

Summary
Leukemias exhibit a dysregulated developmental program mediated through both genetic and epigenetic mechanisms. Although IKZF2 is expressed in hematopoietic stem cells (HSCs), we found that it is dispensable for mouse and human HSC function. In contrast to its role as a tumor suppressor in hypodiploid B-Acute Lymphoblastic Leukemia, we find that IKZF2 is required for myeloid leukemia. IKZF2 is highly expressed in leukemic stem cells (LSCs) and its deficiency results in defective LSC function. IKZF2 depletion in AML cells reduced colony formation, increased differentiation and apoptosis, and delayed leukemogenesis. Gene expression, chromatin accessibility and direct IKZF2 binding in MLL-AF9 LSCs demonstrate that IKZF2 regulates a HOXA9 self-renewal gene expression program and inhibits a C/EBP-driven differentiation program. Ectopic HOXA9 expression and CEBPE depletion rescued the effects of IKZF2 depletion. Thus, our study shows that IKZF2 regulates the AML LSC program and provides a rationale to therapeutically target IKZF2 in myeloid leukemia.
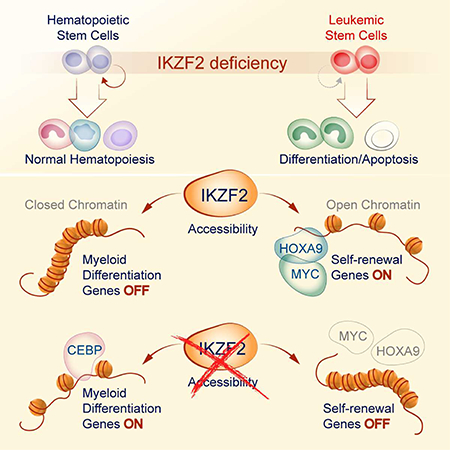
Graphical Abstract
eTOC blurb
Park et al. finds that IKZF2 regulates chromatin accessibility of self-renewal and differentiation genes in leukemic stem cells. Depletion of IKZF2 has preferential effect in leukemic stem cells compared to normal hematopoietic stem cells providing a new strategy for targeting leukemic stem cells.
Introduction
Acute myeloid leukemia (AML) is an aggressive bone marrow malignancy with uncontrolled expansion of immature myeloid cells coupled with a block in differentiation (Estey and Dohner, 2006). This disease is caused by various mutations and chromosomal translocations that target epigenetic regulators (Suva et al., 2013). Translocations of histone methyltransferases such as the mixed-lineage leukemia (MLL) gene give rise to one of the most aggressive subtypes of AML. MLL translocation occurs with more than 60 partners resulting in loss or alteration of its inherent histone methyltransferase activity (Ernst et al., 2004; Hess, 2004; Meyer et al., 2009). Among these fusion partners is the MLL-AF9 translocation t(9;11)(p22;q23), which has been associated with poor therapy response and poor prognosis (Krivtsov and Armstrong, 2007).
Studies have shown that AMLs induced by MLL-AF9 are organized in a cellular hierarchy with phenotypically distinct progenitor-like leukemia stem cells (LSCs) at the apex that self-renew to generate more LSCs and also give rise to differentiated progeny cells (Krivtsov et al., 2006; Somervaille and Cleary, 2006). Clinical studies have also shown that LSCs are resistant to chemotherapy and are responsible for relapse (Ishikawa et al., 2007; Jordan et al., 2006). Thus, understanding the underlying mechanisms of how LSCs undergo self-renewal and block differentiation could provide novel therapeutic strategies.
We and others identified a requirement for the MSI2 RNA binding protein (RBP) in LSC function (Kwon et al., 2015; Park et al., 2015). Previously, we determined that MSI2 maintains the MLL self-renewal program by interacting and retaining efficient translation of critical MLL regulated transcription factors (TFs) including Hoxa9, Myc and Ikzf2. Gene expression analysis identified Ikzf2 as the most downregulated gene in Msi2-deficient LSCs, which have a self-renewal defect (Park et al., 2015). Ikzf2, a member of the Ikaros TF family controls lymphocyte development, promotes quiescence and maintains inhibitory function of regulatory T cells, and is frequently deleted in hypodiploid B-ALLs (Grzanka et al., 2013; Iacobucci and Mullighan, 2017; Kim et al., 2015). Other studies have identified the expression of dominant negative isoforms in T-ALL patients (Asanuma et al., 2013; Tabayashi et al., 2007). These reports suggest a role for Ikzf2 as a tumor suppressor in lymphoid malignancies (Zhang et al., 2007). In contrast, our previous study demonstrated that IKZF2 is expressed in a murine MLL-AF9 leukemia and others have shown that it is a direct MLL fusion transcriptional target (Guenther et al., 2008; Mullighan et al., 2007; Park et al., 2015). Thus, we investigate IKZF2 role in regulating the balance of self-renewal and differentiation by modulation of chromatin accessibility in LSCs and its function in leukemogenesis.
Results
IKZF2 is required for leukemogenesis but dispensable for normal hematopoiesis
Based on previous studies indicating that Ikzf2 is expressed in HSCs, we decided to determine if it is required for normal hematopoietic function (Papathanasiou et al., 2009; Rebollo and Schmitt, 2003). To examine the role of IKZF2 in HSCs, we assessed different populations of the hematopoietic system in Ikzf2f/f and Ikzf2Δ/Δ littermates. Ikzf2 deletion was confirmed by genotyping PCR in bone marrow cells and by qPCR in sorted Lin− Sca1+c-Kit+ (LSK) cells (Figure S1A,B). In Ikzf2 hematopoietic deficient mice there was no significant difference in the frequency and absolute number of phenotypic HSCs, hematopoietic progenitors, mature myeloid, B-cells, T-cells or early erythroid progenitors (Figure S1C-L). To assess the in vitro functional output of HSPCs, we sorted LSK cells from Ikzf2f/f and Ikzf2Δ/Δ mice and performed colony assays, which resulted in comparable number of myeloid progenitor colonies in primary and secondary plating (Figure 1A-B). Furthermore, transplantation of bone marrow cells from Ikzf2f/f and Ikzf2Δ/Δ mice resulted in equivalent chimerism in all hematopoietic cells analyzed at different time points post-transplantation in primary and secondary transplanted mice (Figure S1M-P). These data suggest that Ikzf2 is dispensable for normal hematopoiesis.
Figure 1. IKZF2 is required for leukemogenesis but dispensable for normal hematopoiesis.

(A) Experimental scheme for investigating normal and transformed LSK cells. Leukemia initiation experiment was performed using the murine MLL-AF9 model. (B) Ikzf2 is dispensable for colony-forming ability of normal stem cells. Colony Assay was performed with LSK cells from Ikzf2f/f n=4 and Ikzf2Δ/Δ n=4 mice (C) Ikzf2 deletion in MLL-AF9 transformed LSK cells reduced colony formation. MLL-AF9 transformed LSK cells from Ikzf2f/f n=3 and Ikzf2Δ/Δ n=3 mice were used for colony assay. Mean +/− S.E.M of three independent experiments, Student’s t test **, p<0.01. (D) Deletion of Ikzf2 delays leukemia progression in MLL-AF9 model. Survival analysis is from the result of three combined transplants with genotypes Ikzf2f/f n=13 and Ikzf2Δ/Δ n=28 mice. **, p<0.01 logrank test. (E,F) Ikzf2Δ/Δ deleted leukemic mice have reduced disease burden. (E) Spleen and (F) liver weights of moribund Ikzf2f/f and Ikzf2Δ/Δ mice with proper deletion. Result is from Ikzf2f/f n=12 and Ikzf2Δ/Δ n=8 mice. **P < 0.01, unpaired Student’s t test. (G) QPCR of Ikzf2 in bone marrow leukemic cells from Ikzf2f/f n=12 and Ikzf2Δ/Δ n=8 mice. ****p<0.0001. unpaired Student’s t test. (H) Bone marrow leukemic cells from Ikzf2Δ/Δ mice have reduced IKZF2 expression. Frequency of IKZF2High cells in primary leukemic mice was examined by intracellular flow cytometry for IKZF2. Result from Ikzf2f/f n=12 and Ikzf2Δ/Δ n=10 mice is shown. ***, p<0.001. unpaired Student’s t test.
Our previous studies demonstrated that MSI2 directly binds to Ikzf2 mRNA and modulates its protein abundance in mouse MLL-AF9 LSCs (Park et al., 2015). Moreover, our data suggested that IKZF2 may have a functional requirement in myeloid leukemia. Thus, to investigate the role of IKZF2 in leukemia initiation and maintenance of AML, we utilized the MLL-AF9 retroviral transduction system (Figure 1A). LSK cells transduced with MLL-AF9 virus from Ikzf2f/f and Ikzf2Δ/Δ mice resulted in a significant block in colony formation in the Ikzf2Δ/Δ LSK cells (Figure 1C). Transplantation of transformed LSK cells showed a delayed leukemia progression (67 versus 48 days latency) and reduced disease burden in the Ikzf2-deficient transplanted mice compared to the control (Figure 1D-F). Ikzf2 deletion was also further verified by qPCR and intracellular flow analysis (Figure 1G-H). Of note the remaining leukemia cells from these mice did not upregulate the expression of other Ikaros family members (Ikzf1, Ikzf3 or Ikzf4), (Figure S1Q-S).
IKZF2 is required for leukemia maintenance in vitro and in vivo
For leukemia maintenance experiments, wildtype cells or Ikzf2f/f MLL-AF9 cells were transduced with a retrovirus expressing tamoxifen-controlled Cre recombinase (Cre-ER). (Figure 2A). Ikzf2 deletion was verified at 24hrs after 4-OHT treatment (Figure 2B). Acute deletion of Ikzf2 in established leukemia cells led to reduced colony formation and cell growth compared to control cells (Figure 2C and D). Ikzf2 deletion also resulted in rapid increase in apoptosis 24hrs after 4-OHT treatment, based on Annexin-V and 7-AAD staining compared to control cells (Figure 2E). Additionally, Ikzf2 deficiency led to a drastic increase in differentiation assessed by frequency and Median Fluorescence Intensity (MFI) of myeloid markers Mac-1, Gr-1, F4/80 and CD115 (Figure 2F-G and Figure S2A-B) and cellular morphology (Figure S2C) at 24hrs post treatment. In agreement, leukemic mice treated in vivo with tamoxifen demonstrated delayed leukemogenesis in the Ikzf2f/f group compared to the wildtype group (44 days versus 22 days in 4-OHT treated Ikzf2f/f versus wildtype) (Figure 2H) indicating that IKZF2 plays an important role in maintaining MLL-AF9 leukemia. The effect was specific to IKZF2 loss as overexpression could rescue the increase in differentiation after acute IKZF2 ablation (Figure 2I and S2D). The role for IKZF2 in leukemia was also validated in AML-ETO9a transformed murine leukemia cells (Wang et al., 2011). IKZF2 depleted with two shRNAs in the AML-ETO9a cells resulted in reduced colony formation, proliferation, and delayed leukemogenesis (Figure S2E-I). Most importantly, in the mice that succumbed to leukemia, we were able to detect IKZF2 expression indicating selection against IKZF2 depletion (Figure S2J). Taken together, we demonstrate that IKZF2 contributes to leukemogenesis in two independent murine models of myeloid leukemia.
Figure 2. IKZF2 is required for leukemia maintenance in vitro and in vivo.

(A) Experimental scheme for investigating IKZF2 role in leukemia maintenance in vitro and in vivo. (B) QPCR analysis showing Ikzf2 is acutely deleted by 4-OHT treatment in MLL-AF9 Ikzf2f/f cre-ER cells. (C) Ikzf2 deletion reduces colony forming ability in MLL-AF9 Ikzf2f/f cre-ER cells. (D) Ikzf2 deletion reduces cell growth. (E) Deletion of Ikzf2 increases apoptosis. Left, the percentage of apoptotic cells was determined at 24hr after 4-OHT treatment. Cells were stained for Annexin V and 7-AAD, and quantified by flow cytometry. Right, representative cytometric flow plot showing Annexin V and 7-AAD staining. (F-G) Myeloid differentiation is increased in Ikzf2 deleted leukemic cells. Frequency of population with high myeloid differentiation markers, (F) Mac-1/Gr-1 and (G) CD115 and F4/80 was measured by flow cytometry. Representative flow plots for these myeloid markers are shown in panels (F, Right and G, Right). Data shown in (B, D-G) were analyzed 24 hrs after MLL-AF9 WT or Ikzf2f/f cre-ER cells were treated with10 nM 4-OHT. Results (shown in (B - G) were combined from at least three independent experiments, using one MLL-AF9 WT cre-ER line and three MLL-AF9 Ikzf2f/f cre-ER lines. Mean +/− SEM Student’s t test *p<0.05, ** p<0.01, *** p<0.001, **** p<0.0001 (H) Ikzf2 deletion after leukemic cells are transplanted in mice, delays progression. Survival analysis of mice transplanted with MLL-AF9 WT or Ikzf2f/f cre-ER leukemic cells, treated with corn oil (vehicle) or 160mg/kg Tamoxifen. Data shown are combined from two independent transplants. Indicated groups WT,WT+TAM, Ikzf2f/f and Ikzf2f/f TAM have n=5, n=5, n=9, n=9 mice respectively. ****, p<0.0001 log-rank test. (I) Overexpression of Flag-CBP-IKZF2 partially rescued the differentiation phenotype of MLL-AF9 Ikzf2f/f leukemic cells deleted of endogenous Ikzf2. FACS analysis of F480 and CD115 expression in MLL-AF9 Ikzf2f/f leukemic cells treated with or without 20 nM 4-OHT for 24 hrs. n = 3 independent experiments; error bars, S.E.M *P < 0.05, **P < 0.01, two-tailed t test.
IKZF2 is highly expressed in leukemic stem cells and is required for maintaining leukemic stem cell activity
Previously, we found that MSI2 is required for the LSC program and since IKZF2 is a downstream target of MSI2, we examined IKZF2 level in primary leukemic cells from the MLL-AF9 model. IKZF2 was found to be highly expressed in the c-KitHigh cells, which are enriched for the LSC fraction both at the level of mRNA and protein, suggesting that IKZF2 could contribute to LSC function (Figure 3A-C and Figure S3A). Interestingly, the frequency of the leukemic-GMPs (L-GMP; c-KitHigh FcgRIIb+ CD34+) and the expression of CD34 marker was modestly reduced in the primary Ikzf2Δ/Δ transplanted mice compared to control mice (Figure S3B-C). The reduction of LSC frequency also correlated with the reduced plating efficiency from the Ikzf2-deficient leukemia cells compared to the control obtained from primary transplanted animals, (Figure 3D). More strikingly, serial in vivo transplantation from the primary transplanted mice from the initiation experiment (secondary, tertiary and quaternary) resulted in a progressive delay of leukemogenesis in the Ikzf2-deficient cells compared to the control mice (Figure 3E-G). In the quaternary 66% of the Ikzf2-deficient transplanted mice survived without any signs of leukemia suggesting that IKZF2 was required to maintain LSC function. We then directly assessed the functional LSC frequency from the primary transplanted leukemic mice from the initiation experiment by performing a limiting dilution transplantation assay (Figure 3H and Figure S3D). The leukemia initiating frequency in the Ikzf2-deficient cells was greatly reduced compared to the control by approximately 63-fold (1:7,697 versus 1:122 cells). The LSC defect was also observed when Ikzf2 was deleted after leukemic transformation as observed by a delay in secondary transplants of cells from mice that were prior treated with tamoxifen compared to untreated mice (Figure 3I). Moreover, mice that were treated with tamoxifen in the primary transplant experiment did not have good deletion except for one mouse in which all secondarily transplanted mice from this donor no longer had Ikzf2 deletion at point of death, indicating a selective pressure for loss of Ikzf2 deletion in these leukemic mice (Figure S3E). These results indicate that IKZF2 is required for LSC function in MLL-AF9 leukemia.
Figure 3. IKZF2 is highly expressed in LSCs and is required for maintaining LSC activity.

(A) Ikzf2 mRNA is highly expressed in LSCs, cells enriched in c-KitHigh cells compared to c-KitLow cells. Ikzf2 qPCR was performed in sorted c-KitHigh and c-KitLow cells from bone marrow of MLL-AF9 Ikzf2f/f leukemic mice n=4, Student’s t test * p<0.05. (B) Representative flow plot showing the gating for c-KitHigh (top 15%) and c-KitLow (bottom 40%) cells, and also the gating for IKZF2 High cells shown in (C). (C) c-KitHigh cells have higher expression of IKZF2 compared to c-KitLow cells. Frequency of IKZF2High population was measured in c-KitHigh and c-KitLow cells of MLL-AF9 leukemic mice. Mean and S.E.M of experiments from leukemic bone marrow cells isolated from Ikzf2f/f n=15 mice ***, Student’s t test p<0.001.
(D) Colony Assay was performed with bone marrow leukemic cells from primary transplanted Ikzf2f/f n=3 and Ikzf2Δ/Δ n=3 mice. Student’s t test. *p<0.05. (E) Delay in survival between Ikzf2f/f and Ikzf2Δ/Δ mice is increased in secondary transplant of cells from (Fig 1D), compared to primary transplant. Result is from two combined transplants using four Ikzf2f/f and five Ikzf2Δ/Δ donors with recipients Ikzf2f/f n=19 and Ikzf2Δ/Δ n=26 mice. ***p<0.001. log-rank test. (F) Survival Curve of tertiary transplant of leukemic bone marrow cells from (E) shows worse LSC function when Ikzf2 is deleted. Transplant is shown with Ikzf2f/f n=5 and Ikzf2Δ/Δ n=5 mice ***p<0.001. log-rank test. (G) Survival Curve of quarternary transplant exhibits exhaustion of LSCs in Ikzf2Δ/Δ mice. Three donors of each genotype from (F) were transplanted into recipients n=13 and n=15 mice for Ikzf2f/f and Ikzf2Δ/Δ, respectively. **p<0.01. log-rank test. (H) Ikzf2 deleted leukemic mice have reduced LSC frequency. Limiting dilution experiment assay performed with bone marrow cells from primary leukemic mice is shown. Left panel, table shows different number of cells and mice used for the transplant.. Right panel, graph showing the frequency of LSCs in Ikzf2f/f and Ikzf2Δ/Δ leukemic mice. ELDA software was used to calculate the frequency of the LSCs (Hu and Smyth, 2009). (I) Survival analysis for secondary transplant of the maintenance experiment is shown. Result is from transplanting Ikzf2f/f nonTAM=10 and Ikzf2 f/f prior TAM n=10 mice, with two and one donors respectively. **p<0.01.
IKZF2 is dispensable for normal human CD34+ HSPC cells but required for human leukemia cell survival
Since we found that IKZF2 did not contribute to normal murine HSC function, we then tested if it was dispensable in human HSPCs. We utilized two independent shRNAs for human IKZF2 in normal CD34+ enriched cord blood cells and found comparable colony forming ability and myeloid differentiation with no increase in cell death (Figure 4A-B and S4A-C). To understand IKZF2 function in human AML cells, we first measured protein levels in various Chronic Myeloid Leukemia-Blast Crisis and AML cell lines. We found a spectrum of IKZF2 protein expression in 7 out of 8 myeloid leukemia cell lines spanning various subtypes of AML disease (Figure 4C). We initially focused on MOLM-13 cells because they harbor an MLL translocation. To evaluate the effects of IKZF2 depletion, we first confirmed that shRNAs specific for IKZF2 led to reduced IKZF2 expression by western blot analysis in MOLM-13 cells. Similar to what was observed in mouse MLL-AF9 cells, IKZF2 depletion resulted in reduced colony formation, proliferation and increased apoptosis and differentiation (Figure 4D-H and S4D). Transplantation of MOLM-13 cells into NSG mice resulted in delayed leukemogenesis in the IKZF2 shRNA transplanted groups compared to scramble group (Figure 4I). Additionally, the role for IKZF2 in myeloid leukemia was not specific only to MOLM-13 cells as shRNA depletion in KCL-22 (BCR-ABL), Kasumi-1 (AML1-ETO) and NOMO1 (MLL-rearranged) also resulted in similar effects of reduced proliferation, increased apoptosis and differentiation (Figure S4E-Q). We also demonstrated that the IKZF2 shRNA effect was on-target as overexpression of a shRNA resistant form of IKZF2 could rescue the apoptosis and differentiation effects in another AML cell line NB4 (PML-RAR) cells (Figure S4R-U). To determine if IKZF2 plays a role in primary AML, we examined IKZF2 protein levels in nine heterogeneous primary AML patients (Table S1) by intracellular flow cytometry and found IKZF2 expression is enriched in the CD34+CD38− fraction compared to the CD34− fraction (Figure 4J). We could also detect the expression of IKZF2 in the bulk AML cells from five out of the eight patients and of which three patients had increased abundance compared to normal human CD34+ enriched cord blood cells (Figure S4V). Depletion of IKZF2 in four primary patients resulted in reduced CD34+CD38− frequency (Figure 4K-L). We also found that IKZF2 loss contributed to reduced colony forming ability in two AML patient samples correlating with the efficiency of knockdown (Figure 4M and S4W). Utilizing five AML cell lines and four primary patient samples, we demonstrate that IKZF2 is required for human AML cells.
Figure 4. IKZF2 is required for human leukemia cell survival.

(A) Western blot analysis showing depletion of IKZF2 in human cord blood CD34+ (HSPCs) at day 4 post transduction with lentivirus expressing either Scramble control or two independent IKZF2 shRNAs. (B) Colony assay was performed with HSPCs in (A). 104 cells were plated at day 4 post transduction and different colonies were counted two weeks later. Result is from n=3 independent experiments. (C) Western blot of IKZF2 was performed in whole cell lysates from eight cell lines, using ACTIN as loading control. (D) Western blot analysis showing IKZF2 depletion in MOLM-13 cells after four days post-transduction with lentivirus expressing IKZF2 shRNAs. ACTIN was used as loading control. (E) Depletion of IKZF2 in MOLM-13 cells reduced colony formation. Colonies were measured a week after plating shRNA transduced-MOLM-13 cells. (F) IKZF2 knockdown increases apoptosis in MOLM-13 cells. Apoptosis was measured by flow cytometry using Annexin V-PE and 7-AAD staining at day 7 post-transduction. (G-H) IKZF2 knockdown leads to increased differentiation. Flow cytometry measuring myeloid markers (G) CD13/CD14 and (H) CD11b/CD33 in MOLM-13 cells at day 7 post-transduction. Results in (B-H) are from more than three independent experiments using triplicates are shown. Mean +/− S.E.M Student’s t test p value. *p<0.05, **p<0.01 and ***p<0.001. (I) IKZF2 knockdown prolongs leukemia-free survival in mice. MOLM-13 cells transduced with scramble or IKZF2 shRNA virus were counted at day 4 post-transduction and transplanted into sub-lethally irradiated NSG mice. Results from four combined transplants with total scram n=20, Ikzf2 shRNA-3 n=18, and Ikzf2 shRNA-5 n=20 mice are shown. log-rank test. **p<0.01 and ***p<0.001. (J) IKZF2 is highly expressed in CD34+CD38− population in AML patients. Left, IKZF2 intracellular flow cytometry data showing IKZF2 expression in CD34− and CD34+CD38− populations in nine AML patients. Right, representative flow plot showing gating of CD34− and CD34+CD38− populations. (K) IKZF2 Intracellular flow cytometry data in AML patients cells transduced with lentivirus expressing Scram control or IKZF2 shRNA at day 4 post transduction. Results are from experiments using cells from four AML patients. Mean +/− S.E.M Student’s t test p value. *p<0.05, (L) CD34+CD38− population is reduced when IKZF2 is knocked down in AML patient cells. Leukemic stem cell population was analyzed at day 4 post transduction with Scram or IKZF2 shRNA. Results are from four AML patients. Mean +/− S.E.M Student’s t test p value. *p<0.05. (M) IKZF2 depletion reduces colony formation in AML patient cells. Cells from Patient #1 were transduced with lentivirus expressing Scramble or IKZF2 shRNA and sorted for GFP at day 4 post transduction. Left, graph showing number of colonies scored 2 weeks after plating. Right, flow plot of intracellular IKZF2 staining in patient cells.
IKZF2 loss leads to increased accessibility of differentiation genes and decreased accessibility of self-renewal genes in LSCs
To obtain genome-wide insights into how IKZF2 regulates the MLL self-renewal program, we performed RNA-sequencing of MLL-AF9 Ikzf2f/f and Ikzf2Δ/Δ LSCs from the initiation experiment. We found 217 differentially expressed genes with 138 upregulated and 81 downregulated in Ikzf2Δ/Δ compared to Ikzf2f/f LSCs (FDR-corrected p < 0.05, Table S2) (Figure S5A). Gene Set Enrichment Analysis (GSEA)(Subramanian et al., 2005) was performed using the rank list of differentially expressed genes from the Ikzf2f/f and Ikzf2Δ/Δ LSCs (Table S2). As a result, GSEA identified signatures consistent with loss of the self-renewal program and increase in myeloid differentiation with Ikzf2 deletion (Figure S5B,C and Table S2). Interestingly, the top ranked gene sets also include loss of enrichment of MLL-AF9 direct targets, loss of MYC program and enrichment of HOXA9 and Meis1 downregulated genes in the Ikzf2Δ/Δ LSCs (Figure S5D-F). Surprisingly, we also found loss of enrichment of MSI2 direct RNA targets when Ikzf2 is lost suggesting that there is a link between the transcription regulation by IKZF2 and translation regulation by MSI2 (Figure S5G).
With previous reports implicating the role of IKZF2 in chromatin remodeling, we performed genome-wide ATAC-sequencing in MLL-AF9 LSCs to understand the global chromatin structure when IKZF2 is lost. IKZF2 loss led to substantial changes in chromatin accessibility with a gain of 127 peaks (FDR<0.2) and loss of 235 accessible peaks (FDR<0.2), (Figure 5A and Table S3). Annotation of the changes in the ATAC-seq peaks in the Ikzf2Δ/Δ LSCs indicate that a substantial portion of the decreased accessibility changes occur in the intronic regions (34.65% for open peaks compared with 45.95% for closed peaks) whereas more promoter regions are opened than closed (21.26% for open peaks; 12.77% for closed peaks) in the Ikzf2Δ/Δ LSCs (Figure 5B).
Figure 5. IKZF2 loss leads to increased accessibility of differentiation genes and decreased accessibility of self-renewal genes in LSCs.

(A) Scatterplot to show differentially accessible peaks from ATAC-seq analysis on Ikzf2f/f n=2 and Ikzf2Δ/Δ n=2 LSCs (c-Kit High) sorted from bone marrow of primary leukemic mice, under the Benjamini-Hochberg adjusted q-value threshold of 0.2. Red dots represent more accessible and blue represents less accessible peaks. (B) Location of increased accessible (left panel) and decreased accessible (right) ATAC-seq peaks in Ikzf2Δ/Δ LSCs. (C) Chromatin accessibility has positive correlation with gene expression. Cumulative distribution function (CDF) graph shows enrichment of accessibility in upregulated genes (shown in red) from the RNA sequencing data of Ikzf2f/f and Ikzf2Δ/ Δ LSCs. Loss of accessibility is found in downregulated genes (shown in blue). (D) Ikzf2 deletion results in increased accessibility and expression of myeloid differentiation program. Overlap of gene sets from genes with increased accessibility (ATAC-seq data), increased and decreased RNA expression (RNA-seq data) leading to seven gene sets including myeloid development, C/EBPα network, neutrophil maturation and gene set that is increased when Hoxa9 and Meis1 is upregulated. (E) TF motifs enriched in differentially accessible regions in Ikzf2Δ/Δ LSCs. C/EBPε and C/EBPδ motifs are the most enriched motifs in the increased accessible regions whereas HOXA9 motif is the top TF motif for loss of enrichment in decreased accessible regions in Ikzf2 deleted LSCs. Further information is found in STAR methods. (F) Tornado plot of differentially accessible ATAC-seq peaks (left) selected by nominal p ≤ 0.05. 1191 opened and 1523 closed peaks are shown. The genomic domain is defined as the summit ± 3 kb. To show the native binding of IKZF2, the signal of CUT&RUN sequencing at the same loci (right) is shown in parallel. (G) Data analyzed in (E) was further queried by additionally overlapping with genomic regions bound by IKZF2. TF motifs enriched in differentially accessible regions by the nominal p-value cutoff of 0.05 and showing averaged signal of IKZF2 CUT&RUN above 0.5 TPM over the region of summit ± 250 bp, encompassing 9259 peaks. TFs with the K-S test effect size above 0.05 and the odds ratio above 1.3 are highlighted with red (opened) and blue (closed) colors, respectively. The two IKZF2 motifs from TRANSFAC are also highlighted.
This result suggests that reduced accessibility preferentially occurs in intronic enhancers whereas increased accessibility is found in classical promoters, when IKZF2 is lost. Analysis assessing the cumulative distribution function (CDF) of differential accessibility from ATAC-seq data and differentially expressed genes from RNA-seq data revealed that there is a clear correlation between accessibility and expression of genes that are both upregulated and downregulated after Ikzf2 deletion, respectively (Figure 5C). To understand the processes regulated by the accessibility and transcription of genes affected by IKZF2 loss, we overlapped gene sets with differential accessibility and gene expression (Figure 5D, Figure S5H and Table S2–3). Interestingly, overlap of increased accessibility and increased RNA expression identified seven gene sets that include myeloid development, CCAAT/enhancer-binding proteinα (C/EBPα) network and myeloid cell maturation (Figure 5D) indicating the myeloid differentiation program is strongly turned on in the Ikzf2Δ/Δ LSCs.
To determine how Ikzf2 deletion alters the accessibility of specific TF binding sites, we analyzed all the expressed TF motifs. We found that deletion of Ikzf2 led to increased accessibility of fourteen TF motifs whereas only one had significant decreased accessibility. In accordance with IKZF2 acting as a transcriptional suppressor, IKZF2 motifs (Figure 5SI) were found to be among the motifs with increased accessibility. Among the affected motifs, the most increased motifs were the C/EBPδ and C/EBPε while HOXA9 motif became less accessible in the Ikzf2Δ/Δ LSCs (Figure 5E). The ATAC-seq data can be used to predict the direct sites regulated by IKZF2 by examining genomic regions with changes in accessibility that associate with IKZF2 motifs. Using this approach, we were able to identify the C/EBPδ, C/EBPε and HOXA9 binding sites within the peaks that contained the IKZF2 motifs (Figure S5I-J). To directly assess where IKZF2 binds, we utilized the protocol from a published method called cleavage under targets and release using nuclease (Cut and Run) (Skene et al., 2018; Skene and Henikoff, 2017) to identify sites bound by IKZF2 (Figure 5F). This method allows mapping of TF binding sites in the native state without cross-linking and chromatin fragmentation, and produces higher signal to background noise ratio making it ideal for studying challenging chromatin factors that are known to be difficult for chromatin immunoprecipitation. To further analyze the regions with accessibility changes that are directly bound by IKZF2, we overlapped the differential ATAC-seq peaks with the IKZF2 cut and run peaks, and found that CEBP and HOXA9 motifs remained the most affected motifs consistent with the motif assessment from the ATAC-seq data alone (Figure 5F-G). Furthermore, the CDF plot of differential accessibility from the ATAC-seq data, and cut and run suggest that IKZF2 binds to genomic regions that are more open (Figure S5K). More specifically, we found that 14 genes shared IKZF2 and C/EBP motifs, that had increased gene expression (Log2FC>0.75, pval<0.05) coupled with increased accessibility (Log2FC>1, pval<0.05) after Ikzf2 deletion and had direct IKZF2 binding (Figure S5L and Table S4–5).
These data suggest that IKZF2 loss could directly alter the chromatin accessibility of specific genes that are linked to the gain of the differentiation and loss of self-renewal programs. Accordingly, some of these genes have been implicated in immune function and myeloid differentiation including S100a8, S100a9, C3, Fpr2, and Mpo (Figure S5M). These results demonstrate that IKZF2 loss leads to changes in chromatin structure that results in increased accessibility and expression of myeloid differentiation genes.
IKZF2 represses the expression of differentiation transcription factor, C/EBP and maintains expression of self-renewal transcription factors, HOXA9 and MYC.
To understand if IKZF2 was actively maintaining the epigenetic state, we performed a kinetics analysis with the genes we identified in the primary transplanted LSCs. Utilizing the Cre-ER deletion system, we found that Ikzf2 deletion could be verified as early as 14hrs post 4-OHT treatment in the LSCs (Figure 6A). Among the 14 genes tested that demonstrated increased accessibility and increased expression after Ikzf2 was deleted in the LSCs from the primary transplanted mice, we identified a similar rapid increase in mRNA levels of C3, Fpr2, S100a8 and S100a9 (Figure 6B-E and Figure S6A). Myeloid differentiation related genes were also upregulated rapidly after acute Ikzf2 deletion (Figure S6B). In accordance with the C/EBPε motif being the most accessible after Ikzf2 deletion, we found increased mRNA abundance of C/ebpε among the C/ebp family members after acute IKZF2 depletion (Figure 6F and Figure S6C). Additionally, we observed a 50% reduction in the mRNA of the L-GMP marker, Cd34 and a modest increase in FcgR2 supporting a rapid change in the surface phenotype of the LSC (Figure S6D-E). In concordance with our murine leukemia data, primary AML patients’ expression of CD34 also correlated with high IKZF2 expression (top 25%) compared to low expressing patients (bottom 25%) (Figure 6J). Furthermore, C3, CEBPE, CEBPD and CEBPB were significantly reduced in patients with higher IKZF2 expression compared to low expressing patients with a trend toward reduced levels of S100A8 and S100A9 (Figure 6K-M and S6F-H). Altogether these data support that IKZF2 actively represses the differentiation program.
Figure 6. IKZF2 represses the expression of differentiation transcription factor, C/EBP and maintains expression of self-renewal transcription factors, HOXA9 and C-MYC.

(A) QPCR analysis of Ikzf2 mRNA in sorted MLL-AF9 Ikzf2f/f cre-ER c-Kit High cells treated with 4-OHT for different time points. (B-E) Acute deletion of Ikzf2 in LSCs increases genes data showing that acute deletion of Ikzf2 reduces Hoxa9 and c-Myc mRNA levels. (A-H) Results shown are from four different experiments. Student’s t test *p<0.05, ** p<0.01, *** p<0.001, **** p<0.0001. (I) Western blot analysis showing reduction of IKZF2, HOXA9 and C-MYC after Ikzf2 is acutely deleted in MLL-AF9 Ikzf2f/f cre-ER High cells. (J-N) Box and whiskers plot showing correlation of the indicated genes in patients with high (n=24) versus low IKZF2 (n=45) expression in AML patients from TCGA dataset. (J) CD34 (K) C3 (L) CEBPE (M) CEBPD (N) MYC .*P < 0.05, **P < 0.01, ***P < 0.001, two-tailed t test.
Conversely, we also observed a role for IKZF2 in maintaining the c-MYC and HOXA9 gene expression program (Figure 5SE-F). In the primary transplanted leukemia cells deficient for IKZF2, we found reduced mRNA and protein levels of c-Myc and Hoxa9 compared to the controls (Figure 6SI-J). Similarly, this reduction of c-Myc and Hoxa9 gene expression was also observed after acute deletion at the mRNA levels at early time points and at protein levels after 24hrs post Ikzf2 deletion (Figure 6G-I). MYC levels also correlated with IKZF2 expression in the patients (Figure 6N). These data suggest that IKZF2 directly or indirectly maintains the expression of c-Myc and Hoxa9. Since genes containing HOXA9 motifs share IKZF2 binding and HOXA9 is rapidly lost after Ikzf2 deletion, we tested if reintroducing HOXA9 alone would reverse the cellular affects from depleting IKZF2. In line with HOXA9 being an important downstream mediator of IKZF2 function, forced expression of HOXA9 could partially rescue the differentiation, apoptosis and the reduction of colony formation caused by Ikzf2 deletion (Figure 7A-F). Similarly, CEBPE depletion could rescue the differentiation effects (Figure 7G-H) and reverse the activation of several co-regulated targets including S100a8 and S100a9 when IKZF2 is lost (Figure 7I-L).
Figure 7. CEBPE and HOXA9 can partially rescue phenotypes of Ikzf2 loss.

(A) Differentiation measured by CD115/F480 and (B) apoptosis were partially rescued when Ikzf2 was deleted in HOXA9 overexpressing MLL-AF9 Ikzf2f/f cre-ER cells. n=3 independent experiments. Student’s t test *p<0.05, ** p<0.01, **** p<0.0001. (C) Western blot analysis demonstrating efficient deletion of IKZF2 and overexpression of HOXA9. (D) Colony assay showing rescue of Ikzf2 deletion by HOXA9 overexpression in 4-OHT treated MLL-AF9 Ikzf2f/f cre-ER cells. (E) QPCR showing complete deletion of Ikzf2 in MLL-AF9 Ikzf2f/f cre-ER vector control and HOXA9 overexpressing cells after 4-OHT treatment. (F) QPCR showing expression of Hoxa9 in the vector control and HOXA9 expressing cells after 4-OHT treatment in the MLL-AF9 Ikzf2f/f cre-ER cells. (D-F) All data represent the mean +S.E.M of at least three independent replicates. * p < 0.05, ***p < 0.001, ****p<0.0001 Student’s t test. (G-J) CEBPE depleted cells lacks differentiation induction and IKZF2 targets caused by IKZF2 loss. (G) Differentiation state was measured by flow cytometry using Mac1 and Gr1 as markers in the MLL-AF9 Ikzf2f/f cre-ER cells transduced with lentivirus expressing scramble or Cebpe shRNA and were treated with 4-OHT or left alone. (H) Median Fluorescent Intensity of Mac1 was measured in same experiment in (G). (I) S100a8 (J) S100a9, targets of IKZF2 were measured using qPCR in experiments in (G). (K) Cebpe and (L) Ikzf2 mRNAs were measured to check for depletion in experiment mentioned in (G). (G-L) All data represent the mean +S.E.M of at least three independent replicates. * p < 0.05, **p < 0.01, ****p<0.0001 Student’s t test. (M) Model showing the role of IKZF2 in regulating chromatin accessibility of differentiation and self-renewal program in LSCs. IKZF2 loss leads to increased C/EBPε and chromatin accessibility of regions in differentiation genes containing C/EBP motifs, thereby turning on myeloid genes. HOXA9 expression and chromatin accessibility of HOXA9 motifs are reduced, turning off the self-renewal genes.
Overall, these results demonstrate that IKZF2 differentially maintains the accessibility and expression of the self-renewal and differentiation genes in LSCs. IKZF2 suppresses the accessibility of myeloid differentiation genes containing C/EBP motifs. Additionally, IKZF2 maintains accessibility of genes associated with the self-renewal program including genes with HOXA9 motifs in MLL-AF9 LSCs (Figure 7M).
Discussion
AML is characterized by a gain of self-renewal and a differentiation block. MLL fusion oncogenes found in some of the most aggressive myeloid leukemia establish a dysregulated epigenetic state within a myeloid progenitor that resembles a normal GMP that has acquired self-renewal (Krivtsov et al., 2006). Recently, post-transcriptional regulation in leukemia has been highlighted as a novel way for maintaining the LSC program (Park et al., 2015; Vu et al., 2017). Previously, we demonstrated that RBP MSI2 is required for LSC function by maintaining the translation of a number of MLL targets including Myc, Hoxa9 and Ikzf2 (Park et al., 2015). Post-transcriptional regulation is connected to transcriptional program by positive feedback mechanisms to sustain the LSC program. To understand the mechanistic link that connects the post-transcriptional regulation to the transcriptional program, we focused on one of these transcriptional regulators. Despite extensive studies implicating the importance of Myc and Hoxa9 in leukemia (Collins and Hess, 2016; Hoffman et al., 2002), the functional role for Ikzf2 in myeloid leukemia is unclear. Ikzf2 being the most down-regulated gene in Msi2-deficient LSCs and the high expression of IKZF2 and MSI2 in LSCs suggested that these factors can coordinate to maintain the LSC program. Furthermore, we found that IKZF2 depletion results in reduced expression of MSI2 direct mRNA binding targets (Figure S5G). Additionally, increased expression of IKZF2 was found in a Msi2-deficient leukemia that recovered its ability to transplant, further supporting that IKZF2 is playing an important role downstream of MSI2 (Park et al., 2015). These results suggest that the IKZF2-MSI2 axis can partially integrate the post-transcriptional regulation of RBPs to the transcription program to maintain LSC function.
Ikzf2, a member of the Ikaros TF family has been shown to be required for regulatory T cell functions (Kim et al., 2015; Thornton et al., 2010). The Ikaros family members play different roles in transcriptional regulation depending on the cell-type and differentiation stage, through transcriptional activation and repression, which are in part mediated by chromatin remodeling. Similar to IKZF1, IKZF2 has also been found to interact with chromatin remodeling complexes that contain histone deacetylase (HDAC), the nucleosome-remodeling and histone deacetylase (NuRD) and SIN3 complexes which are involved in gene repression (Sridharan and Smale, 2007; Zhao et al., 2016). Our data supports a dominant role for IKZF2 in repressive complexes in LSCs based on a larger number of significant TF motifs that become more accessible after Ikzf2Δ/Δ deletion which includes the known IKZF2 motif.
Genomic profiling and chromatin accessibility analysis demonstrated that IKZF2 loss led to increased myeloid differentiation. This phenotypic change correlated with induction of myeloid gene expression program. The most significantly increased accessible motifs were C/EBPδ and C/EBPε which are important for cytokinin induced granulocytic differentiation in leukemic cells and terminal differentiation of cells in the myeloid lineage, respectively (Ishii et al., 2005; Ramji and Foka, 2002). The C/EBPα is a prerequisite TF for LSC formation because it allows for differentiation of HSCs to a downstream myeloid progenitor (Ye et al., 2015). Myeloid differentiation was unperturbed in cells lacking IKZF2 but in the leukemic state IKZF2 loss was able to induce reactivation of the terminal differentiation gene expression program. The increased accessibility within the regions that contain both IKZF2 and C/EBP motifs in the Ikzf2Δ/Δ LSCs suggest that Ikzf2 deletion could lead to loss of the repressive complex recruitment or occupancy allowing transcription machinery to be recruited to the C/EBP motifs found in the myeloid genes to turn on transcription. Interestingly, acute deletion of Ikzf2 led to a rapid and significant increase in the transcription of myeloid genes regulated by C/EBPs and also the mRNA of C/ebpε suggesting an autoregulatory mechanism found in C/EBPs (Niehof et al., 2001; Ramji and Foka, 2002).
In contrast to the increased myeloid C/EBP program, IKZF2 loss led to reduced HOXA9 accessibility and loss of self-renewal gene expression. Acute deletion of Ikzf2 led to rapid reduction of Hoxa9 and c-Myc suggesting that it could be a direct effect of IKZF2 loss rather than a downstream effect from the activation of the differentiation program. This notion is strengthened by a recent report that showed that IKZF2 is part of an activator complex with RELA, FoxP3 and KAT5 (Kwon et al., 2017), suggesting that IKZF2 in an activator complex could be important for expression of self-renewal genes such as Hoxa9 and c-Myc. Additionally, a recent study has identified a variant NuRD complex in ES cells that includes the WDR5, a component of the SET/MLL1 complex that contributes to H3K4 trimethlyation (Ee et al., 2017). This MBD3C/NuRD complex can compete with the MLL1 complex for interaction with WDR5, suggesting opposing role to the MLL1 complex (Ee et al., 2017). With the previous described involvement of IKZF2 in the NuRD complex it will be interesting to determine if IKZF2 loss can affect the composition or localization of either of these complexes that leads to the reduction of the self-renewal program. Furthermore, since IKZF2 knockdown results in similar phenotype in human cell lines with different translocations (Figure 4 and S4; MLL-AF9, BCR-ABL, AML1-ETO and PML-RAR) and in two different murine AML models, we speculate that IKZF2 could be working through transcriptional complexes, not limited to MLL rearranged complexes.
Previous studies have been focused on IKZF2 function as a tumor suppressor as it is deleted in many B-ALLs patients (Grzanka et al., 2013; Iacobucci and Mullighan, 2017) and short isoforms acting as dominant negative are found in T-ALL patients (Asanuma et al., 2013; Tabayashi et al., 2007). Interestingly, we found that IKZF2 has an oncogenic role in AML by using primary patient cells, human cell lines and two mouse models. We demonstrated that IKZF2 is required for leukemia cell survival and LSC function by controlling chromatin accessibility of self-renewal and differentiation programs. How IKZF2 can have opposing roles in different cell types is unclear but it could be due to its recruitment to different complexes. Since IKZF2 can form heterodimers with other members of the family, we examined if other members can compensate for IKZF2 loss. We did not observe any significant changes in expression of the other members in Ikzf2-deficient leukemias. The finding that Ikzf2-deficient mice have no gross defect in HSC function and a requirement for LSC function suggests a possible therapeutic window. Despite the availability for drugs (i.e lenalidomide) that have specificity to degrade IKAROS members (IKZF1 and IKZF3), these proteins have an amino acid substitution in the critical region that precludes binding to these imids (Kronke et al., 2014). It might therefore be possible to modify this class of compounds to generate either specific IKZF2 inhibitors or pan IKAROS family degraders. We propose that unraveling how IKZF2 functions through different complexes may also provide insights into LSCs function and new venues for therapeutic intervention.
STAR Methods
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for reagents may be directed to, and will be fulfilled by the corresponding author Michael Kharas (kharasm@mskcc.org).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
6 to 8 weeks old Ikzf2fl/fl vavcre- or Ikzf2 fl/fl vavcre+ (Ikzf2Δ/Δ) mice in the C57BL/6 background strain were generously provided by Dr. Ethan Schevach (Thornton et al., 2010). Ikzf2 fl/fl mice were generated by inserting floxed LoxP sites flanking part of exon 8. 6–8 weeks old female C57BL/6 CD45.1 or CD45.2 mice purchased from Taconic Biosciences and Jackson Laboratories respectively, were used as recipients for transplant experiments. All animal procedures were approved by the Institutional Animal Care and Use Committee at Memorial Sloan Kettering Cancer Center.
Cell Culture and Generation of Stable Cell Lines
Generation of MLL-AF9 leukemic cells
Bone marrow cells were extracted from 6 to 8 weeks old Ikzf2fl/fl or Ikzf2Δ/Δ mice (Thornton et al., 2010). For LSK cell isolation, bone marrow cells were enriched for c-Kit positive cells by incubating with CD117 microbeads and then processed through the autoMACS Pro Separator (Miltenyi Biotec), following the manufacturer’s instructions. C-Kit enriched cells were stained with Lineage antibody cocktail (CD3, CD4, CD8, Gr1, B220, CD19, TER119 conjugated with PeCy5), Sca1-Pac Blue, CD34-FITC, SLAM-APC, CD48-PE, and c-KIT-APC-Cy7. Stained cells were sorted for Lin−Sca+Kit+ cells using a BD FACS Aria II instrument. Sorted LSK cells were incubated overnight in Stemspan SFEM medium (Stem Cell Technologies) with 10 ng/ml IL-3, 10 ng/ml IL-6, 50 ng/ml SCF, 10 ng/ml thrombopoietin (TPO), and 20 ng/ml FLT-3 ligand. Cells were incubated with supernatant containing retrovirus expressing MLL-AF9 together with GFP (a gift from Scott Armstrong, Dana Farber Cancer Institute) on retronectin-coated plates and spun for 1hr at 2500 RPM. Two cycles of spinfection was performed and the cells were grown in M3434 methylcellulose medium (Stem Cell Technologies) for a week. GFP positive cells were sorted and 200,000 GFP positive cells were injected retro-orbitally with 250,000 bone marrow support cells into lethally irradiated 6-week-old C57BL/6 mice.
Generation of MLL-AF9 WT and Ikzf2f/f cre-ER cells
Mouse leukemic bone marrow cells from WT and Ikzf2f/f mice were grown in RPMI (Cellgro) medium containing 10% FBS, 6 ng/ml IL-3, 10 ng/ml SCF, 10 ng/ml IL-6 and 10 ng/ml GM-CSF. Cells were spinfected with high titer retrovirus expressing cre-ER (virus was generated with MSCV-cre-ER-puromycin) twice and selected with 5ug/ml of puromycin.
Isolation, culture and transduction of cord blood-derived CD34+ HSPC cells.
For purification of cord blood CD34+ HSPCs, five to ten mixed CB units (each unit is from a healthy donor) were used. Purified mononuclear cells were obtained from cord blood by using Hespan and Ficoll-Hypaque Plus density centrifugation, followed by positive selection using the Auto MACS Pro Separator and isolation Kit (Miltenyi). CD34+ cells were grown in 80% Iscove’s modified Dulbecco’s medium (IMDM, Cellgro) and 20% BIT 9500 medium (Stem Cell Technologies), supplemented with 100 ng/ml SCF, 10 ng/ml FLT-3 ligand, 20ng/ml IL-6 and 100 ng/ml TPO, which was used as basic media. To differentiate HSPCs into myeloid lineage cells, cells were incubated with myeloid-promoting media that contain 100 ng/ml SCF, 10 ng/ml FLT-3 ligand, 20 ng/ml IL-3, 20 ng/ml IL-6, 20 ng/ml GM-CSF and 20 ng/ml G-CSF. For transduction of CD34+ cells, cells were incubated with high-titer lentiviral supernatant together with 8 μg/ml polybrene (Millipore) and centrifuged at 1600 RPM for 1 hr.
Culture and transduction of AML Patient cells
All primary patient samples, unless otherwise noted, were collected under the Biospecimen collection/banking study 09–141. The use of the samples for research purposes is covered under the Biospecimen research protocol 16–354. Frozen AML patient cells were thawed in warm RPMI containing 20% FBS and penicillin/streptomycin, washed and incubated in IMDM medium containing 20% BIT 9500, 1X Lipoproteins (low density human plasma), beta-mercaptoethanol, 25 ng/ml SCF, 10 ng/ml IL-3, 10 ng/ml IL-7, 10 ng/ml FLT-3, 10 ng/ml GCSF, L-Glutamine, Penicillin and Streptomycin for 5 hrs. Cells were then transduced with concentrated virus expressing scramble or IKZF2 shRNA together with GFP and 10 ug/ml polybrene. Cells were incubated for 6 hrs and new media was added. 24 hrs later cells were spun and media was changed with new media. 72–96 hrs post transduction, cells were sorted for GFP and plated on methylcellulose or analyzed for flow cytometry.
Culture and transduction of human leukemia cell lines
MOLM-13, KCL-22, NOMO-1, Kasumi-1, NB4, KG-1, TF-1 and K562 cells were cultured in RPMI media (Cell gro) supplemented with 10% FBS, glutamine and penicillin/streptomycin and grown at 37°C with 5% C O2. Cell lines were infected with viruses expressing scramble and shRNAs against IKZF2 by spinning cells in RPMI with 10% FBS together with viral supernatant and 10 ug/ml polybrene at 1600 RPM for 1 hr. 24 hrs later, media was changed and 48hrs later, cells were selected with 3 μg/ml puromycin for two days.
METHOD DETAILS
Leukemic transplants
For primary transplant of transformed MLL-AF9 LSK cells in leukemia initiation experiments, 200,000 leukemic cells were injected into 6-week-old C57BL/6 female mice, which were lethally irradiated with 900 rads. In secondary, tertiary and quarternary transplants, cell numbers of 500 000, 100 000 and 5000 bone marrow leukemic cells were transplanted respectively, into C57BL/6 mice sublethally irradiated with 450 rads. MLL-AF9 leukemia maintenance experiments were performed by injecting MLL-AF9 WT and Ikzf2f/f cre-ER cells into sublethally irradiated mice, and treating the mice with 160 mg/kg Tamoxifen or corn oil 3 days post transplant. For secondary transplant of maintenance experiments, 100 000 cells from corn oil treated or Tamoxifen treated primary transplanted mice were selected and injected into sublethally irradiated mice.
For transplant of AML1-ETO9a leukemic cells (Wang et al., 2011), 150 000 cells that were at day 4 post transduced with scram or Ikzf2 shRNA expressing virus and puromycin selected, were injected into sublethally irradiated C57BL/6 mice.
Transplant of human leukemia cell line, MOLM-13 cells were performed by injecting 500,000 cells (at day 4 post transduction with scram or IKZF2 shRNA virus) into NSG mice that were irradiated with 200 rads 24 hrs before the transplant. All mice were transplanted by retro-orbital injection of leukemic cells.
Normal transplants and chimerism assessment
Noncompetitive transplants were performed with 1 million BM cells from 6–8-wk-old Ikzf2fl/fl vavcre- or Ikzf2fl/fl vavcre+ mice, injected into lethally irradiated B6SJL congenic CD45.1 recipients. In the primary transplants, chimerism was checked from 8 to 34 weeks, every 4 weeks by either assessing chimerism via peripheral blood or bone marrow aspirates. Mice were sacrificed 34 weeks after injection for primary transplant and 24 weeks for secondary transplant experiments. For chimerism, either peripheral blood or bone marrow cells was extracted and subjected to red blood cell lysis. For chimerism assessment in mature cells of the different lineages, Abs for following markers were used: Mac1, Gr1, c-Kit, CD71, Ter119, B220, CD19, IgM, CD3, CD4, CD8, CD45.2 and CD45.1. For chimerism assessment in stem cells and progenitor cells, following cocktail panel was used: (Lineage; CD3, CD4, CD8, Gr1, B220, CD19, and TER119 all conjugated with PeCy5), Sca-Pac Blue, CD34-FITC, SLAM-APC, CD48-PE, c-KIT-APC-Cy7, FcgRIIb-PeCy7, CD45.2-Alexa700 and CD45.1-PE Texas-red. Stained cells were then analyzed on the BD Fortessa instrument. Following markers were used to define different cell population : HPCs (Lin−c-Kit+Sca1−), GMPs (LK, FcgRIIbHighCD34+), CMPs (LK, FcgRIIbMid CD34+), MEPs (LK, FcgRIIbLow CD34−), B cells (B220+, CD19, IgM), T cells (CD3+, CD4, CD8) and erythroid (Ter119, CD71).
Colony forming Unit Assay
Colony Assay for murine LSK cells was performed by plating 500 sorted LSK cells on methylcellulose (MethoCult M3434, Stem Cell Technologies). CFU colonies corresponding to erythroid progenitor cells (BFU-E), granulocyte-macrophage progenitor cells (CFU-GM, CFU-G and CFU-M), and multipotent granulocyte, erythroid, macrophage and megakaryocyte progenitor cells (CFU-GEMM) were scored 7 days after seeding. For transformed murine LSK and leukemic cells, 1000 cells were plated and colonies were counted 5 days later. Colony Assay for human cord blood CD34+ HSPCs and AML patient cells were performed by plating 10 000 cells and 3000 cells respectively on methylcellulose (MethoCult H4434 Classic-Stem Cell Technologies). GEMM (Granulocyte/Erythrocyte/Macrophage/Megakaryocyte), GM (Granulocyte/Macrophage), G(Granulocyte), M(Macrophage), CFU-E(Erythroid) and BFU-E(Erythroid) colonies were assessed and counted two weeks after seeding for CD34+HSPCs. Total colonies were counted for AML patient cells after two weeks post plating.
Quantitative RT-PCR
Total RNA was extracted from cells using TRIZOL (Life Technologies) and RNeasy RNA extraction kit (Qiagen). Equal amount of RNA from samples was reverse transcribed into cDNA with iSCRIPT (BioRad), and qPCR was performed using an ABI 7500 sequence detection system using primers listed in Table S6 together with SYBR green master mix (ABI systems).
Intracellular staining and flow cytometry
For IKZF2 intracellular staining, cells were fixed with 1.5% paraformaldehyde and permeabilized with ice-cold methanol. Cells were washed 2 times with PBS and incubated with IKZF2 antibody-PacBlue (Biolegend) together with Mac1-PE and c-Kit-APC-Cy7 in 2% PBS for 1hr at room temperature. Cells were then washed twice with 2% FBS/RPMI and analyzed using BD Fortessa instrument. Murine leukemic cells were stained for Mac1-PB, Gr1-APC, F480-PE-Cy7, CD115-APC and c-Kit-APC-Cy7 and analyzed to assess differentiation status. For human leukemic cells CD13-APC, CD14-PE, CD33-APC and CD11b-PE were used for differentiation analysis. For measuring apoptosis, cells were washed with PBS and incubated with anti-Annexin V-PE (BD Biosciences) in the Annexin-V binding buffer (10 mM HEPES, pH 7.4, 140 mM NaCl, 4 mM KCl, 0.75 mM MgCl2, 1 mM CaCl2) together with 5 μl of 7-AAD in the reaction volume of 100 μl for 15 minutes as recommended by the manufacturer.
Cell growth assay for MLL-AF9 WT and Ikzf2f/f cre-ER cells
For deletion of Ikzf2, cells were treated with 10nM of 4-OHT Tamoxifen (Sigma-Aldrich) or ethanol. For cell growth assay, cells were plated at 300 000 cells per ml and treated with 10nM of 4-OHT or ethanol. Viable cells were counted 24hr later.
Proliferation assay for human cell lines
Two days after puromycin selection, at day 4 of post-transduction, cells were plated at 100,000 cells/ml for proliferation assay in a 12-well plate. Cells were counted for 5 consecutive days.
Western blot analysis
For LSCs, MLL-AF9 Ikzf2f/f cre-ER cells were sorted on the top 15% c-Kit high cells and cultured in RPMI medium containing 10% FBS, 6 ng/ml IL-3, 10 ng/ml SCF, 10 ng/ml IL-6 and 10ng/ml GMSCF for a few days and treated with with10 nM of 4-OHT for deletion. 500 000 cells were treated for 24hrs, washed with PBS and lysed in Laemmli sample buffer. For human cell lines 1 million cells were counted at day 4-post transduction with lentiviral expressing shRNAs, and collected and processed as mentioned above. Lysates were run on 4%–15% gradient SDS-PAGE and transferred to nitrocellulose membrane. Membranes were blotted for different proteins using antibodies listed in the resource table.
Assay for transposase-accessible chromatin sequencing (ATAC-seq) and analysis
10 000 sorted LSCs from Ikzf2fl/fl mice (n = 2) and Ikzf2Δ/Δ mice (n = 2) were processed for ATAC-sequencing. Briefly, nuclei were isolated by washing with ATAC buffer (10 mM Tris pH 7.4, 10 mM NaCl, 3 mM MgCl2) and lysing with 50ul of ATAC lysis buffer (10 mM Tris pH 7.4, 10 mM NaCl, 3 mM MgCl2, 0.1% NP-40 ). 1ml of ATAC buffer was added to dilute lysis buffer. Cell lysates were spun to obtain nuclear pellet. Pellet was subjected to transposase reaction (Illumina Nextera DNA Sample Preparation Kit) following manufacturer’s instructions. Reaction was stopped by adding SDS to final concentration of 0.2%. DNA was purified using the Agencourt AMPure XP beads (Beckman Coulter), barcoded and library was generated (using NEBNext Q5 Hot Start HiFi PCR mix and Nextera primers) for sequencing.
For ATAC-sequencing data analysis, the reads were aligned to mm10 using bowtie2 version 2.2.5 (Langmead and Salzberg, 2012). PCR duplicates were removed using picard MarkDuplicates v1.140 (URL http://broadinstitute.github.io/picard), and MACS2 filterdup step was not used as a result of this step. Trimming for Tn5 transposase artifact was done as described in (Buenrostro et al., 2013; van der Veeken et al., 2016). MACS2 version 20160309 (Zhang et al., 2008)(URL https://github.com/taoliu/MACS) was used to call peaks from aligned reads, with the options extending the reads to both sides, assuming reads after removal of duplicates, and using a permissive p-value threshold (-p 1e-2 --nomodel --shift −100 --extsize 200 --keep-dup all). IDR (Li et al., 2011) ((https://github.com/nboley/idr) was used with the threshold of 0.05 to filter the peaks found reproducibly in both replicates of any sample type. Finally, we arrived at the atlas of 31,626 total peaks reproducibly found in at least one sample type. Each peak in the atlas was annotated to the nearest RefSeq gene. Annotation of peaks was done in a similar way shown in (Gonzalez et al., 2015). Briefly, annotation of promoter was given to peaks that lied within 2 kilobases of known transcription start sites. The peaks that overlapped with exons, were annotated as exons and those that did not overlap with any gene body, were annotated as intergenic. The rest were annotated as introns.
Motif enrichment plot by Kolmogorov-Smirnov test
CIS-BP Mus musculus transcription factor database (http://cisbp.ccbr.utoronto.ca/) was chosen as the starting set. Each ATAC-seq peak in the atlas was associated with TF motifs in the CIS-BP database using FIMO (Grant et al., 2011) of MEME suite (http://meme-suite.org/index.html) using the default p-value cutoff of 1e-4. We chose 137 TFs that had expression above 30 RPKM in at least one of the RNA-seq samples as listed in Table S7. CTCF, a DNA binding protein associated with 3D chromatin structure, was excluded. The shift in the cumulative distribution of chromatin accessibility changes of the subset of the atlas occupied by each TF, compared to that of the full atlas, was measured by a one-sided Kolmogorov-Smirnov (K-S) test in either direction. Binding motifs for IKZF2, in the form of position probability matrix, were acquired from the TRANSFAC database (URL http://www.generegulation.com/pub/databases.html (Matys et al., 2003; Wingender, 2008). In Figure 5B, TF symbol annotations are written where the effect size ≥ 0.05 and odds ratio (circle size) ≥ 1.3; the odds ratio was defined as the ratio of foreground occurrence over background occurrence. The foreground occurrence is the number of peaks containing a particular TF motif within the group of differentially upregulated or downregulated peaks, respectively. The background occurrence is the number of peaks containing a particular TF motif found among all the peaks in all samples. The list of significant motifs is in Table S7.
Cleavage under Targets and Release Using Nuclease (CUT&RUN) Sequencing and analysis
Briefly, 250K MLL-AF9 leukemic cells harboring the Nras G12D (RN2 cells; kind gift from Dr. Vakoc, Cold Spring Harbor Laboratory) were transduced with retrovirus expressing FLAG-murine IKZF2 together with BFP. BFP positive cells were sorted and cultured for one week. Cells were washed twice with wash buffer (20 mM HEPES-KOH, 150 mM NaCl, 0.5 mM Spermidine, protease inhibitor) and incubated with concanavalin conjugated magnetic beads for 10 min at RT. Buffer was removed and 50 ul of Antibody buffer containing anti-IKZF2 Ab or control Rabbit Ab was added and incubated overnight at 4° C. Samples were washed with buffer wash containing 0.1% digitonin. Buffer was removed and 50ul digitonin wash buffer containing 700 ng/ml pA-MNase were added and incubated for 1 hr at 4° C. After washing with wash buffer containing digitonin, CaCl2 was added to initiate digestion and incubated for 30 min on ice. Digestion was terminated by adding stop solution (340 mM NaCl, 20 mM EDTA, 4 mM EGTA, 0.02% Digitonin, 50 ug/ml RNASE, 50 ug/ml Glycogen) containing heterologous spike-in DNA. Reaction was incubated at 37° C for 10 min for fragmented chromatin release. DNA was extracted using spin column and barcoded library for Illumina sequencing was generated using with Tru-Seq adapters. 60 million reads using paired end Illumina sequencing was performed. Reads were aligned to mm10 reference genome using bowtie2 version 2.2.5 (Langmead and Salzberg, 2012). Local alignment was performed, and the range of insert size was allowed between 10 and 700 bp, with the parameters --no-mixed --no-discordant --local -I 10 -X 700 -N 1 --mm. In order to generate more sharply defined regions, only the properly aligned read pairs with the insert size less than 120 were used, as suggested in(Skene et al., 2018), (Skene and Henikoff, 2017). MACS2 callpeak (Zhang et al., 2008) command was executed to find the genomic regions with enrichment, using all the combined treatment data and the combined input data and the parameters of -f BAMPE -B --SPMR -p 1e-2 --keep-dup auto. For each ATAC-seq peak, the neighboring CUT&RUN peaks were associated if they were within 3 kb from the summit. Finally, in the conditional motif enrichment analysis of differentially accessible ATAC-seq peaks, the ATAC-seq atlas was filtered by the average signal of CUT&RUN above 0.5 TPM around summit ± 250 bp, leaving 9259 peaks of 31626 in the atlas.
Drawing sequence tracks
R package r tracklayer(Lawrence et al., 2009) was used to load .bigWig coverage track files, and the signal was smoothed by moving average of ± 20 bp. Replicates of the Ikzf2f/f signal and Ikzf2Δ/Δ signal were merged together and represented in the unit of counts per million. Motif binding sites of IKZF2 and C/EBP were displayed, if any.
RNA sequencing and Gene Set Enrichment Analysis
Sorted c-KitHigh cells from Ikzf2 fl/fl mice (n = 2) and Ikzf2Δ/Δ mice (n = 2) were processed for RNA extraction using TRIZOL and RNeasy RNA extraction kit. Samples were prepared with a standard Illumina kit using the TruSeq RNA SamplePrep Guide (Illumina). mRNA fragments with a length of 200–300 bp were selected for library construction. Sequencing was performed on a HiSeq 2000 platform using a standard paired-end protocol. Paired-end RNA-seq reads were first processed to remove TruSeq adaptor sequences and bases with quality scores below 20, and reads with less than 30 remaining bases were discarded. Trimmed reads were then aligned to mm10 genome with the STAR spliced-read aligner version 2.3.0 (Dobin et al., 2013) with default parameters. For each gene from the RefSeq annotations, the number of uniquely mapped reads overlapping with the exons was counted with HTSeq (http://www-huber.embl.de/users/anders/HTSeq/). Differentially expressed genes were called with the negative binomial regression model using DESeq2 (Love et al., 2014) with the Benjamini-Hochberg corrected q-value cutoff of 0.05. GSEA was performed using the rank list of differentially expressed genes from the Ikzf2f/f and Ikzf2Δ/Δ LSCs (Table S2) on all curated gene sets in the Molecular Signatures Database (MSigDB, http://www.broadinstitute.org/msigdb; 3,256 gene sets) (Liberzon et al., 2011) combined with an additional set of relevant gene sets (92 gene sets from our experimentally derived or published hematopoietic self-renewal and differentiation signatures).
Analysis of TCGA Data from AML Patients
The RNA-sequencing data from a cohort of 200 AML patient specimens were obtained from The Cancer Genome Atlas (TCGA) project (https://portal.gdc.cancer.gov). For gene expression, RPKM values were used. IKZF2 high and low patient groups were determined by analyzing patients with 25% of the top and bottom RPKM values of IKZF2. Gene expression of genes of interest was analyzed by comparing the RPKM of genes between the IKZF2 high (n=24) and low (n=45) patient groups.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical parameters for each data are reported in Figure Legends. The 2-tailed Student’s t test was used for significance testing in the bar graphs, except where stated otherwise. P values less than 0.05 were considered significant. Error bars reflect the SEM, except where stated otherwise. Survival probabilities were estimated using the Kaplan-Meier method and compared with the log-rank test. All statistical analyses were carried out using GraphPad Prism 7.0 and the R statistical environment.
DATA AND SOFTWARE AVAILABILITY
ATAC-sequencing data and RNA-sequencing data in this study have been deposited into the National Center for Biotechnolongy Information (NCBI) Gene Expression Omnibus (GEO) with the accession code GSE108367. Cut and Run data has been deposited with accession code GSE120623.
Supplementary Material
Highlights.
IKZF2 is a chromatin remodeler, highly expressed in leukemic stem cells
IKZF2 maintains self renewal and inhibits differentiation in leukemic stem cells
Cut and Run Assay shows that IKZF2 binds to IKZF2 motifs adjacent to HOXA9 and CEBPD/E motifs
IKZF2 regulates opened accessibility for HOXA9 motifs and closed accessibility for CEBPD/E motifs regulating self-renewal and differentiation gene expression
Acknowledgments
We would like to thank Alex Kentsis, Stephen Nimer and Ross Levine for their critical advice and helpful suggestions. M.G.K. was supported by the US National Institutes of Health National Institute of Diabetes Digestive and Kidney Diseases Career Development Award, NIDDK NIH R01-DK101989–01A1, NCI 1R01CA193842–01, Kimmel Scholar Award, V-Scholar Award, Geoffrey Beene Award, Leukemia Lymphoma Society Career Development Award, Starr Cancer Consortium and Alex’s Lemonade Stand A Award. EMS and AMT were supported by the Intramural Research Program of the National Institute of Allergy and Infectious Diseases, National Institutes of Health. This work was also supported by NIH grants CA176745 and CA066996 to SAA.
Footnotes
Declaration of Interests
S.A.A. consults for Epizyme Inc, Imago Biosciences, Cyteir Therapeutics, C4 Therapeutics, Syros Pharmaceuticals and Accent Therapeutics. S.A.A. receives research support from Janssen, Novartis, and AstraZeneca.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Asanuma S, Yamagishi M, Kawanami K, Nakano K, Sato-Otsubo A, Muto S, Sanada M, Yamochi T, Kobayashi S, Utsunomiya A, et al. (2013). Adult T-cell leukemia cells are characterized by abnormalities of Helios expression that promote T cell growth. Cancer science 104, 1097–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro JD, Giresi PG, Zaba LC, Chang HY, and Greenleaf WJ (2013). Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nature methods 10, 1213–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins CT, and Hess JL (2016). Role of HOXA9 in leukemia: dysregulation, cofactors and essential targets. Oncogene 35, 1090–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ee LS, McCannell KN, Tang Y, Fernandes N, Hardy WR, Green MR, Chu F, and Fazzio TG (2017). An Embryonic Stem Cell-Specific NuRD Complex Functions through Interaction with WDR5. Stem cell reports 8, 1488–1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst P, Fisher JK, Avery W, Wade S, Foy D, and Korsmeyer SJ (2004). Definitive hematopoiesis requires the mixed-lineage leukemia gene. Developmental cell 6, 437–443. [DOI] [PubMed] [Google Scholar]
- Estey E, and Dohner H (2006). Acute myeloid leukaemia. Lancet 368, 1894–1907. [DOI] [PubMed] [Google Scholar]
- Gonzalez AJ, Setty M, and Leslie CS (2015). Early enhancer establishment and regulatory locus complexity shape transcriptional programs in hematopoietic differentiation. Nature genetics 47, 1249–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant CE, Bailey TL, and Noble WS (2011). FIMO: scanning for occurrences of a given motif. Bioinformatics 27, 1017–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grzanka J, Leveson-Gower D, Golab K, Wang XJ, Marek-Trzonkowska N, Krzystyniak A, Wardowska A, Mills JM, Trzonkowski P, and Witkowski P (2013). FoxP3, Helios, and SATB1: roles and relationships in regulatory T cells. International immunopharmacology 16, 343–347. [DOI] [PubMed] [Google Scholar]
- Guenther MG, Lawton LN, Rozovskaia T, Frampton GM, Levine SS, Volkert TL, Croce CM, Nakamura T, Canaani E, and Young RA (2008). Aberrant chromatin at genes encoding stem cell regulators in human mixed-lineage leukemia. Genes & development 22, 3403–3408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess JL (2004). MLL: a histone methyltransferase disrupted in leukemia. Trends in molecular medicine 10, 500–507. [DOI] [PubMed] [Google Scholar]
- Hoffman B, Amanullah A, Shafarenko M, and Liebermann DA (2002). The protooncogene c-myc in hematopoietic development and leukemogenesis. Oncogene 21, 3414–3421. [DOI] [PubMed] [Google Scholar]
- Hu Y, and Smyth GK (2009). ELDA: extreme limiting dilution analysis for comparing depleted and enriched populations in stem cell and other assays. Journal of immunological methods 347, 70–78. [DOI] [PubMed] [Google Scholar]
- Iacobucci I, and Mullighan CG (2017). Genetic Basis of Acute Lymphoblastic Leukemia. J Clin Oncol 35, 975–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii Y, Kasukabe T, and Honma Y (2005). Induction of CCAAT/enhancer binding protein-delta by cytokinins, but not by retinoic acid, during granulocytic differentiation of human myeloid leukaemia cells. British journal of haematology 128, 540–547. [DOI] [PubMed] [Google Scholar]
- Ishikawa F, Yoshida S, Saito Y, Hijikata A, Kitamura H, Tanaka S, Nakamura R, Tanaka T, Tomiyama H, Saito N, et al. (2007). Chemotherapy-resistant human AML stem cells home to and engraft within the bone-marrow endosteal region. Nature biotechnology 25, 1315–1321. [DOI] [PubMed] [Google Scholar]
- Jordan CT, Guzman ML, and Noble M (2006). Cancer stem cells. N Engl J Med 355, 1253–1261. [DOI] [PubMed] [Google Scholar]
- Kim HJ, Barnitz RA, Kreslavsky T, Brown FD, Moffett H, Lemieux ME, Kaygusuz Y, Meissner T, Holderried TA, Chan S, et al. (2015). Stable inhibitory activity of regulatory T cells requires the transcription factor Helios. Science 350, 334–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krivtsov AV, and Armstrong SA (2007). MLL translocations, histone modifications and leukaemia stem-cell development. Nature reviews Cancer 7, 823–833. [DOI] [PubMed] [Google Scholar]
- Krivtsov AV, Twomey D, Feng Z, Stubbs MC, Wang Y, Faber J, Levine JE, Wang J, Hahn WC, Gilliland DG, et al. (2006). Transformation from committed progenitor to leukaemia stem cell initiated by MLL-AF9. Nature 442, 818–822. [DOI] [PubMed] [Google Scholar]
- Kronke J, Udeshi ND, Narla A, Grauman P, Hurst SN, McConkey M, Svinkina T, Heckl D, Comer E, Li X, et al. (2014). Lenalidomide causes selective degradation of IKZF1 and IKZF3 in multiple myeloma cells. Science 343, 301–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon HK, Chen HM, Mathis D, and Benoist C (2017). Different molecular complexes that mediate transcriptional induction and repression by FoxP3. Nature immunology 18, 1238–1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon HY, Bajaj J, Ito T, Blevins A, Konuma T, Weeks J, Lytle NK, Koechlein CS, Rizzieri D, Chuah C, et al. (2015). Tetraspanin 3 Is Required for the Development and Propagation of Acute Myelogenous Leukemia. Cell stem cell 17, 1521–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nature methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence M, Gentleman R, and Carey V (2009). rtracklayer: an R package for interfacing with genome browsers. Bioinformatics 25, 1841–1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li JJ, Jiang CR, Brown JB, Huang H, and Bickel PJ (2011). Sparse linear modeling of next-generation mRNA sequencing (RNA-Seq) data for isoform discovery and abundance estimation. Proceedings of the National Academy of Sciences of the United States of America 108, 19867–19872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome biology 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matys V, Fricke E, Geffers R, Gossling E, Haubrock M, Hehl R, Hornischer K, Karas D, Kel AE, Kel-Margoulis OV, et al. (2003). TRANSFAC: transcriptional regulation, from patterns to profiles. Nucleic acids research 31, 374–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer C, Kowarz E, Hofmann J, Renneville A, and Zuna J (2009). New insights to the MLL recombinome of acute leukemias. Leukemia : official journal of the Leukemia Society of America, Leukemia Research Fund, UK. [DOI] [PubMed] [Google Scholar]
- Mullighan CG, Kennedy A, Zhou X, Radtke I, Phillips LA, Shurtleff SA, and Downing JR (2007). Pediatric acute myeloid leukemia with NPM1 mutations is characterized by a gene expression profile with dysregulated HOX gene expression distinct from MLL-rearranged leukemias. Leukemia 21, 2000–2009. [DOI] [PubMed] [Google Scholar]
- Niehof M, Kubicka S, Zender L, Manns MP, and Trautwein C (2001). Autoregulation enables different pathways to control CCAAT/enhancer binding protein beta (C/EBP beta) transcription. Journal of molecular biology 309, 855–868. [DOI] [PubMed] [Google Scholar]
- Papathanasiou P, Attema JL, Karsunky H, Hosen N, Sontani Y, Hoyne GF, Tunningley R, Smale ST, and Weissman IL (2009). Self-renewal of the long-term reconstituting subset of hematopoietic stem cells is regulated by Ikaros. Stem cells 27, 3082–3092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SM, Gonen M, Vu L, Minuesa G, Tivnan P, Barlowe TS, Taggart J, Lu Y, Deering RP, Hacohen N, et al. (2015). Musashi2 sustains the mixed-lineage leukemia-driven stem cell regulatory program. The Journal of clinical investigation 125, 1286–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramji DP, and Foka P (2002). CCAAT/enhancer-binding proteins: structure, function and regulation. The Biochemical journal 365, 561–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebollo A, and Schmitt C (2003). Ikaros, Aiolos and Helios: transcription regulators and lymphoid malignancies. Immunology and cell biology 81, 171–175. [DOI] [PubMed] [Google Scholar]
- Skene PJ, Henikoff JG, and Henikoff S (2018). Targeted in situ genome-wide profiling with high efficiency for low cell numbers. Nat Protoc 13, 1006–1019. [DOI] [PubMed] [Google Scholar]
- Skene PJ, and Henikoff S (2017). An efficient targeted nuclease strategy for high-resolution mapping of DNA binding sites. Elife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somervaille TC, and Cleary ML (2006). PU.1 and Junb: suppressing the formation of acute myeloid leukemia stem cells. Cancer cell 10, 456–457. [DOI] [PubMed] [Google Scholar]
- Sridharan R, and Smale ST (2007). Predominant interaction of both Ikaros and Helios with the NuRD complex in immature thymocytes. The Journal of biological chemistry 282, 30227–30238. [DOI] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, and Mesirov JP (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences of the United States of America 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suva ML, Riggi N, and Bernstein BE (2013). Epigenetic reprogramming in cancer. Science 339, 1567–1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabayashi T, Ishimaru F, Takata M, Kataoka I, Nakase K, Kozuka T, and Tanimoto M (2007). Characterization of the short isoform of Helios overexpressed in patients with T-cell malignancies. Cancer science 98, 182–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornton AM, Korty PE, Tran DQ, Wohlfert EA, Murray PE, Belkaid Y, and Shevach EM (2010). Expression of Helios, an Ikaros transcription factor family member, differentiates thymic-derived from peripherally induced Foxp3+ T regulatory cells. Journal of immunology 184, 3433–3441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Veeken J, Gonzalez AJ, Cho H, Arvey A, Hemmers S, Leslie CS, and Rudensky AY (2016). Memory of Inflammation in Regulatory T Cells. Cell 166, 977–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vu LP, Prieto C, Amin EM, Chhangawala S, Krivtsov A, Calvo-Vidal MN, Chou T, Chow A, Minuesa G, Park SM, et al. (2017). Functional screen of MSI2 interactors identifies an essential role for SYNCRIP in myeloid leukemia stem cells. Nature genetics 49, 866–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Gural A, Sun XJ, Zhao X, Perna F, Huang G, Hatlen MA, Vu L, Liu F, Xu H, et al. (2011). The leukemogenicity of AML1-ETO is dependent on site-specific lysine acetylation. Science 333, 765–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wingender E (2008). The TRANSFAC project as an example of framework technology that supports the analysis of genomic regulation. Briefings in bioinformatics 9, 326–332. [DOI] [PubMed] [Google Scholar]
- Ye M, Zhang H, Yang H, Koche R, Staber PB, Cusan M, Levantini E, Welner RS, Bach CS, Zhang J, et al. (2015). Hematopoietic Differentiation Is Required for Initiation of Acute Myeloid Leukemia. Cell stem cell 17, 611–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, and Liu XS (2008). Model-based analysis of ChIP-Seq (MACS). Genome biology 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Swindle CS, Bates JT, Ko R, Cotta CV, and Klug CA (2007). Expression of a non-DNA-binding isoform of Helios induces T-cell lymphoma in mice. Blood 109, 2190–2197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao S, Liu W, Li Y, Liu P, Li S, Dou D, Wang Y, Yang R, Xiang R, and Liu F (2016). Alternative Splice Variants Modulates Dominant-Negative Function of Helios in T-Cell Leukemia. PloS one 11, e0163328. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
ATAC-sequencing data and RNA-sequencing data in this study have been deposited into the National Center for Biotechnolongy Information (NCBI) Gene Expression Omnibus (GEO) with the accession code GSE108367. Cut and Run data has been deposited with accession code GSE120623.