

Abstract
Human whole-genome-sequencing reveals about 4 000 000 genomic variants per individual. These data are mostly stored as VCF-format files. Although many variant analysis methods accept VCF as input, many other tools require DNA or protein sequences, particularly for splicing prediction, sequence alignment, phylogenetic analysis, and structure prediction. However, there is no existing webserver capable of extracting DNA/protein sequences for genomic variants from VCF files in a user-friendly and efficient manner. We developed the SeqTailor webserver to bridge this gap, by enabling rapid extraction of (i) DNA sequences around genomic variants, with customizable window sizes and options to annotate the splice sites closest to the variants and to consider the neighboring variants within the window; and (ii) protein sequences encoded by the DNA sequences around genomic variants, with built-in SnpEff annotator and customizable window sizes. SeqTailor supports 11 species, including: human (GRCh37/GRCh38), chimpanzee, mouse, rat, cow, chicken, lizard, zebrafish, fruitfly, Arabidopsis and rice. Standalone programs are provided for command-line-based needs. SeqTailor streamlines the sequence extraction process, and accelerates the analysis of genomic variants with software requiring DNA/protein sequences. It will facilitate the study of genomic variation, by increasing the feasibility of sequence-based analysis and prediction. The SeqTailor webserver is freely available at http://shiva.rockefeller.edu/SeqTailor/.
INTRODUCTION
In humans, whole-genome sequencing (WGS) and whole-exome sequencing (WES) generate large amounts of genomic data, generally revealing for each individual studied about 4 000 000 variants by WGS and 80 000 variants by WES, with both identifying about 20 000 coding variants. According to the 125 748 WES data released by gnomAD database (1), there are ∼40% variants are non-synonymous (missense, stop-gained, stop-lost, start-lost, and frameshift), 17% are synonymous, 35% are intronic variants, whereas ∼5% of the WES variants are in the essential splice sites. These variant data are usually recorded in variant call format (VCF), which includes structured fields for chromosome (CHROM), position in reference genome sequence (POS), reference allele (REF), alternative allele (ALT) and others (e.g. QUAL, FILTER, INFO, etc.). The major human genomic variation databases (e.g. gnomAD/ExAC (1), 1000 Genomes Project (2)), the inherited disease mutation databases (e.g. HGMD (3), and ClinVar (4)), and most research laboratories and publications present and manage genomic variants in VCF format. For further variant annotations, predictions and visualizations, most of the leading tools use VCF as input (e.g. SnpEff (5), VEP (6), ANNOVAR (7), CADD (8) and PopViz (9)).
However, as the DNA sequence around the variant site presents richer information than the variant alone, many popular genomic software require DNA sequences as input, particularly for splicing prediction (e.g. Human Splicing Finder (10), NetGene2 (11), Spliceman (12)), sequence homology search (e.g. BLAST (13), BLAT (14)), multiple sequence alignment (e.g. MUSCLE (15), Clustal (16)), and phylogenetic analysis (e.g. PAML (17), IQ-TREE (18)). In addition, many tools use amino-acid sequences for protein domain identification and functional annotation (e.g. Pfam (19), PolyPhen-2 (20)), protein structure prediction (SWISS-MODEL (21), HMMER (22)), protein feature calculation (e.g. POSSUM (23), PROFEAT (24)), as well as protein sequence homology search (13) and alignment (15). Hence, knowledge of the reference protein sequence and the protein sequences altered by genomic variants would make it possible to evaluate their possible effects on protein domains, structures and functions.
Users wishing to analyze genomic variation data in VCF files with software that requires DNA or protein sequence must therefore extract the corresponding reference and alternative sequences. However, the available tools (e.g. UCSC genome browser (25), BEDTools-getfasta (26), Samtools-faidx (27), IGV (28)) can be used to extract the reference sequences, but not to generate the mutated alternative sequences. UCSC genome browser provides web interface with pages of user-defined parameters, BEDTools and samtools require script knowledge, while IGV is impractical for high-throughput applications (see detailed comparison in Discussion). Thus, there is currently no tool available for simple extraction of reference and alternative DNA/protein sequences directly from VCF files. We therefore developed the SeqTailor webserver, which offers a user-friendly, efficient and standardized approach for streamlining DNA and protein sequence extraction from the genomic variant data in VCF files in human and another 10 model organisms, with an array of useful and straightforward features to implement.
MATERIALS AND METHODS
Webserver
The SeqTailor webserver is run by Apache HTTP (version 2.2.15), on a Red Hat Enterprise Linux Server (version 6.9), with 8 Intel CPU processors@2.4 GHz and 48GB RAM. The website interface is designed and presented in HTML, PHP, CSS and JavaScript. The data are stored in MySQL tables and plain text files. The sequence extraction programs are coded in Python 2.7.
Data collection and pre-processing
The reference genome sequences, gene annotations, coding sequences (CDS) and protein sequences for 11 species (human (GRCh37/GRCh38), chimpanzee, mouse, rat, cow, chicken, lizard, zebrafish, fruit fly, Arabidopsis and rice) were collected from Ensembl Database release 95 (29). We extracted the reference genome sequences of autosomal chromosomes, sex chromosomes and mitochondria for SeqTailor DNA sequence extraction. In addition, since the key feature of human GRCh38 assembly is to provide alternate loci for a more robust representation of human population variations (including the highly variant MHC region and the divergent haplotypes (30)), we also supported DNA sequence extraction from 329 alternate loci and scaffolds in GRCh38 assembly (Supplementary Table S1) according to the Ensembl Database. Moreover, we extracted the transcripts of 15 biotypes in the categories of protein coding and pseudogenes (Supplementary Table S2) to compose a collection of ‘all transcripts’ in SeqTailor. We further selected the transcripts labeled as protein-coding biotype only, and identified the longest protein-coding transcript for each gene, to compose a collection of ‘canonical transcripts’. The genomic positions of all exons were extracted, and hence the donor splice sites (5′ end of the intron) and the acceptors splice sites (3′ end of the intron) were obtained and used to annotate the nearest splice sites for genomic variants. The workflow of data collection and pre-processing is shown in Figure 1, and the supported 11 species are summarized in Table 1.
Figure 1.

The workflow of data collection and pre-processing in the SeqTailor webserver.
Table 1.
List of organisms supported by SeqTailor webserver, including the assembly version of each reference genome, the supported genome regions, and the number of genes, transcripts, splice sites and protein sequences for all transcripts and canonical transcripts respectively
| All Transcripts | Canonical Transcripts | |||||||
|---|---|---|---|---|---|---|---|---|
| Organism | Genome assembly | Genome regions | # Genes | # Transcripts | # Splice sites | # Proteins | # Genes /transcripts /proteins | # Splice sites |
| Human | GRCh37 | Chr1–22, X, Y, MT | 35 091 | 138 613 | 2 018 124 | 95 304 | 20 676 | 413 814 |
| Human | GRCh38 | Chr1–22, X, Y, MT, 329 alternate loci | 36 100 | 155 341 | 2 259 292 | 107 498 | 20 528 | 421 390 |
| Chimpanzee | Pan-tro-3.0 | Chr1–22, X, Y, MT | 17 381 | 41 504 | 947 134 | 41 468 | 17 348 | 373 978 |
| Mouse | GRCm38 | Chr1–19, X, Y, MT | 36 148 | 94 462 | 1 350 332 | 65 679 | 22 504 | 420 830 |
| Rat | Rnor_6.0 | Chr1–20, X, Y, MT | 23 671 | 31 110 | 588 376 | 28 897 | 21 935 | 404 576 |
| Cow | ARS-UCD1.2 | Chr1–29, X, MT | 16 515 | 31 214 | 785 250 | 31 188 | 16 489 | 353 780 |
| Chicken | GRCg6a | Chr1–33, W, Z, MT | 12 303 | 22 158 | 573 360 | 22 158 | 12 303 | 278 736 |
| Lizard | AnoCar2.0 | Chr1–6, MT | 6105 | 6321 | 155 556 | 6321 | 6105 | 149 500 |
| Zebrafish | GRCz11 | Chr1–25, MT | 25 864 | 49 308 | 908 190 | 45 633 | 24 568 | 483 174 |
| Fruitfly | BDGP6 | Chr2–4, X, Y, MT | 14 226 | 30 804 | 363 118 | 30 478 | 13 926 | 119 752 |
| Arabidopsis | TAIR10 | Chr1–5, MT | 12 702 | 23 376 | 336 118 | 23 376 | 12 702 | 159 070 |
| Rice | IRGSP-1.0 | Chr1–12, MT | 2779 | 12 455 | 134 960 | 12 452 | 2 776 | 36 886 |
Built-in SnpEff variant annotation
SnpEff v4.3 was integrated in SeqTailor, for variant annotation and consequence prediction (5), with multi-threading configuration. Based on the previously identified collections of all transcripts and canonical transcripts, we extracted the corresponding gene annotations, CDS sequences and protein sequences respectively. These data were then employed to build the customized SnpEff databases on all transcripts and canonical transcripts separately. The customized SnpEff database of canonical transcripts allows 3–4× faster variant annotation, compared to annotating variants on all transcripts. The genomic variants are then filtered to retain 13 types of the consequences, which have direct impact on protein sequences, including: missense, stop-gained, synonymous, frameshift, in-frame insertion/deletion, conservative/disruptive in-frame insertion/deletion, and feature/transcript/gene ablation.
Runtime evaluation
The runtime evaluation was performed by applying four input data sizes (10, 100, 1000 and 10 000 genomic variants) on human reference genome GRCh37. We generated 50 VCF files for each input data size, by randomly selecting variants from the gnomAD database (1), and retaining only the first five columns of information (CHROM, POS, ID, REF, ALT). We therefore used a total of 200 VCF files with different numbers of human genomic variants to evaluate the runtime performance of SeqTailor for DNA and protein sequence extraction.
RESULTS
We developed the SeqTailor webserver to provide a user-friendly and efficient approach for the rapid extraction of DNA and protein sequences for genomic variants (single nucleotide variants and indels) in VCF format, with user-defined window sizes. Additionally, users can choose to annotate the nearest splice sites and to consider the neighboring variants within the given window for DNA sequence extraction. SeqTailor accepts input from either the webpage textbox or a user-uploaded file with maximum 10 000 input data, and outputs the extracted sequences in the browser and in a downloadable FASTA file, as well as a report file to inform the users of any error encountered in the input data. The framework of SeqTailor webserver is provided in Figure 2, and the exception handlings are summarized in Supplementary Table S3. The standalone programs are coded in Python 2.7, and the command line instructions are available in the Documentation page of the SeqTailor website.
Figure 2.

The framework of the SeqTailor webserver for DNA sequence extraction (upper) and protein sequence extraction (lower).
DNA sequence extraction
For DNA sequence extraction, SeqTailor supports 12 reference genomes of 11 species based on the Ensembl Database (29), 1/0-based genomic coordinate indexing, and the choice of the both/forward/reverse strand(s). It accepts genomic variants in VCF format with 5 standard mandatory fields (CHROM, POS, ID, REF, ALT) where ID is not used and can be filled with a dot ‘.’, and outputs the extracted DNA sequences in FASTA format. Once it has received the query for DNA sequence extraction, SeqTailor first pre-processes all input data, and then handles the exceptions by writing the relevant messages (comments, actions taken, and submitted data) into a report file, such that the users will be properly informed of the errors that have occurred. Next, the pre-processed data are submitted for DNA sequence extraction, to generate the wild-type reference (ref.) and mutated alternative (alt.) DNA sequences, with user-defined up/downstream (±) window sizes (in base pairs), on the selected strand(s). When the users opt to annotate the nearest splice sites, SeqTailor will compute the distance between each given variant and its closest splice site, check if the distance is within the user-defined window sizes, followed by appending the nearest splice site information (distance, gene symbol, transcript ID, exon number and acceptor/donor site) to the header line of the output FASTA file. Moreover, SeqTailor offers an option to consider the neighboring variants within the given window of sequence extraction, thereby making multiple changes to the extracted reference sequence accordingly. The neighboring variants are then appended in the header line of the output FASTA file, and also provided in the report file.
To demonstrate the input, output and functionality of this module, we selected five variants in the gene IFNAR1 (on forward strand) from the gnomAD database. In this example (Figure 3), SeqTailor extracts the ref./alt. DNA sequences on the forward strand, with window size ±25 base pairs on Human GRCh37 assembly, considering the neighboring variants within the given window, and annotating the nearest splice sites. In the output sequence shown below, the three neighboring variants (chr21–34713283-A-G, chr21–34713304-G-T, and chr21–34713315-G-T) were identified, and the multiple resulting nucleotide changes were indicated in the reference sequence accordingly. Moreover, a splice site (ENST00000270139; exon_3; acceptor_site) was identified in the extracted DNA sequences for these three variants, and the distances from the splice site to these three variants were given as +22, +1 and –10 bp, respectively. The variants (chr21–34713447-TATGAGGTTGACTC-T and chr21–34715874-A-ATT) led to deletion and insertion as shown, and no splice sites were identified within their extraction window. The nucleotide changes are highlighted in Figure 3.
Figure 3.
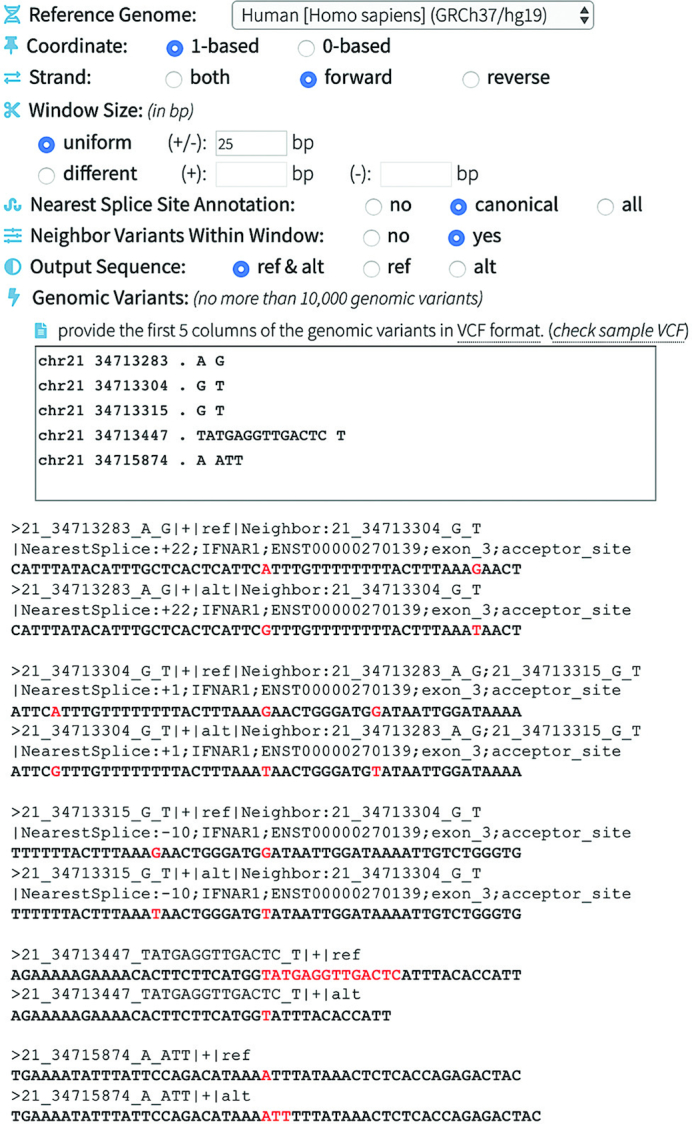
An example showing the input, output and functionality in extracting DNA sequence for genomic variants.
Additionally, SeqTailor also accepts genomic ranges in BED format with the mandatory fields (CHROM, START, END) for DNA sequence extraction. It generates a report file for exception handling and a FASTA file containing the extracted DNA sequences within the given intervals on the selected strand(s), exactly from the START position to the END position.
Protein sequence extraction
For protein sequence extraction, SeqTailor firstly inspects all input data, and generates a report file, before sending for built-in SnpEff variant annotation. It retains 13 types of variant consequences for protein sequence extraction and alteration. For missense variants, SeqTailor directly alters the reference protein sequences based on the annotated amino acid changes. For stop-gained variants, SeqTailor terminates the reference protein sequences on the annotated position, and appends a ‘*’ character to the new last amino acid. For synonymous variants, the protein sequences are unchanged. For variants annotated as frameshift, in-frame deletion or in-frame insertion, SeqTailor alters the reference CDS sequences, followed by running amino acid translation according to the genetic code table (Supplementary Tables S4–S7) to generate the alternative protein sequences. The translation stops when a stop codon is encountered, and a ‘*’ character is appended to the new last amino acid. If no stop codon is encountered in the altered CDS sequence, SeqTailor will output the altered protein sequence ending with ‘…’. This is based on the assumption that no aberrant splicing events will result from this variation. For variants annotated as feature/transcript/gene ablation, SeqTailor outputs ‘!’ as the alternative protein sequence. SeqTailor offers the option to extract the entire protein sequence, or the partial protein sequence within a customized window size (in amino acids). Finally, it provides the reference and altered protein sequences in FASTA format, with gene symbols, transcript IDs and amino-acid changes given in the header line. The SnpEff annotation file is also available for download in the result page. Because the UCSC Genome Browser (25) well handles the extraction of protein sequences from genomic ranges, we did not develop this function in SeqTailor.
To illustrate the input, output, and functionality of this module, we identified four variants of different consequences in the gene STAT1 from the gnomAD database. In this example (Figure 4), SeqTailor annotates the input variants on canonical transcripts of Human GRCh37 assembly, and then extracts the ref. / alt. protein sequences with window size +/−25 amino acids. The missense variant (chr2–191862962-A-G) changed the 205th amino acid from M (Met) to T (Thr); the frameshift variant (chr2–191863003-A-AG) changed the 192th amino acid from Q (Gln) to S (Ser) thereby terminating the protein sequence at 16 amino acids downstream; the in-frame insertion variant (chr2–191863010-T-TTTGCCACACCATTGG) inserted five duplicated amino acids TNGVA from position 184 to 188; and the stop-gained variant (chr2–191873724-G-A) changed the 80th amino acid from Q (Gln) to a stop codon to terminate the protein sequence. The amino acid changes and the sequence terminations are highlighted in Figure 4.
Figure 4.
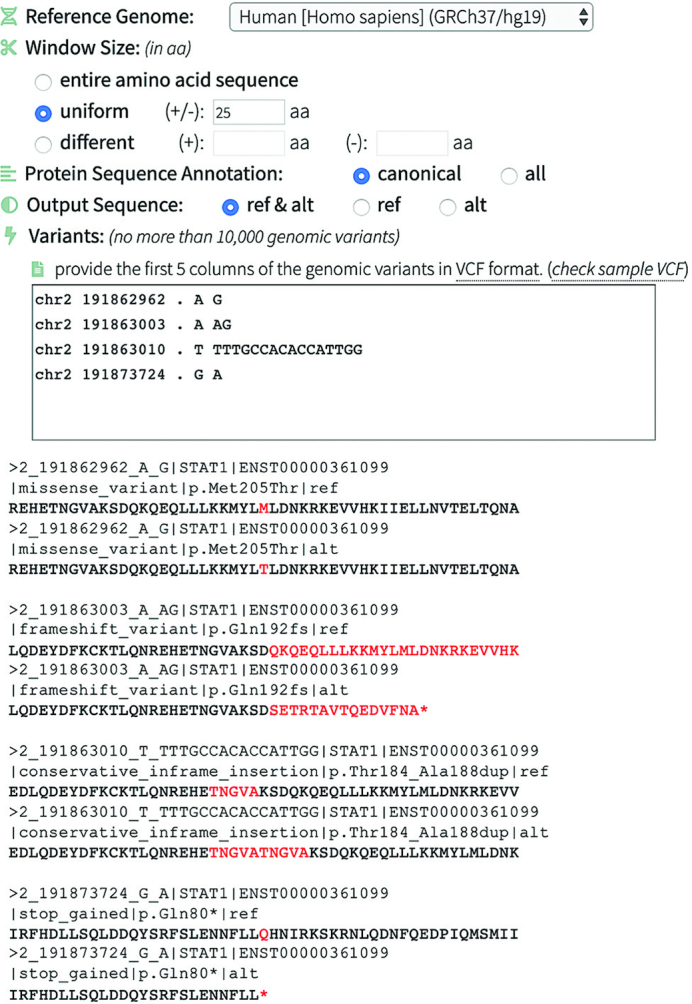
An example showing the input, output and functionality in extracting protein sequence for genomic variants.
SeqTailor standalone programs
We also provided standalone programs for users who wish to run SeqTailor DNA sequence extraction in a command-line based manner. The programs are coded in Python 2.7, and users should have Biopython library installed (31). Currently, we provided three standalone programs: (i) ‘Seqtailor_DNA_VCF_independent.py’ for extracting DNA sequences for genomic variants independently in VCF files; (ii) ‘SeqTailor_DNA_VCF_neighborhood.py’ for extracting DNA sequences for genomic variants in VCF files with the consideration of the neighboring variants falling inside the given window size and (iii) ‘SeqTailor_DNA_BED.py’ for extracting DNA sequences from genomic ranges in BED files. The feature to annotate the nearest splice sites in DNA sequence extraction, and the module for protein sequences extraction are not yet available for the standalone version, at this moment. These programs and the command line instructions are available in the Documentation page of the SeqTailor website, and further upgrades or fixes will be posted accordingly on the website.
SeqTailor bridges genomic variants with sequence-based tools
We further exhibited a case study on pathogenic genetic variants with different effects and different clinical consequences identified in five human genes (MSH2, BRAF, GJB2, BRCA2 and IL2RG) by both the HGMD professional database (3) and the ClinVar database (4) (Table 2). We used SeqTailor to extract the DNA sequences and protein sequences for these variants, and directly applied the output sequences with a number of tools for splicing prediction, protein domain search, and functional prediction. The details of the case studies and their bridged applications are described in the Supplementary Data. These examples demonstrated the practical power of SeqTailor to bridge the gap between genomic variant data and sequence-based tools for downstream analyses and predictions. SeqTailor makes it efficient to further investigate the genomic variant data and renders sequence-based software more accessible.
Table 2.
List of pathogenic genomic variants with different consequences used in the case study
| ClinVar submission ID | Gene | Genomic variant in VCF | Effects | Diseases | |||
|---|---|---|---|---|---|---|---|
| SCV000107433.2 | MSH2 | chr2 | 47635062 | T | G | Intronic, new donor site | Lynch syndrome |
| SCV000616361.3 | BRAF | chr7 | 140481402 | C | T | Missense | Cardio-facio-cutaneous syndrome |
| SCV000840535.3 | GJB2 | chr13 | 20763554 | AG | G | Deletion, frameshift | Nonsyndromic hearing loss and deafness |
| SCV000635728.2 | BRAC2 | chr13 | 32954282 | GG | TA | Essential splicing | Hereditary breast-ovarian cancer |
| SCV000637244.1 | IL2RG | chrX | 70330553 | T | C | Intronic, new acceptor site | X-linked severe combined immunodeficiency |
SeqTailor runtime evaluation
The SeqTailor webserver is designed for the rapid extraction of DNA and protein sequences from genomic variant data in VCF files, in a user-friendly manner. The runtime evaluation was performed by applying 50 VCF files for each of the input data sizes (10, 100, 1000 and 10 000 genomic variants) on human GRCh37 genome. The online version was tested on the computer server hosting SeqTailor (8 CPUs and 48GB RAM), and the standalone version was tested on a desktop (20 CPUs and 128 RAM). The SeqTailor runtime performance is shown in Figure 5.
Figure 5.

Runtime performance in extracting DNA sequences (left: online webserver, middle: standalone program), and protein sequences (right: online webserver), from varying sizes of input VCF data.
To extract DNA sequences from genomic variants independently, the SeqTailor webserver takes 6, 11, 12 and 13 seconds on average for 10, 100, 1000 and 10 000 genomic variants, respectively. When the option to consider the neighboring variants within the window is enabled, the average runtime is 6, 12, 13 and 105 seconds for 10, 100, 1000 and 10 000 genomic variants. The runtime grows significantly when the input data size is over 1000, because SeqTailor then needs to search iteratively for neighboring variants for each variant. The option to annotate the nearest splice site does not noticeably increase the runtime.
The standalone programs were tested by using the same VCF files and the same parameters as previously used. The program ‘Seqtailor_DNA_VCF_independent.py’ takes approximately 12, 13, 13 and 14 s, and the program ‘Seqtailor_DNA_VCF_neighborhood.py’ takes approximately 12, 13, 13 and 66 s to extract DNA sequences for 10, 100, 1000 and 10 000 genomic variants, respectively. Since the online version only loads the genome sequences on the given chromosomes, it takes slightly shorter time to load the sequences and complete the sequence extraction, if the input data size is small (∼10). When the input data size is between 100 to 1000, the runtime is very close between the online version and the standalone version. In the case when considering the neighboring variants in the DNA sequence extraction, the standalone version performs much faster (66 seconds on average) than the online version (104 s on average). This is because the neighboring variants are identified iteratively for each variant, and a better configured hardware makes it faster.
For protein sequence extraction, the input variants are first sent for build-in SnpEff annotation on canonical transcripts or all transcripts. SeqTailor webserver runtime is approximately 9, 10, 10 and 11 s for the option of canonical transcripts, and 31, 33, 33 and 36 s for the option of all transcripts, to extract protein sequences for 10, 100, 1000 and 10 000 genomic variants, respectively. Currently, we do not supply a standalone version for protein sequence extraction.
In addition, the only comparable common feature between SeqTailor-standalone, BEDTools-getfasta and SAMtools-faidx is the script-based extraction of DNA reference sequences from genomic ranges in BED format file (or slightly modified BED format for samtools). More details about BEDTools and samtools are discussed in the Discussion section. We ran a runtime comparison of these tools, by applying 50 BED files for each of the input data sizes (10, 100, 1000 and 10 000 genomic ranges) on human GRCh37genome, on a desktop with 20 CPUs and 128 RAM. Here, as shown in Supplementary Figure S1, SeqTailor takes 12, 13, 13 and 14 s on average, and samtools takes 1, 2, 5 and 44 s on average to complete the DNA sequence extraction for 10, 100, 1000 and 10 000 genomic ranges, respectively. Meanwhile, BEDTools is able to finish running 10 000 genomic ranges within 2 s.
DISCUSSION
Three major sequence extraction methods are currently available: UCSC Genome Browser (25), BEDTools-getfasta (26), and SAMtools-faidx (27). UCSC Genome Browser converts genomic ranges in BED/position format to DNA/protein sequences without supporting genomic variant data in VCF format. Users are required to go through several pages and define a number of parameters (annotation track, gene table, field table, retrieval region, etc.) to obtain the results. Comparing the DNA sequence extraction exactly from the START position to the END position in SeqTailor, the UCSC Genome Browser always provides an overlap between the input genomic ranges and certain genomic regions (exons, introns, UTRs, etc.), and outputs the sequences segregated into different regions. BEDTools-getfasta and SAMtools-faidx provide script-based options. However, BEDTools-getfasta is intended mainly for extracting reference sequences within the given genomic ranges in BED files. If the inputs are VCF files, BEDTools only outputs the reference alleles on the given genomic positions. Users have to write scripts to generate genomic ranges for genomic variants, before extracting the reference sequence around each variant. SAMtools-faidx does not accept BED or VCF format directly, so users have to transform data into the ‘CHROM:START-END’ format for sequence extraction. Importantly, these tools do not generate the mutated alternative DNA sequences based on the variation data, and they do not support the extraction of protein sequences encoded by the DNA sequences around genomic variants. For users who have script knowledge and who wish to extract reference sequences only, BEDTools and SAMtools are recommended. For non-programmers, SeqTailor webserver can serve for this purpose.
Alternatively, IGV software (28) can be used to locate the position of the variant on the reference genome, by manually copying and pasting to extract the desired DNA sequences or the corresponding protein sequences one at a time, which would be very time-consuming, error-prone, and impractical in high-throughput data applications. The popular GALAXY online platform (32) and Biopython library (31) do not offer such options for DNA or protein sequence extraction. Moreover, none of the existing tools provide a function to annotate the nearest splice sites for genomic variants, or to consider the neighboring variants within the given window for sequence alteration. A comparison of these approaches with SeqTailor is presented in Table 3.
Table 3.
Comparison of SeqTailor with other existing relevant tools
| Tool Name | Interface | Ref. genome | Window size | Input (format) | Output | Splice site Annotation | neighboring Variation |
|---|---|---|---|---|---|---|---|
| SeqTailor | Webserver | Built-in 11 species, or user-defined in standalone | Scalable | Genomic variants (VCF), or genomic ranges (BED) | ref./alt. DNA or protein sequences, with user-defined window size, in browser and to a file | Yes | Yes |
| UCSC Genome Browser | Webserver | Built-in >50 species | Scalable | Genomic ranges (BED) | ref. DNA or protein sequences overlapped with defined genomic regions, in the browser and to a file | No | No |
| BEDTools | Script | User-defined | Scalable | Genomic variants (VCF), or genomic ranges (BED) | ref. DNA alleles or sequences to a file | No | No |
| samtools | Script | User-defined | Scalable | Genomic ranges (BED) | ref. DNA sequences to a file | No | No |
| IGV | Software | User-defined | Scalable | variant positions | Copy-paste to extract DNA or protein sequences from IGV, one at a time | No | No |
Overall, the SeqTailor webserver is the first tool to allow user-friendly and rapid extraction of DNA and protein sequences for genomic variants in VCF files. It presents an efficient and customizable approach for this purpose, requiring only succinct input parameters and no knowledge of script programming. SeqTailor promises to make the analyses of genomic variants with DNA or protein sequence-based tools more rapid and efficient than is currently possible.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Y. Nemirovskaya and D. Papandrea for administrative support, M. Woollett and A. Gall for manuscript editing and polishing, and Z. Yang for the artwork of website design.
Notes
Present address: Peng Zhang, St. Giles Laboratory of Human Genetics of Infectious Diseases, The Rockefeller University, New York, NY 10065, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
The Rockefeller University, Howard Hughes Medical Institute, Charles Bronfman Institute for Personalized Medicine, Icahn School of Medicine at Mount Sinai. Funding for open access charge: Howard Hughes Medical Institute.
Conflict of interest statement. None declared.
REFERENCES
- 1. Lek M., Karczewski K.J., Minikel E.V., Samocha K.E., Banks E., Fennell T., O’Donnell-Luria A.H., Ware J.S., Hill A.J., Cummings B.B. et al.. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016; 536:285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Genomes Project, C. Auton A., Brooks L.D., Durbin R.M., Garrison E.P., Kang H.M., Korbel J.O., Marchini J.L., McCarthy S., McVean G.A. et al.. A global reference for human genetic variation. Nature. 2015; 526:68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Stenson P.D., Mort M., Ball E.V., Evans K., Hayden M., Heywood S., Hussain M., Phillips A.D., Cooper D.N.. The Human Gene Mutation Database: towards a comprehensive repository of inherited mutation data for medical research, genetic diagnosis and next-generation sequencing studies. Hum. Genet. 2017; 136:665–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Landrum M.J., Lee J.M., Riley G.R., Jang W., Rubinstein W.S., Church D.M., Maglott D.R.. ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 2014; 42:D980–D985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cingolani P., Platts A., Wang le L., Coon M., Nguyen T., Wang L., Land S.J., Ruden D.M.. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin). 2012; 6:80–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. McLaren W., Gil L., Hunt S.E., Riat H.S., Ritchie G.R., Thormann A., Flicek P., Cunningham F.. The ensembl variant effect predictor. Genome Biol. 2016; 17:122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wang K., Li M., Hakonarson H.. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010; 38:e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kircher M., Witten D.M., Jain P., O’Roak B.J., Cooper G.M., Shendure J.. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 2014; 46:310–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zhang P., Bigio B., Rapaport F., Zhang S.Y., Casanova J.L., Abel L., Boisson B., Itan Y.. PopViz: a webserver for visualizing minor allele frequencies and damage prediction scores of human genetic variations. Bioinformatics. 2018; 34:4307–4309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Desmet F.O., Hamroun D., Lalande M., Collod-Beroud G., Claustres M., Beroud C.. Human Splicing Finder: an online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 2009; 37:e67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Brunak S., Engelbrecht J., Knudsen S.. Prediction of human mRNA donor and acceptor sites from the DNA sequence. J. Mol. Biol. 1991; 220:49–65. [DOI] [PubMed] [Google Scholar]
- 12. Lim K.H., Fairbrother W.G.. Spliceman–a computational web server that predicts sequence variations in pre-mRNA splicing. Bioinformatics. 2012; 28:1031–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Altschul S.F., Madden T.L., Schaffer A.A., Zhang J., Zhang Z., Miller W., Lipman D.J.. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997; 25:3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kent W.J. BLAT–the BLAST-like alignment tool. Genome Res. 2002; 12:656–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Edgar R.C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004; 32:1792–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sievers F., Wilm A., Dineen D., Gibson T.J., Karplus K., Li W., Lopez R., McWilliam H., Remmert M., Soding J. et al.. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011; 7:539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yang Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007; 24:1586–1591. [DOI] [PubMed] [Google Scholar]
- 18. Trifinopoulos J., Nguyen L.T., von Haeseler A., Minh B.Q.. W-IQ-TREE: a fast online phylogenetic tool for maximum likelihood analysis. Nucleic Acids Res. 2016; 44:W232–W235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Finn R.D., Coggill P., Eberhardt R.Y., Eddy S.R., Mistry J., Mitchell A.L., Potter S.C., Punta M., Qureshi M., Sangrador-Vegas A. et al.. The Pfam protein families database: towards a more sustainable future. Nucleic Acids Res. 2016; 44:D279–D285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Adzhubei I.A., Schmidt S., Peshkin L., Ramensky V.E., Gerasimova A., Bork P., Kondrashov A.S., Sunyaev S.R.. A method and server for predicting damaging missense mutations. Nat. Methods. 2010; 7:248–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Waterhouse A., Bertoni M., Bienert S., Studer G., Tauriello G., Gumienny R., Heer F.T., de Beer T.A.P., Rempfer C., Bordoli L. et al.. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 2018; 46:W296–W303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Potter S.C., Luciani A., Eddy S.R., Park Y., Lopez R., Finn R.D.. HMMER web server: 2018 update. Nucleic Acids Res. 2018; 46:W200–W204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wang J., Yang B., Revote J., Leier A., Marquez-Lago T.T., Webb G., Song J., Chou K.C., Lithgow T.. POSSUM: a bioinformatics toolkit for generating numerical sequence feature descriptors based on PSSM profiles. Bioinformatics. 2017; 33:2756–2758. [DOI] [PubMed] [Google Scholar]
- 24. Zhang P., Tao L., Zeng X., Qin C., Chen S.Y., Zhu F., Yang S.Y., Li Z.R., Chen W.P., Chen Y.Z.. PROFEAT Update: A protein features web server with added facility to compute network descriptors for studying omics-derived networks. J. Mol. Biol. 2017; 429:416–425. [DOI] [PubMed] [Google Scholar]
- 25. Karolchik D., Hinrichs A.S., Furey T.S., Roskin K.M., Sugnet C.W., Haussler D., Kent W.J.. The UCSC Table Browser data retrieval tool. Nucleic Acids Res. 2004; 32:D493–D496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Quinlan A.R., Hall I.M.. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010; 26:841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R. Genome Project Data Processing, S . The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009; 25:2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Thorvaldsdottir H., Robinson J.T., Mesirov J.P.. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief. Bioinform. 2013; 14:178–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zerbino D.R., Achuthan P., Akanni W., Amode M.R., Barrell D., Bhai J., Billis K., Cummins C., Gall A., Giron C.G. et al.. Ensembl 2018. Nucleic Acids Res. 2018; 46:D754–D761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Schneider V.A., Graves-Lindsay T., Howe K., Bouk N., Chen H.C., Kitts P.A., Murphy T.D., Pruitt K.D., Thibaud-Nissen F., Albracht D. et al.. Evaluation of GRCh38 and de novo haploid genome assemblies demonstrates the enduring quality of the reference assembly. Genome Res. 2017; 27:849–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Cock P.J., Antao T., Chang J.T., Chapman B.A., Cox C.J., Dalke A., Friedberg I., Hamelryck T., Kauff F., Wilczynski B. et al.. Biopython: freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics. 2009; 25:1422–1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Afgan E., Baker D., Batut B., van den Beek M., Bouvier D., Cech M., Chilton J., Clements D., Coraor N., Gruning B.A. et al.. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res. 2018; 46:W537–W544. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.