


Abstract
Even in the era of next generation sequencing, in which bioinformatics tools abound, annotating transcriptomes and proteomes remains a challenge. This can have major implications for the reliability of studies based on these datasets. Therefore, quality assessment represents a crucial step prior to downstream analyses on novel transcriptomes and proteomes. DOGMA allows such a quality assessment to be carried out. The data of interest are evaluated based on a comparison with a core set of conserved protein domains and domain arrangements. Depending on the studied species, DOGMA offers precomputed core sets for different phylogenetic clades. We now developed a web server for the DOGMA software, offering a user-friendly, simple to use interface. Additionally, the server provides a graphical representation of the analysis results and their placement in comparison to publicly available data. The server is freely available under https://domainworld-services.uni-muenster.de/dogma/. Additionally, for large scale analyses the software can be downloaded free of charge from https://domainworld.uni-muenster.de.
INTRODUCTION
As sequencing technologies improve and become increasingly more affordable, the rate at which genomes are being sequenced and assembled is increasing. Even genomes which may previously have been deemed unattainable, for example due to size or repeat content, can now be sequenced and assembled within a reasonable time and financial budget. These advancements have led to new demands on algorithms processing the resulting reads. As a consequence, many different programs and pipelines have been developed to assemble (e.g. ALLPATHS-LG (1), Canu (2)) and annotate (e.g. AUGUSTUS (3), MAKER (4)) sequencing data. Thus, depending on the chosen programs and parameters, the resulting assembly and gene annotations may vary substantially. A correct quality assessment of the results is therefore critical. This helps to ensure a comparability and reliability of findings and avoids possible artefacts in downstream analyses due to the usage of low quality proteomes or transcriptomes.
One important factor for judging the quality of a proteome or transcriptome is its completeness score, indicating whether all proteins or transcripts have been annotated. A common approach to measuring the completeness of a sequence set is to compare it to proteomes of known high quality from closely related species. For this purpose, as a first step, a core set of conserved features (e.g. genes or domains that are shared across all included species) is extracted from the high-quality dataset. In a second step the conserved features are looked up in the novel sequence set. The amount of missing features from the conserved core set serves as an indicator for the completeness of the whole sequence set.
A high proportion of missing features indicates a potential problem with the generated data. The first programs utilizing this approach were CEGMA (5) and BUSCO (6), for which genes served as the conserved units. Both are also available as a web server, implemented from an independent group (7). Another recently published study uses a combination of conserved proteins and DNA fragments to assess the quality of fungal genomes (8). In contrast to the before mentioned programs, DOGMA (9) compares protein domains and domain arrangements as conserved elements.
Protein domains are conserved, functional or structural units, well suited for this purpose. The set of domains in one protein is called a domain arrangement, which is defined by the order of the domains in the sequence. Computationally, domains are usually modeled using Hidden Markov Models (HMMs) built from sequence profiles. Programs from the HMMER (10) or HHsuite (11) can be used to identify domains in unknown sequences. Although domains can be recombined to form new arrangements, the majority of them are conserved across a whole phylogenetic clade and can be used as molecular markers to evaluate the completeness and quality of a sequence set.
DOGMA
DOGMA compares a set of precomputed conserved domain arrangements (CDAs), the so called ‘core set’, to the proteome or transcriptome of interest. Absent domain arrangements are then determined and the completeness of the analyzed data is calculated (see Figure 1).
Figure 1.

Example use case and work-flow of DOGMA. A newly sequenced genome is assembled and annotated with proteins and domains. It is compared to a set of conserved single domains and domain arrangements extracted from a set of reference species. The completeness score then reflects the fraction of missing domains in the new annotation compared to the core set.
DOGMA can be applied to proteomes as well as transcriptomes, which, due to inherent differences in the data, are processed in slightly different ways. For proteomes, the core set contains the number of conserved occurrences of a domain or domain arrangement and this number is included in the evaluation. In transcriptome mode, on the other hand, only presence or absence is checked. The proteome mode offers slightly more information but requires the sequence input to be isoform free. While this is unproblematic for proteomes (if necessary, isoforms can be easily removed to keep only the longest isoform), transcriptomes are usually not free of isoforms. In both modes, consecutive repeats of the same protein domain are collapsed into a single domain (e.g.: A-B-B-C → A-B-C) as it has been shown that a different number of repeats can occur even in closely related species (12).
Naturally, the composition of the core set heavily depends on the chosen reference species and therefore influences the calculated completeness score. Our precomputed core sets incorporate between five and six high-quality proteomes. CDAs are extracted from these proteomes, which are then used for measuring completeness scores for other proteomes. We consider a domain arrangement to be conserved when it appears in all species at least once and if the difference in number of duplicates is not higher than two. The greater the relatedness between species, the more common domain arrangements they possess. Therefore, a core set based on a set of closely related species is larger (and therefore potentially more accurate) than a set that is computed from less closely related species. However, a more specific core set can only be applied to a smaller set of species. For this reason, we have precomputed 11 core sets to cover various phylogenetic clades (eukaryotes, vertebrates, mammals, arthropods, insects, plants, eudicots, monocots, fungi, bacteria and archaea). It is recommended to always use the core set of the most specific clade containing the species of interest as this provides the most realistic assessment of the data although the score will potentially be lower compared to using a more general core set.
In the latest DOGMA version we have added a partial domain score. It has been shown (13) that partial domains (domains that were not annotated to the full length) are seldom a biological reality (e.g. due to a specific isoform, destroying that domain). More common is that a partial domain is an artefact, for example due to an assembly problem or a wrong gene annotation. To account for this, we have added the fraction of all domains that have a length of <50% of the HMM that was used for their annotation. This calculation can be performed on all annotated domains and does not need any precomputed data. The partial domain analysis is therefore not limited to a specific subset. The partial score thereby provides a general score on all gene models in the dataset. While a low amount of partial domains is to be expected (due to biological reasons or the threshold settings of the HMMs) a high fraction of partial domains would indicate a possible problem with the annotated gene models.
To test its accuracy, we compared DOGMA to another quality assessment program, BUSCO (Figure 2). The comparison was based on a set of 153 eukaryotic species from the ENSEMBL database in version 94 (14) with the provided eukaryotes core set in both programs. From DOGMA the total score was taken, while for BUSCO the percentage of complete BUSCO’s has been used. A calculated pearson correlation of 0.96 for these two scores shows a high agreement in the quality assessments of the analyzed data independent of the implemented method.
Figure 2.
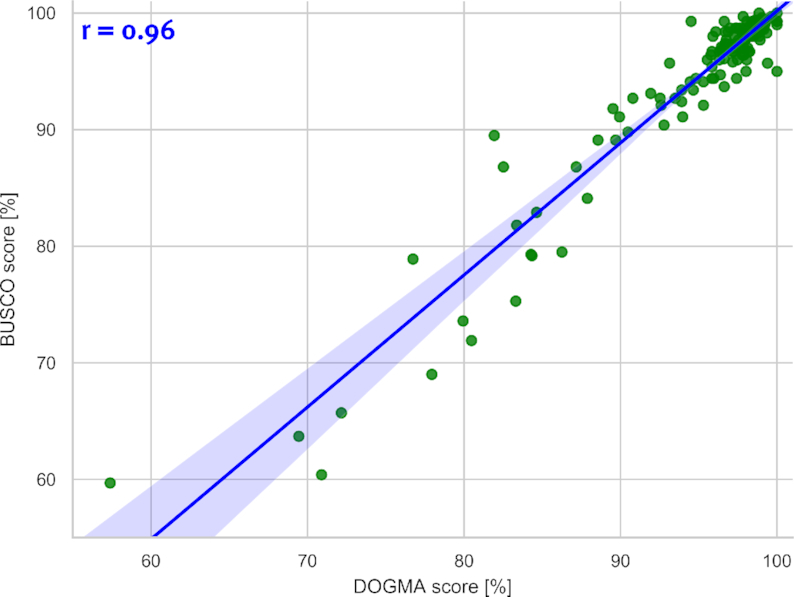
Comparison of DOGMA and BUSCO quality scores. The quality estimations of both programs show a very high correlation, with a Pearson correlation coefficient of 0.96.
BUSCO and DOGMA both address the problem of quality assessment and can be used side-by-side (e.g. (15,16)). Both have some advantages depending on the data to be analyzed. BUSCO is able to assess genomes as well, DOGMA on the other hand has an advantage when analyzing fast evolving species as HMMs are usually more sensitive and should be able to find the domains even if the sequences are already quite distant from the core set.
DOGMA WEB SERVER
Implementation
The web server is written in Python 3 and uses the Django Web framework. The Celery software is used together with the RabbitMQ message broker system for the deployment of the queuing system. The violin plots are created using the Python plotting library matplotlib. Apache is used for the deployment of the web server.
Web server features
The web server, beside providing the actual quality scores, also compares the results to precomputed quality scores of other proteomes. This allows users to assess the quality of their tested data in relation to other species from the same phylogenetic clade (Figure 3). The comparative data contain DOGMA scores for a large number of species, mostly taken from the ENSEMBL (14) database. As there is no specific ENSEMBL data base for archaea, proteomes for this clade have been taken from the web page of the protein quality index (17).
Figure 3.
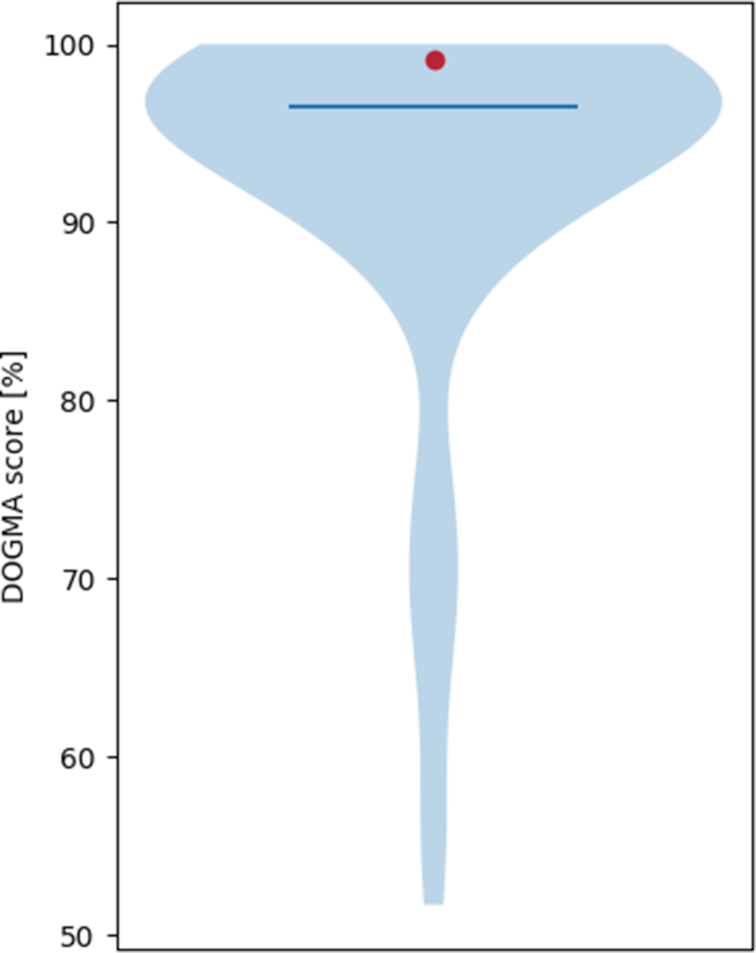
Comparison of the DOGMA score of the high-quality Pan troglodytes proteome (red dot) to 89 mammalian proteomes (violin plot). The dark blue line represents the median DOGMA score of the mammalian proteomes. In general, the better the quality of the analyzed data the higher should be the DOGMA score. The web server also computes a similar image for the partial score.
Independent of the computation mode (proteome/transcriptome) used to calculate the DOGMA score, in the violin plots the result is always compared to data based on the proteome mode. We made this decision as proteomes are generally more complete than transcriptomes, as they are independent of tissue specificity. If the user wants to compare a tissue specific transcriptome to other comparable samples, the stand-alone DOGMA version still offers the possibility to test against self-made core sets.
Furthermore, the web server provides general statistics about the input (e.g. number of domains and domain arrangements) as well as graphical representations of the missing CDAs in the dataset. The missing CDAs are sorted by size and, in the proteome mode the number of missing arrangements is also provided. The domains are hyperlinked to the Pfam (18) database allowing the user, with one mouse click, to obtain further information on the missing domains and their functionality (Figure 4).
Figure 4.

Example listing of missing CDAs of length three from the Pan troglodytes proteome quality assessment. The output shows the number of CDAs found compared to the expected number. CDAs which were found in the expected number of occurrences are not displayed. Domains are links to the Pfam database, allowing the user to obtain further information on the function of the missing domains.
Use case
A typical use case for DOGMA is when a new genome or transcriptome has been sequenced and annotated. In this case its quality needs to be verified to ensure its suitability for further analyses. Another scenario is when analyzing/comparing several proteomes. If the proteomes are of different quality this might affect the analysis and results in technical artefacts. In both cases the quality of the proteome/transcriptome should be checked with DOGMA before any downstream analyses take place, to ensure reliability of further findings.
The following list is a description with more detailed steps of the first use case:
assemble and annotate genome using the chosen pipeline
remove short isoforms if necessary (proteome mode)
annotate sequences with Pfam domains (e.g. using pfam_scan.pl provided by the Pfam database)
run DOGMA to perform a quality assessment of the proteome/transcriptome
depending on the completeness and partial scores improve the annotation and reassess the quality. The annotation might be improved using additional RNA-seq data, changing the used parameters or software. One might additionally want to check the genome assembly for problems, which can be done using a software like BUSCO.
given a good quality of the annotation continue with downstream analyses
CONCLUSION
The DOGMA web server allows a user to assess the completeness of a proteome or transcriptome. Furthermore, the partial score can provide information about the quality of the existing gene models, without being limited to a specific conserved gene set, an advantage compared to other existing analysis tools. Additionally, the web server provides a direct graphical comparison against other species. The web server allows a fast and easy use of DOGMA without the need to install it or knowledge about how to use the command line. It also provides additional information and links to the Pfam database allowing a user to quickly and easily obtain additional information on the missing domain arrangements.
ACKNOWLEDGEMENTS
We thank Mark Harrison for proof reading and improving the manuscript. We would also like to thank all the testers of our web server for their valuable feedback.
FUNDING
Funding for open access charge: Publication charges will be paid from the household funds of the principal investigator.
Conflict of interest statement. None declared.
REFERENCES
- 1. Gnerre S., Maccallum I., Przybylski D., Ribeiro F.J., Burton J.N., Walker B.J., Sharpe T., Hall G., Shea T.P., Sykes S. et al.. High-quality draft assemblies of mammalian genomes from massively parallel sequence data. Proc. Natl. Acad. Sci. U.S.A. 2011; 108:1513–1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Koren S., Walenz B.P., Berlin K., Miller J.R., Bergman N.H., Phillippy A.M.. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017; 27:722–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Keller O., Kollmar M., Stanke M., Waack S.. A novel hybrid gene prediction method employing protein multiple sequence alignments. Bioinformatics. 2011; 27:757–763. [DOI] [PubMed] [Google Scholar]
- 4. Cantarel B.L., Korf I., Robb S.M.C., Parra G., Ross E., Moore B., Holt C., Alvarado A.S., Yandell M.. MAKER: an easy-to-use annotation pipeline designed for emerging model organism genomes. Genome Res. 2008; 18:188–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Parra G., Bradnam K., Korf I.. CEGMA: a pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics. 2007; 23:1061–1067. [DOI] [PubMed] [Google Scholar]
- 6. Simão F.A., Waterhouse R.M., Ioannidis P., Kriventseva E.V., Zdobnov E.M.. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics. 2015; 31:3210–3212. [DOI] [PubMed] [Google Scholar]
- 7. Nishimura O., Hara Y., Kuraku S.. gVolante for standardizing completeness assessment of genome and transcriptome assemblies. Bioinformatics. 2017; 33:3635–3637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cissé O.H., Stajich J.E.. FGMP: assessing fungal genome completeness. BMC Bioinformatics. 2019; 20:184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Dohmen E., Kremer L. P.M., Bornberg-Bauer E., Kemena C.. DOGMA: domain-based transcriptome and proteome quality assessment. Bioinformatics. 2016; 32:2577–2581. [DOI] [PubMed] [Google Scholar]
- 10. Eddy S.R. Accelerated Profile HMM Searches. PLoS Comput. Biol. 2011; 7:e1002195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Remmert M., Biegert A., Hauser A., S’oding J.. HHblits: lightning-fast iterative protein sequence searching by HMM-HMM alignment. Nat. Methods. 2011; 9:173–175. [DOI] [PubMed] [Google Scholar]
- 12. Ekman D., Bj’orklund A.K., Elofsson A.. Quantification of the elevated rate of domain rearrangements in metazoa. J. Mol. Biol. 2007; 372:1337–1348. [DOI] [PubMed] [Google Scholar]
- 13. Triant D.A., Pearson W.R.. Most partial domains in proteins are alignment and annotation artifacts. Genome Biol. 2015; 16:99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zerbino D.R., Achuthan P., Akanni W., Amode M.R., Barrell D., Bhai J., Billis K., Cummins C., Gall A., Girón C.G. et al.. Ensembl 2018. Nucleic Acids Res. 2018; 46:D754–D761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sablok G., Hayward R.J., Davey P.A., Santos R.P., Schliep M., Larkum A., Pernice M., Dolferus R., Ralph P.J.. SeagrassDB: an open-source transcriptomics landscape for phylogenetically profiled seagrasses and aquatic plants. Sci. Rep. 2018; 8:2749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Thomas G.W., Dohmen E., Hughes D.S., Murali S.C., Poelchau M., Glastad K., Anstead C.A., Ayoub N.A., Batterham P., Bellair M. et al.. The Genomic Basis of Arthropod Diversity. 2018; bioRxiv doi: 10.1101/382945, 04 August 2018, preprint: not peer reviewed. [DOI]
- 17. Zaucha J., Stahlhacke J., Oates M.E., Thurlby N., Rackham O.J.L., Fang H., Smithers B., Gough J.. A proteome quality index. Environ. Microbiol. 2015; 17:4–9. [DOI] [PubMed] [Google Scholar]
- 18. El-Gebali S., Mistry J., Bateman A., Eddy S.R., Luciani A., Potter S.C., Qureshi M., Richardson L.J., Salazar G.A., Smart A. et al.. The Pfam protein families database in 2019. Nucleic Acids Res. 2019; 47:D427–D432. [DOI] [PMC free article] [PubMed] [Google Scholar]