


Abstract
Network theory is now a method of choice to gain insights in understanding protein structure, folding and function. In combination with molecular dynamics (MD) simulations, it is an invaluable tool with widespread applications such as analyzing subtle conformational changes and flexibility regions in proteins, dynamic correlation analysis across distant regions for allosteric communications, in drug design to reveal alternative binding pockets for drugs, etc. Updated version of NAPS now facilitates network analysis of the complete repertoire of these biomolecules, i.e., proteins, protein–protein/nucleic acid complexes, MD trajectories, and RNA. Various options provided for analysis of MD trajectories include individual network construction and analysis of intermediate time-steps, comparative analysis of these networks, construction and analysis of average network of the ensemble of trajectories and dynamic cross-correlations. For protein–nucleic acid complexes, networks of the whole complex as well as that of the interface can be constructed and analyzed. For analysis of proteins, protein–protein complexes and MD trajectories, network construction based on inter-residue interaction energies with realistic edge-weights obtained from standard force fields is provided to capture the atomistic details. Updated version of NAPS also provides improved visualization features, interactive plots and bulk execution. URL: http://bioinf.iiit.ac.in/NAPS/
INTRODUCTION
Over the past decade, concepts of graph theory have been widely applied to numerous problems in protein science, viz., identify residues critical for structural stability, protein folding, identify repeats and domains, allosteric regulation, etc. (1–4). With increasing number of high-resolution structures of biomolecules including protein and nucleic acid structures and their complexes, this alternative approach has gained popularity as it reduces the complex 3D organizations to a mathematical entity retaining all the connectivity information in the structure. Molecular dynamics (MD) simulations of these biomolecules provide wealth of information related to their dynamics and the conformational space associated with a given state. Using network theory in combination with dynamical information from conformational ensembles have been shown to provide valuable insights in understanding allosteric regulation, analysis of protein–nucleic acid interface, identifying critical residues for structural stability, active/binding sites and protein folding and function, to name a few (5–8). Similar to protein contact network analysis, graph representations of RNAs reduce drastically the conformational space and can help in structure-function analysis. Most of these approaches use network representations of RNA secondary structure such as RAG-3D (9), which is a 3D search tool that exploits graph representations of RNAs for searching similar 3D structural fragments. To the best of our knowledge there are no network based tools for the analysis of RNA tertiary structures that capture non-covalent interactions between the nucleotides similar to residue interaction networks of proteins. Some resources are available that provide analysis of nucleic acids and their complexes by capturing non-covalent interactions (10,11), such as types of interactions present between the nucleotide residues/interface, long range interactions between secondary structure elements, etc. Further, it has been observed that cellular functions are carried out by proteins, nucleic acids and their complexes in an orchestrated manner. Thus, analysis of protein–nucleic acid complexes can provide meaning insights into the functional relationships of these macromolecules.
Some of the recently developed resources for network analysis of protein structures are RING 2.0 (12) and protein contact atlas (13). RING constructs a residue interaction network by capturing a range of physicochemical interactions between amino acid residues including covalent and non-covalent interactions. Protein contact atlas captures all the non-covalent interactions within a protein or protein complex and displays them at different scales ranging from atomic level to the entire complex level with various interactive visual features. The Cytoscape (14) plugins for network analysis of proteins such as RINalyzer (15), structuctViz2 (16) and CyToStruct (17) are also extensively used as the networks constructed using these plugins can be analysed by other Cytoscape applications. With the advancements in computing power, large systems are being captured over longer time scales in molecular dynamics (MD) simulations. Comprehensive analysis of the huge MD data is challenging and requires computationally efficient approaches for easy interpretation of the data. To the best of our knowledge, apart from MDN (18), majority of the resources for network analysis of MD trajectories are standalone software packages such as xPyder (19), PyInteraph (20), MD-TASK (21), gRINN (22) and PSN-Ensemble (23). xPyder is a PyMOL (https://www.pymol.org) plugin that constructs the network based on dynamic cross correlation between amino acid residues integrated with various features of PyMOL. PyInterraph and gRINN provide network construction based on interaction energy of the residues while MD-TASK provides Cβ network representation. PSN-Ensemble is also a standalone application which provides network construction based on residue side-chain interaction strength with options to analyse the hubs, clusters, cliques, and shortest paths in the network. MDN is a web portal for network analysis of MD trajectories. It allows construction of networks based on inter-residue interaction energy and supports analysis of betweeness centrality and network coupling, a measure of efficiency of signal propagation through the network. Apart from limited topological analysis, it also has limitations on the size of the input trajectory file (100 MB).
The earlier version of NAPS (24) supported network analysis of static structures of single protein and protein–protein complex (two chains). The workflow of the updated NAPS showing all its features is given in Figure 1 with the enhancements depicted in ‘red’. It is now updated to an integrated platform that supports network analysis of a wide range of macromolecules, viz., protein molecules and their complexes (protein–protein/DNA/RNA), molecular dynamics trajectories and 3D-RNA structures. Depending on molecule/analysis type, user may choose an appropriate network construction method from a range of options provided. Major enhancements include:
Analysis of trajectories from molecular dynamics simulations
Analysis of protein–nucleic acid complexes
Analysis of RNA tertiary structures
Supporting analysis of up to four chains simultaneously in the protein–protein/DNA/RNA complexes
Four new types of network construction incorporated: (i) energy network, (ii) dynamic cross-correlation (DCC) network, (iii) average ensemble network and (iv) bipartite network.
Interactive plots for visual analysis
Bulk upload
Figure 1.

The NAPS workflow for the construction and analysis of protein, protein–protein/DNA/RNA complex, RNA and Molecular Dynamics trajectories. The network construction and analyses options available for the input types are marked by superscript numbers. New features introduced in updated NAPS are highlighted in ‘red’ colour and ‘*’ indicates that the option is available for all 6 input types.
PROTEINS AND PROTEIN-PROTEIN COMPLEXES
In the previous version of NAPS webserver, extensive network analysis of a single protein structure and a complex of two proteins was provided (24). Taking the structural information in PDB format as input, Protein Contact Network (PCN) or Residue Interaction Network (RIN) of the proteins was constructed based on the type of analysis desired. In this section, we briefly discuss various features of the previous version of NAPS for protein structure analysis and highlight the enhancements incorporated in the updated version.
Network construction
Various methods are provided for constructing protein contact networks (PCNs) in NAPS. These include methods based on geometry and energy considerations with weighted/unweighted edges, both for proteins and protein–protein complexes. The geometry-based methods attempt to capture the overall protein connectivity and 3D topology of the protein by identifying main-chain/side-chain/centroid/atom-pair contacts based on spatial proximity, defined by a cut-off distance. In network representation, the basic unit of protein, amino acid residue, is considered as node, and possible interaction between two residues is represented by an edge. For example, in the Cα network, an edge is drawn between two amino acids if the backbone Cα–Cα distance is within a cut-off distance (7 Å) (24). This captures the topology of the protein structure very well and has been extensively studied to understand protein folding (25,26), intra-molecular communication (27), identification of structural repeats and domains (28,29), etc. A more realistic network realisation is obtained by capturing the side-chain interactions, wherein an edge is drawn between two amino acids if the Cβ–Cβ distance (Cβ network) or distance between the centre of mass of two amino acids (centroid network) is within a cut-off distance (7 Å) (24). These networks have been useful in understanding protein dynamics (30), binding cavities (31) and identification of important residues for protein function (32). The number of non-bonded interactions between two residues is captured in atom-pair contact network and interaction strength networks. These network types provide fine-grained analysis useful in understanding protein folding mechanism (33), allosteric regulation (34), thermo-stability (3) and DNA binding mechanism (35). However, both these networks are computationally intensive as they require calculating Euclidean distance between all atom pairs of the two residues (24). For each of these network representations, edges may be considered between residues that are sequentially separated by 10–12 residues along the protein backbone to capture long range interactions. All the networks discussed above efficiently capture the topology and associated properties of proteins; however the chemistry is not captured. In the updated version, network construction based on inter-residue interaction energies, with realistic edge-weights obtained from standard force fields, called energy network, is provided and its construction is given below.
Energy network
In this network representation, amino acid residues (nodes) are connected by an edge if the non-bonded interaction energy (Eij) between two residues i and j, given by
 |
(1) |
is lower than the user defined threshold (default < 0 kJ/mol) (36). The van der Waals component (VLJ) is given by Lennard–Jones potential and the electrostatic energy component (VC) is given by Coulombic potential:
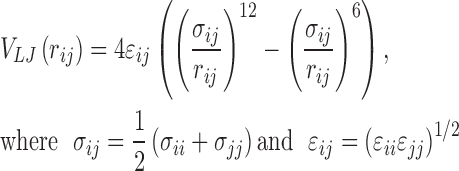 |
(2) |
 |
(3) |
The user is provided with two options for the calculation of interaction energies, Eij, CHARMM36m (37) and AMBER (38). Although it is the most computationally intensive method of network construction, the realistic edge weights obtained from standard force fields capture residue-residue interactions at the atomistic level.
Network analysis
A number of network topological parameters that capture general as well as specific features of proteins and protein complexes are provided in NAPS. For all the six network types, one can carry out an in-depth network analysis by computing global properties (such as number of nodes and edges, diameter, clustering coefficient, average degree and average path length), various centrality measures, eigen spectra, shortest paths and k-cliques. These have been integrated with interactive visualization of the network and 3D protein structure facilitating analysis of structure-function relationships (24).
Centrality measures such as degree, closeness, betweenness, clustering coefficient, eccentricity and average nearest neighbour degree provide ranking of nodes based on various topological properties and quantify the importance of a residue towards the stability of structure and intra-molecular communications in the network. Edge betweenness is a centrality measure that indicates how central an edge is to the various communication pathways in a network. Eigen spectra of adjacency matrix provides information regarding the contribution of each node to the network by capturing the connectivity of a node, its neighbour's connectivity, that of its neighbour's neighbours connectivity, and so on (2), while the eigen spectra of Laplacian matrix provides clustering information, i.e., nodes with similar eigenvector values belong to the same cluster (39). Analysis of shortest paths through the network is useful in identifying the importance of residues involved in long range communications in the protein. It has been shown in numerous studies that allosteric communications where inter-molecular signal propagates from one functional site to a distant site within the protein are along the shortest path (34). The densely connected k-clique component in a protein structure represents compact structural regions that are likely to be involved in providing structural stability to the protein (40). Thus various network metrics provided in NAPS aid in functional analysis of proteins.
Visual analysis
Five interactive visual features are provided in NAPS for protein analysis—3D network and structure views, 2D contact map, distance matrix and domain views. The 3D network and structure views are integrated with centrality, graph spectra, shortest path and k-clique analyses. Important nodes identified by these analyses are highlighted in both the network and 3D structure views for ease in analysing the results. User can also highlight the residues based on their physic-chemical properties (hydrophobic, hydrophilic and charged) to analyse the nature of interactions involved. For example, cluster of hydrophobic residues that form the buried core of the protein are known to be important in stabilizing the structure of the protein, while these residues on the surface play a role in protein–protein and protein–nucleic acid interactions (41). Similarly, identifying cluster of charged residues can help in elucidating the function of the protein as these are mainly present at the active site or metal binding sites (42).
NETWORK ANALYSIS OF MD TRAJECTORIES
Various network representations are provided for the analysis of molecular dynamics simulation data of protein molecules in NAPS. The analysis can be carried out by constructing the networks (Cα, Cβ or Energy) for every time-step (stride), or, a single average network capturing the complete ensemble of trajectories. Analysis of the atomic motions and their collective behaviour is central to the understanding of biological function of proteins. In NAPS, the dynamic cross-correlation heat map and network can be constructed to analyse the correlated motions between atom pairs.
Time-step network
The structural conformation of a protein (or complex) at intermediate time-steps of the simulation can be analysed by constructing individual networks for each time-step. Three network types, Cα, Cβ or energy network are provided for analysis (as discussed in protein network analysis section). Network analysis at various time-steps can provide insight to the dynamical behaviour of proteins in different conformational states, e.g., bound and unbound states (43), active and inactive states in allosteric regulation (7), folded and unfolded states (5), etc.
Average network
An average network (a single network) representing the ensemble of trajectories by a single network. It captures the average representation of any of the three network types (Cα, Cβ or energy) constructed for intermediate time-steps. In this case, an amino acid residue is considered as node and an edge is drawn if two nodes share an edge in a fixed proportion (default 60%) of intermediate time-step networks.
Dynamic cross-correlation network
The dynamic cross correlation (DCC) analysis is a popular method for analyzing the trajectories of molecular dynamics (MD) simulations. It captures the collective behaviour of the atoms, i.e., the degree to which they move together. The correlation values vary between –1 and 1, where ‘1’ corresponds to complete correlation, ‘–1’, complete anti-correlation, and ‘0’ as no correlation. The DCC network is constructed by considering an amino acid residue as node and an edge is drawn if the DCC value between a residue pair is within a threshold (default ≥ |0.5|). It is a measure of similarity of two amino acid residues as a function of their relative displacement throughout the trajectory and is given by
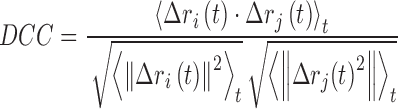 |
(4) |
where, ri(t) denotes the vector of the ith atom's coordinates as a function of time t, <⋅>t is the time ensemble average and  .
.
Analysis
Specialized analysis options have been incorporated for detailed network analyses of MD simulation trajectories. Various visualization, centrality and shortest path analyses are also applicable to the network corresponding to any intermediate time-step of the trajectory or the ensemble average network, similar to those provided for protein network analysis (discussed in the section on network analysis of proteins). Below some salient features for MD simulation data analysis are listed.
Visual analysis
One can carry out comparison of all the network topological features between any two time-steps. In the 2D network view (Figure 2A) and 3D network view (Figure 2B), the common edges between two time-steps being compared are shown in ‘grey’ while edges specific to the two time-steps are coloured in ‘blue’ and ‘green’ respectively. Thus, any variation in the connectivity of nodes in the two conformations is easily identified. The 3D structure view (Figure 2C) depicts the superposition of two conformations, complementing the network view and provides a platform for the user to analyse the structural differences along with the change in the connectivity.
Figure 2.

Network analysis of ubiquitin simulation. (A)–(D) show comparative analysis of two intermediate time-steps of the simulation. (A) 2D contact map showing common edges in ‘grey’ while edges specific to the time-steps 1 and 2 are shown in ‘blue’ and ‘green’ colours respectively. (B) 3D network view showing all nodes and common edges in the two time-steps in ‘grey’ while the edges specific to time-steps 1 and 2 are shown in ‘blue’ and ‘green’ colours respectively. (C) 3D molecular view using NGL showing the structures of the two time-steps in ‘blue’ and ‘green’ colours. (D) Overlap of the betweenness centrality plot for the two time-steps is shown in ‘blue’ and ‘green’ colours. (E) 2D visualization of the DCC matrix, colour-coded based on pairwise correlation values.
Centrality analysis
The centrality measures are displayed in tabular format along with the network and 3D molecular views. For comparative analysis of centrality measures at two time-steps, an interactive plot is displayed, with the centrality profiles overlapped on the same plot in two different colours to highlight the residues exhibiting variation. For illustration, the betweenness plot shown in Figure 2D clearly indicates that conformational changes in the protein may result in variation in the number of shortest paths passing through the nodes, (e.g. X38 and X51, in this case).
Shortest path analysis
Conformational changes can also lead to variation in shortest paths between a pair of residues. Paths common in the two time steps network are listed along with those unique to each time-step. The analysis of unique paths in intermediate time-steps can be useful in understanding intramolecular communication causing allosteric regulation in proteins as the protein undergoes conformational changes (8).
Dynamic cross correlation map
The DCC matrix obtained from the MD trajectories is represented as a 2D plot coloured based on correlation values where positively and negatively correlated pairs are shown in ‘blue’ and ‘red’ colours, respectively, for easy visual interpretation (Figure 2E). The DCC map can be computed for all the three network representations of MD trajectories.4
NETWORK ANALYSIS OF NUCLEIC ACIDS AND PROTEIN NUCLEIC ACID COMPLEXES
One of the important enhancements in the second version of NAPS is the network analysis of RNA structures and protein–nucleic acid complexes. With increasing number of tertiary structures of RNA molecules available in PDB, graph representation of these structures is desirable to gain insight into structure-function relationships. Though most graph based approaches proposed for analysis of RNA molecules have been for secondary structure analysis, recently network analysis of 3D structures is being studied, but in our knowledge no tools are available for the same.
RNA network
RNA molecules form modular, hierarchical three-dimensional structures and carry out important regulatory and catalytic functions in various cellular processes such as transcription, splicing, translation, etc., and are also involved in protein binding and RNA–RNA binding interactions (44,45). Analysis of the topological features of complex RNA structures can provide information about the interactions between nucleotides and the nature of these interactions (46). Since the function of RNA molecule is governed by its tertiary structure, insights into important residues for function, structural stability, RNA folding and long-range interaction pathways in the molecule can be achieved by network analysis. In NAPS, complex RNA structure is represented by atom-pair contact network, wherein a nucleotide is considered as a node and an edge drawn if the distance between any two atoms of the corresponding nucleotide pair is within a threshold distance (default 5 Å) to capture the non-covalent interactions. Various analysis options provided for protein contact network are also available for RNA atom-pair contact network. In Figure 3A, the 3D network and structure view of M-box riboswitch (PDB: 2QBZ, chain X) responsible for the regulation of intracellular magnesium is shown (47). Visual inspection of degree centrality analysis is depicted in the figure with highly connected nodes (hubs) responsible for forming the compact structure of the self-assembly coloured in ‘red’.
Figure 3.

(A) 3D network and structure view of M-box riboswitch (PDB: 2QBZ, chain X) shown. The nodes are coloured based on their connectivity (i.e. degree) with highly connected nodes depicted in ‘red’ colour. (B) 3D network and structure view of protein-DNA interface of human TATA box binding protein (PDB: 1CDW). The interface component with amino acids ‘KVFP’ (highlighted in red) is characteristic of β sheet–DNA interaction interface.
Protein nucleic acid complex
For the analysis of protein–nucleic acid complexes, NAPS provides two types of network representations, namely, atom-pair contact network (for the whole complex) and bipartite interaction strength network (for the interface).
Atom-pair contact network
For protein-DNA/RNA complex, it is constructed by considering the amino acid residues and nucleotide residues as nodes and an edge is drawn if any two atoms of a residue pair are within a threshold distance (default 5 Å). The edges in the network represent interaction between two amino acids, two nucleotides, or an amino acid with nucleotide residue. Analysis options for protein–nucleic acid complexes are the same as those provided for protein–protein complexes—various centrality analyses, shortest paths, k-cliques, graph spectral analyses and visual analyses.
Bipartite interface network
In this case, the amino acid residues and nucleotides are treated as different node types and the network is constructed only for the interface. The non-bonded interactions between amino acid side chains and nucleotides of a protein–DNA/RNA at the interface are represented by the interaction strength network (35).
Network construction
Since amino acid residues at the protein-DNA/RNA interface interact differently with the three components (phosphate, sugar and base) of the nucleotide, which are captured by three separate bipartite networks are constructed in this case (35). A protein–phosphate bipartite network (Pp) is constructed by considering amino acid residues as one node set and nucleotides as the other node set, and an edge is drawn between them if the interaction strength, given by Iij= [nij/(Ni*Nj)]*100, is ≥threshold (default 4%). Here, nij is the number of side chain atoms of amino acid i present within 4.5Å distance of the phosphate group atoms of nucleotide j, and Ni and Nj are the normalization values of the corresponding amino acid and phosphate group respectively. In a similar fashion, protein–sugar (Ps) and protein-base (Pb) networks are constructed (35). The normalization values for amino acids and different components of the nucleotide are computed by considering the average non-covalent contacts observed for the residue in a non-redundant set of protein-nucleotide complexes (35,48). Thus, Iij corresponds to the interaction of the protein with the three components of nucleotides, phosphate, deoxyribose and the base, respectively, in the three bipartite networks for a protein–DNA/RNA complex.
The analyses options are available for the sparsely connected bipartite network of the protein–nucleic acid interface are:
Hubs: Amino acid residues connected to ≥ k number of nucleotides (default k = 4) or vice versa are defined as hubs. These are highly connected nodes in the network and represent the most important residues having maximal interaction at the interface. For example, Phenylalanine as a hub is predominantly observed in the β-sheet–DNA interface in protein-base bipartite networks (35). The hub nodes can be coloured nodes based on their physicochemical properties to analyse the nature of interaction.
Connected components (clusters): A connected set of amino acids and nucleotide components is identified using the Depth First Search (DFS) algorithm (49). These connected components represent cluster of strongly interacting residues at the interface and are responsible for the stability of the complex. It has been observed that the different protein-DNA complexes such as β-sheet group, β-hairpin group, helix-turn-helix (HTH), zipper type, zinc coordinating group, etc. exhibit different interaction patterns with the nucleotide components (35). For example, component comprising of amino acids ‘KVFP’ is commonly observed in β sheet–DNA interface (35), which can be identified by connected component analysis in NAPS (Figure 3B). Also, in the study of influence of iron on metalloprotein-DNA complex, the difference in the mechanism of binding in metallated and non-metallated system could be explained by the difference in cluster formation pattern at the interface (43).
IMPLEMENTATION DETAILS
The NAPS portal can be used for the network analysis of large macromolecular structures (determined experimentally by X-Ray, NMR, etc. or computationally modelled) such as protein, RNA and protein–protein/DNA/RNA complex; or MD simulations of a protein or protein–protein complex. The portal takes the structural coordinates of the macromolecule in PDB format along with the chain information representing the macromolecule and its interacting partners. For the analysis of MD data, the trajectory is accepted in DCD format along with the option to select start, end and interval (stride) of timesteps. The size of the MD trajectory (usually in the order of gigabytes) is the major challenge for web based analysis tools, which is addressed by carrying out pre-processing of data at client site and optimizing the data upload process. The optimal information required for the analysis is parsed at the client site (usually less than 1 MB) from binary DCD trajectory file using computationally efficient JavaScript API and sent to the server to minimize the bottleneck step of data transfer between the server and client site. Depending on the type of analysis, the user may select the network parameters such as network type, threshold, weighted or unweighted, force field (for energy network) and residue separation along the backbone.
The backend computation for network construction is carried out using python scripts, and network analyses are performed by C and python programs using the graph libraries igraph (50) and Networkx (51). Similar to the previous version, the results are displayed using interactive HTML and PHP pages implementing browser independent JavaScript libraries and the visualization using WebGL API which utilize the client site system resources. The workflow of the portal and data transfer protocol allows analyses of large structures (or complexes) and simulations with ease, where the maximum size of the biological system that can be analysed is dependent on the system configuration and internet bandwidth of the user. The interactive 3D molecular visualization features have been improved in the updated NAPS by incorporating computationally efficient and scalable molecular NGL viewer (52), which can quickly create visual representation of large complexes. The centrality measures are displayed as interactive plots to facilitate analysis of large structures. The 2D contact map and distance matrix, 3D network and macromolecule structure views and centrality plots can be downloaded in high resolution (1200*1200 pixels) PNG and JPEG formats, while all the analyses results such as edgelist, global parameters, centrality values, shortest paths, k-cliques and graph spectra can be downloaded as tab separated text files. Bulk execution mode can be initiated for up to 50 proteins (or complexes) by providing a list of PDB ids and chain(s) to the webserver. Python API can be used to programmatically execute the backend codes for analyses of large number of macromolecules. The edgelist, global properties and centrality for all the proteins can be downloaded in compressed zip format in the bulk mode execution.
Exhaustive functional testing of the portal is carried out on protein structures (and complexes) with different structural class (α, β and αβ), sizes (up to 3751 residues and 14426 edges in 3J8E(C)) and domain architecture (single and multi-domain). The centrality values have been tested against Cytoscape (14) and Centibin (53), k-clique with Mclique app of Cytoscape (http://apps.cytoscape.org/apps/mclique) and shortest path with Pesca (54). The energy values calculated for the construction of energy network are compared with INTAA webserver (55). The network analysis of MD trajectory has been tested for a maximum of 20 ns simulation generating a trajectory file of 20.2 GB.
CONCLUSION
NAPS is an integrated platform for the construction and analysis of network representations of protein, protein complexes, RNA structure and MD trajectory data with no restrictions on the upload size of trajectory file. It is browser independent platform and does not require any installations and pre-processing of the input data. The graph centrality measures capturing the topological properties of the structure and the shortest path analysis capturing the intra-molecular information flow can be analysed for a static structure (or complex) and comparative analysis between time-steps of the trajectory. The network analyses is integrated with interactive visual analysis of the network (2D contact map and 3D network view), macromolecular structure, cross-correlation matrix and centrality plots. For the static macromolecular structure, analyses of highly connected subgraph components through k-cliques and clustering through graph spectra analysis can also be analysed. Thus, multiple modules available for computing various network parameters allows the users to make a judicious choice from an array of options based on the biological question being addressed. The portal is regularly updated based on advances in the field and we intend to extend the network analysis of MD trajectory to nucleic acids and protein–nucleic acid complexes in future.
ACKNOWLEDGEMENTS
BC acknowledges the support of Department of Biotechnology, Government of India for DBT BINC PhD fellowship.
FUNDING
Funding for open access charge: The open access publication charge for this paper has been waived by Oxford University Press - NAR.
Conflict of interest statement. None declared.
REFERENCES
- 1. Bagler G., Sinha S.. Assortative mixing in Protein Contact Networks and protein folding kinetics. Bioinformatics. 2007; 23:1760–1767. [DOI] [PubMed] [Google Scholar]
- 2. Chakrabarty B., Parekh N.. PRIGSA: protein repeat identification by graph spectral analysis. J. Bioinform. Comput. Biol. 2014; 12:1442009. [DOI] [PubMed] [Google Scholar]
- 3. Brinda K.V., Vishveshwara S.. A network representation of protein structures: implications for protein stability. Biophys. J. 2005; 89:4159–4170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Taylor N.R. Small world network strategies for studying protein structures and binding. Comput. Struct. Biotechnol. J. 2013; 5:e201302006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Vijayabaskar M.S., Vishveshwara S.. Insights into the fold organization of TIM barrel from interaction energy based structure networks. PLoS Comput. Biol. 2012; 8:e1002505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bhattacharyya M., Upadhyay R., Vishveshwara S.. Interaction signatures stabilizing the NAD(P)-binding Rossmann fold: a structure network approach. PLoS ONE. 2012; 7:e51676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Nussinov R., Tsai C.-J., Csermely P.. Allo-network drugs: harnessing allostery in cellular networks. Trends Pharmacol. Sci. 2011; 32:686–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ghosh A., Vishveshwara S.. A study of communication pathways in methionyl- tRNA synthetase by molecular dynamics simulations and structure network analysis. Proc. Natl. Acad. Sci. U.S.A. 2007; 104:15711–15716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zahran M., Sevim Bayrak C., Elmetwaly S., Schlick T.. RAG-3D: a search tool for RNA 3D substructures. Nucleic Acids Res. 2015; 43:9474–9488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lu X.-J., Bussemaker H.J., Olson W.K.. DSSR: an integrated software tool for dissecting the spatial structure of RNA. Nucleic Acids Res. 2015; 43:e142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lindow N., Baum D., Leborgne M., Hege H.C.. Interactive Visualization of RNA and DNA Structures. IEEE Trans Vis Comput Graph. 2018; doi:10.1109/TVCG.2018.2864507. [DOI] [PubMed] [Google Scholar]
- 12. Piovesan D., Minervini G., Tosatto S.C.E.. The RING 2.0 web server for high quality residue interaction networks. Nucleic Acids Res. 2016; 44:W367–W374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kayikci M., Venkatakrishnan A.J., Scott-Brown J., Ravarani C.N.J., Flock T., Babu M.M.. Visualization and analysis of non-covalent contacts using the Protein Contacts Atlas. Nat. Struct. Mol. Biol. 2018; 25:185–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Shannon P., Markiel A., Ozier O., Baliga N.S., Wang J.T., Ramage D., Amin N., Schwikowski B., Ideker T.. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003; 13:2498–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Doncheva N.T., Klein K., Domingues F.S., Albrecht M.. Analyzing and visualizing residue networks of protein structures. Trends Biochem. Sci. 2011; 36:179–182. [DOI] [PubMed] [Google Scholar]
- 16. Morris J.H., Huang C.C., Babbitt P.C., Ferrin T.E.. structureViz: linking Cytoscape and UCSF Chimera. Bioinformatics. 2007; 23:2345–2347. [DOI] [PubMed] [Google Scholar]
- 17. Nepomnyachiy S., Ben-Tal N., Kolodny R.. CyToStruct: augmenting the network visualization of cytoscape with the power of molecular viewers. Structure. 2015; 23:941–948. [DOI] [PubMed] [Google Scholar]
- 18. Ribeiro A.A.S.T., Ortiz V.. MDN: a web portal for network analysis of molecular dynamics simulations. Biophys. J. 2015; 109:1110–1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Pasi M., Tiberti M., Arrigoni A., Papaleo E.. xPyder: a PyMOL plugin to analyze coupled residues and their networks in protein structures. J. Chem. Inf. Model. 2012; 52:1865–1874. [DOI] [PubMed] [Google Scholar]
- 20. Tiberti M., Invernizzi G., Lambrughi M., Inbar Y., Schreiber G., Papaleo E.. PyInteraph: a framework for the analysis of interaction networks in structural ensembles of proteins. J. Chem. Inf. Model. 2014; 54:1537–1551. [DOI] [PubMed] [Google Scholar]
- 21. Brown D.K., Penkler D.L., Sheik Amamuddy O., Ross C., Atilgan A.R., Atilgan C., Tastan Bishop Ö.. MD-TASK: a software suite for analyzing molecular dynamics trajectories. Bioinformatics. 2017; 33:2768–2771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Serçinoglu O., Ozbek P.. gRINN: a tool for calculation of residue interaction energies and protein energy network analysis of molecular dynamics simulations. Nucleic Acids Res. 2018; 46:W554–W562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bhattacharyya M., Bhat C.R., Vishveshwara S.. An automated approach to network features of protein structure ensembles. Protein Sci. 2013; 22:1399–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chakrabarty B., Parekh N.. NAPS: network analysis of protein structures. Nucleic Acids Res. 2016; 44:W375–W382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Patra S.M., Vishveshwara S.. Backbone cluster identification in proteins by a graph theoretical method. Biophys. Chem. 2000; 84:13–25. [DOI] [PubMed] [Google Scholar]
- 26. Paola L.D., Paci P., Santoni D., Ruvo M.D., Giuliani A.. Proteins as sponges: a statistical journey along protein structure organization principles. J. Chem. Inf. Model. 2012; 52:474–482. [DOI] [PubMed] [Google Scholar]
- 27. Vishveshwara S., Ghosh A., Hansia P.. Intra and inter-molecular communications through protein structure network. Curr. Protein Pept. Sci. 2009; 10:146–160. [DOI] [PubMed] [Google Scholar]
- 28. Chakrabarty B., Parekh N.. Identifying tandem Ankyrin repeats in protein structures. BMC Bioinformatics. 2014; 15:6599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yalamanchili H.K., Parekh N.. Rajasekaran S. Graph Spectral Approach for Identifying Protein Domains. Bioinformatics and Computational Biology, Lecture Notes in Computer Science. 2009; Berlin, Heidelberg: Springer; 437–448. [Google Scholar]
- 30. Atilgan A.R., Akan P., Baysal C.. Small-world communication of residues and significance for protein dynamics. Biophys. J. 2004; 86:85–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Estrada E. Universality in protein residue networks. Biophys. J. 2010; 98:890–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cusack M.P., Thibert B., Bredesen D.E., Del Rio G.. Efficient identification of critical residues based only on protein structure by network analysis. PLoS One. 2007; 2:e421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Greene L.H., Higman V.A.. Uncovering network systems within protein structures. J. Mol. Biol. 2003; 334:781–791. [DOI] [PubMed] [Google Scholar]
- 34. del Sol A., Fujihashi H., Amoros D., Nussinov R.. Residues crucial for maintaining short paths in network communication mediate signaling in proteins. Mol. Syst. Biol. 2006; 2:2006.0019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sathyapriya R., Vijayabaskar M.S., Vishveshwara S.. Insights into protein-DNA interactions through structure network analysis. PLoS Comput. Biol. 2008; 4:e1000170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Vijayabaskar M.S., Vishveshwara S.. Interaction energy based protein structure networks. Biophys. J. 2010; 99:3704–3715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Huang J., Rauscher S., Nawrocki G., Ran T., Feig M., de Groot B.L., Grubmüller H., MacKerell A.D.. CHARMM36m: an improved force field for folded and intrinsically disordered proteins. Nat. Methods. 2017; 14:71–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Maier J.A., Martinez C., Kasavajhala K., Wickstrom L., Hauser K.E., Simmerling C.. ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 2015; 11:3696–3713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Vishveshwara S., Brinda K.V., Kannan N.. Protein structure: insights from graph theory. J. Theor. Comput. Chem. 2002; 01:187–211. [Google Scholar]
- 40. Deb D., Vishveshwara S., Vishveshwara S.. Understanding protein structure from a percolation perspective. Biophys. J. 2009; 97:1787–1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Dao-pin S., Anderson D.E., Baase W.A., Dahlquist F.W., Matthews B.W.. Structural and thermodynamic consequences of burying a charged residue within the hydrophobic core of T4 lysozyme. Biochemistry. 1991; 30:11521–11529. [DOI] [PubMed] [Google Scholar]
- 42. Karlin S., Zhu Z.Y.. Classification of mononuclear zinc metal sites in protein structures. Proc. Natl. Acad. Sci. U.S.A. 1997; 94:14231–14236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ghosh S., Chandra N., Vishveshwara S.. Mechanism of iron-dependent repressor (IdeR) activation and DNA binding: a molecular dynamics and protein structure network study. PLoS Comput. Biol. 2015; 11:e1004500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Mortimer S.A., Kidwell M.A., Doudna J.A.. Insights into RNA structure and function from genome-wide studies. Nat. Rev. Genet. 2014; 15:469–479. [DOI] [PubMed] [Google Scholar]
- 45. Miao Z., Westhof E.. RNA structure: advances and assessment of 3D structure prediction. Annu. Rev. Biophys. 2017; 46:483–503. [DOI] [PubMed] [Google Scholar]
- 46. Wang K., Jian Y., Wang H., Zeng C., Zhao Y.. RBind: computational network method to predict RNA binding sites. Bioinformatics. 2018; 34:3131–3136. [DOI] [PubMed] [Google Scholar]
- 47. Ramesh A., Wakeman C.A., Winkler W.C.. Insights into metalloregulation by M-box riboswitch RNAs via structural analysis of manganese-bound complexes. J. Mol. Biol. 2011; 407:556–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kannan N., Vishveshwara S.. Identification of side-chain clusters in protein structures by a graph spectral method. J. Mol. Biol. 1999; 292:441–464. [DOI] [PubMed] [Google Scholar]
- 49. Tarjan R. Depth-first search and linear graph algorithms. 12th Annual Symposium on Switching and Automata Theory (swat 1971). 1971; 114–121. [Google Scholar]
- 50. Csardi G., Tamas N.. The igraph software package for complex network research. InterJournal, Complex Systems. 2006; 1695:1–9. [Google Scholar]
- 51. Hagberg A., Schult D., Swart P.. Exploring Network Structure, Dynamics, and Function using NetworkX. 7th Python in Science conference (SciPy 2008). 2008; 11–15. [Google Scholar]
- 52. Rose A.S., Bradley A.R., Valasatava Y., Duarte J.M., Prlic A., Rose P.W.. NGL viewer: web-based molecular graphics for large complexes. Bioinformatics. 2018; 34:3755–3758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Junker B.H., Koschützki D., Schreiber F.. Exploration of biological network centralities with CentiBiN. BMC Bioinformatics. 2006; 7:219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Scardoni G., Tosadori G., Pratap S., Spoto F., Laudanna C.. Finding the shortest path with PesCa: a tool for network reconstruction [version 2; peer review: 2 approved, 2 approved with reservations]. F1000Research. 2016; 4:484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Galgonek J., Vymetal J., Jakubec D., Vondrášek J.. Amino Acid Interaction (INTAA) web server. Nucleic Acids Res. 2017; 45:W388–W392. [DOI] [PMC free article] [PubMed] [Google Scholar]