


Abstract
The study of contact residues and interfacial waters of antibody–antigen (Ab-Ag) structures could help in understanding the principles of antibody–antigen interactions as well as provide guidance for designing antibodies with improved affinities. Given the rapid pace with which new antibody–antigen structures are deposited in the protein databank (PDB), it is crucial to have computational tools to analyze contact residues and interfacial waters, and investigate them at different levels. In this study, we have developed AppA, a web server that can be used to analyze and compare 3D structures of contact residues and interfacial waters of antibody–antigen complexes. To the best of our knowledge, this is the first web server for antibody–antigen structures equipped with the capability for dissecting the contributions of interfacial water molecules, hydrogen bonds, hydrophobic interactions, van der Waals interactions and ionic interactions at the antibody–antigen interface, and for comparing the structures and conformations of contact residues. Various examples showcase the utility of AppA for such analyses and comparisons that could help in the understanding of antibody–antigen interactions and suggest mutations of contact residues to improve affinities of antibodies. The AppA web server is freely accessible at http://mspc.bii.a-star.edu.sg/minhn/appa.html.
INTRODUCTION
Antibodies with their ability to recognize an almost infinite array of protein antigens are of great interest as therapeutics. Understanding the underlying principles and mechanisms of antibody–antigen interactions are necessary for antibody engineering to facilitate the development of antibody-based therapeutics. Computational methods have been used as important tools for engineering antibodies and optimizing their affinities for antigens (1–3). A major effort towards developing these computational methods has been focused on understanding the physico-chemical characteristics of the interacting regions of antibody–antigen structures (paratopes and epitopes). However, these computational methods have examined the available datasets which have been small, consisting of only 111, 53 and 107 antibody–antigen complexes in the studies of Peng et al., Kringelum et al. and Ramaraj et al. (2,4,5), respectively. With the increasing number of antibody–antigen complexes (2135 structures deposited in the PDB as of 22 April 2019), it is essential to develop tools/web servers to efficiently analyze interfacial/contact residues for all these structures. In this study, we present the development of a new web server, called AppA, to provide a user-friendly interface to dissect the contributions of hydrogen bonds, van der Waals interactions, hydrophobic interactions, ionic interactions, and interfacial waters at the antibody–antigen interfaces, as well as compare the 3D structures of contact residues for all the antibody–antigen structures available in the PDB and the models deposited. AppA has been designed such that it will automatically identify contact residues and interfacial waters, and calculate the hydrogen bonds, van der Waals interactions, hydrophobic interactions, ionic interactions at the interfaces of any new antibody–antigen structures that are deposited in the PDB every week.
Previous studies have shown that interfacial waters mediate interactions at the interfaces of molecules that do not have optimal shape complementarity (6). The importance of interfacial water mediated hydrogen bonds in the interactions between HyHEL-10 Fv antibody and HEL (hen egg white lysozyme) was demonstrated in the study of Yokota et al. (7). We have previously demonstrated the apparently critical role of interfacial waters in mediating the interactions of antibodies and antigens (8). In this study, AppA identifies and details the hydrogen bonds made between the interfacial waters and the contact residues for all the antibody–antigen structures available in the PDB.
Furthermore, compared to the other two popular web servers for antibody structures, SAbDab (9) and PyIgClassify (10) that only highlight information and classification for antibody CDR (Complementarity Determining Region) conformations, AppA provides analysis and comparison of structures/conformations of contact residues not only in the antibody CDR but also in framework regions, and provides a comprehensive analysis of interfacial waters. Such analyses could suggest mutations of contact residues of antibodies to improve affinity, especially since antibodies are currently the fastest growing class of therapeutics (3).
We demonstrate the utility of our web server through various examples that highlight the capabilities of AppA for analyzing characteristics of contact residues and interfacial waters of antibody–antigen structures, and comparing/superimposing their 3D structures. The results from AppA could make contributions in understanding how an antibody interacts with an antigen, and give insights into binding specificities of paratopes and epitopes.
IMPLEMENTATION
Program overview
In our study, the contact residues are identified as residues of an antibody (antigen) structure whose solvent accessible surface areas (ASA) changes upon the formation of its antibody–antigen complex and they are within 6 Å of the complexed epitope (paratope) (8). The algorithm of Richmond and Richards is used for calculating solvent accessible surface areas (11). A contact residue is buried if its side chain solvent accessibility <8%, intermediate if its side chain solvent accessibility is between 8% and 30%, or exposed otherwise (12). The DSSP algorithm is used to assign secondary structures of contact residues (13). The Chothia, Kabat, IMGT and Martin annotated sequences are used to identify CDR regions of contact residues of antibodies (14). Hydrogen bonds, van der Waals interactions, hydrophobic interactions, and ionic interactions of contact residues and interfacial water molecules are identified by using the computational methods outlined in our previous study (8). The detailed definitions of contact residues and hydrogen bonds, van der Waals interactions, hydrophobic interactions, and ionic interactions of contact residues and interfacial water molecules are provided in Supplementary.
AppA compares the 3D structures of contact residues by optimizing the CLICK algorithm (12). CLICK has been extensively benchmarked and compared to other popular methods for protein structural alignments (15,16) as well as for the comparison of binding sites of biological macromolecules (12,17–21). In our study, a pair of 3D structures A and B of contact residues is superimposed by matching cliques based on the superimposition of their Cartesian coordinates with 3D least squares fitting. Here, cliques are optimal groupings of representative Cα atoms of contact residues within a certain spatial proximity (10 Å). Clique matching identifies equivalent residues in the two structures A and B. Using these equivalences, a final 3D least squares fit is performed to superimpose A and B. Since it is possible to generate multiple superimpositions, the chosen superimposition is the one that maximizes structure overlap (12). The CLICK program also produces a Z-score to assess the reliability of comparing 3D structures of contact residues, and we had previously established that a score of 2.0 and above was indicative of a significant comparison (22).
Server description
Input
Input antibody–antigen structures can be submitted by specifying the four-letter code for an existing antibody–antigen structure deposited in the PDB, or by uploading a 3D model/structure in the PDB format.
In addition to the four-letter code, the user can specify antibody (heavy and/or light chain) and antigen chain for the query structure. The detailed chain names for all the antibody–antigen structures available in the PDB can be found at (http://mspc.bii.a-star.edu.sg/minhn/Ab_Ag_chains.html). The list of antibody–antigen structures is updated every week from the PDB.
The detailed explanation of input antibody–antigen structures for analysis and comparison is provided in the help page of AppA (http://mspc.bii.a-star.edu.sg/minhn/help_appa.html).
Output
A 3D rendition of the query structure with the associated antibody, antigen chains and interfacial water molecules is displayed using JSMol (http://www.jmol.org/) (Figures 1 and 2). The contact residues of antibody and antigen are highlighted in sticks. The list and number of contact residues, and the number of hydrogen bonds, van der Waals interactions, hydrophobic interactions, and ionic interactions and interfacial waters that each contact residue is involved in, are displayed in the output tables for antibody and antigen (Figures S2A, B and C in Supplementary). The table of interfacial water molecules and their hydrogen bonds with contact residues of antibody and antigen is also shown (Figure S2D in Supplementary).
Figure 1.

AppA displays the 3D rendition of the heavy chain of a therapeutic monoclonal antibody (blue color) with the associated antigen of human interferon alpha-2A (salmon color) (PDB code: 4YPG) using JSMol (http://www.jmol.org/). The contact residues of antibody and antigen are highlighted in sticks. As seen, the structure (conformations) of the antigen of human interferon alpha-2A is mainly composed of alpha-helices.
Figure 2.

The 3D rendition of the heavy chain of antibody drug Avastin (blue color) with antigen VEGF (salmon color) (PDB code: 1BJ1) and interfacial waters (red balls) using AppA. The contact residues of heavy chain of Avastin and VEGF are highlighted in sticks. As seen, the structure (conformations) of the antigen VEGF is mainly composed of beta-sheets.
The detailed visualizations of contact atoms and interfacial waters are displayed by using the ‘Highlight Contact Residues’ and ‘Zoom In’ functions of AppA (Figures 5 and 6). For each contact residue and interfacial water, their detailed hydrogen bonds, van der Waals interactions, hydrophobic interactions and ionic interactions are shown when users click on any contact residue and interfacial water in their output tables (Figure 6, and Figure S4E in Supplementary). In addition to these output tables, the detailed interactions of contact residues/atoms and the structures of contact residues are downloadable in text format and in PDB format, respectively. The hydrogen bonds made by the interfacial waters with contact residues, and the structures of interfacial waters are also downloadable. In addition, we have used Chothia, Kabat, IMGT and Martin annotated sequences to identify CDR regions of contact residues, and provided this comprehensive information as well as ASA values in the output files for analyzing contact residues in the CSV format (Figure S6 in Supplementary).
Figure 5.

Interfacial water molecules (red balls) are observed at the interface of the heavy chain of antibody drug Lucentis and the antigen VEGF (PDB code: 1CZ8) using AppA. Two interfacial waters (HOH457 and HOH202) bridge the sidechain of Asn52 of Avastin and the main chain of His86 of VEGF.
Figure 6.

The detailed visualization of interfacial waters and contact atoms is displayed using the ‘Highlight Contact Residues’ and ‘Zoom In’ functions of AppA. As seen, the atom O of interfacial water HOH202 makes a hydrogen bond with the main chain atom N of His86 of antigen VEGF, and another hydrogen bond with the atom O of interfacial water HOH457. The atom O of HOH457 makes another hydrogen bond with the sidechain atom ND2 of Asn52 of Lucentis.
For comparison of a pair of 3D structures A and B of contact residues between antibody and antigen, the 3D rendition of the structural superimposition of A and B and their interfacial water molecules is displayed using JSMol (Figure 3, and Figures S3F, S5C, S5D in Supplementary). Statistics relevant to the comparison including structure overlap, RMSD (root mean square deviation), Z-score, number of contact residues of structures A and B, the matched contact residues, and the number of identical matched residues between A and B are listed in a table (Figure 3). The table of matched contact residues is also displayed, and the identical matched residues are highlighted in red (Figure 4). The comparison is downloadable in text format that shows one matched contact residue between structures A and B per line. The coordinates of the superimposed structures A and B are downloadable in the PDB format. AppA also provides detailed information of mismatched residues for comparing 3D structures of contact residues. The information relating to mismatched contact residues are displayed when users click on the ‘Mismatched contact residues’ function in the output of AppA for comparison of contact residues (Figure S1C in Supplementary).
Figure 3.
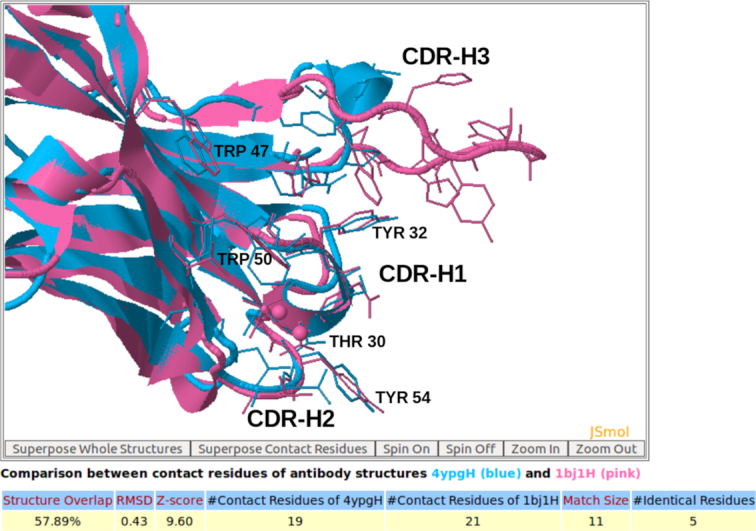
The 3D rendition of the structural superimposition of contact residues of the heavy chain of the therapeutic monoclonal antibody (4YPG) and Avastin (1BJ1) with their antigens from human interferon alpha-2A and VEGF using AppA. As seen, contact residues Thr30, Tyr32 in the CDR-H1 region, Trp47 and Trp50 in the framework region, and Tyr54 in CDR-H2 of 4YPG and Avastin are conserved and occupy identical spatial locations.
Figure 4.

AppA displays the matched contact residues of the heavy chain of the therapeutic monoclonal antibody (4YPG) and Avastin (1BJ1) for structure comparison. The identical matched residues are highlighted in red color.
Furthermore, AppA has useful features to browse existing antibody–antigen structures from the PDB using different cutoffs of number of hydrogen bonds, van der Waals interactions, hydrophobic interactions, ionic interactions, interfacial waters, and contact residues (Figure S7 in Supplementary). It also has a toggle option to show the full superimposed antibody/antigen structures for comparing the 3D structures of contact residues (Figure 3).
The detailed examples of output for analysis and comparison of antibody–antigen structures are also provided in the help page of AppA (http://mspc.bii.a-star.edu.sg/minhn/help_appa.html).
RESULTS
Case study 1: Analysis and comparison of contact residues of antibody drug Avastin and a therapeutic monoclonal antibody
In this example, we analyze and compare contact residues of the heavy chain of a therapeutic monoclonal antibody bound to its antigen, the human interferon alpha-2A (PDB code: 4YPG) and those of Avastin (generic name: bevacizumab, PDB code: 1BJ1) with antigen VEGF (Human Vascular Endothelial Growth Factor) (23,24). Avastin approved by US Food and Drug Administration (FDA) in 2004, 2006, 2008, 2009 is currently widely used for the treatment of various cancers.
As seen in Figures 1 and 2, the structures (conformations) of the antigens of human interferon alpha-2A (alpha-helices) and VEGF (beta-sheets) are completely different. Interestingly, the superimposition of contact residues of the heavy chains of 4YPG and Avastin indicates that five residues, Thr30 and Tyr32 in CDR-H1, Trp47 and Trp50 in the framework region, and Tyr54 in CDR-H2 are not only conserved between the two antibodies, but also occupy identical spatial locations (Figures 3 and 4). This suggests that although CDR-H1 and CDR-H2 contain fewer number of contact residues than CDR-H3 (Figures S1A and S1B in Supplementary), the contact residues of CDR-H1 and CDR-H2 and their conformations are more conserved (Figures 3 and 4). In addition, Figure S1C in Supplementary displaying the mismatched contact residues of heavy chains of 4YPG and Avastin also identifies that the CDR-H3 regions have the most number of mismatched contact residues. These analyses also begin to shed light on how antibody paratopes with relatively little structural variations could recognize antigens with immense structural diversity.
Case study 2: Analysis of contact residues and interfacial waters of antibody drug Lucentis and antigen VEGF
Lucentis (generic name: ranibizumab, PDB code: 1CZ8) was approved by FDA in 2006 for the treatment of age-related wet macular degeneration (25). As seen in Figures S2A and S2B in Supplementary, 26 contact residues of heavy and light chains of Lucentis are involved in making 15 hydrogen bonds, 198 van der Waals interactions, 75 hydrophobic interactions, 0 ionic interaction with antigen VEGF and 4 hydrogen bonds with interfacial waters. The heavy chain of Lucentis makes significantly higher number of hydrogen bonds, van der Waals interactions, hydrophobic interactions, and ionic interactions with VEGF compared to the light chain (Figures S2A and S2B in Supplementary). These numbers indicate that the heavy chain of Lucentis contributes substantially to the interactions with VEGF.
In addition, interfacial water networks are observed at the interface of Lucentis and VEGF (Figure S2D in Supplementary Materials). Two interfacial water molecules (HOH457 and HOH202) bridge the sidechain of Asn52 in the heavy chain of Lucentis and the main chain of His86 of antigen VEGF (Figures 5 and 6). As seen in Figure 6, the atom O of interfacial water HOH202 makes a hydrogen bond with the main chain atom N of His86 of antigen VEGF, and another hydrogen bond with the atom O of interfacial water HOH457. The atom O of HOH457 also contributes another hydrogen bond with the sidechain atom ND2 of Asn52 of Lucentis (Figure 6). This example suggests that the interfacial water molecules mediate interactions between Lucentis and antigen VEGF.
Case study 3: Analysis and comparison of contact residues and interfacial waters of antibody drugs Avastin and Lucentis with antigen VEGF
From the analysis of AppA, 26 contact residues of heavy and light chains of Avastin are involved in making 10 hydrogen bonds, 139 van der Waals interactions, 60 hydrophobic interactions, 2 ionic interaction with antigen VEGF and 3 hydrogen bonds with interfacial waters (Figures S3A and S3B in Supplementary) that are much smaller in number than those made by Lucentis with VEGF (as shown in the case study 2). These analyses agree well with the binding experiments that have demonstrated that the affinity of Lucentis for VEGF is higher than that of Avastin for VEGF (25).
From Figure S3A in Supplementary displaying the contact residues of the heavy chain of Avastin, it appears that the residues Thr30, Asn31, Thr55, His101, Tyr103, Ser105 and His107 could be mutated to improve affinity as these residues are located at coil (loop) and exposed regions, do not make hydrogen bond interactions and are involved in small number of van der Waals, hydrophobic and ionic interactions with VEGF, compared to the other contact residues. Indeed, Lucentis contains Asn31His, His101Tyr and Ser105Thr and has been shown to have improved affinity (25). Comparison of the contact residues of Lucentis and Avastin (Figures S3F and S3G in Supplementary) shows that the sidechains of contact residues His31 and Tyr101 of Lucentis are closer to VEGF than Asn31 and His101 of Avastin, resulting in an increased number of interactions between Lucentis and VEGF.
Furthermore, four interfacial water molecules (HOH421, HOH116 of Avastin and HOH457, HOH202 of Lucentis) are seen to bridge the heavy chains of both antibodies to VEGF (Figures 5, 6 and Figure S3E in Supplementary). Superimposition of the contact residues of Avastin and Lucentis indicates that these waters are in identical spatial locations (Figure S3F in Supplementary) and hence likely make important contributions to the interactions of both Avastin and Lucentis with VEGF.
Case study 4: Analysis of contact residues of 3D model of antibody drug Avastin and antigen VEGF
In this example, AppA shows the analysis of contact residues for the 3D model of antibody drug Avastin (Figure S4A in Supplementary). This 3D model was built by using MODELLER (26) with Ser at position 100B of the heavy chain of Avastin (PDB code: 1BJ1) mutated to Ala.
As seen in Figures S4B and S4C in Supplementary, 26 contact residues of heavy chain and light chain of this 3D model are involved in making 6 hydrogen bonds, 135 van der Waals interactions, 57 hydrophobic interactions, and 4 ionic interaction with VEGF that are lower in number than those of Avastin with VEGF (as shown in the case study 3). This analysis agrees well with the binding experiments that have demonstrated that the affinity of Avastin with the mutation Ser_100B_Ala for VEGF is lower than that of Avastin for VEGF (24).
Case study 5: Analysis and comparison of contact residues of antibody drugs Pertuzumab (Perjeta) and Trastuzumab (Herceptin) with the antigen HER2
In this example, AppA has been used to analyze and compare of the contact residues of antibody drugs Pertuzumab (PDB code: 1S78) and Trastuzumab (PDB code: 6BGT). Both these antibody drugs target the same antigen HER2 (human epidermal growth factor receptor 2) but their binding regions (epitopes) are different (Figures S5A and S5B in Supplementary). As seen in Figure S5C in Supplementary, the comparison of contact residues of antibody drugs Pertuzumab and Trastuzumab shows that their CDR regions (CDR-L1, CDR-L2, CDR-L3, CDR-H1 and CDR-H2) are structurally similar with Z-score of 2.48 while the structures of binding interfaces of their antigens are different with Z-score of 0.85 (Figure S5D in Supplementary). This study as well as the case study 1 of Avastin and the therapeutic monoclonal antibody (PDB code: 4YPG) indicate that the contact residues of CDR-H1 and CDR-H2 and their conformations are more conserved compared to those of CDR-H3.
CONCLUSION
In this study, we have developed the AppA web server for comprehensive analysis, comparison, and visualization of contact residues and interfacial waters of antibody–antigen structures and models. AppA provides a user-friendly interface to identify and dissect the contributions of hydrogen bonds, van der Waals interactions, hydrophobic interactions and ionic interactions of contact residues and the interactions of interfacial waters, and compare their 3D structures for all the antibody–antigen structures available in the PDB.
We have previously highlighted the important role of interfacial water molecules in mediating the interactions between antibodies and antigens (8). In this study, AppA provides the detailed hydrogen bonds made between the interfacial water molecules and the contact residues for all the antibody–antigen structures from the PDB. In addition, AppA will automatically identify interfacial waters and contact residues, and calculate their hydrogen bonds, van der Waals interactions, hydrophobic interactions, and ionic interactions for any new antibody–antigen structures that are deposited in the PDB every week.
Furthermore, we have demonstrated the utility of AppA through various examples in contributing towards understanding antibody–antigen interactions, suggesting mutations of contact residues to improve affinity, as well as give insights into binding specificities of paratopes and epitopes. In the future, we will take into account the study of the role of glycans that can also be critical in antibody–antigen interactions.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Dr M.S. Madhusudhan and Dr M.R. Pradhan for valuable comments and insights. We also offer special thanks to Yong Taipang and Chew Chee Siang for help in setting up, maintaining and improving the server.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Research Foundation, Prime Minister's Office, Singapore under its BIOLOGICAL DESIGN TOOLS AND APPLICATIONS Programme (BDTA) Award [NRF2013-THE001-070]; Biomedical Research Council (BMRC)-Economic Development Board (EDB) Industry Alignment Fund [IAF311017G, H18/01/a0/B14 to M.N.N.]; A*STAR, Singapore for funding. Funding for open access charge: National Research Foundation [NRF2013-THE001-070].
Conflict of interest statement. Chandra S. Verma is the founder/scientific consultant of Sinopsee Therapeutics, a biotechnology company developing molecules for therapeutic purposes; the current work has no conflict with the company.
REFERENCES
- 1. Olimpieri P.P., Chailyan A., Tramontano A., Marcatili P.. Prediction of site-specific interactions in antibody–antigen complexes: the proABC method and server. Bioinformatics. 2013; 29:2285–2291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Peng H.P., Lee K.H., Jian J.W., Yang A.S.. Origins of specificity and affinity in antibody-protein interactions. Proc. Natl. Acad. Sci. U.S.A. 2014; 111:E2656–E2665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Fischman S., Ofran Y.. Computational design of antibodies. Curr. Opin. Struct. Biol. 2018; 51:156–162. [DOI] [PubMed] [Google Scholar]
- 4. Kringelum J.V., Nielsen M., Padkjaer S.B., Lund O.. Structural analysis of B-cell epitopes in antibody:protein complexes. Mol. Immunol. 2013; 53:24–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ramaraj T., Angel T., Dratz E.A., Jesaitis A.J., Mumey B.. Antigen-antibody interface properties: composition, residue interactions, and features of 53 non-redundant structures. Biochim. Biophys. Acta. 2012; 1824:520–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Barakat K., Issack B.B., Stepanova M., Tuszynski J.. Effects of temperature on the p53-DNA binding interactions and their dynamical behavior: comparing the wild type to the R248Q mutant. PLoS One. 2011; 6:e27651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yokota A., Tsumoto K., Shiroishi M., Kondo H., Kumagai I.. The role of hydrogen bonding via interfacial water molecules in antigen-antibody complexation. The HyHEL-10-HEL interaction. J. Biol. Chem. 2003; 278:5410–5418. [DOI] [PubMed] [Google Scholar]
- 8. Nguyen M.N., Pradhan M.R., Verma C., Zhong P.. The interfacial character of antibody paratopes: analysis of antibody–antigen structures. Bioinformatics. 2017; 33:2971–2976. [DOI] [PubMed] [Google Scholar]
- 9. Dunbar J., Krawczyk K., Leem J., Baker T., Fuchs A., Georges G., Shi J., Deane C.M.. SAbDab: the structural antibody database. Nucleic Acids Res. 2014; 42:D1140–D1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Adolf-Bryfogle J., Xu Q., North B., Lehmann A., Dunbrack R.L. Jr. PyIgClassify: a database of antibody CDR structural classifications. Nucleic Acids Res. 2015; 43:D432–D438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Richmond T.J., Richards F.M.. Packing of alpha-helices: geometrical constraints and contact areas. J. Mol. Biol. 1978; 119:537–555. [DOI] [PubMed] [Google Scholar]
- 12. Nguyen M.N., Madhusudhan M.S.. Biological insights from topology independent comparison of protein 3D structures. Nucleic Acids Res. 2011; 39:e94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kabsch W., Sander C.. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers. 1983; 22:2577–2637. [DOI] [PubMed] [Google Scholar]
- 14. Dunbar J., Deane C.M.. ANARCI: antigen receptor numbering and receptor classification. Bioinformatics. 2016; 32:298–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Nguyen M.N., Tan K.P., Madhusudhan M.S.. CLICK–topology-independent comparison of biomolecular 3D structures. Nucleic Acids Res. 2011; 39:W24–W28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Brown P., Pullan W., Yang Y., Zhou Y.. Fast and accurate non-sequential protein structure alignment using a new asymmetric linear sum assignment heuristic. Bioinformatics. 2016; 32:370–377. [DOI] [PubMed] [Google Scholar]
- 17. Garma L.D., Medina M., Juffer A.H.. Structure-based classification of FAD binding sites: a comparative study of structural alignment tools. Proteins. 2016; 84:1728–1747. [DOI] [PubMed] [Google Scholar]
- 18. Nguyen M.N., Sim A.Y., Wan Y., Madhusudhan M.S., Verma C.. Topology independent comparison of RNA 3D structures using the CLICK algorithm. Nucleic Acids Res. 2017; 45:e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kumar A.P., Nguyen M.N., Verma C., Lukman S.. Structural analysis of protein tyrosine phosphatase 1B reveals potentially druggable allosteric binding sites. Proteins. 2018; 86:301–321. [DOI] [PubMed] [Google Scholar]
- 20. Maurer-Stroh S., Krutz N.L., Kern P.S., Gunalan V., Nguyen M.N., Limviphuvadh V., Eisenhaber F., Gerberick G.F.. AllerCatPro - prediction of protein allergenicity potential from the protein sequence. Bioinformatics. 2019; doi:10.1093/bioinformatics/btz029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Pradhan M.R., Siau J.W., Kannan S., Nguyen M.N., Ouaray Z., Kwoh C.K., Lane D.P., Ghadessy F., Verma C.S.. Simulations of mutant p53 DNA binding domains reveal a novel druggable pocket. Nucleic Acids Res. 2019; 47:1637–1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Nguyen M.N., Sen N., Lin M., Joseph T.L., Vaz C., Tanavde V., Way L., Hupp T., Verma C.S., Madhusudhan M.S.. Discovering putative protein targets of small Molecules: A study of the p53 activator nutlin. J. Chem. Inf. Model. 2019; 59:1529–1546. [DOI] [PubMed] [Google Scholar]
- 23. Oganesyan V., Peng L., Woods R.M., Wu H., Dall’Acqua W.F.. Structural insights into the neutralization properties of the fully human, anti-interferon monoclonal antibody sifalimumab. J. Biol. Chem. 2015; 290:14979–14985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Muller Y.A., Chen Y., Christinger H.W., Li B., Cunningham B.C., Lowman H.B., de Vos A.M.. VEGF and the Fab fragment of a humanized neutralizing antibody: crystal structure of the complex at 2.4 A resolution and mutational analysis of the interface. Structure. 1998; 6:1153–1167. [DOI] [PubMed] [Google Scholar]
- 25. Chen Y., Wiesmann C., Fuh G., Li B., Christinger H.W., McKay P., de Vos A.M., Lowman H.B.. Selection and analysis of an optimized anti-VEGF antibody: crystal structure of an affinity-matured Fab in complex with antigen. J. Mol. Biol. 1999; 293:865–881. [DOI] [PubMed] [Google Scholar]
- 26. Sali A., Blundell T.L.. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993; 234:779–815. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.