


Abstract
Secondary metabolites produced by bacteria and fungi are an important source of antimicrobials and other bioactive compounds. In recent years, genome mining has seen broad applications in identifying and characterizing new compounds as well as in metabolic engineering. Since 2011, the ‘antibiotics and secondary metabolite analysis shell—antiSMASH’ (https://antismash.secondarymetabolites.org) has assisted researchers in this, both as a web server and a standalone tool. It has established itself as the most widely used tool for identifying and analysing biosynthetic gene clusters (BGCs) in bacterial and fungal genome sequences. Here, we present an entirely redesigned and extended version 5 of antiSMASH. antiSMASH 5 adds detection rules for clusters encoding the biosynthesis of acyl-amino acids, β-lactones, fungal RiPPs, RaS-RiPPs, polybrominated diphenyl ethers, C-nucleosides, PPY-like ketones and lipolanthines. For type II polyketide synthase-encoding gene clusters, antiSMASH 5 now offers more detailed predictions. The HTML output visualization has been redesigned to improve the navigation and visual representation of annotations. We have again improved the runtime of analysis steps, making it possible to deliver comprehensive annotations for bacterial genomes within a few minutes. A new output file in the standard JavaScript object notation (JSON) format is aimed at downstream tools that process antiSMASH results programmatically.
INTRODUCTION
Bacterial and fungal natural products constitute a key source of scaffolds for the development of antimicrobials and other drugs (1), and mediate ecological interactions between organisms in various ways (2).
Mining genomic data for the presence of biosynthetic pathways that enable organisms to produce such molecules, which are also referred to as secondary or specialized metabolites, have become an essential approach that complements activity- and chemistry-guided isolation and identification approaches (3). Several computational tools, such as CLUSEAN (4) or PRISM (5), have been developed to support scientists with this task. The ‘antibiotics and secondary metabolites analysis shell’, antiSMASH, is a pioneer amongst these tools. Initially released in 2011 (6), it has since been further extended and improved (7–12), and is currently used by thousands of academic and industrial scientists worldwide to identify so called secondary metabolite ‘biosynthetic gene clusters’ (BGCs) in their genomes of interest. In 2017, a database component was added to the antiSMASH framework, which provides instant access to thousands of pre-computed antiSMASH genome mining results of publicly available genomes (13,14). Furthermore, several independent tools, such as the mass-spectrometry guided peptide mining tool Pep2Path (15), the ‘Antibiotic Resistance Target Seeker’ ARTS (16), the sgRNA design tool CRISPy-web (17), a reverse-tailoring tool to match finished NRPS/PKS structures to antiSMASH-predicted core structures (18) and the BGC clustering and classification platform BiG-SCAPE (19) were developed that directly interact with and interpret results generated by antiSMASH and provide information that is outside the scope of a core-antiSMASH analysis.
Here, we present version 5 of antiSMASH, which contains many improvements. In addition to many features visible to the end users, such as extended and improved BGC detection and analysis capabilities and a modernized and improved User Interface (see below), antiSMASH version 5 was completely rewritten in Python version 3 and the code was restructured to increase performance, reliability and ease of maintenance. This has led to a significant speed increase of the pipeline. A complete list of antiSMASH 5 features is included in the antiSMASH documentation https://docs.antismash.secondarymetabolites.org/antiSMASH5features/.
NEW FEATURES AND UPDATES
New gene cluster classes and refinement of cluster detection rules
The most widely used and recommended mode to detect BGCs in genomic data is via manually curated and validated gene cluster rules. These are based on identifying co-occurring conserved core enzymes in the genome using HMM-profiles that were derived from Pfam (20), SMART (21), BAGEL (22) or Yadav et al. (23), or that were created specifically for antiSMASH. While antiSMASH version 4 supported the rule-based detection of 44 different biosynthetic types, antiSMASH 5 now includes rules for 52 different BGC types. In version 5, new rules were added to detect BGCs encoding the biosynthesis of N-acyl amino acids (24), β-lactones (25), polybrominated diphenyl ethers (26), C-nucleosides (27), pseudopyronines (28), fungal RiPPs (29–31) and RaS-RiPPs (32,33). Furthermore, a new ‘nrps-like’ rule was defined for NRPS-fragments, i.e. atypical NRPSs that don’t have the typical C-A-T module architecture. The previous ‘otherks’ rule was split into two rules to individually assign heterocyst glycolipid synthase-like clusters and other atypical PKSs. In addition, some rules were improved based on user case reports. The rules describing lanthipeptides and trans-AT type I PKS were refined to reduce the number of false positive hybrid calls on other cluster types. For trans-AT- type I PKS and type II PKS, we increased the size of the cluster cutoffs to capture previously missed tailoring enzymes in published clusters. The rule for linear azole/azoline-containing peptides was made more generic to better cover the range of described clusters.
The rule describing microcin clusters was removed, as microcins are a class of RiPPs defined via their production in Enterobacteriaceae, and are already captured by one of our other specific RiPP cluster rules, depending on their respective biosynthesis pathway (e.g. microcin J25-like RiPPs were previously covered by the old microcin cluster rules but chemically are lasso peptides, while microcin B17 is a linear azol(in)e-containing peptide).
Improved type II PKS prediction
Bacterial type II PKS BGCs code for the biosynthesis of aromatic polyketides, such as the antibiotic tetracycline or the anti-tumour drug doxorubicin. From the beginning, antiSMASH has had rules that were able to detect type II PKS BGCs by checking for the presence of the KSα and KSβ/CLF component of the minimal PKS. However, no detailed prediction methods had been added since antiSMASH’s first version. In antiSMASH 5, we introduce a new PKS II analysis module (12), which uses a collection of manually curated HMMs to predict potential starter units, the number of elongation cycles (and thus a rough estimation of the putative molecular weight of the core compound), cyclization patterns and some conserved type II PKS specific tailoring reactions. This module is automatically triggered whenever a type II PKS BGC is detected.
Annotation of resistance genes via Resfams
The Resfams database (34) is a curated database of protein families with confirmed antibiotic resistance function. antiSMASH 5 uses the profile Hidden Markov Models (pHMMs) from Resfams to annotate potential resistance genes found in predicted gene regions. Potential resistance gene-hits are displayed in the ‘gene details’ panel along with other functional annotations.
GO-term annotations
The Gene Ontology (GO) is a controlled vocabulary for describing biological processes, molecular functions and cellular components in a consistent way to enable comparison of these between different species. Amongst its wide range of uses, the GO has been used to predict gene clusters in eukaryotes and bacteria (35) and, in conjunction with antiSMASH, to refine cluster boundaries in antiSMASH output for Aspergillus species (36).
To facilitate these and other GO-based analyses, antiSMASH 5 includes an option to automatically annotate GO terms on Pfam domains. This functionality makes use of the fact that GO terms may be linked not only to specific gene products, but also to other means of classification in so-called ‘mappings’ (http://geneontology.org/page/download-mappings). As antiSMASH can automatically annotate Pfam domains, the GO annotation functionality makes use of the Pfam to GO mapping supplied by the Gene Ontology Consortium’s website (37). If the ID of a predicted Pfam domain in an antiSMASH record is present in the Pfam to GO mapping, the respective GO terms are assigned and presented in the ‘gene details’ panel.
Link to the antiSMASH database
antiSMASH provides options to search for similar gene clusters in public datasets. As already implemented in previous versions of the software, the KnownClusterBlast functionality searches each identified region against the manually curated MIBiG (38) repository. The KnownClusterBlast and ClusterBlast search functions use an algorithm first described in antiSMASH 1 (6), which also is in use in a generalized version in MultiGeneBlast (39). In the previous versions of antiSMASH, the ClusterBlast database was generated by scripts that used the antiSMASH BGC detection logic on sequences downloaded from the NCBI Genbank/RefSeq databases. As version 2 of the antiSMASH database now also contains BGCs of draft genomes (14), starting with antiSMASH 5 the ClusterBlast databases will be directly generated from the new antiSMASH database and complemented with individual BGC records that were submitted to NCBI outside of whole-genome submissions. This provides several advantages: The abundance of entries for selected genera/species in the public databases (and thus also in the previous ClusterBlast database) is strongly skewed towards clinically or industrially relevant organisms. There are, for example, more than 15 000 assemblies for Escherichia coli deposited at NCBI. For the antiSMASH database, a sequence-based dereplication workflow was established (14) that reduced the number of redundant entries with very high sequence similarity. Thus, the updated ClusterBlast database contains fewer entries than the previous release, despite the increase in publicly available sequence data. This decrease has resulted in reduced computation times, while simultaneously providing more relevant hits. Furthermore, as the entries of the ClusterBlast database are directly related to the BGCs in the antiSMASH database, a link to the respective BGC is now included for all ClusterBlast hits, promptly directing the user to the detailed report of the similar gene clusters.
New ‘region’ concept
In previous versions, antiSMASH referred to all co-located, hybrid and independent BGCs with the single label ‘cluster’. In many cases, this led to confusing structure predictions when distinct BGCs are encoded side-by-side. For example, many Streptomyces plasmids exist for which all BGCs lie so close to each other that all were joined into a single large ‘cluster’. In order to better distinguish the different biological options that lead to BGCs, antiSMASH 5 introduces some new terminology.
The definitions now used in antiSMASH 5 are:
Core: The minimum area containing one or more genes that code for enzymes for a single BGC type that are detected by the manually curated detection rules. These genes do not have to be contiguous, but can be within a certain cutoff distance as defined by the detection rule for the BGC type in question.
Neighbourhood: Distance up- and downstream of the cluster core that is used to find tailoring genes/enzymes; the neighbourhood distances for the individual biosynthetic types were empirically determined and defined in the detection rules.
Protocluster: Contains core + neighbourhoods at both sides of the core; each protocluster always will have one single product type (for example, NRPS). Protoclusters may overlap partially or completely with other protoclusters. In the result webpage, protoclusters are displayed as boxes above the gene arrows. The cores are shown as solid colour boxes, the neighbourhoods are the half-transparent areas around the cores.
Candidate cluster: Contains one or more protoclusters; the candidate clusters are defined as described below. These definitions better allow modelling of hybrid clusters, such as PKS/NRPS hybrids, which combine two or more different biosynthetic classes (as identified in the detection rules), or cases where one class is used to biosynthesize a precursor for a second class. An example of the latter is found in glycopeptide biosynthesis, where one of the amino acids is synthesized by a type III PKS, which is then incorporated into the product by a NRPS. Candidate clusters may overlap partially or completely with other candidate clusters. In the result webpage, candidate clusters are shown as boxes above the protoclusters.
Region: Contains one or more candidate clusters; The regions in antiSMASH 5 correspond to the entities called ‘clusters’ in antiSMASH 1 – 4 and now constitute what is displayed on a page of the results webpage. Sometimes, a region will contain multiple mutually exclusive candidate clusters; in such cases, comparative genomic analysis and/or experimental work is required to assess which of these candidate clusters constitute actual BGCs. Regions will not overlap with each other. At least one of the contained candidate clusters will cover the full length of the region.
There are four kinds of candidate clusters: chemical hybrids, interleaved, neighbouring and single.
Chemical hybrid candidate clusters contain at least two protoclusters that share at least one gene that codes for enzymes of two or more separate BGC types (e.g. a single gene coding for type I PKS and NRPS modules) (Figure 1A). An example of this type are hybrid PKS/NRPSs. Please note that this type of candidate cluster can also include protoclusters within that shared range that do not share a coding sequence provided that they are completely contained within the candidate cluster.
Figure 1.
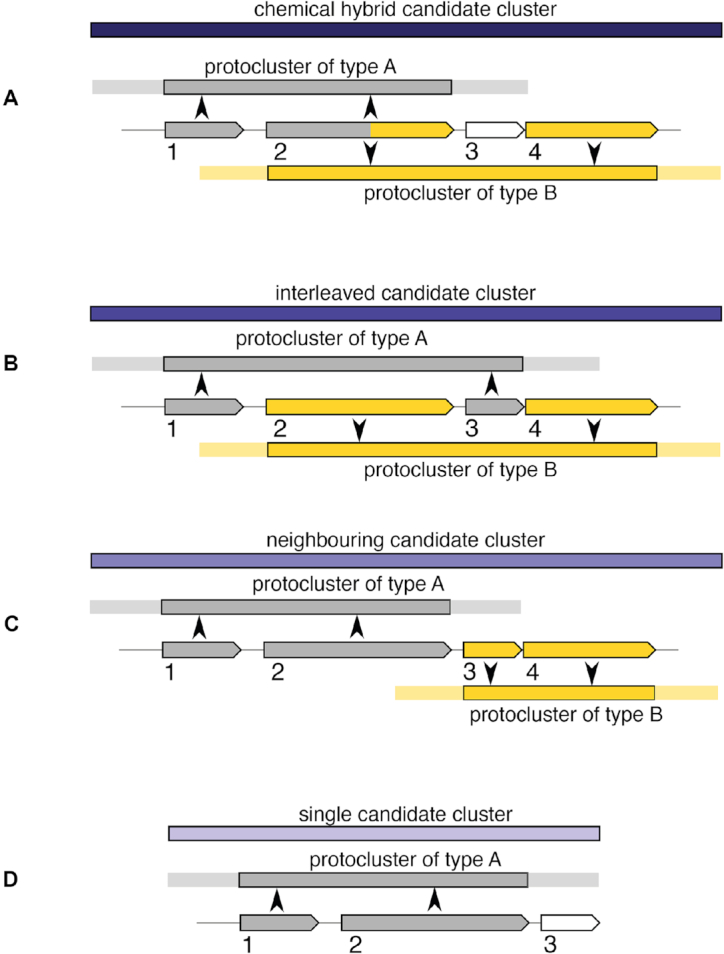
Candidate cluster types. 1,2,3,4: Grey/yellow: gene involved in protocluster A/B. (A) Chemical Hybrids. Since cluster type A and cluster type B share a CDS that defines those protoclusters, they are classified as ‘chemical hybrid’. (B) Interleaved: Since none of the protoclusters share any defining CDS with any other protocluster, it is not annotated as a chemical hybrid, even though the biosynthetic product may or may not be. The two protoclusters form an interleaved candidate clusters, since the core of A overlaps with the core of B. (C) Neighbouring: Neighbouring candidate clusters are defined if the neighbourhoods of two protoclusters but not their cores overlap. (D) Singles: If protoclusters don’t have any overlap/relation with other protoclusters, the term single candidate cluster is assigned.
Interleaved candidate clusters contain protoclusters that do not share cluster-type-defining coding sequences, but their core locations overlap (Figure 1B).
Neighbouring candidate clusters contain protoclusters which transitively overlap in their neighbourhoods (Figure 1C).
Single candidate clusters (Figure 1D) exist for consistency of access, they contain only a single protocluster. Note that individual protoclusters can be contained by more than one candidate cluster (typically a neighbouring candidate cluster and one of single, interleaved or chemical hybrid).
Each candidate cluster assignment is transitive, for example if a protocluster would form a chemical hybrid with each of two neighbouring protoclusters, but these neighbours would not form a chemical hybrid on their own, all three together will still form a chemical hybrid candidate cluster.
Improved user interface
A central aim of antiSMASH is to provide very detailed and specific information via an easy to use and understand user interface (UI). The UI remained principally unchanged from the initial release of antiSMASH in 2011, despite the increased functionality added with each new version. In this version, we have modernized the UI using updated web technologies that allow a better structuring of the result-content of the antiSMASH results pages. For redesigning the UI, it was important that the reliable and well-established look-and-feel was conserved, while also retaining the ability to download the whole web-based results folder and to display it locally in a variety of web-browsers.
We and others (such as (40)) have realized that antiSMASH results using the heuristic ClusterFinder algorithm (41) were, more often than not, wrongly interpreted. At the same time, ClusterFinder contributed significantly to the computational workload. For these reasons, we decided to remove this feature from the pubic antiSMASH web server. It is, of course, still included in the download version of antiSMASH and can be enabled via the command line.
In the Regions overview section (Figure 2), a graphical overview showing the location of the identified regions on the chromosome/plasmid/scaffolds/contig is displayed. In the detailed view, regions that are located on contig-borders are now clearly labelled. This often indicates that parts of the BGC are missing or that several sections of a BGC are located on different contigs and are therefore reported individually (for a more detailed discussion on this phenomenon, please see (42)). For the first time, antiSMASH 5 now offers interactive browsing of the BGCs, including selection of ‘functional’ units, i.e. core enzymes, transporters, etc., zooming to individual genes or regions/candidate clusters/protoclusters. Details of the selection are now provided in side panels instead of pop-up windows, using a hierarchical view of the analysis summaries (which can be expanded by clicking ‘+’) to provide additional details. For the display of the PKS/NRPS domain organization, the user now can choose whether to limit the shown domains to the currently selected genes or just display the results of the selected gene(s)/enzyme(s). Furthermore, the information is now organized in ‘tabs’ that do not require scrolling down along an often very long results page.
Figure 2.

Screenshot of the antiSMASH 5 user interface (example: NCBI-acc: Y16952; balhimycin BGC). The new region overview now allows panning/zooming. The candidate cluster and protocluster boxes are explained in the ‘new region concept’ section above. Information about the currently selected gene are displayed at the right ‘Gene details’ panel. For PKS or NRPS regions, the detailed domain annotation is displayed; by pressing the tabs, users can select the domain overview (shown) or the ClusterBlast, KnownClusterBlast or SubClusterBlast results. At the right, the structure prediction and details of specificity predictions are displayed upon selecting the plus sign.
CODE REFACTORING AND SPEED-UP
Large parts of the pre-antiSMASH 5 code base were still derived from antiSMASH version 1, which was released in 2011. In order to maintain future compatibility, the antiSMASH code base had to be migrated from python 2.7, which will reach end-of-life in 2020, to the current versions 3.5–3.7. As this transition required significant modification to the antiSMASH code, we decided to take this as a chance to completely rewrite the software with a special consideration on runtime, code stability and code maintainability. A unit test and integration test framework was implemented that covers most parts of the antiSMASH 5 code allowing a much easier debugging and—most importantly—extension of the code while at the same time ensuring that new features do not negatively impact the results of existing modules. For some of the externally contributed modules (Sandpuma, trans-AT PKS comparisons, terpene PrediCAT), our contributors are currently preparing updated and compliant versions, which will be added to antiSMASH 5 in minor releases once they are finished and tested. Like the earlier antiSMASH versions, antiSMASH 5 provides the analysis results in an interactive webpage and richly annotated GenBank-format files for the whole genome and individual clusters. As a new feature in version 5, all data are also available as a computer readable JSON container, which allows third party tools to easily process antiSMASH annotations. This JSON output has superseded some other output types, such as BioSynML and XLS.
In addition to the advantages mentioned above, the code refactoring and cleanup has also led to a significant speed increase of the new version by a factor of 4-11× (depending on genome and selected options); instead of waiting times of several hours, antiSMASH results are now usually delivered within 30–40 min after the start of the job for a typical submission at the public web server.
CONCLUSIONS AND FUTURE PERSPECTIVES
With the help of software like antiSMASH, genome mining for specialized metabolites has established itself as a complementary approach for the identification of novel metabolites, which is routinely used within the natural products research community and increasingly applied in related fields such as metagenomics, environmental biology or metabolic engineering. With the improvements to the antiSMASH user interface and performance, we keep pace with these developments. Furthermore, the complete refactoring of the antiSMASH 5 code base will allow us to increasingly use antiSMASH as a tool that provides analysis data on which other software can perform additional analyses.
DATA AVAILABILITY
antiSMASH is available from https://antismash.secondarymetabolites.org/ (bacterial version) or https://fungismash.secondarymetabolites.org/ (fungal version). The antiSMASH documentation, including a PDF user guide, is available from https://docs.antismash.secondarymetabolites.org. These websites are free and open to all users and there is no login requirement. The antiSMASH source code is available from https://github.com/antismash/antismash. antiSMASH is also available via Docker.
ACKNOWLEDGEMENTS
We thank Justin J.J. van der Hooft for critical comments on the manuscript and providing documentation and Emilia Palazzotto and Tetiana Gren for helpful discussions and user testing of the new features.
Contributor Information
Kai Blin, The Novo Nordisk Foundation Center for Biosustainability, Technical University of Denmark, Kemitorvet bygning 220, 2800 Kgs. Lyngby, Denmark.
Simon Shaw, The Novo Nordisk Foundation Center for Biosustainability, Technical University of Denmark, Kemitorvet bygning 220, 2800 Kgs. Lyngby, Denmark.
Kat Steinke, German Centre for Infection Research (DZIF), Interfaculty Institute of Microbiology and Infection Medicine, Auf der Morgenstelle 28, University of Tübingen, 72076 Tübingen, DE, Germany.
Rasmus Villebro, The Novo Nordisk Foundation Center for Biosustainability, Technical University of Denmark, Kemitorvet bygning 220, 2800 Kgs. Lyngby, Denmark.
Nadine Ziemert, German Centre for Infection Research (DZIF), Interfaculty Institute of Microbiology and Infection Medicine, Auf der Morgenstelle 28, University of Tübingen, 72076 Tübingen, DE, Germany.
Sang Yup Lee, The Novo Nordisk Foundation Center for Biosustainability, Technical University of Denmark, Kemitorvet bygning 220, 2800 Kgs. Lyngby, Denmark; Department of Chemical and Biomolecular Engineering (BK21 Plus Program) and BioInformatics Research Center, Korea Advanced Institute of Science and Technology, 291 Daehak-ro, Yuseong-gu, Daejeon 34141, South Korea.
Marnix H Medema, Bioinformatics Group, Wageningen University, Droevendaalsesteeg 1, 6708PB Wageningen, the Netherlands.
Tilmann Weber, The Novo Nordisk Foundation Center for Biosustainability, Technical University of Denmark, Kemitorvet bygning 220, 2800 Kgs. Lyngby, Denmark.
FUNDING
Novo Nordisk Foundation [NNF10CC1016517 to S.Y.L.,T.W.; NNF16OC0021746 to T.W.]; Center for Microbial Secondary Metabolites (CeMiSt), Danish National Research Foundation [DNRF137 to T.W.]; Reinhold and Maria Teufel Foundation (to K.S.). Funding for open access charge: The Novo Nordisk Foundation.
Conflict of interest statement. None declared.
REFERENCES
- 1. Newman D.J., Cragg G.M. Natural products as sources of new drugs from 1981 to 2014. J. Nat. Prod. 2016; 79:629–661. [DOI] [PubMed] [Google Scholar]
- 2. van der Meij A., Worsley S.F., Hutchings M.I., van Wezel G.P. Chemical ecology of antibiotic production by actinomycetes. FEMS Microbiol. Rev. 2017; 41:392–416. [DOI] [PubMed] [Google Scholar]
- 3. Ziemert N., Alanjary M., Weber T. The evolution of genome mining in microbes - a review. Nat. Prod. Rep. 2016; 33:988–1005. [DOI] [PubMed] [Google Scholar]
- 4. Weber T., Rausch C., Lopez P., Hoof I., Gaykova V., Huson D.H., Wohlleben W. CLUSEAN: a computer-based framework for the automated analysis of bacterial secondary metabolite biosynthetic gene clusters. J. Biotechnol. 2009; 140:13–17. [DOI] [PubMed] [Google Scholar]
- 5. Skinnider M.A., Merwin N.J., Johnston C.W., Magarvey N.A. PRISM 3: expanded prediction of natural product chemical structures from microbial genomes. Nucleic Acids Res. 2017; 45:W49–W54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Medema M.H., Blin K., Cimermancic P., de Jager V., Zakrzewski P., Fischbach M.A., Weber T., Takano E., Breitling R. antiSMASH: rapid identification, annotation and analysis of secondary metabolite biosynthesis gene clusters in bacterial and fungal genome sequences. Nucleic Acids Res. 2011; 39:W339–W346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Blin K., Medema M.H., Kazempour D., Fischbach M.A., Breitling R., Takano E., Weber T. antiSMASH 2.0—a versatile platform for genome mining of secondary metabolite producers. Nucleic Acids Res. 2013; 41:W204–W212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Weber T., Blin K., Duddela S., Krug D., Kim H.U., Bruccoleri R., Lee S.Y., Fischbach M.A., Müller R., Wohlleben W. et al. antiSMASH 3.0—a comprehensive resource for the genome mining of biosynthetic gene clusters. Nucleic Acids Res. 2015; 43:W237–W243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Blin K., Wolf T., Chevrette M.G., Lu X., Schwalen C.J., Kautsar S.A., Suarez Duran H.G., de Los Santos E.L.C., Kim H.U., Nave M. et al. antiSMASH 4.0-improvements in chemistry prediction and gene cluster boundary identification. Nucleic Acids Res. 2017; 45:W36–W41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kautsar S.A., Suarez Duran H.G., Blin K., Osbourn A., Medema M.H. plantiSMASH: automated identification, annotation and expression analysis of plant biosynthetic gene clusters. Nucleic Acids Res. 2017; 45:W55–W63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Blin K., Kazempour D., Wohlleben W., Weber T. Improved lanthipeptide detection and prediction for antiSMASH. PLoS One. 2014; 9:e89420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Villebro R., Shaw S., Blin K., Weber T. Sequence-based classification of type II polyketide synthase biosynthetic gene clusters for antiSMASH. J. Ind. Microbiol. Biotechnol. 2019; 46:469–475. [DOI] [PubMed] [Google Scholar]
- 13. Blin K., Medema M.H., Kottmann R., Lee S.Y., Weber T. The antiSMASH database, a comprehensive database of microbial secondary metabolite biosynthetic gene clusters. Nucleic Acids Res. 2017; 45:D555–D559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Blin K., Pascal Andreu V., de Los Santos E.L.C., Del Carratore F., Lee S.Y., Medema M.H., Weber T. The antiSMASH database version 2: a comprehensive resource on secondary metabolite biosynthetic gene clusters. Nucleic Acids Res. 2019; 47:D625–D630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Medema M.H., Paalvast Y., Nguyen D.D., Melnik A., Dorrestein P.C., Takano E., Breitling R. Pep2Path: automated mass spectrometry-guided genome mining of peptidic natural products. PLoS Comput. Biol. 2014; 10:e1003822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Alanjary M., Kronmiller B., Adamek M., Blin K., Weber T., Huson D., Philmus B., Ziemert N. The Antibiotic Resistant Target Seeker (ARTS), an exploration engine for antibiotic cluster prioritization and novel drug target discovery. Nucleic Acids Res. 2017; 45:W42–W48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Blin K., Pedersen L.E., Weber T., Lee S.Y. CRISPy-web: An online resource to design sgRNAs for CRISPR applications. Synth. Syst. Biotechnol. 2016; 1:118–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Shirley W.A., Kelley B.P., Potier Y., Koschwanez J.H., Bruccoleri R., Tarselli M. Unzipping natural products: improved natural product structure predictions by ensemble modeling and fingerprint matching. 2018; ChemRxiv doi: 26 July 2018, preprint: not peer reviewed http://doi:10.26434/chemrxiv.6863864.
- 19. Navarro-Muñoz J., Selem-Mojica N., Mullowney M., Kautsar S., Tryon J., Parkinson E., De Los Santos E., Yeong M., Cruz-Morales P., Abubucker S. et al. A computational framework for systematic exploration of biosynthetic diversity from large-scale genomic data. 2018; bioRxiv doi: http://doi:10.1101/445270, 17 October 2018, preprint: not peer reviewed.
- 20. Finn R.D., Coggill P., Eberhardt R.Y., Eddy S.R., Mistry J., Mitchell A.L., Potter S.C., Punta M., Qureshi M., Sangrador-Vegas A. et al. The Pfam protein families database: towards a more sustainable future. Nucleic Acids Res. 2016; 44:D279–D285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Letunic I., Bork P. 20 years of the SMART protein domain annotation resource. Nucleic Acids Res. 2018; 46:D493–D496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. de Jong A., van Heel A.J., Kok J., Kuipers O.P. BAGEL2: mining for bacteriocins in genomic data. Nucleic Acids Res. 2010; 38:W647–W651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yadav G., Gokhale R.S., Mohanty D. Towards prediction of metabolic products of polyketide synthases: an in silico analysis. PLoS Comput. Biol. 2009; 5:e1000351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Craig J.W., Cherry M.A., Brady S.F. Long-chain N-acyl amino acid synthases are linked to the putative PEP-CTERM/exosortase protein-sorting system in Gram-negative bacteria. J. Bacteriol. 2011; 193:5707–5715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Robinson S.L., Christenson J.K., Wackett L.P. Biosynthesis and chemical diversity of β-lactone natural products. Nat. Prod. Rep. 2018; 36:458–475. [DOI] [PubMed] [Google Scholar]
- 26. Agarwal V., Blanton J.M., Podell S., Taton A., Schorn M.A., Busch J., Lin Z., Schmidt E.W., Jensen P.R., Paul V.J. et al. Metagenomic discovery of polybrominated diphenyl ether biosynthesis by marine sponges. Nat. Chem. Biol. 2017; 13:537–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sosio M., Gaspari E., Iorio M., Pessina S., Medema M.H., Bernasconi A., Simone M., Maffioli S.I., Ebright R.H., Donadio S. Analysis of the Pseudouridimycin biosynthetic pathway provides Insights into the formation of C-nucleoside antibiotics. Cell Chem. Biol. 2018; 25:540–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bauer J.S., Ghequire M.G.K., Nett M., Josten M., Sahl H.-G., De Mot R., Gross H. Biosynthetic origin of the antibiotic pseudopyronines A and B in Pseudomonas putida BW11M1. Chembiochem. 2015; 16:2491–2497. [DOI] [PubMed] [Google Scholar]
- 29. Luo H., Hallen-Adams H.E., Scott-Craig J.S., Walton J.D. Ribosomal biosynthesis of α-amanitin in Galerina marginata. Fungal Genet. Biol. 2012; 49:123–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Nagano N., Umemura M., Izumikawa M., Kawano J., Ishii T., Kikuchi M., Tomii K., Kumagai T., Yoshimi A., Machida M. et al. Class of cyclic ribosomal peptide synthetic genes in filamentous fungi. Fungal Genet. Biol. 2016; 86:58–70. [DOI] [PubMed] [Google Scholar]
- 31. Ding W., Liu W.-Q., Jia Y., Li Y., van der Donk W.A., Zhang Q. Biosynthetic investigation of phomopsins reveals a widespread pathway for ribosomal natural products in Ascomycetes. Proc. Natl. Acad. Sci. U.S.A. 2016; 113:3521–3526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bushin L.B., Clark K.A., Pelczer I., Seyedsayamdost M.R. Charting an unexplored streptococcal biosynthetic landscape reveals a unique peptide cyclization motif. J. Am. Chem. Soc. 2018; 140:17674–17684. [DOI] [PubMed] [Google Scholar]
- 33. Caruso A., Bushin L.B., Clark K.A., Martinie R.J., Seyedsayamdost M.R. A radical approach to enzymatic β-Thioether bond formation. J. Am. Chem. Soc. 2019; 141:990–997. [DOI] [PubMed] [Google Scholar]
- 34. Gibson M.K., Forsberg K.J., Dantas G. Improved annotation of antibiotic resistance determinants reveals microbial resistomes cluster by ecology. ISME J. 2015; 9:207–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yi G., Sze S.H., Thon M.R. Identifying clusters of functionally related genes in genomes. Bioinformatics. 2007; 23:1053–1060. [DOI] [PubMed] [Google Scholar]
- 36. Inglis D.O., Binkley J., Skrzypek M.S., Arnaud M.B., Cerqueira G.C., Shah P., Wymore F., Wortman J.R., Sherlock G. Comprehensive annotation of secondary metabolite biosynthetic genes and gene clusters of Aspergillus nidulans, A. fumigatus, A. niger and A. oryzae. BMC Microbiol. 2013; 13:91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. The Gene Ontology Consortium Expansion of the Gene Ontology knowledgebase and resources. Nucleic Acids Res. 2016; 45:D331–D338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Medema M.H., Kottmann R., Yilmaz P., Cummings M., Biggins J.B., Blin K., de Bruijn I., Chooi Y.H., Claesen J., Coates R.C. et al. Minimum information about a biosynthetic gene cluster. Nat. Chem. Biol. 2015; 11:625–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Medema M.H., Takano E., Breitling R. Detecting sequence homology at the gene cluster level with MultiGeneBlast. Mol. Biol. Evol. 2013; 30:1218–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Baltz R.H. Natural product drug discovery in the genomic era: realities, conjectures, misconceptions, and opportunities. J. Ind. Microbiol. Biotechnol. 2018; 46:281–299. [DOI] [PubMed] [Google Scholar]
- 41. Cimermancic P., Medema M.H., Claesen J., Kurita K., Wieland Brown L.C., Mavrommatis K., Pati A., Godfrey P.A., Koehrsen M., Clardy J. et al. Insights into secondary metabolism from a global analysis of prokaryotic biosynthetic gene clusters. Cell. 2014; 158:412–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Blin K., Kim H.U., Medema M.H., Weber T. Recent development of antiSMASH and other computational approaches to mine secondary metabolite biosynthetic gene clusters. Brief. Bioinform. 2017; doi:10.1093/bib/bbx146. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
antiSMASH is available from https://antismash.secondarymetabolites.org/ (bacterial version) or https://fungismash.secondarymetabolites.org/ (fungal version). The antiSMASH documentation, including a PDF user guide, is available from https://docs.antismash.secondarymetabolites.org. These websites are free and open to all users and there is no login requirement. The antiSMASH source code is available from https://github.com/antismash/antismash. antiSMASH is also available via Docker.