burden >30% of the total) in >80% of patients [120,121].
In the trial by Porta and colleagues it was reported that median time to progression in patients harboring EGFR mutations treated with erlotinib was 11.7 months compared to 5.8 months for control patients whose EGFR mutational status had not been assessed (P < 0.05). Overall survival times were 12.9 months and 3.1 months (P < 0.001), respectively [120]. Other studies have confirmed benefit of these agents in those who carry this mutation, with increased overall progression-free survival (PFS) of 15.2 months compared to 4.4 months in those patients who do not carry the mutation [122]. These findings have been reproduced and expanded with additional inhibitors, such as osimertinib, which doubled PFS from 4.2 months of patients receiving chemotherapy to 8.5 months in those treated with the targeted therapy in a Phase Ill study with brain metastasis patients [123]. The importance of BBB penetration is illustrated by crizotinib, the first approved TKI that poorly penetrates the brain (CSF/serum ratio of <0.1% to 0.26%), in patients with ALK-translocated lung cancer [124,125]. Patients receiving crizotinib had a high rate of brain metastasis relapse of 45% [6]. which might be a consequence of suboptimal intracranial drug concentration. However, next-generation TKls targeting translocated ALK have improved brain penetration and demonstrated ability to control brain relapse after crizotinib therapy, with intracranial responses ranging from 45% (ceritinib) [126], 42–67% (brigatinib) [127], to 64% (alectinib) [128]. In patients treated with ceritinib the median duration of response was 9.2 months and the median PFS was 5.4 months [114]. In the patients treated with brigatinib, the median intracranial PFS was 15.6 months (treated with 90 mg daily) and 12.8 months (treated with 180 mg daily) [115]. In the alectinib study, CNS duration of response was 11.1 months [116].
Melanoma brain metastases also benefit from targeted therapies. In addition to immunotherapy (discussed below), targeted therapies such as the BRAF V600E TKI dabrafenib have induced 39% intracranial response in asymptomatic brain metastases [129], reaching 58% in studies that used a combination of dabrafenib and trametinib [130]. The duration of response or median intracranial PFS in these studies was 5–6 months. Response rates of 18% have been reported in those treated with vemurafenib [131]. In this trial patients were divided into cohort 1 (previously untreated brain metastasis) and cohort 2 (previously treated brain metastasis). Median PFS (brain only, investigator-assessed) was 3.7 months in cohort 1 and 4.0 months in cohort 2. Median overall survival (OS) was 8.9 months in cohort 1 and 9.6 months in cohort 2, respectively [131]. Interestingly, novel melanoma models have shown that BRAF V600E tumors reproducibly and in high proportion generate spontaneous brain metastasis in an AKT-dependent manner [43]. These models suggest novel combination therapies to complement current targeted approaches.
The relative higher incidence of brain metastasis of HER2+ breast cancer patients treated with the blocking antibody trastuzumab in comparison to those patients who did not received this therapy [132] reinforced the need to develop alternative brain-penetrant strategies for these patients. Lapatinib, a TKI targeting HER2 and EGFR, only when complemented with capecitabine, generated a 66% intracranial response rate with a median intracranial time to progression of 5.5 months in a Phase II study of radiation-naive HER2-positive breast cancer patients with brain metastases [133–135]. Similar findings apply to neratinib [136], another TKI targeting the same receptors as lapatinib, also in combination with capecitabine. Experimental models of intracranial breast cancer cells have also confirmed that combination strategies for HER2+ brain metastasis including trastuzumab, lapatinib, and the VEGFR2 blocking antibody DC101 allowed better control than single therapies. Interestingly, these combination strategies were not required to control mammary fat pad implantation of the same cancer cells, since monotherapies were already effective [137].
In general, targeted therapies do not achieve complete responses in the brain. Several molecular strategies have been proposed to explain this phenomenon [138]. To address this essential clinical question in the brain (mechanisms of resistance to targeted therapies), brain metastasis experimental models receiving targeted therapies are needed. These include patient-derived xenografts, cancer cell lines, and mouse models with clinically relevant genetic alterations (Figure 1 and Box 1) such as the ALK-translocated model of lung cancer recently reported [139]. Pioneering studies have exploited these experimental approaches (Figure 1). For instance, NSCLC human brain metastases were successfully established as patient-derived xenografts upon intracranial or intracardiac implantation in immunodeficient mice. Five of these human samples were grown as low-passage primary spheres and were screened for their sensitivity to a panel of 20 different drugs, which emphasized dramatic difference of more than 100-fold in their IC50 values, providing novel opportunities for personalized treatments [140].
lmmunotherapy
The normal brain has limited infiltration of leucocytes [141]. However, when affected by injury a strong response takes place involving the infiltration of non-resident cells. Human brain metastases typically show moderate to dense mixed immune cell infiltration, including CD3+, CD4+, CD8+, FoxP3+, and CD45RO+ lymphocytes, natural killer (NK) cells, and cells of the macrophage lineage [9,142]. This local inflammatory environment also applies to immunocompetent brain metastasis models [48,143,144]. The amount of tumor-infiltrating leucocytes (TLs) was found to positively correlate with patient survival and with the amount of peritumoral brain edema [9], suggesting that although Tlls are unable to stop tumor progression, some components of the infiltrate are able to offer resistance to cancer cells (Figure 6A).
Figure 6. Potential Approaches for lmmunotherapy in Brain Metastasis.
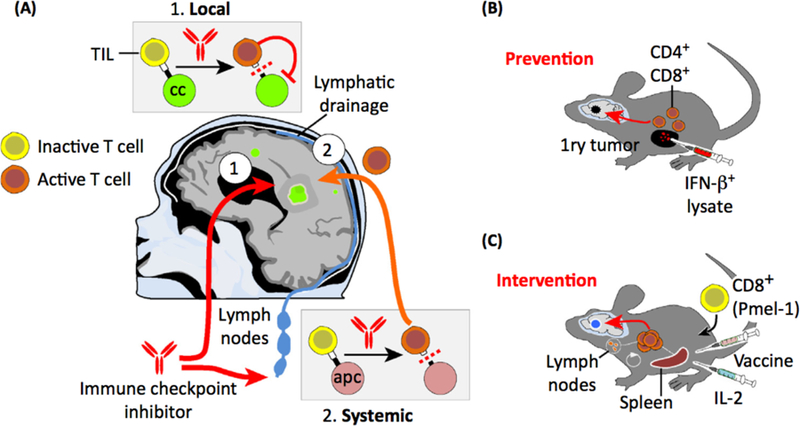
(A) Immune checkpoint inhibitors have been used in brain metastasis patients, and positive responses were reported. The potential mechanisms of action of this therapeutic approach include local and systemic effects. Locally derived effects include access of blocking antibodies to the brain parenchyma at therapeutic levels. In a complementary scenario, blocking antibodies would impair the checkpoint between antigen-presenting cells and lymphocytes in regional lymph nodes or other organs. Activated lymphocytes then access the brain to target cancer cells. (B,C) Experimental cell transfer immunotherapies that have been report for brain metastasis. (B) In a prevention setting, I FN- β-stimulated CD4+/CD8+. lymphocytes were induced in cancer-free mice into which metastatic cells were later inoculated, and T cells were able to prevent the development of brain metastasis. (C) After brain metastasis developed, a combined therapy including CDs+ Pmel-1 together with IL-2 and a gp-100 vaccine efficiently induced an initial expansion of T cells in the spleen and regional lymph nodes that later targeted cancer cells in the brain. Abbreviations: Ape, antigen-presenting cell; CC, cancer cell; TIL, tumor-infiltrating lymphocyte.
Research on the immune system associated with brain metastasis is an exciting emerging field that might also have relevance in view of the limited lymphatic drainage exclusively localized in the meningeal surface (Figure 6A) [145–147].
Interestingly, several clinical trials have applied immune checkpoint inhibitors to patients harboring brain metastases. Expression of immunomodulatory molecules such as PD-L1, PD-L2, and various cytokines is commonly found in brain metastases from lung cancer (NSCLC and SCLC), melanoma, renal cancer, and breast cancer [148–150]. In lung cancer, significant differences between the inflammatory microenvironment of paired primary tumors and brain metastases were detected, with discrepancies in tumor cell PD-L 1 expression in 14% of cases and TIL PD-L 1 expression in 26% of cases [151]. In the discrepant cases, the brain metastases lacked PD-L1 expression, tumor lymphocyte infiltration, or both, even though they were present in the primary lung cancer specimens.
A Phase II trial using the CTLA-4 inhibitor ipilimumab in patients with brain metastases has reported encouraging results [152]. Of patients who were asymptomatic at baseline, 24% attained disease control, with median PFS of 2.7 months and median OS of 7 months. Importantly, OS at 24 months was 26%, indicating that a subset of patients did have prolonged benefit. Outcomes were overall inferior for patients who were symptomatic and on steroids at baseline, but even in this group 10% of patients werealive at 24 months. Because the PD-1 axis inhibitors have demonstrated significant and durable activity in a subset of patients with melanoma, lung cancer, bladder cancer, and many other malignancies, their activity has begun to be studied in patients with brain metastases. The PD-1 inhibitor pembrolizumab was studied in a Phase II trial of patients with asymptomatic, progressing brain metastases from melanoma or NSCLC [153]. An interim analysis demonstrated activity in the CNS in both diseases: an intracranial response was achieved in four of 18 patients with melanoma and in six of 18 patients with NSCLC (including four patients with complete response in the brain). All but one patient who had a systemic response also responded in the CNS, and most responses were durable and ongoing at the time of data analysis. Nivolumab, another PD-1 inhibitor, has also been studied in patients with NSCLC and untreated CNS metastases. An initial report demonstrated an intracranial response in two of 12 patients, one of whom displayed a complete response ongoing after 10.5 months [154].
In summary, although positive but limited results have been obtained for immunotherapy in patients with brain metastases, basic questions remain. Further research into the mechanism of action of these therapies in the brain will determine whether they work locally or as a consequence of systemic immune activation (Figure 6A). Expanding the preclinical observations regarding the potential antitumor effects of immunotherapies (Figure 6B,C) will certainly contribute to our understanding of how to improve the benefits from this class of agents, as well as to develop more effective approaches.
Prevention of Brain Metastasis
Considering all the difficulties that are associated with the treatment of established brain metastasis, it appears logical to develop strategies that prevent their occurrence altogether. While studies on prophylactic WBRT in lung cancer have consistently shown that such a preventive intervention can be very effective [155,156], its use is associated with considerable neurotoxicity. Therefore, it would be a breakthrough if a long-term, well-tolerated, non-neurotoxic, and affordable drug could be developed to be given to patients at particularly high risk of brain metastasis, preventing their future occurrence. In principle, many targeted therapies would fulfill these criteria, for example second-generation ALK inhibitors in ALK-rearranged lung cancer [6]. Other patients with a cumulative risk of brain metastasis above 40% should be also considered for preventive strategies (Figure 7A) [18,19].
Figure 7. Potential Strategies To Prevent Brain Metastasis.
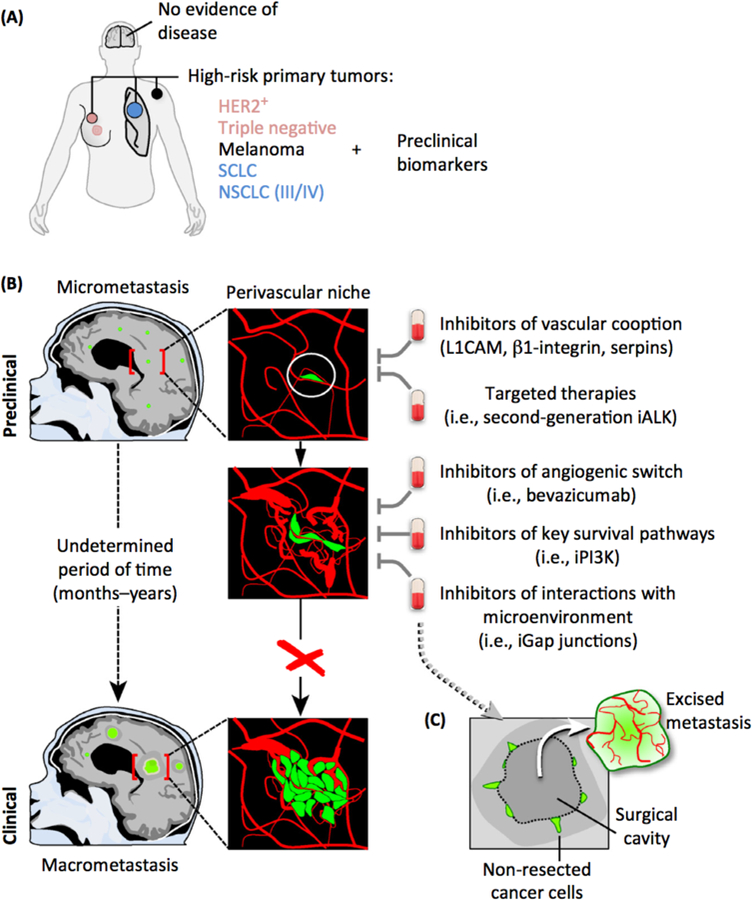
(A) Patients with HER2+ or triple-negative breast cancer, melanoma, small-cell lung cancer (SCLC), and stage IIV/IV non-scuamous non-small cell lung cancer (NSCLC) are at high risk for the development of brain metastasis during the progression of the disease. To guide clinical decisions in initiating brain metastasis-preventive trials, the discovery of biomarkers in preclinical models that could be translated to this group of patients will be a valuable resource. (B) Although clinically undetectable, brain micrometastasis might be present in asymptomatic patients with these high-risk tumors. Experimental findings suggest the importance of the interaction with the vasculature to allow metastasis-initiating cells to progress. Metastatic cells initially interact with pre-existing vessels (vascular cooption), and drugs targeting key mediators of this process will therefore impede their outgrowth. The efficacyof ALK inhibitors (iALK) that cross the blood-brain barrier (BBB) will allow targeting of cancer cells before they are detectable by imaging. Some micrometastases may have proliferated and started to influence the microenvironment, and at this point a VEGF-dependent switch from vascular cooption to angiogenesis is necessary to support the outgrowth of the metastatic lesion derived from lung cancer. Preventive trials with BBB-permeable Pl3K inhibitors might be considered because there is experimental evidence for their efficacy [41]. In addition, interaction with astrocytes could be targeted with blockers of gap junction communication (iGap junctions). (C) An alternative option will be to apply preventive therapies after neurosurgical resection, given the known ability of brain metastatic cells to infiltrate the tissue [57] and the likelihood that some cancer cells remain in surgical margins. In this sense the infiltrative phenotype of brain metastasis has been linked to reduced survval [167]. During reinitiation of local growth, the metastasis might follow the same principles and molecular regulation as during the inltial stages.
A preventive strategy should ideally be based on the accurate identification of patients at high risk for brain metastasis. Multiple biomarkers of brain metastasis have been reported in preclinical models; several were also validated in primary tumor samples [8,21–28], and a plethora of molecular factors relevant for brain-specific tumor cell-host interactions have been found. Repeatedly, and perhaps consistently in preclinical models and patient material, deregulation of the PTEN/Pl3K and HER2/3 pathways in brain-tropic tumors has been reported [32,157,158], and patient serum levels of carcinoembryonic antigen (CEA) ≥40 ng/ml have also been reported [159]. However, none of those putative predictive biomarkers has been independently validated in a controlled clinical study, and therefore more clinical research is needed before patients can be stratified according to biomarkers in future brain metastasis prevention trials.
Most drugs tested in preclinical studies have been validated in a ‘prophylactic’ setting [160,161]. Many of them might have failed when translated to the clinic given that they were tested in ‘intervention settings, in which efficiency is evaluated by the reduction in the size of established metastasis [162,163].
One strategy for preventing brain metastasis in preclinical studies leading to increased survival is to target brain metastasis-initiating cells and micrometastasis at the perivascular niche, given that this location supports survival and is required for proliferation (Figure 7B) [7,8,28,52,113,164,165]. This strategy might even provide benefit to prevent local relapse after neurosurgery, given that cancer cells left behind might survive in this specific niche (Figure 7B) [57].
Another possibility could be to prevent a crucial and early angiogenic switch by subclinical doses of the antiangiogenic drug bevacizumab, although this may only be applicable to NSCLC brain micrometastasis, as shown by a retrospective analysis of a large Phase III trial in this cancer type (Figure 7B) [7,166].
Together, these preclinical and clinical data demonstrate four important points: (i) potential antimetastatic effects of systemic drugs should be investigated for their specific brain activity, (ii) the specifics of the brain metastatic cascade in each different tumor type need to be taken into account, (iii) many preclinical models can faithfully predict effectiveness (or lack thereon in patients, and (iv) it might be possible to reduce the established doses of drugs many-fold to achieve strong brain metastasis-preventive effects.
The following general considerations for such a trial appear relevant.
(i) Study Design.
Ideally a stage II randomized clinical trial with incidence of brain metastasis as the brain-specific primary endpoint, and OS and quality of life as secondary endpoints.
(ii) Disease Stage.
Either patients without detectable brain metastasis, as documented by magnetic resonance imaging (MRI) at study entry, or patients who received definite treatment (surgery or radiosurgery) for 1–3 brain metastases, given that resistant cells left behind would, at some point, regrow (Figure 7B) [57,167]. No active disease outside the brain.
(iii) Drug Considerations.
Brain-penetrant strategies must be selected. Brain metastasis-preventive drug administration is allowed to continue even if non-CNS progression occurs, provided that patients and doctors agree.
Given the abundant preclinical positive results regarding opportunities to effectively prevent brain metastasis, efforts to evaluate how to translate these simple experimental approaches to the infinitely more challenging situation in cancer patients should be considered.
Concluding Remarks
The high morbidity and mortality associated with brain metastases have historically led to therapeutic and scientific nihilism. However, recent work in both clinical and basic research sphere has uncovered novel biology that has translated into innovative approaches. These have already been translated into better clinical management of the disease. Current challenges include understanding the molecular basis for different clinical entities, including patients at high risk of developing metastatic spread to their brain, single or limited active-progressing brain metastasis, local relapse after surgery or stereotactic radiosurgery, and multiple progressing lesions incompatible with local treatments. Each of these represents a complex clinical management decision reflecting a complex oncogenic process. It is thus crucial to integrate the progress made on the biology of brain metastasis with the many remaining clinical challenges. Emerging evidence from basic and clinical research suggests that brain-specific approaches might be a future therapeutic strategy. In this sense the brain microenvironment is being consolidated as a crucial compartment that defines the viability of cancer cells in this organ, and therapies targeting crosstalk between cancer and non-cancer cells in the microenvironment might become a reality in the years to come. A recurrent concern is whether available experimental models (Box 1) reliably recapitulate the human disease. In addition to the use of xenotransplants, immunocompetent models are available, and recent reports have shown that spontaneous brain metastasis can be modeled in genetically engineered mouse models (GEMMs) (Figure 1C). In this sense, the incorporation of CRISPR/Cas9 technology will provide elegant ways to model the human disease in mice. Nevertheless, because brain metastases often occur in patients pretreated with multiple lines of therapy, different therapies must be incorporated into experimental brain metastasis models. Surprisingly, this is rarely the case. Going forward, our efforts to improve therapies must take into account the essential functions of the unique target organ. This obvious consideration takes on a new urgency because life expectancy increases significantly after modern therapies. Safer and more effective therapies are urgently needed to treat brain metastasis patients. The various fronts open for discovery (see Outstanding Questions) have created a fertile soil in which promising opportunities make us confident that significant progress will be achieved.
Highlights.
Increasing evidence from basic and clinical research suggests that colonization of the brain involves specific requirements that might not be needed extracranially.
The brain microenvironment is crucial to understanding the biology of brain metastasis, and could be the source of novel therapeutic targets.
New targeted therapies that cross the blood-brain barrier have improved disease control and survival of selected patients with brain metastasis.
Small clinical trials suggest that immunotherapies may become another strategy to target brain metastasis.
Management of brain metastasis includes maintenance of neurocognitive functions, and this questions the use of some techniques as standardof-care (i.e., whole-brain radiation therapy).
Preclinical evidences suggest that preventing experimental brain metastasis is feasible with several therapeutic approaches; however, this strategy has yet to be translated into patients.
Outstanding Questions.
Can preventive therapies reported in brain metastasis experimental models be translated to patients?
Do brain metastases cluster in speciflC subgroups according to additional genomic alterations besides those described for their primary tumor source?
Could brain-specific therapies, targeting crosstalk with the microenvironment or brain-specific genomic alterations, provide signif1Cant benefrts for patients?
Can experimental models be improved by incorporating therapies (i.e., surgery, radiation, systemic therapies), modeling patient-relevant genomic alterations, and interrogating single or multiple components of the microenvironment?
What is the biology behind the positive responses seen in brain metastasis by using immune checkpoint blockers? Do they have local access to TIL. or do they act extracranially? Are there strategies to improve the number of patients responding to immunotherapies in the brain?
Should we implement behavioral science in experimental models to understand the biology of neurocognition during brain metastasis colonization and therapeutic intervention?
Acknowledgements
We thank members of our laboratories for helpful discussions. We are grateful for funding from the National Cancer Institute 2P30CAOOB748–48, to A. B.), Mundipharma (E. L.R.), Amgen (E. L.R.), German Cancer Aid (Deutsche Krebshilfe), Translational Oncology Program, the Prevent Brain Metastasis Consortium (F.W.), MINECO-Retos (SAF2014–57243-R, to M.V.), MINECO-Europa Excelencia (SAF2015–62547-ERC, to M.V.), IX FERO Grant for Research in Oncology (M.V.), Bristol-Myers Squibb Melanoma Research Alliance Young Investigator Award 2017 (M.V.), and the Beug Foundation Prize for Metastasis Research 2017 (M.V.). M.V. is a Ramón y Cajal Investigator (RYC-2013–13365).
References
- 1.Bamholtz-Sloan JS et al. (2004) Incidence proportions of brain metastases in patients diagnosed (1973 to 2001) to in the Metropolitan Detroit Cancer Surveillance System. J. Din. Oneal 22, 2865–2872 [DOI] [PubMed] [Google Scholar]
- 2.Schouten LJ. et al. (2002) Incidence of brain metastases in a cohort of patients with carcinoma of the breast, colon, kidney, and lung and melanoma. Cancer 94, 2698–2705 [DOI] [PubMed] [Google Scholar]
- 3.Smedby KE et al. (2009) Brain metastases admissions in Sweden between 1987 and 2006. Br. J. Cancer 101, 1919–1924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gállego Perez-Larraya J, and Hildebrand J (2014) Brain metastases. Handbook Clin. Neurol 121, 1143–1157 [DOI] [PubMed] [Google Scholar]
- 5.Guérin A et al. (2016) The economic burden of brain metastasis among lung cancer patients in the United States. J. Med. Econ 19, 526–536 [DOI] [PubMed] [Google Scholar]
- 6.Peters S et al. (2017) Aleclinib versus crizotinib in untreated ALK-positive non-smail-cell lung cancer. N. Engl. J. Med 377, 829–838 [DOI] [PubMed] [Google Scholar]
- 7.Kienast Y et al. (2010) Reai-time imaging reveais the single steps of brain metastasis formation. Nat. Med 16, 116–122 [DOI] [PubMed] [Google Scholar]
- 8.Valiente M et al. (2014) Serpins promote cancer cell survival and vascular co-option in brain metastasis. Cell 156, 1002–1016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Berghoff AS. et al. (2016) Density of tumor-infiltrating lymphocytes correlates with extent of brain edema and overall survival time in patients with brain metastases. Oncoimmunology 5, e1057388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sloot S et al. (2018) Improved survival of patients with melanoma brain metastases in the era of targeted BRAF and immune checkpoint therapies. Cancer 124, 297–305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nayak L et al. (2012) Epidemiology of brain metastases. Curr. Oneal. Rep 14, 48–54 [DOI] [PubMed] [Google Scholar]
- 12.Bachmann C et al. (2015) CNS metastases in breast cancer patients: prognostic implications of tumor subtype. Med. Oncol 32, 400. [DOI] [PubMed] [Google Scholar]
- 13.Gugger A et al. (2016) Cutaneous meomoma with brain metastasis report of 193 patients with new observations. PLoS One 11, e0156115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cagney DN. et al. (2017) Incidence and prognosis of patients with brain metastases at diagnosis of systemic malignancy: a population-based study. Neuro Oncol 19, 1511–1521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Martin AM. et al. (2017) Brain metastases in newly diagnosed breast cancer: a population-based study. JAMA Oneal 3, 1069–1077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ramakrishna N et al. (2014) Recommendations on disease management for patients with advanced human epidermal growth factor receptor 2-posttive breast cancer and brain metastases: Amertcan Society of Clinical Oncciogy clinical practice guideline. J. Clin. Oneal 32, 2100–2108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Spagndo F et al. (2016) Survial of patients with metastatic melanoma and brain metastases in the era of MAP-kinase inhibitors and immunologic checkpoint biockade antibodies: a systematic review. Cancer Treat. Rev 45, 38–45 [DOI] [PubMed] [Google Scholar]
- 18.Marnon HJ et al. (2005) High risk of brain metastases in surgically staged IIIA non-small-cell lung cancer patients treated with surgery, chemotherapy, and radiation. J. Clin. Oneal 23, 1530–1537 [DOI] [PubMed] [Google Scholar]
- 19.Steeg PS et al. (2011) Brain metastases as preventive and therapeutic targets. Nat. Rev. Cancer 11, 352–363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sperduto PW et al. (2017) Estimating survival in patients with lung cancer and brain metastases: an update of the graded prognostic assessment for lung cancer using molecular markers (Lung-molGPA). JAMA Oneal 3, 827–831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bos PD et al. (2009) Genes that mediate breast cancer metastasis to the brain. Nature 459, 1005–1009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nguyen DX et al. (2009) WNT/TCF signaling through LEF1 and HOXB9 mediates lung adenocarcinoma metastasis. Cell 138, 51–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Martínez-Aranda A et al. (2015) FN14 and GRP94 expression are prognostic/predictive biomarkers of brain metastasis outcome that open up new therapeutic strategies. Oncotarget 6, 44254–44273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li B et al. (2013) Elevated PLGF contributes to small-cell lung cancer brain metastasis. Oncogene 32, 2952–2962 [DOI] [PubMed] [Google Scholar]
- 25.Sevenich L et al. (2014) Analysis of tumour- and stroma-supplied proteolytic networks reveals a brain-metastasis-promoting role for cathepsin S. Nat. Cell Biol 16, 876–888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wrage M et al. (2015) Identification of HERC5 and its potential role in NSCLC progression. Int. J. Cancer 136, 2264–2272 [DOI] [PubMed] [Google Scholar]
- 27.Jilaveanu LB et al. (2015) PLEKHA5 as a biomarker and potential mediator of melanoma brain metastasis. Clin. Cancer Res 21, 2138–2147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen Q et al. (2016) Carcinoma-astrocyte gap junctions promote brain metastasis by cGAMP transfer. Nature 533, 493–498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vanharanta S and Massagué J (2013) Orgins of metastatic traits. Cancer Cell 24, 410–421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jacob LS. et al. (2015) Metastatic competence can emerge with selection of preexisting oncogenic alleles without a need of new mutations. Cancer Res 75, 3713–3719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brastianos PK et al. (2015) Genomic: characterization of brain metastases reveals branched evolution and potential therapeutic targets. Cancer Discov 5, 1164–1177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Paik PK et al. (2015) Next-generation sequencing of stage IV squamous cell lung cancers reveals an association of Pl3K aberrations and evdence of clonal heterogenetty in patients with brain metastases. Cancer Discov 5, 610–621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Saunus JM et al. (2015) Integrated genomk: and transcrtptomic analysis of human brain metastases identifies alterations of potential clinical sgnificance. J. Pathol 237, 363–378 [DOI] [PubMed] [Google Scholar]
- 34.De Mattos-Arruda L et al. (2015) Cerebrospinal fluid-dertved circulating tumour DNA better represents the genomic alterations of brain tumours than plasma. Nat. Commun 6, 8839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee JY et al. (2015) Mutational profiling of brain metastasis from breast cancer: matched pair analysis of targeted sequencing between brain metastasis and primary breast cancer. Oncotarget 6, 43731–43742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pentsova EI et al. (2016) Evaluating cancer of the central nervous system through next-generation sequencing of cerebrospinal fluid. J. Clin. Oncol 34, 2404–2415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Prtedigkeit N et al. (2017) lntrtnsic subtype switching and acquired ERBB2/HER2 amplifications and mutations in breast cancer brain metastases. JAMA Oncol 3, 666–671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen G et al. (2014) Molecular profiling of patient-matched brain and extracranial melanoma metastases implicates the Pl3K pathway as a therapeutic: target. Clin. Cancer Res 20, 5537–5546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ding L et al. (2010) Genome remodelling in a basal-like breast cancer metastasis and xenograft. Nature 464, 999–1005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dagogo-Jack L et al. (2016) Brain metastasis: clinical implications of branched evolution. Trends Cancer 2, 332–337 [DOI] [PubMed] [Google Scholar]
- 41.Nanni P et al. (2012) Multiorgan metastasis of human HER-2+ breast cancer in Rag2−/−;ll2rg−/− mice and treatment with PI3K inhibitor. PLoS One 7, e39626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ni J et al. (2016) Combination inhibition of Pl3K and mTORC1 yields durable, remissions in mice bearing orthotopic patientderived xenografts of HER2-positMl breast cancer brain metastases. Nat. Med 22, 723–726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cho JH et al. (2015) AKT1 activation promotes development of melanoma metastases. Cell Rep 13, 898–905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang L et al. (2015) Microenvironment-induced PTEN loss by exosomal microRNA primes brain metastasis outgrowth. Nature 527, 100–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Duchnowska R et al. (2012) Conversion of epidermal growth factor receptor 2 and hormone receptor expression in breast cancer metastases to the brain. Breast Cancer Res 14, R119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Quail DF and Joyce JA (2017) The microenvironmental landscape of brain tumors. Cancer Cell 31, 326–341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Charles NA et al. (2012) The brain tumor microenvironment. Glia 60, 502–514 [DOI] [PubMed] [Google Scholar]
- 48.Lorger M. and Felding-Habermann B (2010) Capturtng changes in the brain microenvironment during initial steps of breast cancer brain metastasis. Am. J. Pathol 176, 2958–2971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sofroniew MV (2015) Astrocyte barriers to neurotoxic inflammation. Nat. Rev. Neurosci 16, 249–263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang X et al. (2013) Astrocytic Fas ligand expression is required to induce T-cell apoptosis and recovery from experimental autoimmune enceph lomyelitis. Eur. J. Immunoi 43, 115–124 [DOI] [PubMed] [Google Scholar]
- 51.Wanner IB et al. (2013) Glial scar borders are formed by newly proliferated, elongated astrocytes that interact to corral inflammatory and fibrotic cells via STAT3-dependent mechanisms after spinal cord injury. J. Neurosci 33, 12870–12886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Carbonell WS et al. (2009) The vascular basement membrane as ‘soil’ in brain metastasis. PLoS One 4, e5857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rafii S et al. (2016) Angiocrine functions of organ-specific endothel al cells. Nature 529, 316–325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Loulier K et al. (2009) Beta1 integrin maintains integrity of the embryonic neocortical stem cell niche. PLoS Biol 7, e1000176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shen Q et al. (2008) Adult SVZ.stem cells lie in a vascular niche: a quantitative analysis of niche celkcell interactions. Cell Stem Call 3, 289–300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Holash J et al. (1999) Vessel cooption, regression, and growth in tumors mediated by angiopoietins and VEGF. Science 284, 1994–1998 [DOI] [PubMed] [Google Scholar]
- 57.Berghoff AS et al. (2013) Invasion patterns in brain metastases of solid cancers. Neuro Oncol 15, 1664–1672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bentolila LA. et al. (2016) Imaging ofangiotropism/vascular co-option in a murine model of brain melanoma: implications for melanoma progression along extravascular pathways. Sci. Rep 6, 23834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Garcia MA et al. (2017) Discovery of additional brain metastases on the day of stereotactic radiosurgery: risk factors and outcomes. J. Neurosurg 126, 1756–1763 [DOI] [PubMed] [Google Scholar]
- 60.Xing F et al. (2013) Reactive astrocytes promote the metastatic growth of breast cancer stem-like cells by activating Notch signalling in brain. EMBO Mol. Med 5, 384–396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lin Q et al. (2010) Reactive astrocytes protect melanoma cells from chemotherapy by sequestering intracellular calcium through gap junction communication channels. Neoplasia 12, 748–754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Stoletov K et al. (2013) Role of connexins in metastatic breast cancer and melanoma bran colonization. J. Cell Sci 126, 904–913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kim S-J et al. (2011)Astrocytes upregulate survival genes in tumor cells and induce protection from chemotherapy. Neoplasia 13, 286–298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Adkins CE et al. (2016) Characterization of passive permeability at the blood-tumor barrier in five preclinical models of brain metastases of breast cancer. Clin. Exp. Metastasis 33, 373–383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lockman PR et al. (2010) Heterogeneous blood-tumor barrier permeability determines drug efficacy in experimental brain metastases of breast cancer. Clin. Cancer Res 16, 5664–5678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lyle LT. et al. (2016) Alterations in pericyte subpopulations are associated with elevated blood-tumor barrier permeability in experimental brain metastasis of breast cancer. Clin. Cancer Res 22, 5287–5299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Dickens AM. et al. (2017) Astrocyte-shed extracellular vesicles regulate the peripheral leukocyte response to inflammatory brain lesions. Sci. Signal 10, eaaai7696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Schwartz H et al. (2016) Incipient melanoma brain metastases instigate astrogliosis and neuroinflammation. Cancer Res 76, 4359–4371 [DOI] [PubMed] [Google Scholar]
- 69.Rietkötter E et al. (2015) Anti-CSF-1 treatment is effective to prevent carcinoma invasion induced by monocyte-derived cells but scarcely by microglia. Oncotarget 6, 15482–15493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Rietkötter E et al. (2013) Zoledronic acid inhibits macrophage/microglia-assisted breast cancer cell invasion. Oncotarget 4, 1449–1460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Pukrop T et al. (2010) Microglia promote colonization of brain tissue by breast cancer cells in a Wnt-dependent way. Glia 58, 1477–1489 [DOI] [PubMed] [Google Scholar]
- 72.Brantley EC et al. (2010) Nitric oxide-mediated tumoricidal activity of murine microglial cells. Transl. Oncol 3, 380–388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Louie E et al. (2013) Neurotrophin-3 modulates breast cancer cells and the microenvironment to promote the growth of breast cancer bran metastasis. Oncogene 32, 4064–4077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bowman RL et al. (2016) Macrophage ontogeny underlies differences in tumor-specific education in brain malignancies. Cell Rep 17, 2445–2459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hara M et al. (2017) Interaction of reactive astrocytes with type I collagen induces astrocytic scar formation through the integrin-N-cadherin pathway after spin l cord injury. Nat. Med 23, 818–828 [DOI] [PubMed] [Google Scholar]
- 76.Keren-Shaul H et al. (2017) A unique microgliatype associated with restricting development of Alzheimer’s disease. Cell 169, 1276–1290 [DOI] [PubMed] [Google Scholar]
- 77.Anderson MA et al. (2016) Astrocyte scar formation aids central nervous system axon regeneration. Nature 532, 195–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Liddelow SA et al. (2017) Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541, 481–487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Soreq L et al. (2017) Major shifts in glial regional identity are a transcriptional hallmark of human brain aging. Cell Rep 18, 557–570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Masgrau R et al. (2017) Should we stop saying ‘glia’ and ‘neuroinflammation’? Trends Mol. Med 23, 486–500 [DOI] [PubMed] [Google Scholar]
- 81.Clarke JL et al. (2010) Leptomeningeal metastases in the MRI era. Neurology 74, 1449–1454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Brower JV et al. (2016) Management of leptomeningeal metastases: prognostic factors and associated outcomes. J. Can. Neurosd 27, 130–137 [DOI] [PubMed] [Google Scholar]
- 83.Chamberlain M et al. (2017) Leptomeningeal metastases: a RANO proposal for response criteria. Oncal 19, 484–492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kokkoris CP (1983) Leptomeningeal carcinomatosis. How does cancer reach the pia-arachnoid? Cancer 51, 154–160 [DOI] [PubMed] [Google Scholar]
- 85.Boire A et al. (2017) Complement component 3 adapts the cerebrospinal fluid for leptomeningeal metastasis. Cell 168, 1101–1113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Glover RL et al. (2014)Teaching neurolmages: leptomeningeal lung carcinoma. Neurology 82, e183–4 [DOI] [PubMed] [Google Scholar]
- 87.Boyle R et al. (1980) Diffuse involvement of the leptomeninges by tumour - a clinical and pathological study of 63 cases. Postgrad. Med. J 56, 149–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Nayak L et al. (2013) Rare cell capture technology for the diagnosis of leptomeningeal metastasis in solid tumors. Neurology 80, 1598–1605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kesari S and Batchelor TT (2003) Leptomeningeal metastases. Neurol Clin 21, 25–66 [DOI] [PubMed] [Google Scholar]
- 90.Gwak H-S et al. (2013) Analysis of treatment outcomes of intraventricular chemotherapy in 105 patients for leptomeningeal carcinomatosis from non-small-cell lung cancer. J. Thorac. Oncal 8, 599–605 [DOI] [PubMed] [Google Scholar]
- 91.Scott BJ and Kesari S (2013) Leptomeningeal metastases in breast cancer. Am. J. Cancer. Res 3, 117–126 [PMC free article] [PubMed] [Google Scholar]
- 92.Morris PG et al. (2012) Leptomeningeal metastasis from non-small cell lung cancer: survivel and the impact of whole brain radiotherapy. J. Thorac. Oncol 7, 382–385 [DOI] [PubMed] [Google Scholar]
- 93.Wu Y-L et al. (2016) Intrathecal chemotherapy as a treatment for leptomeningeal metastasis of non-small cell lung cancer: A pooled analysis. Oncol. Lett 12, 1301–1314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Scott BJ et al. (2016) Leptomeningeal metastasis in breast cancer - a systematic review. Oncotarget 7, 3740–3747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Le Rhun E et al. (2017) EANO-ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up of patients with leptomeningeal metastasis from solid tumours. Ann. Oncol 28, iv84–iv99 [DOI] [PubMed] [Google Scholar]
- 96.Tan C-S et al. (2017) Treatment options for EGFR mutant NSCLC with CNS involvement - can patients BLOOM with the use of next generation EGFR TKls? Lung Cancer 108, 29–37 [DOI] [PubMed] [Google Scholar]
- 97.McTyre E et al. (2013) Whole brain radiotherapy for brain metastasis. Surg. Neurol. Int 4, S236–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Neman J et al. (2013) Co-evolution of breast-to-brain metastasis and neural progenitor cells. Clin. Exp. Metastasis 30, 753–768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Neman J et al. (2014) Human breast cancer metastases to the brain display GABAergic properties in the neural niche. Proc. Natl. Acad. Scl. U. S. A 111,984–989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.DeAngelis LM et al. (1989) Radiation-induced dementia in patients cured of brain metastases. Neurology 39, 789–796 [DOI] [PubMed] [Google Scholar]
- 101.Chang EL et al. (2009) Neurocognition in patients with brain metastases treated with radiosurgery or radiosurgery plus whole-brain irradiation: a randomised controlled trial. Lancet Oncal 10, 1037–1044 [DOI] [PubMed] [Google Scholar]
- 102.Brown PD et al. (2016) Effect of radiosurgery alone vs radio-surgery with whole brain radiation therapy on cognitive function in patients with 1 to 3 brain metastases: a randomized clinical trial. JAMA 316, 401–409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Soffetti R et al. (2013) A European Organisation for Research and Treatment of Cancer phase Ill trial of adjuvant whole-brain radiotherapy versus observation in patients with one to three brain metastases from solid tumors after surgical resection or radiosurgery: quality-of-life results. J. Clin Oncol 31, 65–72 [DOI] [PubMed] [Google Scholar]
- 104.Aoyama H et al. (2007) Neurocognitive function of patients with brain metastasis who received either whole brain radiotherapy plus stereotactic radiosurgery or radiosurgery alone. Int. J. Radat. Oncol. Biol. Phys 68, 1388–1395 [DOI] [PubMed] [Google Scholar]
- 105.Satyamitra MM et al. (2016) Understanding the pathophysiology and challenges of development of medical countermeasures for radiation-induced vascular/endothelial cell injuries: report of a NIAD workshop, August 20, 2015. Radiat. Res 186,99–111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Monje ML et al. (2002) Irradiation induces neural precursor-cell dysfunction. Nat Med 8, 955–962 [DOI] [PubMed] [Google Scholar]
- 107.Brown PD et al. (2013) Memantine for the prevention of cognitive dysfunction in patients receiving whole-brain radio-therapy: a randomized, double-blind, placebo-controlled trial. Neuro Oncol 15, 1429–1437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Rothman SM and Olney JW (1987) Excitotoxity and the NMDA receptor. Trends Neurosci 10, 299–302 [DOI] [PubMed] [Google Scholar]
- 109.Suh JH (2014) Hippocampal-avoidance whole-brain radiation therapy: a new standard for patients with brain metastases? J. Clin. Oncol 32, 3789–3791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Gondi V et al. (2014) Preservation of memory with conformal avoidance of the hippocampal neural stem-cell compartment during whole-brain radiotherapy for brain metastases (RTOG 0933): a phase II multi-institutional trial. J. Clin. Oncol 32, 3810–3816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Rapp SR et al. (2015) Donepezil for irradiated brain tumor survivors: a phase Ill randomized placebo-controlled clinical trial. J. Qin. Oncol 33, 1653–1659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Puhalla S et al. (2015) Unsanctifying the sanctuary: challenges and opportunities with brain metastases. Neuro Oncol 17, 639–651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Osswald M et al. (2016) Impact of blood-brain barrier integrity on tumor growth and therapy response in brain metastases. Clin. Cancer Res 22, 6078–6087 [DOI] [PubMed] [Google Scholar]
- 114.luchi T et al. (2015) Frequency of brain metastases in non-small-cell lung cancer, and their association with epidermal growth factor receptor mutations. Int J. Clin. Oncol 20, 674–679 [DOI] [PubMed] [Google Scholar]
- 115.Nicoś M et al. (2018) Screening for ALK abnormalities in central nervous system metastases of non-small-cell lung cancer. Brain Pathol 28, 77–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Carlino MS et al. (2014) Correlation of BRAF and NRAS mutation status with outcome, site of distant metastasis and response to chemotherapy in metastatic melanoma. Br. J. Cancer 111, 292–299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Lynch TJ et al. (2004) Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-smallcell lung cancer to gefitinib. N. Engl. J. Med 350, 2129–2139 [DOI] [PubMed] [Google Scholar]
- 118.Hotta K et al. (2004) Effect of gefitinib (‘lressa’, ZD1839) ZD on brain metastases in patients with advanced non-small-cell lung cancer. Lung Cancer 46, 255–261 [DOI] [PubMed] [Google Scholar]
- 119.Namba Y et al. (2004) Gefitinib in patients with brain metastases from non-small-cell lung cancer: review of 15 clinical cases. Clin. Lung Cancer 6, 123–128 [DOI] [PubMed] [Google Scholar]
- 120.Porta R et al. (2011) Brain metastases from lung cancer responding to eriotinib: the importance of EGFR mutation. Eur. Respir. J 37, 624–631 [DOI] [PubMed] [Google Scholar]
- 121.luchi T et al. (2013) Phase II trial of gefitinib alone without radiation therapy for Japanese patients with brain metastases from EGFR-mutant lung adenocarcinoma. Lung Cancer 82, 282–287 [DOI] [PubMed] [Google Scholar]
- 122.Wu YL et al. (2013) Erlotinib as second-line treatment in patients with advanced non-small-cell lung cancer and asymptomatic brain metastases: a phase II study (CTONG-0803). Ann. Oncol 24, 993–999 [DOI] [PubMed] [Google Scholar]
- 123.Mok TS et al. (2017) Osimertinib or platinum-pemetrexed in EGFR T790M-positive lung cancer. N. Engl. J. Med 376, 629–640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Metro G et al. (2015) CSF concentration of crizotinib in two ALK-positive non-small-cell lung cancer patients with cns metastases deriving clinical beneit from treatment. J. Thorac. Oncol 10, e26–a27 [DOI] [PubMed] [Google Scholar]
- 125.Costa DB et al. (2011) CSF concentration of the anaplastic lymphoma kinase inhibitor crizotinib. J. Clin. Oncol 29, e443–e445 [DOI] [PubMed] [Google Scholar]
- 126.Crinò L et al. (2016) Multicenter Phase II study of whole-body and intracranial activity with ceritinib in patients with ALK-rear-rearranged non-small-cell lung cancer previously treated with chemotherapy and crizotinib: results from ASCEND-2. J. Clin. Oncol 34, 2866–2873 [DOI] [PubMed] [Google Scholar]
- 127.Kim D-W et al. (2017) Brigatinib in patients with crizotinib-refractory anaplastic lymphoma kinase-positive non-small-cell lung cancer: a randomized, multicenter Phase II trial. J. Clin. Oncol 35, 2490–2498 [DOI] [PubMed] [Google Scholar]
- 128.Gadgeel SM et al. (2016) Pooled analysis of CNS response to alectinib in two studies of pretreated patients with ALK-positive non-small-cell lung cancer. J. Clin. Oncol 34, 4079–4085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Long GV et al. (2012) Dabrafenib in patients with Val600Glu or Val600Lys BRAF-mutant melanoma metastatic to the brain (BREAK-MB): a mul icentre, opon-label, phase 2 trial. Lancet Oncol 13, 1087–1095 [DOI] [PubMed] [Google Scholar]
- 130.Davies MA et al. (2017) Dabrafenib plus trametinib in patients with BRAF(V600)-mutant melanoma brain metastases (COMBI-MB): a multicentre, multicohort, open-label, phase 2 trial. Lancet Oncol 18, 863–873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.McArthur GA et al. (2017) Vemurafenib in metastatic melanoma patients with brain metastases: an open-label, single-arm, phase 2, multicentre study. Ann. Oncol 28, 634–641 [DOI] [PubMed] [Google Scholar]
- 132.Bendell JC et al. (2003) Central nervous system metastases in women who receive trastuzumab-based therapy for metastatic breast carcinoma. Cancer 97, 2972–2977 [DOI] [PubMed] [Google Scholar]
- 133.Lin NU et al. (2008) Phase II trial of lapatinib for brain metastases in patients with human epidermal growth factor receptor 2-positive breast cancer. J. Qin. Oncol 26, 1993–1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Lin NU et al. (2009) Multicenter phase II study of lapatinib in patients with brain metastases from HER2-positive breast cancer. Clin. Cancer Res 15, 1452–1459 [DOI] [PubMed] [Google Scholar]
- 135.Bachelot T et al. (2013) Lapatinib plus capecitabine in patients with previously untreated brain metastases from HER2-positive metastatic breast cancer (LANDSCAPE): a single-group phase 2 study. Lancet Oncol 14, 64–71 [DOI] [PubMed] [Google Scholar]
- 136.Freedman RA et al. (2016) Translational Breast Cancer Research Consortium (TBCRC) 022: A Phase II trial of neratinib for patients with human epidermal growth factor receptor 2-positive breast cancer and brain metastases. J. Oin. Oncol 34, 945–952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Kodack DP et al. (2012) Combined targeting of HER2 and VEGFR2 for effective treatment of HER2-amplified breast cancer brain metastases. Proc. Natl.Acad. Sci. U. S. A 109, E3119–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Holohan C et al. (2013) Cancer drug resistance: an evolving paradigm. Nat Rev. Cancer 13, 714–726 [DOI] [PubMed] [Google Scholar]
- 139.Maddalo D et al. (2014) In vivo engineering of oncogenic chromosomal rearrangements with the CRISPR/Cas9 system. Nature 516, 423–427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Lee HW et al. (2015) Patient-derived xenografts from non-small cell lung cancer brain metastases are valuable translational platforms for the development of personalized targeted therapy. Qin. Cancer Res 21, 1172–1182 [DOI] [PubMed] [Google Scholar]
- 141.Kipnis J (2016) Multifaceted interactions between adaptive immunity and the central nervous system. Science 353, 766–771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Berghoff AS. et al. (2013) Characterization of the inflammatory response to solid cancer metastases in the human brain. Clin. Enp. Metastasis 30, 69–61 [DOI] [PubMed] [Google Scholar]
- 143.Amit M et al. (2013) Characterization of the melanoma brain metastatic niche in mice and humans. Cancer Med 2, 155–163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Sugihara AQ. et al. (2009) Regulatory T cells actively infiltrate metastatic brain tumors. Int. J. Oncol 34, 1533–1540 [DOI] [PubMed] [Google Scholar]
- 145.Louveau A et al. (2015) Structural and functional features of central nervous system lymphatic vessels. Nature 523, 337–341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Aspelund A et al. (2015) A dural lymphatic vascular system that drains brain interstitial fluid and macromolecules. J. Enp. Med 212, 991–999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Absinta M et al. (2017) Human and nonhuman primate meninges harbor lymphatic vessels that can be visualized noninva-sively by MRI. elite 6, e29738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Wyler L et al. (2014) Brain metastasis in renal cancer patients: metastatic pattern, tumour-associated macrophages and che-mokine/chemoreceptor expression. Br. J. Cancer 110, 686–694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Berghoff AS. et al. (2016) Tumor infiltrating lymphocytes and PD-L1 expression in brain metastases of small cell lung cancer (SCLC). J. Neurooncol 130, 19–29 [DOI] [PubMed] [Google Scholar]
- 150.Duchnowska R et al. (2016) Immune response in breastcancer brain metastases and their microenvironment: the role of the PD-1/PD-L axis. Breast Cancer Res 18, 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Mansfield AS et al. (2016)Temporaland spatial discordance of programmed cell death-ligand 1 expression and lymphocyte tumor infiltration between paired primary lesions and brain metastases in lung cancer. Ann. Oncol 27, 1953–1958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Margolin K et al. (2012) lpilimumab in patients with melanoma and brain metastases: an open-label, phase 2 trial. Lancet Oncol 13, 459–465 [DOI] [PubMed] [Google Scholar]
- 153.Goldberg SB et al. (2016) PembrOizumab for patients with melanoma or non-small-cell lung cancer and untreated brain metastases: early analysis of a non-randomised, open-label, phase 2 trial. Lancet Oncol 17, 976–983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Goldman JW et al. (2016) P2. 36: nivolumab (nivo) in patients (pts) with advanced (adv) NSCLC and central nervous system (CNS) metastases (mets): track: immunotherapy. J. Thorac. Oncol 11, S238–S239 [Google Scholar]
- 155.Slotman B et al. (2007) Prophylactic cranial irradiation in extensive small-cell lung cancer. N. Engl. J. Med 357, 664–672 [DOI] [PubMed] [Google Scholar]
- 156.Gore EM et al. (2011) Phase Ill comparison of prophylactic cranial irradiation versus observation in patients with locally advanced non-small-cell lung cancer: primary analysis of Radiation Therapy Oncology Group study RTOG 0214. J. Clin. Oncol 29, 272–278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Weidle UH et al. (2016) Dissection of the Process of Brain metastasis reveals targets and mechanisms for molecular-based intervention. Cancer Genomics Proteomics 13, 245–258 [PubMed] [Google Scholar]
- 158.Le Rhun E et al. (2017) Identification of single nucleotide polymorphisms of the PI3K-AKT-mTOR pathway as a risk factor of central nervous system metastasis in metastatic breast cancer. Eur. J. Cancer 87, 189–198 [DOI] [PubMed] [Google Scholar]
- 159.Arrieta O et al. (2009) Brain metastasis development and poor survival associated with carcinoembryonic antigen (CEA) level in advanced non-small cell lung cancer: a prospective analysis. BMC Cancer 9, 119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Palmieri D et al. (2014) Profound prevention of expermental brain metastases of breast cancer by temozolomide in an MGMT-dependent manner. Clin. Cancer Res 20, 2727–2739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Lin NU et al. (2013) CNS metastases in breast cancer: old challenge, new frontiers. Clin. Cnrcer Res 19, 6404–6418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Steeg PS (2012) Perspective: the right trials. Nature 485, S58–S59 [DOI] [PubMed] [Google Scholar]
- 163.Francia G et al. (2011) Mouse models of advanced spontaneous metastasis for experimental therapeutics. Nat Rev. Cancer 11, 135–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Winkler F (2015) The brain metastatic niche. J. Mo/. Med 93, 1213–1220 [DOI] [PubMed] [Google Scholar]
- 165.Winkler F (2014) The brain microenvironment: friend or foe for metastatic tumor cells? Neuro Oncd 16, 1565–1566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.llhan-Mu lu A et al. (2016) Bevacizumab prevents brain metastases formation in lung adenocarcinoma. Mot. CancerTiler 15, 702–710 [DOI] [PubMed] [Google Scholar]
- 167.Siam L et al. (2015) The metastatic infiltration at the metastasis/brain parenchyma-interface is very heterogeneous and has a significant impact on survival in a prospective study. Oncotarget 6, 29254–29267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Pencheva N et al. (2014) Broad-spectrum therapeutic suppression of metastatic melanoma through nuclear hormone receptor activation. Cell 156, 986–1001 [DOI] [PubMed] [Google Scholar]
- 169.Morsi A et al. (2013) Development and characterization of a clinically relevant mouse model of melanoma brain metastasis. Pigment Cell Melanoma Res 26, 743–745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Malladi S et al. (2016) Metastatic latency and Immune evasion through autocrine inhibition of WNT. Cell 165, 45–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Wall BA et al. (2015) Riluzole isa radio-sensitizing agent in an in vivo model of brain metastasis derived from GRM1 expressing human melanoma cells. Pigment Cell Melanoma Res 28, 105–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Contreras-Zárate MJ et al. (2017) Development of novel patient-derived xenografts from breast cancer brain metastases. Front. Oncol 7, 252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Kato M et al. (1998) Transgenic mouse model for skin malignant melanoma. Oncogene 17, 1885–1888 [DOI] [PubMed] [Google Scholar]
- 174.Meuwissen R et al. (2003) lnduction of small cell lung cancer by somatic inactivation of both Trp53 and Rb1 in a conditional mouse model. Cancer Cell 4, 181–189 [DOI] [PubMed] [Google Scholar]