


Abstract
As more and more high-throughput data has been produced by next-generation sequencing, it is still a challenge to classify RNA transcripts into protein-coding or non-coding, especially for poorly annotated species. We upgraded our original coding potential calculator, CNCI (Coding-Non-Coding Index), to CNIT (Coding-Non-Coding Identifying Tool), which provides faster and more accurate evaluation of the coding ability of RNA transcripts. CNIT runs ∼200 times faster than CNCI and exhibits more accuracy compared with CNCI (0.98 versus 0.94 for human, 0.95 versus 0.93 for mouse, 0.93 versus 0.92 for zebrafish, 0.93 versus 0.92 for fruit fly, 0.92 versus 0.88 for worm, and 0.98 versus 0.85 for Arabidopsis transcripts). Moreover, the AUC values of 11 animal species and 27 plant species showed that CNIT was capable of obtaining relatively accurate identification results for almost all eukaryotic transcripts. In addition, a mobile-friendly web server is now freely available at http://cnit.noncode.org/CNIT.
INTRODUCTION
Numerous studies show that non-coding RNAs (ncRNAs) have critical roles in diverse biological processes from plants to animals (1–4), such as sponging by microRNAs (5), cell development (6), acting as modular scaffolds (7) and regulating epigenetic inheritance (8). Despite the increasing number of high-throughput data produced by next-generation sequencing, the classification of protein-coding or non-coding transcripts remains a challenge, especially for poorly annotated species. For instance, existing software available for annotating plants is rare and/or of low accuracy and plants are important resource for novel drug leads (9). The study of plant long non-coding RNA is still in its infancy, and the biological functions and mechanisms of plant non-coding RNAs are mainly focused on model plants such as rice and Arabidopsis. The first step is to identify lncRNA with effective identification software at the beginning of new research, so as to determine the research method and direction for the functional delineation of the newly discovered RNA. At present, few existing software programs can be used to identify plant non-coding RNA, and in general, the accuracy of identification has not been verified by a large number of data sets.
To overcome these shortcomings and make it easier for users to distinguish transcripts, we updated our CNCI algorithm (10) to create CNIT. In comparison with CNCI, CNIT runs ∼200 times faster than CNCI and exhibits higher accuracy, especially for plants, when using human and Arabidopsis data as training sets. Because CNIT, similar to CNCI, classifies protein-coding and non-coding RNAs solely based on intrinsic sequence composition, it is potentially applicable to a variety of species lacking a whole-genome sequence or with poorly annotated information. In addition, we constructed a mobile-friendly web server for researchers, making CNIT now freely available at http://cnit.noncode.org/CNIT/ as both a web server and a downloadable stand-alone package.
MATERIALS AND METHODS
Dataset processing
In order to construct and validate the CNIT model, we downloaded and filtered protein-coding and non-coding sequence data of 11 animal and 26 plant species from Refseq and Ensembl (Supplementary materials and methods and Supplementary Table S2). Animal protein-coding and non-coding transcripts were from the RefSeq database (11). For plants, coding transcripts were obtained from the Refseq, and noncoding transcripts were from Ensembl Plants (v37) database (12). A total of 19752 coding RNAs and 19752 non-coding RNAs of human origin (GRCH38) were selected for training and testing. In addition, 2588 coding RNAs and 2588 non-coding RNAs of Arabidopsis thaliana species (EnsemblPlants-v37) were used to build the plant model. Among the above total coding and non-coding transpcripts dataset, 70% were selected for training and 30% for testing. To evaluate the cross-species performance of CNIT, the rest of 10 animal species and 25 plant species were used for validation. These training and testing datasets collected by CNIT can be obtained from the download page (http://cnit.noncode.org/CNIT/download). In recent years, small open reading frame (sORF, length of sequence less than 300nt) has been studied continuously, but still has not formed a well-organized known database (13–15). Therefore, the existence of sORF was not considered in all the above lncRNA data sets. Moreover, in order to compare the performance of identifying mRNAs with sORFs, we then extracted the human mRNAs data set which contains sORFs.
To evaluate the performance of CNIT compared with other software, we further downloaded independent test datasets from CPC2 datasets (http://cpc2.cbi.pku.edu.cn/help/data_set.php), including human, mouse, zebrafish, fruitfly, worm and Arabidopsis thaliana datasets, for validation and comparison, which met strict standards and were high-quality (16).
Model construction
Consistent with CNCI (10), we first constructed a comparison frequency matrix of adjoining nucleotide triplets (ANT) using the training dataset (lncRNA sequence & coding domain sequence (CDS). Based on the comparison frequency matrix, a sub-sequence as most-like CDS (MLCDS) with highest summation of ANT frequency were found in each reading frame. Six MLCDSs were obtained from six open reading frames. Among them, the MLCDS with a maximal score (summation of ANT frequency) was termed as MMLCDS. Based on the six MLCDSs, the MMLCDS score, standard deviation of six MLCDS scores, standard deviation of six MLCDS lengths, and MMLCDS codon frequency (4*4*4 = 64 dimensions) with a total of 67 features were finally used to construct the XGBoost models (Supplementary materials and methods).
RESULTS
CNIT identification performance and comparison with existing tools
CPC2 and CNCI in the existing tools can be used to compare the performance in a wide range of species. The data were collected from test data downloaded from the CPC2 website (http://cpc2.cbi.pku.edu.cn/help/data_set.php, Figure 1A). The data of six species were identified by the five software programs: CNIT, CNCI, CPC2, CPAT (It only can identify four species) (17) and PLEK (18) software, and the accuracy was calculated (Figure 1B). Compared with CNCI, CNIT has higher accuracy. In terms of computing time, CNIT is ∼200 times faster than CNCI, as evaluated by calculating the average running time ratio of CNCI and CNIT in the six species when both were in single thread mode. Moreover, CNIT is almost better than that of CPC2, except for the identification of worm sequences (accuracy: 0.915 versus 0.975). CNIT also showed a better performance in more species than the CPAT and PLEK with more accuracy. Then, we used the above five software programs to identify mRNAs with sORFs (Supplementary Table S3). According to the results, compared with CNCI, CPAT and CPC2, CNIT has a higher accuracy in mRNAs with sORFs sequence identification, while PLEK has the highest accuracy.
Figure 1.
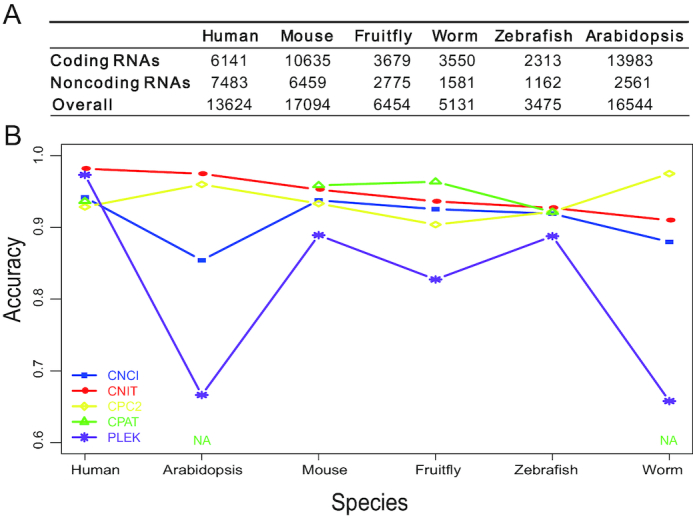
Evaluation of the accuracy of CNIT, CNCI, CPC2, CPAT and PLEK software. Overall comparison data (A) and detailed accuracy (B) in the six organisms from the CPC2 website.
For all downloaded animals and plants sequence data, CNIT identified them one by one and drew AUC curves to see the identification effect. For animal species, CNIT achieved a very high AUC value for mammals, amphibians, reptiles, birds, fish and invertebrates, indicating that it can distinguish coding from non-coding RNA. Similarly, for plants, CNIT also obtained high AUCs for monocotyledons, dicotyledons, bryophytes, ferns, Chlorophyta and red algae, especially monocotyledons and dicotyledons. CNIT validates plants and animals that cover most of the genera of the order family. Although not very rigorously, CNIT can identify most eukaryotic RNA as coding or non-coding RNA. Here, we show the prediction of CNIT for 37 species (11 animal and 26 plant species) with the corresponding AUC value (Figure 2). We also compared the performance of CNIT and CPC2 using the above datasets and showed the prediction accuracy of 37 species in Supplementary Table S2, meanwhile macro-averaged F1 statistic was performed for imbalanced datasets. The relevant comparison showed that CNIT’s ability to recognize coding transcripts outperformed CPC2 (Supplementary Figure S1A). Although CPC2 could identify non-coding sequences better (Supplementary Figure S1B), CNIT has more advantages in identifying sequences including coding and non-coding ones synchronously with higher macro-averaged F1, especially for plant species (Supplementary Figure S1C).
Figure 2.

Global prediction by ROC analysis for CNIT across 37 species.
Web server introduction
It is convenient for users to access the CNIT web portal at http://cnit.noncode.org/CNIT/. The web tool accepts RNA transcripts in FASTA format as input and outputs assess coding potential of the sequences. CNIT provides two identification methods, the simplest is to enter a single or multiple FASTA format RNA sequence through the website home page, and click RUN to submit the identification task. In addition, one can also submit the RNA sequence files in FASTA format on the ‘Batch' page for batch identification. However, if the sequence contains too many Ns or something else (more than 10% of the sequence), it may not produce results. In addition to web-side identification, users can download CNIT software packages and install them under the Linux system. For installation and usage, see the ‘Download’ page.
When the identification program finishes running, the identified results will appear on the results page. CNIT results give an overview of the coding status of the input sequences. Each row corresponds to one input sequence. The columns show the transcript ID, the coding/noncoding classification label (Index), the coding probability score (CNIT Score: where greater than 0 indicates coding RNA, less than 0 indicates non-noncoding RNA; the larger the score, the greater the coding possibility). Users can further click ‘View’ to enter the identification detail page. A unique job ID is assigned to each job by the web server. Users can use job-ID to track the job progress and retrieve the results, which will be saved on the server for one week.
EXAMPLE
We took human coding gene L1 cell adhesion molecule transcript variant 1 (L1CAM: NM_000425.4) (19) as an example and used online CNIT for identification. CNIT predicted that it was a coding transcript, with a CNIT score = 0.88 (Figure 3A).
Figure 3.

Screenshot of the CNIT web server. (A) Summary html view output with coding probability. (B, C) Graphical view of the ‘Details’ page.
‘View’ in the last column can be clicked to display more detailed information. The details page is divided into three parts. A description of L1CAM summarizing its coding probability and features is presented at the top (Figure 3B). In the middle of the page, an interactive visualization of three supporting features, including sequence length, MLCDS start and MLCDS end, are provided. In addition, the sequence detail of L1CAM is noted in the middle of the page (Figure 3C). In the CNIT Score Detail Plot, the red line represents the correct transcriptional reading frame out of other colored lines, such as the identification result for human coding gene L1CAM (Figure 4A). By contrast, CNIT analysis of human non-coding transcript HOX transcript antisense RNA transcript variant 2 (HOTAIR: NR_003716) (20) did not identify a coding sequence (CNIT Score = −0.31, Figure 4B). At the bottom of the ‘details’ page, you can blast your sequence in the NONCODE database in this page directly.
Figure 4.

Examples of CNIT analysis of transcripts for coding RNA L1CAM (A) and non-coding RNA HOTAIR (B). CNIT score distribution of the six reading frames for each transcript is the left y-axis and sequence length is normalized to nucleotide triplets in the x-axis. Red line represents the correct transcription reading frame and the other five lines (blue) represent the other five reading frames.
SUMMARY
Non-coding RNAs have emerged as major components of the eukaryotic transcriptome. Genome-wide analyses have revealed the existence of thousands of long noncoding RNAs (lncRNAs) in several species, and a growing number of lncRNAs have been found to be implicated in human disease (21–23) and plant growing and breeding (4,24–26). Despite the increasing number of high-throughput data produced by next-generation sequencing, the classification of protein-coding or noncoding transcripts remains a challenge, especially for poorly annotated species. In other words, the existing software available for annotating plants is rare or of low accuracy.
CNCI published in 2013 is widely used by worldwide researchers and has been cited >200 times (Web of Science) in the past 5 years (10). To better serve researchers and make it easier for users to distinguish transcripts, we updated our CNCI algorithm to CNIT. Because CNIT classifies protein-coding and non-coding RNAs solely based on intrinsic sequence composition, as does CNCI, it is particularly well suited for transcriptome analysis of not well-studied species with high accuracy, robustness and consistency, to help researchers validate coding or noncoding hypotheses for further functional studies. Moreover, in comparison with CNCI, CNIT runs∼200 times faster than CNCI and exhibits higher accuracy, especially for plants (0.98 versus 0.94 in humans, 0.95 versus 0.93 in mice, 0.93 versus 0.92 in zebrafish, 0.93 versus 0.92 in the fruit fly, 0.92 versus 0.88 in worms, and 0.98 versus 0.85 in Arabidopsis). The current CNIT can be further applied to species with incomplete genome annotations, such as Artemisia annua (Qing Hao), Astragalus membranaceus (Huang Qi), Ginseng (Ren Shen), etc.
Moreover, we constructed a user-friendly web server that is freely available at website: http://cnit.noncode.org/CNIT/. As a result, it will be easy for users to employ this online tool in batches or single sessions rather than just under the Linux system. Thus, CNIT is a handy and useful tool, not only for predicting protein-coding or non-coding sequences generated by high-throughput sequencing data, but also for analyzing the sequence features across species as either an online or offline tool.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Dr Stanley Li Lin from the Department of Cell Biology and Genetics of Shantou University Medical College for assistance in revising the manuscript.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Natural Science Foundation of China [91740113], National Natural Science Foundation for Young Scholars of China [31701141, 31701149, 31501066]; Innovation Project for Institute of Computing Technology, CAS [20186060]. Funding for open access charge: National Natural Science Foundation of China.
Conflict of interest statement. None declared.
REFERENCES
- 1. Eddy S.R. Non-coding RNA genes and the modern RNA world. Nat. Rev. Genet. 2001; 2:919–929. [DOI] [PubMed] [Google Scholar]
- 2. Fu X.D. Non-coding RNA: a new frontier in regulatory biology. Natl. Sci. Rev. 2014; 1:190–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Fang S., Zhang L., Guo J., Niu Y., Wu Y., Li H., Zhao L., Li X., Teng X., Sun X. et al.. NONCODEV5: a comprehensive annotation database for long non-coding RNAs. Nucleic Acids Res. 2018; 46:D308–D314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wan Q., Guan X., Yang N., Wu H., Pan M., Liu B., Fang L., Yang S., Hu Y., Ye W. et al.. Small interfering RNAs from bidirectional transcripts of GhMML3_A12 regulate cotton fiber development. New Phytol. 2016; 210:1298–1310. [DOI] [PubMed] [Google Scholar]
- 5. Salmena L., Poliseno L., Tay Y., Kats L., Pandolfi P.P.. A ceRNA hypothesis: the Rosetta Stone of a hidden RNA language. Cell. 2011; 146:353–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Winkle M., Kluiver J.L., Diepstra A., van den Berg A.. Emerging roles for long noncoding RNAs in B-cell development and malignancy. Crit. Rev. Oncol. Hematol. 2017; 120:77–85. [DOI] [PubMed] [Google Scholar]
- 7. Sun T.T., He J., Liang Q., Ren L.L., Yan T.T., Yu T.C., Tang J.Y., Bao Y.J., Hu Y., Lin Y. et al.. LncRNA GClnc1 promotes gastric carcinogenesis and may act as a modular scaffold of WDR5 and KAT2A complexes to specify the histone modification pattern. Cancer Discov. 2016; 6:784–801. [DOI] [PubMed] [Google Scholar]
- 8. Wang Z., Yang B., Zhang M., Guo W., Wu Z., Wang Y., Jia L., Li S. Cancer Genome Atlas Research, N. . Cancer Genome Atlas Research, N. Xie W. et al.. lncRNA epigenetic landscape analysis identifies EPIC1 as an oncogenic lncRNA that interacts with MYC and promotes cell-cycle progression in cancer. Cancer Cell. 2018; 33:706–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wu Y., Zhang F., Yang K., Fang S., Bu D., Li H., Sun L., Hu H., Gao K., Wang W. et al.. SymMap: an integrativedatabase of traditional Chinese medicine enhanced by symptom mapping. Nucleic Acids Res. 2019; 47:D1110–D1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sun L., Luo H., Bu D., Zhao G., Yu K., Zhang C., Liu Y., Chen R., Zhao Y.. Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts. Nucleic Acids Res. 2013; 41:e166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. O’Leary N.A., Wright M.W., Brister J.R., Ciufo S., Haddad D., McVeigh R., Rajput B., Robbertse B., Smith-White B., Ako-Adjei D. et al.. Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2016; 44:D733–D745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zerbino D.R., Achuthan P., Akanni W., Amode M.R., Barrell D., Bhai J., Billis K., Cummins C., Gall A., Giron C.G. et al.. Ensembl 2018. Nucleic Acids Res. 2018; 46:D754–D761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ingolia N.T., Brar G.A., Stern-Ginossar N., Harris M.S., Talhouarne G.J., Jackson S.E., Wills M.R., Weissman J.S.. Ribosome profiling reveals pervasive translation outside of annotated protein-coding genes. Cell Rep. 2014; 8:1365–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Somers J., Poyry T., Willis A.E.. A perspective on mammalian upstream open reading frame function. Int. J. Biochem. Cell Biol. 2013; 45:1690–1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Anderson D.M., Anderson K.M., Chang C.L., Makarewich C.A., Nelson B.R., McAnally J.R., Kasaragod P., Shelton J.M., Liou J., Bassel-Duby R. et al.. A micropeptide encoded by a putative long noncoding RNA regulates muscle performance. Cell. 2015; 160:595–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kang Y.J., Yang D.C., Kong L., Hou M., Meng Y.Q., Wei L., Gao G.. CPC2: a fast and accurate coding potential calculator based on sequence intrinsic features. Nucleic Acids Res. 2017; 45:W12–W16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wang L., Park H.J., Dasari S., Wang S., Kocher J.P., Li W.. CPAT: Coding-Potential Assessment Tool using an alignment-free logistic regression model. Nucleic Acids Res. 2013; 41:e74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Li A., Zhang J., Zhou Z.. PLEK: a tool for predicting long non-coding RNAs and messenger RNAs based on an improved k-mer scheme. BMC Bioinformatics. 2014; 15:311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Guo J.C., Xie Y.M., Ran L.Q., Cao H.H., Sun C., Wu J.Y., Wu Z.Y., Liao L.D., Zhao W.J., Fang W.K. et al.. L1CAM drives oncogenicity in esophageal squamous cell carcinoma by stimulation of ezrin transcription. J. Mol. Med. (Berl.). 2017; 95:1355–1368. [DOI] [PubMed] [Google Scholar]
- 20. Woo C.J., Kingston R.E.. HOTAIR lifts noncoding RNAs to new levels. Cell. 2007; 129:1257–1259. [DOI] [PubMed] [Google Scholar]
- 21. Guo X., Gao L., Liao Q., Xiao H., Ma X., Yang X., Luo H., Zhao G., Bu D., Jiao F. et al.. Long non-coding RNAs function annotation: a global prediction method based on bi-colored networks. Nucleic Acids Res. 2013; 41:e35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Liao Q., Xiao H., Bu D., Xie C., Miao R., Luo H., Zhao G., Yu K., Zhao H., Skogerbo G. et al.. ncFANs: a web server for functional annotation of long non-coding RNAs. Nucleic Acids Res. 2011; 39:W118–W124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Guo J.C., Wu Y., Chen Y., Pan F., Wu Z.Y., Zhang J.S., Wu J.Y., Xu X.E., Zhao J.M., Li E.M. et al.. Protein-coding genes combined with long noncoding RNA as a novel transcriptome molecular staging model to predict the survival of patients with esophageal squamous cell carcinoma. Cancer Commun. (Lond.). 2018; 38:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zhao X., Li J., Lian B., Gu H., Li Y., Qi Y.. Global identification of Arabidopsis lncRNAs reveals the regulation of MAF4 by a natural antisense RNA. Nat. Commun. 2018; 9:5056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wang Y., Luo X., Sun F., Hu J., Zha X., Su W., Yang J.. Overexpressing lncRNA LAIR increases grain yield and regulates neighbouring gene cluster expression in rice. Nat. Commun. 2018; 9:3516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Golicz A.A., Bhalla P.L., Singh M.B.. lncRNAs in plant and animal sexual reproduction. Trends Plant Sci. 2018; 23:195–205. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.