


Abstract
The frequency by which genes are studied correlates with the prior knowledge accumulated about them. This leads to an imbalance in research attention where some genes are highly investigated while others are ignored. Geneshot is a search engine developed to illuminate this gap and to promote attention to the under-studied genome. Through a simple web interface, Geneshot enables researchers to enter arbitrary search terms, to receive ranked lists of genes relevant to the search terms. Returned ranked gene lists contain genes that were previously published in association with the search terms, as well as genes predicted to be associated with the terms based on data integration from multiple sources. The search results are presented with interactive visualizations. To predict gene function, Geneshot utilizes gene–gene similarity matrices from processed RNA-seq data, or from gene–gene co-occurrence data obtained from multiple sources. In addition, Geneshot can be used to analyze the novelty of gene sets and augment gene sets with additional relevant genes. The Geneshot web-server and API are freely and openly available from https://amp.pharm.mssm.edu/geneshot.
INTRODUCTION
Biomedical researchers that explore the molecular composition of the human cell rely heavily on search engines that retrieve relevant documents from massive corpora of biomedical text such as PubMed. In this way, researchers integrate knowledge about genes and proteins to form new hypotheses that are ultimately tested in controlled bench experiments. This approach for performing research has some drawbacks, for example: (i) Research that describes the functions and interactions of genes and proteins has strong biases toward studying popular genes while ignoring most others (1,2); (ii) It is also common that researchers are overwhelmed with the growing volume of publications, and this leads to pursuing hypotheses that are not fully informed by prior published studies. To mitigate the latter, text mining approaches have been widely applied to biomedical text to help researchers obtain an overview of the information embedded within thousands of related documents (3). Methods such as word2vec (4) and other recent named entity recognition (NER) methods (5) such as Tagger (6) have been increasingly effective in detecting different types of relevant biomedical terms embedded within abstracts and full-text research papers. Gene names are one of those key entity terms that such text mining methods can commonly and effectively detect. Systems that attempt to build networks of genes based on their co-occurrence in publications have been widely applied and used (7). Beyond constructing networks of genes based on their co-occurrence in publications, text mining methods that detect gene names in biomedical documents can be utilized to generate annotated gene sets. Reanalysis and integration of themed collections of gene sets from past studies can produce new insights and lead investigators toward the most promising direction of new biomedical research. For example, curated gene sets can serve as a database for matching user submitted gene sets with annotated and curated gene sets, which are organized into gene set libraries for gene set enrichment analysis (8–10). Several tools have been developed to identify gene sets given arbitrary PubMed search terms. For example, the tool Gene List Automatically Derived for You (GLAD4U) (11) uses the PubMed API to return a ranked list of genes based on any PubMed search. Another related tool, PALM-IST (12), builds protein interaction networks and pathways based on free text searches. Similarly, FACTA+ (13) is a search engine that returns genes, drugs, diseases, symptoms, enzymes and compounds for any search term. In addition, MyGeneFriends (14) is an interesting application that connects investigators to genes and diseases based on social media interactions. These are only representative examples out of a sea of related tools. With some similarity to these previously published tools, Geneshot converts PubMed identifiers (PMIDs) returned for arbitrary search terms to ranked lists of genes using gene-publication associations such as those encoded within Gene References into Function (GeneRIF) (15), a manually curated resource maintained by the National Center for Biotechnology Information (NCBI). As an alternative to GeneRIF, Geneshot also utilizes an automated method to associate genes with publications that we termed AutoRIF. AutoRIF simply harvests all PMIDs returned from searches of gene names while removing entries for genes with ambiguous names. A third resource to associate genes with PMIDs is created with Tagger (6), an NER tool that scans full-text articles. However, Geneshot takes the approach of converting search terms to gene sets a step further by utilizing the genes identified in the original PubMed search, as well as gene–gene similarity matrices created from these three sources and other sources, to produce predicted gene sets. Gene–gene co-expression data from the ARCHS4 RNA-seq resource (16) and gene–gene co-occurrence from Enrichr queries (9,10) are used to assess the relevancy of the supplementary genes. In addition, Geneshot supports systematic gene function predictions with the aforementioned resources as well as gene set augmentation and novelty assessment. We benchmarked the different five resources to evaluate the quality of their ability to predict gene function and demonstrate how the Geneshot approach can be used to generate many novel types of annotated gene sets. In addition, Geneshot provides gene set novelty assessment and gene set augmentation by proposing additional genes that are likely relevant to the user input gene set. These are just few implementations that demonstrate how Geneshot opens the door to many creative applications that can facilitate automated hypothesis generation for biomedical research.
MATERIALS AND METHODS
Mining gene–publication associations
Gene–publication associations are encoded within the GeneRIF resource (15). We processed the GeneRIF file available on the NCBI Gene database FTP site. From this file, only the human and mouse gene–PMID associations were obtained. One drawback with GeneRIF is that it is incomplete. GeneRIF only covers a small fraction of gene mentions in publications listed on PubMed. For human and mouse genes, there are currently (April 2019) 1 015 165 gene–PMID entries. Each entry in GeneRIF is marked with a date when the gene–PMID pair was entered into the GeneRIF database. We updated these dates with the date of the publication using the PubMed API. By plotting the cumulative counts of PMID dates for individual genes, we observed that during certain time intervals there are missing entries (Supplementary Figure S1). The cause of these gaps is unclear. To compensate with the incompleteness of GeneRIF, we built an initial alternative version of a dataset that associates genes with publications. We termed this new resource AutoRIF. To compile AutoRIF, we queried PubMed with all human gene symbols using the PubMed API. For each human gene (Ensembl genome annotation 87), all PMIDs were retrieved with the corresponding publication dates. This procedure yielded 8 097 696 PMID–gene pairs for 5 127 253 unique PMIDs. About 1 579 304 PMIDs match more than one gene symbol. About 677 175 PMIDs share more than two gene symbols. To further improve the accuracy of automatically matching genes with publications, we downloaded the data produced by Tagger (6) available from the Jensen Lab website. Tagger was applied to identify genes in PubMed abstracts and full-length open publications with an NER algorithm. Tagger uses official gene symbols as well as their synonyms as the background dictionary. The Tagger file contains 9 353 632 gene–publication pairs. The entries in the Tagger output contain Ensembl IDs. For converting these Ensembl IDs to gene names, BioMart (17), circBase (18) and HUGO Gene Nomenclature Committee (HGNC) (19) resources were used. Ensembl stable protein IDs were converted into gene names using BioMart. circRNAs names were converted into circRNAs IDs using the circBase ID cross-reference file for humans (hg19_circID_to_name.txt) and then converted into gene names using the circBase all Homo sapiens circRNAs file (hsa_hg19_circRNA.txt). All gene names were then cross-referenced with HGNC-approved symbols and any gene synonyms were converted into approved symbols. Only entries that converted into HGNC-approved symbols were included in the final Tagger processed file. Next, we calculated the intersection between Tagger and AutoRIF. The intersection set has 2 918 803 gene–publication pairs. This intersection is used in the interface as the AutoRIF option in the toggle switch between GeneRIF and AutoRIF. While this approach may contain some false positives, it results in a collection of gene–publication pairs with fewer false negatives while containing seven times more associations than GeneRIF.
Preparing the gene–gene co-occurrence and co-expression matrices
Co-occurrence matrices were created from the Tagger output, AutoRIF and GeneRIF files. The GeneRIF data were filtered to include 1 015 165 gene–publication pairs for 647 803 publications and 16 729 genes. The consolidated AutoRIF dataset contains 14 979 unique genes from 1 784 274 publications. All the retained genes are protein coding genes. The reason we do not have all genes are due to ambiguous names of genes, for example, genes with names such as KIT or ITCH, or genes with few or no publication mentions in abstracts on PubMed. These datasets were converted to a co-occurrence matrix by calculating the observed versus expected ratio as follows:
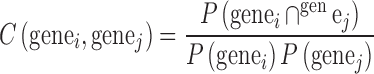 |
(1) |
Similarly, gene–gene co-expression correlations were calculated from the processed data provided by the ARCHS4 resource (16). ARCHS4 contains processed gene expression data derived from RNA-seq experiments deposited in the Gene Expression Omnibus (GEO) (20). For constructing the gene–gene co-expression network, we selected a random set of 4000 human samples across a variety of different tissues and cell types. Next, we quantile normalized the gene counts and calculated the Pearson correlation for all pairwise genes as previously described (16).
To prepare the co-occurance gene-gene similar matrix from Enrichr queries, 1 097 157 unique user-submitted gene sets to the Enrichr tool were dumped from the Enrichr database on 27 October 2017. Lists used for internal testing, lists with >2000 genes, lists with <2 genes and lists from IP addresses that submitted >1000 lists were discarded. Co-occurrence analysis was performed on the remaining 293 747 lists with (Equation 1).
Predicting gene function
By combining annotated gene sets with gene–gene similarity matrices, we can predict novel gene functions. Specifically, we can predict gene functions by combining a gene–gene similarity matrix G with a gene set library GF. The predicted gene set library 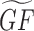 contains scores that quantify the predicted membership of a gene to be part of a gene function. GF can be also considered a bipartite graph with two types of nodes: genes and functions. Functions can be, for example, membership in a pathway, GO term, or membership in a protein complex. This bipartite graph can also be represented as a binary matrix where the rows are the genes, and the columns are the gene functions. The task then is to enhance the edges in GF, by using information from the matrix G, to produce
contains scores that quantify the predicted membership of a gene to be part of a gene function. GF can be also considered a bipartite graph with two types of nodes: genes and functions. Functions can be, for example, membership in a pathway, GO term, or membership in a protein complex. This bipartite graph can also be represented as a binary matrix where the rows are the genes, and the columns are the gene functions. The task then is to enhance the edges in GF, by using information from the matrix G, to produce 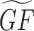 (Equation 2). In our case, we can construct multiple versions for such a
(Equation 2). In our case, we can construct multiple versions for such a 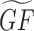 by utilizing the Gs created from AutoRIF, GeneRIF, Tagger, Enrichr co-occurrence or ARCHS4 co-expression. The selection of the matrix GF directs the domain of the predictions that will be performed. The Geneshot website supports gene function prediction from the gene–gene similarity matrices derived from GeneRIF, AutoRIF, Tagger, Enrichr co-occurrence and ARCHS4 co-expression.
by utilizing the Gs created from AutoRIF, GeneRIF, Tagger, Enrichr co-occurrence or ARCHS4 co-expression. The selection of the matrix GF directs the domain of the predictions that will be performed. The Geneshot website supports gene function prediction from the gene–gene similarity matrices derived from GeneRIF, AutoRIF, Tagger, Enrichr co-occurrence and ARCHS4 co-expression.
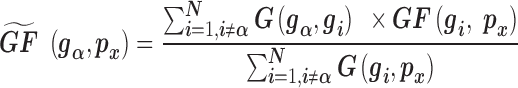 |
(2) |
Benchmarking the gene function predictions
The ability of gene–gene similarity matrices to predict relevant genes for biological terms was benchmarked using 16 gene set libraries downloaded from Enrichr (9,10). For each gene set in each library, the average similarity between each gene and each gene set was calculated and used to rank genes based on their likelihood to be associated with the gene sets (Equation 2). The average area under the curve (AUC) for each gene set library was then calculated by comparing the known gene–term associations with the predicted gene–term associations for each gene set in each library.
Constructing the PI–gene–award association network
A list containing principal investigators (PIs), their respective institutions and the total of NIH funding for 2017 was downloaded from the Blue Ridge Institute for Medical Research (BRIMR) site. Using the PubMed API, the name of each PI was used to query PubMed and the associated PMIDs were collected. The PMIDs for each PI were then converted into genes using Geneshot. Gene sets were then created for each PI. PIs with gene sets >100 genes were truncated at 100 to only include the 100 most occurring genes. PIs with no associated genes and PIs listed under more than one institution were removed to avoid the inclusion of PIs with the same name. The overall NIH award for each gene was calculated by summing up the funding associated with each gene–PI association. A list of dark kinases, dark ion channels and dark GPCRs was obtained from the NIH RFA IDG program announcement RFA-RM-18-021.
Developing the Geneshot web server application
Geneshot is written in Java and is running on a Tomcat 9 server. The interactive front-end elements of Geneshot such as the scatter plot and the histograms are generated using the JavaScript library D3.JS (21). The web application is running in a Docker container (22) and the Docker image is deposited in Docker Hub. Data files are deposited in the AWS S3 cloud storage and loaded during startup of the service. All the functions of Geneshot are also accessible via REST-Endpoint API. The results from the API are returned in JavaScript Object Notation (JSON) format. The site was tested on Chrome, Firefox and Safari on a Mac OS.
RESULTS
Interacting with the Geneshot user interface
The Geneshot user interface for PubMed querying is divided into three parts (Figure 1). The first section contains the user input form. It enables the construction of arbitrary search terms by combining elementary terms with AND and NOT operators. The top search text box is for submitting search terms with a logical AND operator, and the bottom text box is for the NOT terms. The resulting publication set from the AND search is filtered by the publications returned based on the exclusion criteria. Before submitting the search, using a switch, the user can choose between GeneRIF or AutoRIF to identify genes matching the publications. The second section contains the visualization of the returned search results (Figure 1B). After the search completes, an interactive scatter plot displays the genes that are found based on the matching publications. The scatter plot displays the total matching publications for each gene, and a normalized total that is the fraction of matching publications that mention the gene with the search terms over the total publications that mention the gene regardless of whether the search terms were mentioned. More detailed information about each gene can be accessed by clicking on the point that represents each gene. Clicking on the point within the scatter plot invokes a function that loads a histogram that shows publications that are associated with the search term alone over time, as well as publications that also mention the gene. This provides a timeframe that enables a user to visualize when the gene became associated with a research topic. The third section displays the information shown in the scatter plot in an interactive downloadable table (Figure 1C). Near this table, on the right side, another table shows the lists of genes that are predicted to be related to the search term based on GeneRIF, AutoRIF, Tagger or Enrichr gene–gene co-occurence, or the ARCHS4 gene–gene co-expression matrices. Genes from both tables can be submitted to Enrichr for further analysis, or downloaded in various formats.
Figure 1.

Geneshot user interface for the PubMed querying tab. (A) Search engine input section. (B) Scatter plot of all publications that mention both the gene and the search terms against the normalized values (left); gene with and without search terms mentions over time (right). (C) Tables providing ranked lists of relevant genes based on GeneRIF (left), and predictions based on AutoRIF co-occurrence (right).
Predicting gene function
The Geneshot user interface for the gene function prediction requires the user to enter a valid human gene symbol, select a gene set library and select one of the five gene–gene similarity matrices for making the predictions (Supplementary Figure S2A). Once such selection is made, Geneshot produces a table with ranked terms and a ROC curve plot to estimate the quality of the predictions. The ROC curve examines how known functions for the gene are ranked among all terms from the selected gene set library. Known terms are also marked in color in the table (Supplementary Figure S2B).
Benchmarking the gene function predictions
To benchmark the quality of the gene function predictions in Geneshot, we compared the performance of gene function predictions by predicting the content within gene set libraries from Enrichr (9,10). Predictions were made with the gene–gene similarity matrices created from GeneRIF (15), AutoRIF, Tagger (6), Enrichr queries as well as a gene–gene co-expression network derived from ARCHS4 (16) as described in the ‘Materials and Methods’ section. AutoRIF and Tagger outperform all other gene–gene similarity matrices for predicting gene set libraries created by manual curation and are literature based (Figure 2). Hence, gene-set libraries such as GO Biological Process and Reactome utilize information found in the literature, and thus literature-based similarity of genes captures these dependencies, resulting in high predictive performance. One disadvantage of literature based similarity is that while they may unravel novel relationships between genes, they do not include understudied genes with unknown functions. Gene co-expression similarity and gene–gene co-occurrence similarity based on Enrichr queries, on the other hand, is a more data-driven unbiased method to predict gene function. Since RNA-seq gene expression and hundreds of thousands of Enrichr queries cover the whole genome, the gene–gene co-expression matrices created from ARCHS4 and Enrichr are more complete. The similarity matrices from the ARCHS4 gene–gene co-expression matrix outperform the other matrices for predicting libraries created from ARCHS4. However, the Enrichr gene–gene similarity matrix outperforms the literature-based co-occurrence matrices and the ARCHS4 gene–gene similarity matrix when predicting gene functions for all other data-driven libraries such as upstream transcription factors derived from ChIP-seq experiments. Overall, the Enrichr gene–gene similarity matrix performs well across all libraries (Figure 2). This means that the collective knowledge generated by the crowd can be reused for systematic high quality gene function discovery.
Figure 2.

Median area under the receiver operating characteristic curve (AUC) distributions for predicting genes associated with terms from 16 Enrichr gene set libraries. The libraries are labeled as data-driven and manually curated. Predictions were made using four gene–gene similarity matrices created from Tagger, GeneRIF, AutoRIF and ARCHS4.
Retrieving pathway membership
Next, we tested the ability of Geneshot to recover complete pathways by querying Geneshot with pathway terms from the KEGG pathway database (23). We asked whether Geneshot can automatically return the genes that are known members of each pathway. We searched 263 pathway terms with the AutoRIF setting and measured the percentage of successfully recovered genes that are known members of each pathway. This benchmark is meant to simulate a typical use case of an arbitrary search term. On average, we observe that 44% of the pathway member genes are recovered by the Geneshot literature search whereby general pathway terms recover almost all pathway members while specific terms recover only few members (Figure 3A).
Figure 3.

KEGG pathway gene members recovered by Geneshot given only the pathway terms. (A) Fraction of pathway gene members recovered with the Geneshot literature search for all 263 KEGG pathway terms using the AutoRIF settings. (B) Total predicted pathway members recovered using the gene function prediction method with the ARCHS4 gene–gene co-expression correlations. (C) Additional pathways members not recovered by the Geneshot original search but recovered by the ARCHS4 gene–gene co-expression correlations. The input for the predictions was top ranked genes of different sizes returned from the literature search with the AutoRIF settings. Ranking was accomplished by three methods: total counts, normalized counts and a combined score that multiplies the total counts by the normalized counts.
Following, we asked how many pathway members are predicted using the ARCHS4 gene–gene co-expression correlation similarity matrix. To perform the functional prediction, we first ranked the returned genes from the literature search by three different methods. The first method ranks the genes by the absolute publication count matching the search term (absolute count); The second method ranks the genes by the number of publications matching the search term normalized by the total number of publications for the gene (normalized count); and the third method is multiplying the scores of the first two methods as a combined score (combined score). We see that the best method is the combined method in which the gene frequency is multiplied by the total gene count (Figure 3B). The quality of the predictions depends on the number of genes that are submitted from the top ranked lists of genes returned from the literature search, and used as input for performing the predictions. We can see that the performance level saturates at around 25 genes for the combined method. Geneshot returns the 200 most likely associated genes for each KEGG pathway term. The number of genes that could be correctly matched to a KEGG pathway, as a results of the prediction step, but not be retrieved by the AutoRIF search, is shown in Figure 3C.
Gene set novelty and augmentation analysis
The Geneshot user interface for the gene set augmentation and novelty assessment takes as input a list of genes in an entry box, and a background gene–gene matrix to perform the predictions (Supplementary Figure S3A). Once such selection is made, Geneshot returns a bar chart that divides the genes within the gene set into four buckets: rare, uncommon, common and very common based on the number of gene–PMID associations listed in the Tagger dataset. Below is the bar chart displaying two tables. One table lists the entered genes with their publication counts and the other table enlists the additional augmented genes based on their average similarity to the input gene set (Supplementary Figure S3B).
Retrieving gene sets for NIH-funded principal investigators
The Geneshot API opens the opportunity for many applications. To demonstrate one such application, we first obtained a list of all NIH-funded investigators and then used Geneshot to extract the genes that they study based on their prior publications. This enabled us to compute an estimate of how much funds are spent on the study of each gene (Supplementary Figure S4). We observe that well-studied genes are also widely invested in further studying them. To mitigate this trend, the NIH has initiated the Illuminating the Druggable Genome Common Fund program that focuses on a concentrated effort to create new knowledge about genes that have potential to become drug targets from the most known druggable gene families: kinases, GPCRs and ion channels. We see that the lists of kinases, GPCRs and ion channels selected for further study by the NIH are indeed receiving little or no funding.
SUMMARY
Here, we present a new web-server application that enables the systematic generation of gene sets from any biomedical set of terms. Beyond identifying genes associated with publications given any search term(s), Geneshot also predicts genes that may be associated with those search terms, as well as augments the original gene set with predicted genes based on the various gene–gene similarity matrices. We plan to update the site once a year. This decision was made to allow provenance of the results. In other words, we think it is important to have reproducible results so more frequent updates can confuse users. Just because the ARCHS4 based gene–gene co-expression correlation predictions may not perform as well as other libraries in some cases, this does not mean that those predictions are necessarily wrong. It is likely that many highly ranked genes predicted by the co-expression correlations are relevant but not yet discovered. Hence, Geneshot can enable rapid hypothesis generation to direct researchers to the most relevant genes to experimentally perturb in their next set of web-bench experiments. Since Geneshot can be used to produce many new gene sets automatically, Geneshot can be used to significantly expand the collection of gene sets for gene set enrichment analysis tools. In addition, Geneshot's ability to rapidly identify associations between potential drug targets and diseases gives it the potential to enrich the content of resources such as Open Targets (24), Pharos (25) and Harmonizome (26). The PI analysis using Geneshot can be applied to create a network that connects PIs, genes, diseases, drugs and other biomedical terms based on the genes these search terms share. Such a network will connect investigators with other investigators and the areas of research these investigators may overlook. The reason we decided to only query NIH-funded PIs was due to the manageable size of this list of researchers. However, all authors could be connected based on the genes they published to form more comprehensive collaborative networks.
Supplementary Material
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
NIH [U54-HL127624 (LINCS-DCIC), U24-CA224260 (IDG-KMC), T32-GM062754 (Pharmacological Sciences Training Program), OT3-OD025467 (NIH Data Commons)]. Funding for open access charge: NIH [U54-HL127624].
Conflict of interest statement. None declared.
REFERENCES
- 1. Wang Z., Clark N.R., Ma’ayan A.. Dynamics of the discovery process of protein-protein interactions from low content studies. BMC Syst. Biol. 2015; 9:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Oprea T.I., Bologa C.G., Brunak S., Campbell A., Gan G.N., Gaulton A., Gomez S.M., Guha R., Hersey A., Holmes J.. Unexplored therapeutic opportunities in the human genome. Nat. Rev. Drug Discover. 2018; 17:317–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Jensen L.J., Saric J., Bork P.. Literature mining for the biologist: from information retrieval to biological discovery. Nat. Rev. Genet. 2006; 7:119–129. [DOI] [PubMed] [Google Scholar]
- 4. Mikolov T., Chen K., Corrado G., Dean J.. Efficient estimation of word representations in vector space. 2013; arXiv doi:16 January 2013, preprint: not peer reviewedhttps://arxiv.org/abs/1301.3781.
- 5. Wang Z., Lachmann A., Ma’ayan A.. Mining data and metadata from the gene expression omnibus. Biophys. Rev. 2018; 11:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Pletscher-Frankild S., Jensen L.J.. Design, implementation, and operation of a rapid, robust named entity recognition web service. J. Cheminform. 2019; 11:doi:10.1186/s13321-019-0344-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Szklarczyk D., Gable A.L., Lyon D., Junge A., Wyder S., Huerta-Cepas J., Simonovic M., Doncheva N.T., Morris J.H., Bork P.. STRING v11: protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2018; 47:D607–D613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S.. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. 2005; 102:15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chen E.Y., Tan C.M., Kou Y., Duan Q., Wang Z., Meirelles G.V., Clark N.R., Ma’ayan A.. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinform. 2013; 14:128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kuleshov M.V., Jones M.R., Rouillard A.D., Fernandez N.F., Duan Q., Wang Z., Koplev S., Jenkins S.L., Jagodnik K.M., Lachmann A.. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016; 44:W90–W97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jourquin J., Duncan D., Shi Z., Zhang B.. GLAD4U: deriving and prioritizing gene lists from PubMed literature. BMC Genomics. 2012; 13:S20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mandloi S., Chakrabarti S.. PALM-IST: pathway assembly from literature mining-an information search tool. Sci. Rep. 2015; 5:10021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tsuruoka Y., Tsujii J., Ananiadou S.. FACTA: a text search engine for finding associated biomedical concepts. Bioinformatics. 2008; 24:2559–2560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Allot A., Chennen K., Nevers Y., Poidevin L., Kress A., Ripp R., Thompson J.D., Poch O., Lecompte O.. MyGeneFriends: a social network linking genes, genetic diseases, and researchers. J. Med. Internet Res. 2017; 19:e212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Maglott D., Ostell J., Pruitt K.D., Tatusova T.. Entrez Gene: gene-centered information at NCBI. Nucleic Acids Res. 2010; 39:D52–D57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lachmann A., Torre D., Keenan A.B., Jagodnik K.M., Lee H.J., Wang L., Silverstein M.C., Ma’ayan A.. Massive mining of publicly available RNA-seq data from human and mouse. Nat. Commun. 2018; 9:1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Smedley D., Haider S., Ballester B., Holland R., London D., Thorisson G., Kasprzyk A.. BioMart–biological queries made easy. BMC Genomics. 2009; 10:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Glažar P., Papavasileiou P., Rajewsky N.. circBase: a database for circular RNAs. RNA. 2014; 20:1666–1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Povey S., Lovering R., Bruford E., Wright M., Lush M., Wain H.. The HUGO gene nomenclature committee (HGNC). Human Genetics. 2001; 109:678–680. [DOI] [PubMed] [Google Scholar]
- 20. Barrett T., Wilhite S.E., Ledoux P., Evangelista C., Kim I.F., Tomashevsky M., Marshall K.A., Phillippy K.H., Sherman P.M., Holko M.. NCBI GEO: archive for functional genomics data sets—update. Nucleic Acids Res. 2012; 41:D991–D995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bostock M., Ogievetsky V., Heer J.. D³ data-driven documents. IEEE Trans. Visual. Computer Graph. 2011; 17:2301–2309. [DOI] [PubMed] [Google Scholar]
- 22. Boettiger C. An introduction to Docker for reproducible research. ACM SIGOPS Operat. Syst. Rev. 2015; 49:71–79. [Google Scholar]
- 23. Kanehisa M., Goto S.. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000; 28:27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Carvalho-Silva D., Pierleoni A., Pignatelli M., Ong C., Fumis L., Karamanis N., Carmona M., Faulconbridge A., Hercules A., McAuley E.. Open Targets Platform: new developments and updates two years on. Nucleic Acids Res. 2018; 47:D1056–D1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Nguyen D.-T., Mathias S., Bologa C., Brunak S., Fernandez N., Gaulton A., Hersey A., Holmes J., Jensen L.J., Karlsson A.. Pharos: collating protein information to shed light on the druggable genome. Nucleic Acids Res. 2016; 45:D995–D1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rouillard A.D., Gundersen G.W., Fernandez N.F., Wang Z., Monteiro C.D., McDermott M.G., Ma’ayan A.. The harmonizome: a collection of processed datasets gathered to serve and mine knowledge about genes and proteins. Database. 2016; 2016:baw100. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.