


Abstract
Disulfide bonds play a significant role in protein stability, function or regulation but are poorly conserved among evolutionarily related proteins. The Yosshi can help to understand the role of S–S bonds by comparing sequences and structures of homologs with diverse properties and different disulfide connectivity patterns within a common structural fold of a superfamily, and assist to select the most promising hot-spots to improve stability of proteins/enzymes or modulate their functions by introducing naturally occurring crosslinks. The bioinformatic analysis is supported by the integrated Mustguseal web-server to construct large structure-guided sequence alignments of functionally diverse protein families that can include thousands of proteins based on all available information in public databases. The Yosshi+Mustguseal is a new integrated web-tool for a systematic homology-driven analysis and engineering of S–S bonds that facilitates a broader interpretation of disulfides not just as a factor of structural stability, but rather as a mechanism to implement functional diversity within a superfamily. The results can be downloaded as a content-rich PyMol session file or further studied online using the HTML5-based interactive analysis tools. Both web-servers are free and open to all users at https://biokinet.belozersky.msu.ru/yosshi and there is no login requirement.
INTRODUCTION
Disulfide bonds—covalent crosslinks between thiol groups of two cysteine residues—are well-recognized factors of protein stability that can also play a substantial role in function and regulation according to the recent studies (1–6). Various experimental strategies, computational approaches, and empirical design rules were proposed to introduce non-native disulfide bonds into the protein structure in an attempt to improve its properties (7). Some studies rely on a visual expert inspection of a PDB record to predict such hot-spots based on simple proximity estimations between Cα and/or Cβ atoms of the respective residue pairs (8). Computational tools—e.g., Disulfide by Design (9), MODIP (10) and SSBOND (11)—were introduced to rationalize disulfide engineering by systematically selecting all pairs of residues in the 3D-structure of a query protein that could accommodate two cysteines in such a way that their Sγ atoms would be properly oriented to form a crosslink. As stabilizing mutations were most often found in regions of medium to high mobility (7) the predicted disulfides were usually ranked by a B-factor or further studied by molecular modelling to evaluate the impact of a potential crosslink on the flexibility of the corresponding structural fragments (12,13). These computational tools helped to enhance properties of selected proteins/enzymes, but usually predicted a large number of hot-spots to construct S–S bonds while failed to effectively identify the most promising candidates to improve stability or function (7). Perhaps the reason for the poor success rate was that the available computational tools in the vast majority of predictions suggested geometrically correct but artificial S–S bonds which were not found in nature, i.e., did not occur in a superfamily of proteins sharing a common structural framework. As many cases of destabilizing disulfide bonds are being reported (7), it seems that introduction of such non-natural crosslinks into a protein structure can disbalance a complex structure-function relationship which is still poorly understood.
Bioinformatic analysis is increasingly popular in biotechnology and biomedicine providing an opportunity to study the growing sequence and structural data attributed to evolutionarily related proteins with different properties systematically (14–20). Previous studies concluded that disulfide bonds are poorly conserved in structurally similar proteins with different function, stability, and regulation (21,22). Inclusion/exclusion of crosslinks in protein structures could be one of the insufficiently studied mechanisms used to implement diverse properties in members of a superfamily during evolution from a common ancestor. This provides an opportunity to introduce disulfide bonds which naturally occur in some proteins into the structures of their homologs to improve stability or modulate function. Such a homology-driven strategy was successfully used to design robust enzymes and antibodies by inserting stabilizing disulfide bonds in their structures (23–26), as well as to implement novel regulatory mechanisms by introducing allosteric disulfides that alter protein shape and function due to conformational transitions upon reduction/oxidation (1,3). For example, the thermostability of subtilisin E from mesophilic Bacillus subtilis was significantly improved by introducing two cysteines at positions Gly61 and Ser98 that were occupied by an S–S bond in the structure of a homolog from thermophilic Thermus aquaticus (25). In a separate study, a disulfide bridge that was known to regulate both ligand binding and protein reactivity in cytoglobins was introduced at positions Val21 and Val66 of the homologous sperm whale myoglobin. The designed Val21Cys/Val66Cys disulfide bond in myoglobin was shown to alter both structural and functional characteristics of this protein leading to enhanced stability and fine-tuning of O2 binding (3). These examples outline a much broader application of bioinformatic analysis in protein engineering to modulate functional properties of homologs by implementation of disulfide bonds, as opposed to being solely a tool to identify stabilizing hot-spots (as, e.g. Disulfide by Design, MODIP or SSBOND). However, accurate comparative analysis of all available protein sequence and structural data within a superfamily represents a methodological challenge what impedes practical use of bioinformatics at a daily laboratory routine.
We have developed Yosshi—‘Your web-server for S–S bond harvesting’ in protein superfamilies. The Yosshi web-server is integrated with the recently introduced Mustguseal web-server to construct large structure-guided sequence alignments of functionally diverse protein families that can include thousands of proteins based on all available information about their structures and sequences in public databases (27). The integration of web-based bioinformatic tools Yosshi and Mustguseal provides an out-of-the-box easy-to-use solution, first of its kind, to classify and study S–S bonds in protein superfamilies as well as to select hot-spots for disulfide engineering in a user-submitted query protein based on a systematic analysis of disulfides naturally occurring in its homologs. In this paper we discuss the Yosshi + Mustguseal workflow, describe input and output, and conclude that such a strategy can be successfully employed to improve properties of useful proteins/enzymes.
MATERIALS AND METHODS
The Yosshi workflow consists of bioinformatic analysis to search for pairs of cysteine residues in sequences of homologs, and structural filtration to evaluate whether introduction of the selected cysteines at corresponding positions in the user-submitted query protein can result in a formation of a disulfide bond. The input and output can be created, processed, and studied entirely on-line via the web-interface. The results are primarily web-based and viewable on the website, but can also be downloaded to a local computer.
The input
The input to Yosshi is (i) a structure of the query protein in the PDB format and (ii) a multiple alignment of proteins homologous to the query in the FASTA format. Choose the query protein based on your particular task and primary interest. It can be the target protein selected for further experimental design, the most studied member of a superfamily, or the protein which you are most familiar with. The multiple alignment can be automatically prepared for your query protein by the Mustguseal web-server (27). Press the ‘Mustguseal it NOW’ button at the Yosshi submission page. This will redirect you to the Mustguseal web-server and load a preselected set of parameters to automatically construct the alignment of a large representative set of functionally diverse proteins with high structure similarity but low sequence identity to your query protein. Enter PDB ID and chain ID of your query protein structure in the corresponding input box and press ‘Submit’. First, the structure similarity search is used as previously discussed (27) to collect the representative proteins—evolutionarily distant relatives that are expected to represent different protein families (this step is further discussed below). Then, each selected representative protein is used to run the sequence similarity search versus the UniProtKB/Swiss-Prot+TrEMBL databases and collect at most 1000 proteins per search, followed by a set of filters to eliminate redundant entries (at the 100% sequence identity threshold, i.e. if two proteins have the same amino acid sequence then only one would be further considered) and outliers (sharing <0.5 bit score per alignment column with the respective representative protein) to collect evolutionarily close relatives—members of the corresponding families. Finally, superimposition of 3D-structures is implemented to compare evolutionarily distant relatives that have lost sequence similarity during natural selection and specialization from a common ancestor, whereas alignment of sequences is used to match close homologs. The final alignment incorporates the currently known sequence variability within a common structure of the query protein and can be automatically transferred to the Yosshi web-server for further analysis by pressing the ‘Submit to Yosshi’ button at the Mustguseal results page. Mustguseal includes protein accession numbers and names of corresponding source organisms into the FASTA file (i.e. as part of the protein name string) to be further used by Yosshi to automatically generate HTML-links to the respective pages in PDB (28), UniProt (29), and Bacterial Diversity Metadatabase (i.e. BacDive (30)). Illustrated tutorial describing preparation of the input data is available on-line at https://biokinet.belozersky.msu.ru/yosshi-input.
The scope of the Mustguseal alignment is defined by the diversity of representative proteins selected at the ‘Structure similarity search’ step based on the percentage of secondary structure equivalences between the query and target PDB records. Generally speaking, no particular choice of thresholds for this step can be recommended in advance, i.e. they should be selected based on the research objective of the consequent homology-driven analysis, structural organization of the protein family of interest, and availability of the corresponding data in public databases. By default, at least 70% of the target from PDB has to make at least 70% of the query protein to be selected for further consideration. The ‘70–70%’ is a widely used general-purpose pair of thresholds first introduced by the highly popular SSM/PDBeFOLD structure similarity search engine (31) and further adopted by the Mustguseal to collect evolutionarily remote and functionally diverse proteins that still share a sufficient structure similarity to produce a meaningful superimposition. The user can include evolutionarily more distant proteins in the alignment by setting the thresholds below the default values, or limit the scope of the final alignment only to close homologs by setting both thresholds to, e.g., 90%. Non-symmetric threshold can be used to compare proteins with a different domain composition (e.g. sialidases containing a catalytic domain and a various number of lectin domains as discussed in (32)). The guidelines for parameter selection in the Mustguseal protocol are discussed in section ‘The Parameters’ in the Supplementary Material to that paper (27).
The algorithm
The Yosshi workflow contains three major steps (Figure 1).
Figure 1.
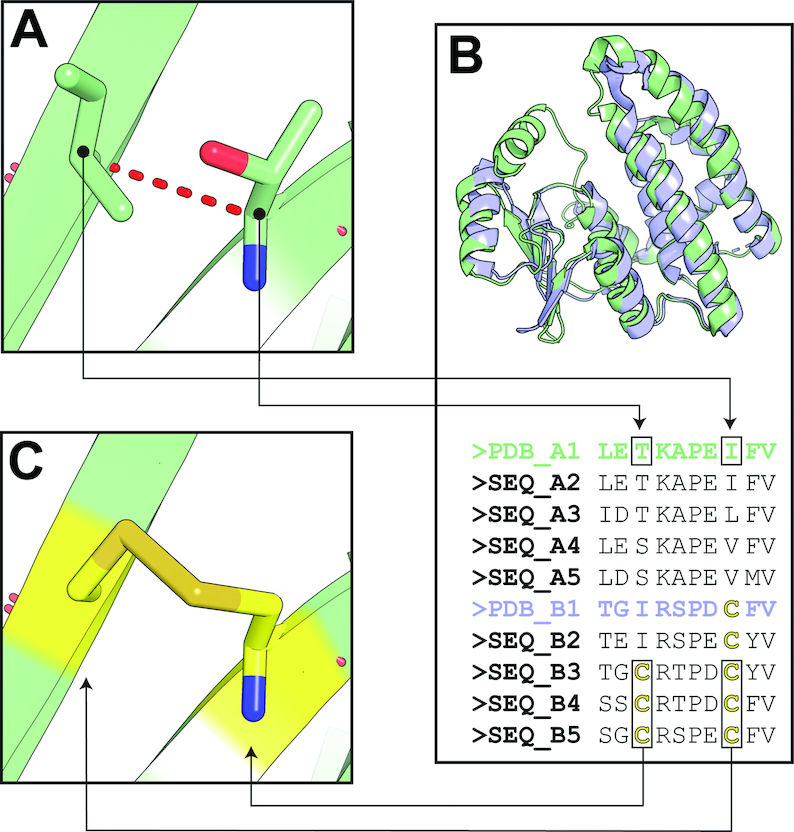
The outline of the Yosshi protocol: (A) the first tier filter; (B) the bioinformatic analysis of protein families; (C) the 3D-motif analysis. See explanation in the text.
Step A: The ‘First tier filter’
The ‘First tier filter’ is applied to dismiss all pairs of positions in the query protein structure the least likely to form a crosslink even if both were mutated to cysteines based on simple estimations of proximity between Cα and Cβ atoms of the respective residue pairs.
Step B: The bioinformatic analysis
The selected pairs of positions are further subjected to bioinformatic analysis of the available protein sequences within a superfamily to identify pairs both occupied by cysteine residues in at least one homolog of the query protein. This key part of the Yosshi protocol requires that a large set of proteins expected to have diverse properties and different disulfide connectivity patterns within a common structural core is collected and aligned, and thus represents a methodological challenge. This step is facilitated by the integrated Mustguseal web-server capable of constructing large structure-guided sequence alignments of functionally diverse protein families automatically (27), as described above (see subsection ‘The input’).
Step C: The 3D-motif analysis
Finally, the 3D-motif analysis is used to evaluate whether the selected pairs of positions in the query protein structure can form a disulfide bond upon mutation to cysteines, and prepare 3D-models of the corresponding mutants. Implementation of 3D-motifs to search for patterns of local structure in protein 3D-records is re-emerging in a wide range of bioinformatic applications (33). In the context of this study, a 3D-motif of a disulfide bond contains 12 atoms of the backbone and side-chain of two covalently linked cysteines observed in a high-quality crystallographic structure (Supplementary Figure S1). Two libraries of 3D-motifs were created from the PDB database and incorporate the current knowledge of the disulfide bond geometry: the ‘Core collection’ featuring 273 most typical configurations of S–S bonds in protein structures, and the ‘Complete collection’ which in addition contains 4748 variants describing the rarely occurring disulfide bonds (see section 1 in the Supplementary Data for details). A pair of positions in the query protein structure selected by the bioinformatic analysis (i.e. both positions were occupied by cysteines in at least one homolog) would be confirmed as a promising site for S–S bond formation if it matches with at least one 3D-motif. In that case, the respective pair of positions in the query protein structure is replaced by the most similar 3D-motif (i.e. selected by the lowest RMSD of the backbone) and this initial three-dimensional model of the mutant is further optimized by a conjugate gradients energy minimization method (34) using AMBER FF14SB parameters for standard residues (35) to remove steric clashes around the inserted cysteines.
Thresholds for structural filtration
There are numerous reports of successfully engineered S–S bonds that violate geometric constraints of the commonly used rigid computational models trained on covalently connected cysteines (7,36). Disulfide engineering experiments demonstrated that introduction of a crosslink into a protein structure is likely to result in a considerable shift of the backbone atoms. Therefore, to select thresholds for the ‘First tier’ structural filtration and the 3D-motif analysis it is important to consider the flexibility of a pair of non-bonded amino acid residues capable of a disulfide bond formation upon mutation to cysteines. In this work, a large non-redundant set of disulfide bonds and their non-bonded equivalences in structures of homologous proteins was collected by bioinformatic analysis of the CATH superfamilies (37). Geometry constraints of the backbone atoms in the collected non-bonded residue pairs were further studied and used to set the structural filtration thresholds within the Yosshi workflow. By default, the cut-off distances between Cα–C′α atoms and Cβ–C′β atoms (i.e. for all residues except Glycines) of the ‘First tier filter’ are set to at most 8.58 and 6.96 Å, respectively. By default, the 3D-motif analysis is applied in the ‘Flexible’ mode to take into account protein backbone flexibility, i.e., a pair of positions in the query protein structure would be confirmed as a promising site for S–S bond formation if it matches with at least one of 273 3D-motifs from the ‘Core collection’ library so that both RMSD values between two pairs of superimposed backbone atoms are within X = 0.70 Å, what corresponds to a P-value of P(x > X) = 0.05 of a normal distribution with μ = 0.39 Å and σ = 0.19 Å, or rejected otherwise. Alternatively, the user can choose to run the 3D-motif analysis in the ‘Rigid’ mode to select only such pairs of positions in the query protein structure that comply with strict geometric criteria of two covalently connected cysteines. In that case, the ‘Complete collection’ of 5021 3D-motifs is used and the RMSD values between two pairs of superimposed backbone atoms are limited to at most X = 0.28 Å, what corresponds to a P-value of P(x > X) = 0.05 of a normal distribution with μ = 0.16 Å and σ = 0.07 Å. The structural filtration in the ‘Flexible’ and ‘Rigid’ modes correctly identified 13 291 and 12 985 out of 13 306 true disulfide bonds in protein structures, for 99.89% and 97.59% success rates, respectively. The details regarding the threshold selection and evaluation of structural filtration are provided in the Supplementary Data (see section 2).
The output
The first output is a list of pairs of positions in the structure of the query protein that can form a disulfide bond assuming both residues are mutated to cysteines, or are already occupied by cysteines that can form a crosslink. The second output is a list of homologs of the query protein, which contain cysteines in equivalent positions for each pair of such residues. For each such homolog and its source organism HTML links to the respective pages in PDB (28), UniProt (29) and BacDive (30) can be automatically created to facilitate further expert analysis (Supplementary Figure S3). The selected pairs are ranked in a descending order of the ‘Disulfide Occurrence’ (DOccur) what is a positive integer equal to the number of times they both were occupied by cysteines in sequences of homologs (i.e. the expected occurrence of the corresponding crosslink in protein families). In addition, the ‘Disulfide Frequency’ (DFreq) is provided for each pair of positions that takes a value from the range 0 to 100% calculated as DOccur divided by the total number of proteins in the multiple alignment. The recommended interpretation of the DOccur metric is discussed in the section ‘Results’. The Yosshi results can be downloaded as a content-rich PyMol session file or further studied online using the built-in interactive analysis tools (Supplementary Figure S3). The online interactivity is implemented in HTML5 and therefore neither plugins nor Java are required (38).
RESULTS
The Yosshi + Mustguseal integrated web-based method provides an intuitive and easy-to-use interface to study the abundance of S–S bonds within a superfamily, compare disulfide connectivity in homologs with different properties, as well as to identify disulfide bridges present in homologs, but not in the query protein that can be introduced to design its stability or function. The homology-driven approach to the analysis and design of disulfide bonds makes Yosshi fundamentally different from the currently available 3D-structure based strategies for disulfide engineering focused on improving protein stability due to artificially created S–S bonds (e.g. Disulfide by Design (9), MODIP (10) and SSBOND (11)). The Yosshi's output is a homology-based annotation of pairs of positions in the structure of a query protein that can form a disulfide bond assuming both residues are mutated to cysteines, or are already occupied by cysteines which can form a crosslink. The selected pairs are ranked in a descending order of the ‘Disulfide Occurrence’ (DOccur), i.e., the expected occurrence of the corresponding crosslinks in protein families. The interpretation of the DOccur metric is equivalent to that of the popular subfamily-specific conservation (19,39). A higher value of the DOccur indicates that a corresponding disulfide is conserved in a larger group of proteins within a superfamily thus suggesting its direct role in a function or property common among these homologs (e.g. in structural stability or regulatory mechanism, as discussed below). Therefore, the purpose of the Yosshi ranking is to show the most conserved disulfides first to facilitate their further analysis in particular proteins. We further conclude that introduction of S–S bonds identified by the bioinformatic analysis into the query protein structure can be successfully employed to improve properties of useful enzymes and design advanced proteins with controllable functions.
The Yosshi + Mustguseal on-line tool with the default setup (in particular, the 3D-motif analysis set to the ‘Flexible’ mode) was applied to study subtilisin E from Bacillus subtilis and its homologs using only a web-browser. The query PDB structure 1SCJ of subtilisin was submitted to the Mustguseal as discussed above (see ‘The input’ in section ‘Materials and Methods’) to automatically construct the alignment of a non-redundant set of 8456 sequences and structures of proteins from the subtilisin-like serine proteases superfamily with high structure similarity but low sequence identity to the query protein, and then subjected to analysis by the Yosshi. In total, 56 pairs of positions in the query protein structure were selected as capable of a disulfide bond formation upon mutation to cysteines and ranked based on the expected abundance of the corresponding crosslinks in homologs. The #1 pair of positions Gly61 and Ser98 in the subtilisin structure was occupied by cysteines in 838 homologs (i.e. 9.9%). Cysteines in both positions were observed in proteins from thermophilic sources, e.g. Aqualysin-1 from Thermus aquaticus (UniProt ID: P08594), as evidenced by the Yosshi's on-line output and the corresponding BacDive entry (BacDive ID: 16714) which includes an annotated thermophilic temperature range for this organism (Supplementary Figure S3). Introduction of this crosslink which naturally occurs in a homolog from thermophilic Thermus aquaticus into the structure of subtilisin E from mesophilic Bacillus subtilis by a double mutation Gly61Cys/Ser98Cys was previously reported to enhance thermostability without changing catalytic efficiency of the enzyme (25). The remaining 55 pairs of positions selected by the bioinformatic analysis in the query protein were found to be substituted by cysteines in up to 701 functionally diverse homologs from different organisms, and thus provide a list of promising hot-spots to further engineer properties of subtilisin and its evolutionary relatives. Full access to the input and output data regarding this example can be obtained by activating the ‘Demo mode’ at the Yosshi submission page.
In a separate example, the on-line tool was used to study the globins superfamily. The PDB structure 1JP6 of sperm whale myoglobin was submitted to the Mustguseal web-server to automatically construct an alignment of a non-redundant set of 5554 proteins. A pair of positions Val21 and Val66 in the myoglobin structure was ranked #1 out of 21 candidates and was substituted by cysteines in 78 protein sequences annotated as cytoglobines. Analysis of the literature (as explained in (3)) showed that the S–S bond between equivalent residues in cytoglobines regulates protein reactivity by reshaping the internal cavities and thus modulating the mechanism of CO escape, although no crystallographic structure with this disulfide in the oxidized (i.e. bonded) form is currently available. Introduction of this naturally occurring crosslink into the structure of sperm whale myoglobin by a double mutation Val21Cys/Val66Cys was previously reported to implement a regulatory mechanism to fine-tune the catalytic reactivity of the protein (3). The input and output data regarding this example are available on-line at https://biokinet.belozersky.msu.ru/yosshi-example.
Other case-studies of disulfide connectivity in different lipases, carbonic anhydrases, xylanases, and members of the ribonuclease A superfamily to reproduce the previously reported disulfide engineering experiments, as well as the comparison with currently available programs are provided in the Supplementary Data (see section 3). In brief, popular 3D-structure based algorithms to improve protein stability by disulfide engineering—Disulfide by Design (9), MODIP (10), and SSBOND (11)—were tested on the same case-studies (Table 1). In contrast to Yosshi that provides a detailed homology-based annotation of disulfides within a common structural fold, the output of these tools is only a list of hot-spots to introduce S–S bonds into the query protein ranked by an energy score, or a B-factor, or not ranked at all. Noteworthy, Disulfide by Design, MODIP and SSBOND, as well as Yosshi with the 3D-motif analysis performed in the ‘Rigid’ mode have failed to predict some mutations mentioned above and in the Supplementary Data. Such a poor performance can probably be explained by the use of strict geometric models trained on covalently connected cysteines to evaluate the candidate positions. The beneficial hot-spots do not always match these constraints as introduction of a crosslink into a protein structure can result in a considerable shift of the backbone atoms, as previously discussed (7,36).
Table 1.
Comparison of the Yosshi with the currently available programs for stability-oriented 3D-structure based disulfide engineering
| Yosshi | Disulfide by Design | ||||||
|---|---|---|---|---|---|---|---|
| PDB | Mutation | Flexible 3D-motif analysis | Rigid 3D-motif analysis | Ranked by energy | Ranked by B-factor | MODIP | SSBOND |
| 1SCJ:A | G61C/S98C | 1 (56) | 1 (31) | − (57) | − (57) | − (97) | − (64) |
| 1JP6:A | V21C/V66C | 1 (21) | − (7) | − (10) | − (10) | − (21) | − (9) |
| 4GW3:A | G181C/S238C | 1 (5) | 1 (1) | 38 (40) | 5 (40) | − (71) | − (42) |
| 5CH8:A | Y22C/G269C | 2 (14) | 1 (5) | 17 (24) | 2 (24) | − (87) | + (42) |
| 2CBA:A | A23C/L203C | 1 (40) | − (9) | − (29) | − (29) | − (70) | − (62) |
| 1BCX:A | S100C/N148C | 2 (17) | 1 (10) | 1 (25) | 12 (25) | A (5) | + (29) |
| V98C/A152C | 6 (17) | 5 (10) | 12 (25) | 15 (25) | B (12) | + (29) | |
| 1XYP:A | S110C/N154C | 2 (18) | 1 (9) | 2 (21) | 9 (21) | A (6) | + (28) |
| 1YNA:A | T3C/T26C | 1 (15) | − (9) | − (27) | − (27) | − (53) | − (38) |
| Q1C/Q24C | − (15) | − (9) | − (27) | − (27) | − (53) | − (38) | |
The case-studies of subtilisin (PDB 1SCJ:A) and myoglobin (PDB 1JP6:A) are discussed in the Main text; results of the bioinformatic analysis of lipases (PDBs: 4GW3:A and 5CH8:A), carbonic anhydrases (PDB 2CBA:A) and xylanases (PDBs 1BCX:A, 1XYP:A, and 1YNA:A) are provided as a Supplementary Data (see section 3). For a correctly identified mutation its rank in the list of predictions is provided, or ‘+’ if the correct prediction was identified, but not ranked in the output, or
‘–’ otherwise. The total number of disulfide bonds predicted in each case (i.e., pairs of hot-spots for disulfide engineering) is shown in parentheses. For a correct prediction by MODIP its grade and the total number of predictions with the same grade are provided according to the ‘ABC’ grading system, i.e., the most promising hot-spots for disulfide engineering are assigned the grade ‘A’ (see (10) for details). The results of the ‘Disulfide by Design’ are ranked in order of increasing bond energy (i.e., the lowest energy is shown first) or decreasing B-factor (i.e. the highest B-factor is shown first).
In all cases, the automatic construction of a large multiple alignment by the Mustguseal web-server with the default setup as discussed in section ‘Materials and Methods’ took at most one and a half hours, and the running time of the Yosshi web-server was within several minutes.
DISCUSSION
The importance of disulfide bridges was mainly considered in the context of protein folding and structural integrity. Assuming that introduction of artificial S–S bonds would benefit protein stability, various computational tools were developed to solve this challenging problem and provide a list of pairs of positions in the query protein structure that would satisfy geometric constraints of a disulfide bond formation upon mutation to cysteines (e.g. Disulfide by Design (9), MODIP (10) and SSBOND (11)). These programs based on the analysis of a single PDB record can be used to improve protein stability by artificially created crosslinks, but can hardly help to study the diverse roles that disulfide bonds are playing within a superfamily. Contrary to expectations, many cases of destabilizing disulfides predicted by such 3D-structure based methods have been reported thus creating a demand for more efficient disulfide engineering strategies (7). The recent studies have shown that S–S bonds can also be important for protein function and regulation (1–6) and are poorly conserved in structurally similar evolutionarily related proteins with different properties (21,22). These findings suggest that formation of disulfide bridges is not just a way to stabilize protein structures, but rather represents a more universal, still insufficiently explored evolutionary instrument to implement functional diversity within a common structural fold of a superfamily. Protein stability was supposed to promote evolvability (40), thus introduction of disulfide bonds has a potential of becoming an effective tool not just to modulate protein stability, but to facilitate design of new functions (41,42). The homology-driven approaches to disulfide engineering based on the comparison of structures/sequences of evolutionarily related proteins with diverse properties were suggested as an alternative to purely 3D-structure based tools (3,23,24,26), but represented methodological and computational challenges to a general biologist—one had to collect sequences and structures of homologs, construct and examine a multiple alignment, and, finally, evaluate if the selected S–S bonds could fit into the query protein structure. As no such automated tools for systematic analysis of disulfides in protein superfamilies were available, the homology-based approaches were rarely used in practice.
We have recently introduced the Mustguseal web-server to construct large structure-guided sequence alignments of functionally diverse protein families and integrated sister web-servers to study the collected data (27). The Yosshi is the fourth web-based bioinformatic method integrated with the Mustguseal protocol and is intended to classify disulfide bonds in diverse protein families, study the roles they play within a common structural fold of a superfamily, and assist to select hot-spots for disulfide engineering by systematically comparing homologs with diverse properties (i.e., stability, function, regulation). The key novelty of Yosshi is implementation of the bioinformatic analysis to search for pairs of cysteine residues in sequences of homologs followed by the 3D-motif analysis to evaluate whether introduction of the selected cysteines at corresponding positions in the user-submitted query protein can result in a formation of a disulfide bond. The output is a list of disulfide bridges present in homologs, but not in the query protein, as well as its own S–S bonds, ranked by their expected occurrence in protein families. Disulfides conserved in larger groups of proteins are shown first suggesting an important role in a common function or property shared by these homologs. The value of crosslinks selected by the bioinformatic analysis can be evaluated by an expert and followed by experimental site-directed mutagenesis (as in the discussed case-studies, see section ‘Results’), or can be studied by molecular modeling to understand the mechanisms affecting protein function or properties due to S–S bond formation (43).
The Yosshi + Mustguseal is a highly automated on-line tool for a systematic homology-driven analysis and engineering of disulfide bonds that can be easily used by a general biologist at a daily laboratory routine. We expect this bioinformatic tool will help to select the most promising hot-spots to implement novel functions, improve stability and evolvability of proteins/enzymes thus promoting the value of S–S bonds in protein engineering.
Supplementary Material
ACKNOWLEDGEMENTS
The work was carried out using equipment of the shared research facilities of HPC computing resources at the Lomonosov Moscow State University supported by the project RFMEFI62117X0011 (44).
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Russian Foundation for Basic Research [18-29-13060]. Funding for open access charge: Russian Foundation for Basic Research [18-29-13060].
Conflict of interest statement. None declared.
REFERENCES
- 1. Chiu J., Hogg P.J.. Allosteric disulfides: Sophisticated molecular structures enabling flexible protein regulation. J. Biol. Chem. 2019; 294:2949–2960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Landeta C., Boyd D., Beckwith J.. Disulfide bond formation in prokaryotes. Nat. Microbiol. 2018; 3:270. [DOI] [PubMed] [Google Scholar]
- 3. Yin L.L., Yuan H., Du K.J., He B., Gao S.Q., Wen G.B., Tan X., Lin Y.W.. Regulation of both the structure and function by a de novo designed disulfide bond: a case study of heme proteins in myoglobin. Chem. Commun. (Camb.). 2018; 54:4356–4359. [DOI] [PubMed] [Google Scholar]
- 4. Plugis N.M., Weng N., Zhao Q., Palanski B.A., Maecker H.T., Habtezion A., Khosla C.. Interleukin 4 is inactivated via selective disulfide-bond reduction by extracellular thioredoxin. Proc. Natl. Acad. Sci. U.S.A. 2018; 115:8781–8786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sun M.A., Wang Y., Zhang Q., Xia Y., Ge W., Guo D.. Prediction of reversible disulfide based on features from local structural signatures. BMC Genomics. 2017; 18:279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Karimi M., Ignasiak M.T., Chan B., Croft A.K., Radom L., Schiesser C.H., Pattison D.I., Davies M.J.. Reactivity of disulfide bonds is markedly affected by structure and environment: implications for protein modification and stability. Sci. Rep. 2016; 6:38572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dombkowski A.A., Sultana K.Z., Craig D.B.. Protein disulfide engineering. FEBS Lett. 2014; 588:206–212. [DOI] [PubMed] [Google Scholar]
- 8. Kanaya S., Katsuda C., Kimura S., Nakai T., Kitakuni E., Nakamura H., Katayanagi K., Morikawa K., Ikehara M.. Stabilization of Escherichia coli ribonuclease H by introduction of an artificial disulfide bond. J. Biol. Chem. 1991; 266:6038–6044. [PubMed] [Google Scholar]
- 9. Craig D.B., Dombkowski A.A.. Disulfide by Design 2.0: a web-based tool for disulfide engineering in proteins. BMC Bioinformatics. 2013; 14:346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dani V.S., Ramakrishnan C., Varadarajan R.. MODIP revisited: re-evaluation and refinement of an automated procedure for modeling of disulfide bonds in proteins. Protein Eng. Des. Sel. 2003; 16:187–193. [DOI] [PubMed] [Google Scholar]
- 11. Hazes B., Dijkstra B.W.. Model building of disulfide bonds in proteins with known three-dimensional structure. Protein Eng. Des. Sel. 1988; 2:119–125. [DOI] [PubMed] [Google Scholar]
- 12. Wijma H.J., Fürst M.J., Janssen D.B.. Bornscheuer U, Höhne M. A computational library design protocol for rapid improvement of protein stability: FRESCO. Protein Engineering. Methods in Molecular Biology. 2018; NY: Humana Press; 69–85. [DOI] [PubMed] [Google Scholar]
- 13. Le Q.A.T., Joo J.C., Yoo Y.J., Kim Y.H.. Development of thermostable Candida antarctica lipase B through novel in silico design of disulfide bridge. Biotechnol. Bioeng. 2012; 109:867–876. [DOI] [PubMed] [Google Scholar]
- 14. Hendrikse N.M., Charpentier G., Nordling E., Syrén P.O.. Ancestral diterpene cyclases show increased thermostability and substrate acceptance. FEBS J. 2018; 285:4660–4673. [DOI] [PubMed] [Google Scholar]
- 15. Buß O., Buchholz P.C., Gräff M., Klausmann P., Rudat J., Pleiss J.. The ω‐transaminase engineering database (oTAED): a navigation tool in protein sequence and structure space. Proteins. 2018; 86:566–580. [DOI] [PubMed] [Google Scholar]
- 16. Beerens K., Mazurenko S., Kunka A., Marques S.M., Hansen N., Musil M., Chaloupkova R., Waterman J., Brezovsky J., Bednar D. et al.. Evolutionary analysis as a powerful complement to energy calculations for protein stabilization. ACS Catal. 2018; 8:9420–9428. [Google Scholar]
- 17. Pellis A., Cantone S., Ebert C., Gardossi L.. Evolving biocatalysis to meet bioeconomy challenges and opportunities. N. Biotechnol. 2018; 40:154–169. [DOI] [PubMed] [Google Scholar]
- 18. Qi F., Motz M., Jung K., Lassak J., Frishman D.. Evolutionary analysis of polyproline motifs in Escherichia coli reveals their regulatory role in translation. PLoS Comput. Biol. 2018; 14:e1005987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Suplatov D., Kirilin E., Švedas V.. Svendsen A. Bioinformatic analysis of protein families to select function-related variable positions. Understanding Enzymes: Function, Design, Engineering, and Analysis. 2016; Singapore: Pan Stanford Publishing; 351–385. [Google Scholar]
- 20. Suplatov D., Voevodin V., Švedas V.. Robust enzyme design: bioinformatic tools for improved protein stability. Biotechnol. J. 2015; 10:344–355. [DOI] [PubMed] [Google Scholar]
- 21. Thangudu R.R., Manoharan M., Srinivasan N., Cadet F., Sowdhamini R., Offmann B.. Analysis on conservation of disulphide bonds and their structural features in homologous protein domain families. BMC Struct. Biol. 2008; 8:55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rubinstein R., Fiser A.. Predicting disulfide bond connectivity in proteins by correlated mutations analysis. Bioinformatics. 2008; 24:498–504. [DOI] [PubMed] [Google Scholar]
- 23. Wieteska L., Ionov M., Szemraj J., Feller C., Kolinski A., Gront D.. Improving thermal stability of thermophilic l-threonine aldolase from Thermotoga maritima. J. Biotechnol. 2015; 199:69–76. [DOI] [PubMed] [Google Scholar]
- 24. Korman T.P., Sahachartsiri B., Charbonneau D.M., Huang G.L., Beauregard M., Bowie J.U.. Dieselzymes: development of a stable and methanol tolerant lipase for biodiesel production by directed evolution. Biotechnol. Biofuels. 2013; 6:70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Takagi H., Takahashi T., Momose H., Inouye M., Maeda Y., Matsuzawa H., Ohta T.. Enhancement of the thermostability of subtilisin E by introduction of a disulfide bond engineered on the basis of structural comparison with a thermophilic serine protease. J. Biol. Chem. 1990; 265:6874–6878. [PubMed] [Google Scholar]
- 26. Saerens D., Conrath K., Govaert J., Muyldermans S.. Disulfide bond introduction for general stabilization of immunoglobulin heavy-chain variable domains. J. Mol. Biol. 2008; 377:478–488. [DOI] [PubMed] [Google Scholar]
- 27. Suplatov D.A., Kopylov K.E., Popova N.N., Voevodin V.V., Švedas V.K.. Mustguseal: a server for multiple structure-guided sequence alignment of protein families. Bioinformatics. 2018; 34:1583–1585. [DOI] [PubMed] [Google Scholar]
- 28. Burley S.K., Berman H.M., Bhikadiya C., Bi C., Chen L., Di Costanzo L., Christie C., Dalenberg K., Duarte J.M., Dutta S. et al.. RCSB Protein Data Bank: biological macromolecular structures enabling research and education in fundamental biology, biomedicine, biotechnology and energy. Nucleic Acids Res. 2019; 47:D464–D474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. UniProt Consortium. UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res. 2019; 47:D506–D515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Reimer L.C., Vetcininova A., Carbasse J.S., Söhngen C., Gleim D., Ebeling C., Overmann J.. Bac Dive in 2019: bacterial phenotypic data for High-throughput biodiversity analysis. Nucleic Acids Res. 2019; 47:D631–D636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Krissinel E., Henrick K.. Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr. D Biol. Crystallogr. 2004; 60:2256–2268. [DOI] [PubMed] [Google Scholar]
- 32. Sharapova Y., Suplatov D., Švedas V.. Neuraminidase a from Streptococcus pneumoniae has a modular organization of catalytic and lectin domains separated by a flexible linker. FEBS J. 2018; 285:2428–2445. [DOI] [PubMed] [Google Scholar]
- 33. Nilmeier J.P., Meng E.C., Polacco B.J., Babbitt P.C.. Rigden DJ. 3D motifs. From Protein Structure to Function with Bioinformatics. 2017; Dordrecht: Springer; 361–392. [Google Scholar]
- 34. Fletcher R., Reeves C.M.. Function minimization by conjugate gradients. Computer J. 1964; 7:149–154. [Google Scholar]
- 35. Maier J.A., Martinez C., Kasavajhala K., Wickstrom L., Hauser K.E., Simmerling C.. ff14SB: improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 2015; 11:3696–3713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Pellequer J.L., Chen S.W.W.. Multi‐template approach to modeling engineered disulfide bonds. Proteins. 2006; 65:192–202. [DOI] [PubMed] [Google Scholar]
- 37. Sillitoe I., Lewis T.E., Cuff A., Das S., Ashford P., Dawson N.L., Furnham N., Laskowski R.A., Lee D., Lees J.G. et al.. CATH: comprehensive structural and functional annotations for genome sequences. Nucleic Acids Res. 2014; 43:D376–D381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hanson R.M., Prilusky J., Renjian Z., Nakane T., Sussman J.L.. JSmol and the next‐generation web‐based representation of 3D molecular structure as applied to proteopedia. Isr. J. Chem. 2013; 53:207–216. [Google Scholar]
- 39. Pleiss J. Systematic analysis of large enzyme families: identification of specificity‐ and selectivity‐determining hotspots. Chem. Cat. Chem. 2014; 6:944–950. [Google Scholar]
- 40. Bloom J.D., Labthavikul S.T., Otey C.R., Arnold F.H.. Protein stability promotes evolvability. Proc. Natl. Acad. Sci. U.S.A. 2006; 103:5869–5874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Finch A., Kim J.. Thermophilic proteins as versatile scaffolds for protein engineering. Microorganisms. 2018; 6:97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Socha R.D., Tokuriki N.. Modulating protein stability–directed evolution strategies for improved protein function. FEBS J. 2013; 280:5582–5595. [DOI] [PubMed] [Google Scholar]
- 43. Childers M.C., Daggett V.. Insights from molecular dynamics simulations for computational protein design. Mol. Syst. Des. Eng. 2017; 2:9–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sadovnichy V., Tikhonravov A., Voevodin V., Opanasenko V.. Vetter JS. Lomonosov: supercomputing at moscow state university. Contemporary High Performance Computing: From Petascale Toward Exascale (Chapman & Hall/CRC Computational Science). 2013; Boca Raton: CRC Press; 283–307. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.