


Abstract
For the best results in quantitative polymerase chain reaction (qPCR) experiments, it is essential to design high-quality primers considering a multitude of constraints and the purpose of experiments. The constraints include many filtering constraints, homology test on a huge number of off-target sequences, the same constraints for batch design of primers, exon spanning, and avoiding single nucleotide polymorphism (SNP) sites. The target sequences are either in database or given as FASTA sequences, and the experiment is for amplifying either each target sequence with each corresponding primer pairs designed under the same constraints or all target sequences with a single pair of primers. Many websites have been proposed, but none of them including our previous MRPrimerW fulfilled all the above features. Here, we describe the MRPrimerW2, the update version of MRPrimerW, which fulfils all the features by maintaining the advantages of MRPrimerW in terms of the kinds and sizes of databases for valid primers and the number of search modes. To achieve it, we exploited GPU computation and a disk-based key-value store using PCIe SSD. The complete set of 3 509 244 680 valid primers of MRPrimerW2 covers 99% of nine important organisms in an exhaustive manner. Free access: http://MRPrimerW2.com
INTRODUCTION
Quantitative polymerase chain reaction (qPCR), also known as real-time PCR, is a widely adopted technique for detecting the mass amplification of specific DNA molecule in real-time. This standard technique is used in a wide range of applications including genetically modified organism (GMO) detection (1), pathogen discovery (2), and validation of changes in expression of interested genes (3). For best results in PCR experiments, optimal primer design is essential. However, it is challenging due to the need to consider both homology test on a huge number of off-target sequences and many filtering constraints. Although many websites are proposed to aid in designing primers, most of them still require an additional step of homology test using BLAST-like tools for the candidate primers satisfying filtering constraints. Moreover, batch design of primers for multi-target qPCR is more complex due to the difficulty of finding a full set of primer pairs that satisfy the same stringent constraints.
There are other four important design features to consider: spanning exon–exon junction, avoiding single nucleotide polymorphism (SNP) sites, supporting input FASTA sequence, and supporting multi-target primer. First, it is useful to design primers across exon–exon junctions such that the designed forward and reverse primers hybridize to different exons which separated by a large intron or intron–exon region to avoid PCR amplification of gDNA (i.e. genomic DNA contamination) as well as to distinguish and quantify splice variants. Second, it is important for the designed primers not to overlie SNP sites because the existence of SNPs within the primers may influence the reaction of PCR experiments (i.e. primer Tm, efficiency of polymerase extension, and target specificity) (4). Third, supporting input FASTA sequence becomes more and more important as the next-generation sequencing (NGS) technology is widely used due to its high accuracy, fast speed and low cost. It can be useful to identify functions and to annotate of new genome sequences obtained by NGS. For example, when a fatal viral infection (e.g. Zika virus) is emerging, primer design for viral sequences with rigorous similarity testing against host sequences in a database can aid in detection and identification of those viruses (5). Fourth, supporting multi-target primer, i.e., designing a pair of primers covering specific multiple target sequences can be useful, in particular, when amplifying multigene family (e.g. olfactory receptors) or many variant genes (e.g. TP53) all at once (23). Multi-target design is different from batch design. The former searches for a pair of primers which amplifies all target sequences, whereas the latter for a set of pairs of primers each of which amplifies each target sequence under the same filtering constraints.
Many websites have been developed to aid in designing primers for qPCR, including Primer3Plus (6,7), BatchPrimer3 (8), Primique (9), QuantPrime (10), Primer-BLAST (11), Oli2go (12) and MRPrimerW (13). There are also many databases containing pre-computed primers, including PrimerBank (14,15) and qPrimerDB (16). Table 1 summarizes a comparison among those websites and databases. Primer3Plus, BatchPrimer3, Primique and QuantPrime focus on supporting user-specific filtering constraints and ranking primer pairs by their penalty scores. BatchPirmer3 and QuantPrime supports batch design of primers with avoiding SNP sites. Primique and QuantPrime perform homology test using a local alignment algorithm in a limited scope, while Primer-BLAST performs homology test using a global alignment algorithm against the entire sequences in a database. Primer-BLAST, however, supports neither batch designing nor multi-target designing. Oli2go supports design of multiplex primers and probes for multiple FASTA sequences. It, however, does not support some important features such as exon spanning, avoiding SNP sites, and ranking. Both PrimerBank and qPrimerDB focus on checking primer specificity. The primers in PrimerBank have been experimentally validated under a uniform condition. The primers in qPrimerDB for 147 livestock and plants have been checked using a thermodynamic-based program, MFPrimer-2.0 (17). Both, however, do not allow users to adjust the filtering constraints and not support the features such as scoring (ranking), TaqMan probes and avoiding SNPs.
Table 1.
Comparison among websites for primer design
| Method | Batch designing | Filtering constraints | Homology test | Scoring (ranking) | TaqMan probes | Exon spanning | Avoiding SNP | Input FASTA sequence | Multi-target designing |
|---|---|---|---|---|---|---|---|---|---|
| Primer3Plus | X | O | X | O | O | X | X | O | X |
| BatchPrimer3 | O | O | X | O | O | X | O | O | X |
| QuantPrime | O | O | △ | O | X | O | O | X | X |
| Primique | O | O | △ | O | X | X | X | O | X |
| Primer-BLAST | X | O | O | △ | O | O | O | O | X |
| Oli2go | O | O | O | X | O | X | X | O | O |
| PrimerBank | X | X | O | X | X | X | X | X | X |
| qPrimerDB | O | X | O | X | X | O | X | △ | X |
| MRPrimerW | O | O | O | O | O | X | X | X | X |
| MRPrimerW2 | O | O | O | O | O | O | O | O | O |
O: fully supported.
△: partially supported.
X: not supported.
MRPrimerW (13) is our previously proposed website which allows users to quickly search for the best valid primer pairs satisfying the same constraints for many target sequences. It extracted a complete set of 341 963 135 valid primers which can amplify only a specific sequence from the human and mouse CCDS databases (https://www.ncbi.nlm.nih.gov/CCDS/) in an exhaustive manner using the large-scale MapReduce program called MRPrimer (18), converted them to a set of indices with annotations, and loaded into the main memory of the website. Then, it performs online processing when users input a set of target sequence IDs and a multitude of filtering constraints as a query. However, MRPrimerW has the major drawbacks that it does not consider sequence variants and SNP sites, does not support other popular model organisms, and does not support designing valid primers for input FASTA sequences against database sequences. Such drawbacks are mainly due to a prohibitive amount of computation and memory space required for supporting the features.
Here, we describe a new website called MRPrimerW2 which solves all the drawbacks of MRPrimerW, but still maintains its advantages such as one-stop searching and high primer specificity. In this update, we changed the template sequence database from CCDS to NCBI Reference Sequence (RefSeq) database (http://www.ncbi.nlm.nih.gov/refseq/) to solve some drawbacks of MRPrimerW, in particular, to support exon spanning and avoiding SNP sites and to provide high-quality primers for other important organisms including human, mouse, rat, zebrafish, cow, pig, thale cress, fruit fly and Caenorhabditis elegans. The MapReduce-based offline processing part of the predecessor, MRPrimer (18), may require several tens of days for extracting valid primers from RefSeq databases in an exhaustive manner, since the size of RefSeq database is much larger than that of CCDS. To overcome the computational limitation, we significantly improved the performance of the offline processing part by exploiting GPU computation, in particular, by performing the most time-consuming step, i.e., homology test, using GPUs. We extracted a complete set of 3 509 244 680 valid primers from RefSeq databases within a day. The set of primers include not only single-target primers, but also multi-target ones. Since the size of the entire valid primers was too large to fit in main memory of the web site, we stored them in a disk-based key-value store, LevelDB (http://leveldb.org/) using high-speed PCIe SSD storage. MRPrimerW2 also supports primer design for input FASTA sequences against database sequences. To support fast ad-hoc homology test, we extracted all possible subsequences of 17–27 bp from RefSeq databases (the total number of subsequences is 13 657 773 610) and stored them in LevelDB. Thus, MRPrimerW2 uses a total of 17 167 018 290 primers or subsequences stored in LevelDB for online processing of primer design. As a result, MRPrimerW2 can design the best valid primer pairs for both database sequences and input FASTA sequences with the new features such as exon spanning, avoiding SNP sites and multi-target primer design.
MATERIALS AND METHODS
MRPrimerW2 provides a total of four search modes depending on the source of target sequences and the number of target sequences: single target in DB sequences, multiple targets in DB sequences, single target in FASTA sequences, and multiple targets in FASTA sequences. Figure 1 illustrates the input interface of MRPrimerW2. There are two tabs, ‘Primer design for DB sequences’ and ‘Primer design for FASTA sequences’. The former tab is for the search modes in DB sequences, and the latter tab for the search modes in FASTA sequences. In both tabs, the default search mode is the single-target mode. If users input multiple gene symbols/IDs or FASTA sequences in a certain tab, MRPrimerW2 performs batch design for them, i.e., searches primers for each target sequence under the same filtering constraints. If users want the multi-target mode, they can do it by checking the option ‘Designing primers amplifying all given sequences’. Batch design and multi-target design are incompatible with each other. Table 2 shows the range of off-target sequences used for homology test in each search mode.
Figure 1.

Input interface of MRPrimerW2. There are two primer design modes, primer design for DB sequences mode and primer design for FASTA sequence mode. MRPrimerW2 takes as input species, query type (NCBI gene symbol, GenBank accession, NCBI Gene ID, aliases, or keyword), and query (a set of target gene symbol/IDs or FASTA format sequences). Also, a user can input their email address to be sent a link of query results. There are five additional options: TaqMan probe design, exon spanning design, SNP avoid design, amplify multiple gene design, and design primers using suggested parameters. In the Advanced Settings, the user can adjust filtering constraints.
Table 2.
Off-targets in each search mode of MRPrimerW2
| Single target (including batch design) | Multiple targets | |
|---|---|---|
| DB sequences | Entire DB sequences except each single target | Entire DB sequences except the multiple targets |
| FASTA sequences | Entire DB sequences | Entire DB sequences |
MRPrimerW2 consists of two parts, offline processing (Figure 2A) and online processing (Figure 2B). Offline processing is independent of user queries and builds two kinds of DBs, DB-1 and DB-2, used for online processing. DB-1 is used for the tab ‘Primer design for DB sequences’ and DB-2 for the tab ‘Primer design for FASTA sequences’. Online processing designs the primers for a given query. It has two different pipelines for two different tabs.
Figure 2.

Overall flow of the MRPrimerW2. MRPrimerW2 consists of two parts, offline processing (A) and online processing (B). (A) Offline processing is independent of user queries and builds two kinds of DBs, ‘DB for valid primers and probes’ and ‘DB for subsequences’, used for online processing. DB for valid primers and probes is used for the tab ‘Primer design for DB sequences’ and DB for subsequences for the tab ‘Primer design for FASTA sequences’. (B) Online processing consists of six steps to design the primers for a given query. It has two different pipelines for two different tabs. In the tab ‘Primer design for DB sequences’, MRPrimerW2 retrieves all candidate primers for a given query and checked filtering constraints on the candidate primers. If a user checks the exon spanning option and/or SNP avoiding option, it evaluates the candidate primers with their exon and/or SNP locus information. In the tab ‘Primer design for FASTA sequences’, MRPrimerW2 extracts all possible subsequences from given FASTA sequence(s) as candidate primers and checks the default filtering constraints for the candidate primers. Then, it performs homology test of the candidate primers using DB for subsequences. Through pair filtering and sorting step, MRPrimerW2 returns the ranked primer pairs.
Offline processing
Offline processing by MRPrimerW2 takes as input a mRNA sequence database and several filtering constraints and returns all valid primers and probes for building DB-1 that satisfy both homology testing and basic filtering constraints. It performs a total of four steps. During Step 1, we extract all possible subsequences and build DB-2 using them. As a source of input mRNA sequence database, we used the NCBI RefSeq database for human, mouse, rat, zebrafish, cow, pig, thale cress, fruit fly, and C. elegans (Table 3). In this update, we selected the RefSeq database as template because the RefSeq database contains much more organisms than CCDS database and provides whole mRNA sequences including UTRs and exons which allows to design exon spanning primers. We adopted dbSNP (https://www.ncbi.nlm.nih.gov/snp, Pre-build 152, the last update was 21 December 2018) for avoiding SNP sites. The most up-to-date RefSeq databases contain 245 013 mRNA sequences for the nine species, where the prefix of GenBank accession number is NM (the last update was 21 November 2018 for human, and 7 November 2018 for others). The result of offline processing includes 3 509 244 680 valid primers, which cover ∼99.9% of genes in RefSeq. Only ∼0.1% of genes in RefSeq do not have any target gene-specific primers. The total size of current databases, DB-1 and DB2, is about 363GB, which will increase by adding new species later.
Table 3.
Statistics of MRPrimerW2 primers
| Total number of genes | Number of covered genes (%) | Number of valid primers* | Number of subsequences** | Size of database (MB) | |
|---|---|---|---|---|---|
| Human (Homo sapiens) | 51 979 | 51 976 (99.9%) | 863 155 888 | 4 026 671 584 | 69 860 |
| Mouse (Mus musculus) | 35 349 | 35 323 (99.9%) | 574 377 223 | 2 461 280 404 | 63 840 |
| Rat (Rattus norvegicus) | 17 639 | 17 637 (99.9%) | 223 541 821 | 910 454 326 | 39 215 |
| Zebrafish (Danio rerio) | 15 876 | 15 834 (99.7%) | 205 306 695 | 757 268 626 | 34 532 |
| Cow (Bos taurus) | 13 382 | 13 382 (100%) | 152 077 285 | 676 441 698 | 31 010 |
| Pig (Sus scrofa) | 4 180 | 4 180 (100%) | 43 265 923 | 169 276 012 | 9 915 |
| Thale cress (Arabidopsis thaliana) | 48 148 | 47 953 (99.6%) | 597 970 532 | 1 876 192 010 | 48 700 |
| Fruit fly (Drosophila melanogaster) | 30 480 | 30 471 (99.9%) | 532 662 088 | 1 918 507 800 | 38 926 |
| C. elegans (Caenorhabditis elegans) | 28 299 | 28 257 (99.9%) | 316 887 225 | 861 681 150 | 27 715 |
| Total | 245 332 | 245 013 (99.9%) | 3 509 244 680 | 13 657 773 610 | 363 713 |
*Valid primer refers to the primer passed the homology test.
**Subsequences are in the range of from 17 to 27 bp (in the ‘Offline’ column in Table 4).
Table 4 shows the list of filtering constraints used in offline and online processing. Four parameters (primer length, melting temperature, GC content, and contiguous residue) are applied during Step 2 of offline processing and most of constraints are checked during online processing which user can adjust in web interface. For TaqMan probes, we used different set of filtering constraints as described in previous publication (10). Homology test is done in Steps 3 and 4. The 5′ cross-hybridization filtering step (Step 3) eliminates the candidate primers that has only a few mismatches (up to four mismatches) at the 5′ end against off-target sequences. The general cross-hybridization filtering step (Step 4) eliminates the candidate primers that have only a few mismatches (up to two mismatches) anywhere against off-target sequences. This step is very time-consuming. We implemented the GPU kernel for general cross-hybridization filtering and run the kernel in a single computer with two Intel Xeon 10-core 2.20 GHz CPUs and eight Geforce GTX 1080ti GPUs. Because this step performs homology tests for every candidate primers and probes against the entire DB sequences in an exhaustive manner, the resultant primers and probes are all single-target gene-specific or multi-target gene-specific (i.e., transcript variants).
Table 4.
List of filtering constraints used in offline and online processing of MRPrimerW2 and qPrimerDB
| Parameter | Value range | |||
|---|---|---|---|---|
| Offline | Online (default)* | qPrimerDB(16) | ||
| each primer | Primer length | 17–27 bp | 19–23 bp | 18–28 bp** |
| melting temperature (TM) | 56–63°C | 58–62°C | 58–64°C** | |
| GC content | 30–70% | 40–60% | 40–60%** | |
| self-complementarity | - | <5-mer | <15-mer | |
| 3′ self-complementarity | - | <4-mer | <15-mer | |
| Contiguous residue | <5-mer | <6-mer | <6-mer | |
| Gibbs free energy (ΔG) | - | ≥-9 kcal/mol | ≥-11 kcal/mol** | |
| Hairpin | - | <3-mer | <9-mer | |
| Primer pair | length difference | - | ≤5-mer | ≤6-mer |
| TM difference | - | ≤3°C | ≤3°C** | |
| product size | - | 100–250 bp | 80–300 bp** | |
| pair-complementarity | - | <5-mer | <14-mer | |
| 3′ pair-complementarity | - | <4-mer | <11-mer | |
-indicates not applicable.
*The value ranges in this column indicate the default setting on the web server. Users can freely adjust these values.
**These values are specified in the qPrimerDB publication (16). The rest of values are calculated with the best primers for human suggested by qPrimerDB.
GPU computation
We performed from Step 1 to Step 3 in Figure 1A using MapReduce as MRPrimerW did, but performed Step 4 using GPUs since it was the most time-consuming step in offline processing. Step 4 checks each candidate primer passed Step 3 whether it occurs in any off-target sequences with only a few mismatches (up to two mismatches). In particular, Step 4 splits not only all candidate primers (output of Step 3) but also all possible subsequences (output of Step 1) into seeds and groups them by using seed+offset+|P| as a grouping key, where |P| means the length of primer (or subsequence) P, and offset the position where seed occurs in P. MRPrimerW (13) performs this grouping using MapReduce shuffle, whereas MRPrimerW2 using hashing in main memory. After grouping, we prepared four arrays for the output of Step 3, P3, P3_offset, SID3, SID3_offset (Supplementary Figure S1A) and four arrays for the output of Step 1, P1, P1_offset, SID1, SID1_offset (Supplementary Figure S1B) in main memory and copied the eight arrays to GPU device memory. Then, we called the GPU kernel with hundreds of thousands GPU threads per GPU where a single thread performs homology test for a single group. Supplementary Figure S1 illustrates two groups of A+0+3 (blue color) and T+1+3 (red color), and so two GPU threads are used. For human RefSeq, a total of 3,309,154 groups are processed when mismatch = 1. To reduce the processing time, we distributed the groups into eight GPUs. We also prepared the output array in GPU memory, which has the same length with P3 array. If a candidate primer in P3 is not similar with any subsequences in the same group of P1 array, the corresponding value in the output array becomes 0 (pass). Otherwise, it becomes 1 (fail). After executing the GPU kernel completely, we copied the output array from GPU memory to main memory.
Building indices
We built two databases using the results of offline processing. Due to the large size of databases, we stored them in a disk-based key-value store LevelDB in this update, while we stored a database in an in-memory database Redis in the predecessor, MRPrimerW. To improve I/O speed to the databases, we used Fusion-io's PCIe SSD storage of 5.2TB for LevelDB. Supplementary Figure S2 illustrates the set of indices stored in the databases.
DB-1 contains a total of six indices (Supplementary Figure S2A–F): annotation index, valid primer index, probe index, metadata index, top-1 primer pair index and SNP locus index (for human only). Annotation data was downloaded from RefSeq ftp (ftp://ftp.ncbi.nlm.nih.gov/refseq/). Although the schemas of all indices except SNP locus index are the same with those of the predecessor, the contents of the indices are quite different from those of the predecessor. For example, the indices of MRPrimerW2 contains the primers for not only single-target design, but also multi-target design. SNP locus data was downloaded from dbSNP ftp (ftp://ftp.ncbi.nih.gov/snp/). Since SNP occurs very frequently, it is difficult to design primers avoiding all SNP sites. Thus, we used the SNP locus data which has the heterozygosity greater than 0 (i.e. frequent SNP occurrence).
DB-2 contains a total of three indices (Supplementary Figure S2G and H), where there are two indices having the same schema (Supplementary Figure S2H). These indices are used to perform homology test for the candidate primers extracted from input FASTA sequences. The first index (Supplementary Figure S2G) is for the 5′ cross-hybridization filtering step (Step 3 in Figure 2B) where the key contains the suffixes of all possible subsequences by removing a prefix of length four. The second and third indices (Supplementary Figure S2H) are for general cross-hybridization filtering step (Step 4 in Figure 2B). In particular, the second index is for a single mismatch, and the third one for two mismatches. The key of these indices contains a set of seeds obtained by splitting each subsequence. A subsequence of length m with at most k mismatches must contain a seed exactly matched of at least 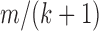 residues (19–21). The value of the indices contains a set of left flank and right flank of each seed within the corresponding subsequence. We illustrate an example of homology test using the indices in Supplementary Figure S3.
residues (19–21). The value of the indices contains a set of left flank and right flank of each seed within the corresponding subsequence. We illustrate an example of homology test using the indices in Supplementary Figure S3.
Online processing
Online processing consists of a total of six steps (Figure 2B). In the tab ‘Primer design for DB sequences’, Step 2 retrieves all candidate primers for a given query using the six indices (Supplementary Figure S2A–F), and the remaining Steps 3–6 are performed on the candidate primers. If a user checks the exon spanning option and/or SNP avoiding option, Step 4 evaluates the candidate primers with their exon and/or SNP locus information. Steps 5–6 are basically the same with the corresponding steps of the predecessor. We, however, reimplemented those steps in a more efficient form to process a much larger number of candidate primers compared with the predecessor still quickly.
In the tab ‘Primer design for FASTA sequences’, Step 1 extracts all possible subsequences of length 19–23 bp from given FASTA sequence(s) as candidate primers. Step 2 checks the default filtering constraints in Table 4 or the constraints adjusted by users for the candidate primers. Steps 3–4 perform homology test of the candidate primers using the three indices (Supplementary Figure S2G and H), that is, homology test against all DB sequences.
Comparative analysis
To compare the specificity and efficiency of the primers designed by MRPrimerW2 with the recent primer database, qPrimerDB, we collected 124,596 thermodynamic-based best primer pairs for six organisms (human: 17 632, mouse: 20 080, rat: 21 530, zebrafish: 25 052, cow: 19 970 and pig: 20 332) from qPrimerDB (https://biodb.swu.edu.cn/qprimerdb/download-primers). For efficiency test, we evaluated the filtering constraints of the collected 17 632 primer pairs for human (‘qPrimerDB’ column in Table 4). They specified only four filtering constraints for each primer and two filtering constraints for primer pairs in their publication (16). Comparing with the default constraints of MRPrimerW2, some of the qPrimerDB primer pairs only satisfy much looser filtering constraints especially in terms of self-complementarity, hairpin and pair-complementarity as in in Table 4. In addition, we checked the specificity of qPrimerDB primer pairs using sequence similarity searching based on RefSeq database. Since qPrimerDB uses the Ensembl database, we cannot evaluate 54% of the best primer pairs which do not exist in the RefSeq database. Figure 3 shows the results of the specificity of the remaining 46% of qPrimerDB primer pairs (human: 13 035 primer pairs). For human, 69% are target-specific, but 28% are non-specific (i.e. may amplify multiple off-targets) and the rest 3% do not amplify, which means each of forward and reverse primer is target-specific, but both are located in different genes so that the pairs may not amplify the target. In total, about 71% of the best primer pairs are target-specific, 27% are non-specific, and 1% of the pairs do not amplify.
Figure 3.
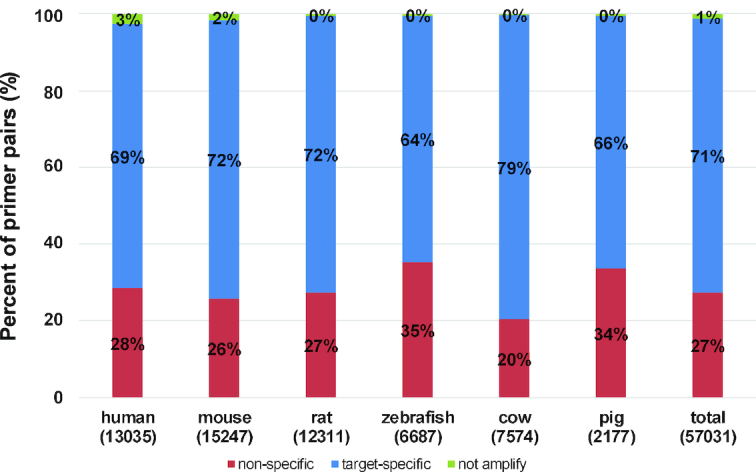
Specificity analysis of qPrimerDB. The distribution of specificity of the collected 124 596 qPrimerDB best primer pairs for six organisms (human, mouse, rat, zebrafish, cow and pig) where the x-axis represents species and the number of primer pairs existed in RefSeq database for each species in parentheses, and the y-axis represents the percent of primer pairs.
We also describe the difference between Oli2go (12) and our MRPrimerW2 in terms of multi-target designing of primers. Oli2go is a multiplex oligonucleotide design tool and so returns multiplex primers to users for given multiple target sequences. It depends on using external tools such as BLAST and BWA (22) for homology test and also requires an additional external tool such as MFEprimer (17) for cross dimer check among multiplex primers, i.e., a set of primer pairs covering the target sequences. Multiplex primer design is a well-known NP-complete problem, and so, the resulting primers may not be target-specific enough. In addition, since Oli2go needs to repeat a series of external tools until finding the feasible and valid primers, its run time may be relatively slow. On the contrary, MRPrimerW2 returns a single primer pair instead of multiplex primers for given multiple target sequences. Since MRPrimerW2 calculates all possible primers for all possible combinations of DB sequences and stores them in the indices, it can find a single primer pair even for multiple targets if it exists, and its run time is inherently faster than Oli2go that uses several external tools repeatedly.
WEB SERVER
The MRPrimerW2 web server is connected to LevelDB (http://leveldb.org/) via PHP-LevelDB (https://github.com/reeze/php-leveldb), which is used for communication between LevelDB and PHP. The web server is also communicated with user's web browser via AJAX (Asynchronous JavaScript and XML). In the web browser, HTML with CSS and bootstrap (http://getbootstrap.com/) are used for generating web pages. JavaScript and jQuery are also used for dynamic HTML behaviour. MRPrimerW2 supports major web browsers including Microsoft Internet Explorer, Google Chrome, Mozilla Firefox, Apple Safari, and Opera. MRPrimerW2 provides six example queries corresponding four search modes and two new features.
Figure 4A illustrates the result of the Example1 query for seven target genes (SAMD11, TNF, IL10, TP53, USH2A, A1CF and HES4 in gene symbols), where species is human, and search type is GenBank accession number. A total of three options, TaqMan probe, exon spanning, and avoiding SNP, are selected. In the result, three of target genes, TNF, IL10 and USH2A, have valid primer pairs which are supposed to amplify each target gene, are spanned exons, and do not overlie SNP sites. One target gene TP53 has primer pairs that amplify the variants of the target gene. The remaining three target genes have no valid primer pairs satisfying the given filtering constraints and options. Specifically, MRPrimerW2 suggests turning off the option of avoiding SNP for the target gene SAMD11, and relaxing the filtering constraints for two target genes, A1CF and HES4, to search for valid primer pairs. Figure 4B shows the top-1 primer pair of the Example4 query for three input FASTA sequences, which are some variations of Influenza A virus (H1N1) nucleocapsid protein (NP) genes. Here, the host species is human, and the option of multi-target design is turned on. The resulting primer pairs are supposed to amplify all the input FASTA sequences at once without amplifying any host sequences. Figure 4C shows the top-1 primer pair of the Example 7 query for the target gene A1CF with turning on the option using suggested parameters. A1CF originally has no result in the Example 1 query due to too strict filtering constraints (i.e., parameters). Using the option, however, adjusts the parameters automatically and returns the valid primer pairs if they exist in DB.
Figure 4.

Output interface of MRPrimerW2. Example result for the tab ‘Primer design for DB sequences’ (A) and (C), and for the tab ‘Primer design for FASTA sequences’ (B). (A) In the tab ‘Primer design for DB sequences’, MRPrimerW2 displays three types of result tables. A user can select the number of the result primer pairs with Top-1, Top-10, and Top-50 option (default is Top-1). Also, the user can download the primer sequences and user-defined filtering constraints using ‘Download data in JSON format’ button on the top of the tables. The first table shows the feasible and valid primer pairs of each target gene. As the user selected the TaqMan probe design option, the table also shows the sequence and position of the probe. If there is no target-specific primer, MRPrimerW2 returns multi-target specific primers. The input query that does not have any valid primers are shown in the third table suggesting values for relaxing conditions and options. (B) In the tab ‘Primer design for FASTA sequences’, MRPrimerW2 displays one result table. The table shows the feasible and valid primer pairs of input target sequence(s). (C) If the user selects the option of designing primers using adjusted parameters, MRPrimerW2 adjusts both the filtering constraints and options such as avoiding SNP automatically so as to find the feasible primer pairs and returns them with additional information about how to change given parameters to new ones.
CONCLUSIONS
Previously we launched the MRPrimerW web server, a tool for designing high-quality primer pairs for multiple target qPCR experiments. It allows users to quickly search for the best valid primer pairs satisfying the same constraints for many target sequences (i.e. batch design). However, it has several major drawbacks that do not consider sequence variants and SNP sites, not support other popular model organisms, not support designing valid primers for new input FASTA sequences against database sequences, and not support designing multi-target primers. In this update, we developed MRPrimerW2 which solves all the drawbacks of the MRPrimerW, but still maintains the advantages of the predecessor such as one-stop searching considering both filtering constraints and homology test and high primer specificity of primers. MRPrimerW2 provides a total of four search modes, while the predecessor only a single search mode among them. To achieve this, we extracted a complete set of 3 509 244 680 valid primers and 13 657 773 610 subsequences from the human, mouse, rat, zebrafish, cow, pig, thale cress, fruit fly, and C. elegans RefSeq databases still in an exhaustive manner. The predecessor cannot extract and utilize such a huge number of primers or subsequences mainly due to a prohibitive amount of computation and memory space required. In MRPrimerW2, we significantly improved the computational performance by exploiting GPUs and also extended the memory space for storing databases by exploiting a disk-based key-value store, LevelDB, using PCIe SSD storage. We believe that MRPrimerW2 will contribute to further increasing the efficiency and specificity of qPCR experiments through supporting input FASTA sequences, supporting multi-target primer design, exon spanning, and avoiding SNP sites.
We consider the follow two directions as future work. The first direction is extending MRPrimerW2 so as to handle more complex cases such as virus family identification and cancer variants amplification. It is usually very difficult to design primers and probes for detecting all variants of a virus or an oncogene at once. In particular, for detecting cancer variants, we need to consider various types of cancer mutations such as SNV, insertion, deletions, and translocations. MRPrimerW2 can be extended to solve the problem by adding a high-level layer on top of the current version and integrating additional DBs such as cancer mutation DBs into primer DBs. The second direction is expanding MRPrimerW2 by integrating it with larger platforms. For doing that, we consider providing the codes that other platforms can easily access and utilizes our DBs as UCSC Genome Browser or Ensembl DB does.
Supplementary Material
Notes
Present address: Min-Soo Kim, Department of Information and Communication Engineering, DGIST, 333, Techno jungang-daero, Hyeonpung-eup, Dalseong-gun, Daegu 42988, Republic of Korea.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) [2018R1A5A1060031]; Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT and Future Planning [2017R1E1A1A01077630]; DGIST R&D Program of the Ministry of Science and ICT [18-BD-0404]; KBRI basic research program through Korea Brain Research Institute funded by Ministry of Science and ICT [19-BR-01-07]. Funding for open access charge: National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) [2018R1A5A1060031].
Conflict of interest statement. None declared.
REFERENCES
- 1. Holst-Jensen A., Rønning S.B., Løvseth A., Berdal K.G.. PCR technology for screening and quantification of genetically modified organisms (GMOs). Anal. Bioanal. Chem. 2003; 375:985–993. [DOI] [PubMed] [Google Scholar]
- 2. Klein D. Quantification using real-time PCR technology: applications and limitations. Trends Mol. Med. 2002; 8:257–260. [DOI] [PubMed] [Google Scholar]
- 3. Wang X., Spandidos A., Wang H., Seed B.. PrimerBank: a PCR primer database for quantitative gene expression analysis, 2012 update. Nucleic Acids Res. 2011; 40:D1144–D1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lefever S., Pattyn F., Hellemans J., Vandesompele J.. Single-nucleotide polymorphisms and other mismatches reduce performance of quantitative PCR assays. Clin. Chem. 2013; 59:1470. [DOI] [PubMed] [Google Scholar]
- 5. Chotiwan N., Brewster C.D., Magalhaes T., Weger-Lucarelli J., Duggal N.K., Rückert C., Nguyen C., Garcia Luna S.M., Fauver J.R., Andre B. et al.. Rapid and specific detection of Asian- and African-lineage Zika viruses. Sci. Transl. Med. 2017; 9:eaag0538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Untergasser A., Nijveen H., Rao X., Bisseling T., Geurts R., Leunissen J.A.M.. Primer3Plus, an enhanced web interface to Primer3. Nucleic Acids Res. 2007; 35:W71–W74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Untergasser A., Cutcutache I., Koressaar T., Ye J., Faircloth B.C., Remm M., Rozen S.G.. Primer3—new capabilities and interfaces. Nucleic Acids Res. 2012; 40:e115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. You F.M., Huo N., Gu Y., Luo M., Ma Y., Hane D., Lazo G.R., Dvorak J., Anderson O.D.. BatchPrimer3: a high throughput web application for PCR and sequencing primer design. BMC Bioinformatics. 2008; 9:253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Fredslund J., Lange M.. Primique: automatic design of specific PCR primers for each sequence in a family. BMC Bioinformatics. 2007; 8:369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Arvidsson S., Kwasniewski M., Riano-Pachon D.M., Mueller-Roeber B.. QuantPrime - a flexible tool for reliable high-throughput primer design for quantitative PCR. BMC Bioinformatics. 2008; 9:465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ye J., Coulouris G., Zaretskaya I., Cutcutache I., Rozen S., Madden T.L.. Primer-BLAST: a tool to design target-specific primers for polymerase chain reaction. BMC Bioinformatics. 2012; 13:134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hendling M., Wolff N., Conzemius R., Pabinger S., Barišić I., Peters K.. Oli2go: an automated multiplex oligonucleotide design tool. Nucleic Acids Res. 2018; 46:W252–W256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kim H., Kang N., An K., Koo J., Kim M.-S.. MRPrimerW: a tool for rapid design of valid high-quality primers for multiple target qPCR experiments. Nucleic Acids Res. 2016; 44:W259–W266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cao H., Shockey J.M.. Comparison of TaqMan and SYBR green qPCR methods for quantitative gene expression in tung tree tissues. J. Agric. Food Chem. 2012; 60:12296–12303. [DOI] [PubMed] [Google Scholar]
- 15. Spandidos A., Wang X., Wang H., Seed B.. PrimerBank: a resource of human and mouse PCR primer pairs for gene expression detection and quantification. Nucleic Acids Res. 2009; 38:D792–D799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lu K., Li T., He J., Chang W., Zhang R., Liu M., Yu M., Fan M., Ma J., Sun W. et al.. qPrimerDB: a thermodynamics-based gene-specific qPCR primer database for 147 organisms. Nucleic Acids Res. 2017; 46:D1229–D1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Qu W., Zhou Y., Zhang Y., Lu Y., Wang X., Zhao D., Yang Y., Zhang C.. MFEprimer-2.0: a fast thermodynamics-based program for checking PCR primer specificity. Nucleic Acids Res. 2012; 40:W205–W208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kim H., Kang N., Chon K.W., Kim S., Lee N., Koo J., Kim M.S.. MRPrimer: a MapReduce-based method for the thorough design of valid and ranked primers for PCR. Nucleic Acids Res. 2015; 43:e130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Baeza-Yates R.A., Perleberg C.H.. Fast and practical approximate string matching. Inform. Process. Lett. 1996; 59:21–27. [Google Scholar]
- 20. Kim M.-S., Whang K.-Y., Lee J.-G.. n-Gram/2L-approximation: a two-level n-Gram inverted index structure for approximate string matching. Comput. Syst. Eng. 2007; 22:26–40. [Google Scholar]
- 21. Kim M.-S., Whang K.-Y., Lee J.-G., Lee M.-J.. n-Gram/2L: a space and time efficient two-level n-Gram inverted index structure. Proceedings of the 31st International Conference on Very Large Data Bases. 2005; 325–336. [Google Scholar]
- 22. Li H., Durbin R.. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics. 2009; 25:1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Srivastava G.P., Hanumappa M., Kushwaha G., Nguyen H.T., Xu D.. Homolog-specific PCR primer design for profiling splice variants. Nucleic Acids Res. 2011; 39:e69. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.