


Abstract
Identifying the transcription factors (TFs) responsible for observed changes in gene expression is an important step in understanding gene regulatory networks. ChIP-X Enrichment Analysis 3 (ChEA3) is a transcription factor enrichment analysis tool that ranks TFs associated with user-submitted gene sets. The ChEA3 background database contains a collection of gene set libraries generated from multiple sources including TF–gene co-expression from RNA-seq studies, TF–target associations from ChIP-seq experiments, and TF–gene co-occurrence computed from crowd-submitted gene lists. Enrichment results from these distinct sources are integrated to generate a composite rank that improves the prediction of the correct upstream TF compared to ranks produced by individual libraries. We compare ChEA3 with existing TF prediction tools and show that ChEA3 performs better. By integrating the ChEA3 libraries, we illuminate general transcription factor properties such as whether the TF behaves as an activator or a repressor. The ChEA3 web-server is available from https://amp.pharm.mssm.edu/ChEA3.
INTRODUCTION
The set of expressed genes defines cell type, and more transiently, cellular response to endogenous and exogenous perturbations. Human intracellular gene expression programs are controlled mainly by ∼1600 putative site-specific transcription factors (TFs). These factors bind and unbind at specific DNA sequences near coding regions to regulate the transcriptional machinery (1). Despite an abundance of gene expression data (e.g. RNA-seq and microarrays), TF–DNA binding data (e.g. ChIP-seq), and TF activity data, we still lack a fundamental global understanding of how observed changes in gene expression are governed by changes in TF activity. This may be due in part to assay-specific limitations, and biases that can limit or confound findings. For example, ChIP-seq assays that target TFs commonly suggest TF localization near many genes that are not functional targets of the TF (2). DNA binding motif analyses also attempt to identify the targets of TF regulation (3), however, such methods produce high level of false positives (2). Some methods use TF–gene co-expression associations to elucidate TF targets and gene regulatory networks (4). However, co-expression associations rely on the mRNA level of TFs, which often do not correlate with TF activity that depends on protein levels, localization, and post-translational modifications. In addition, with co-expression data, it can be difficult to discern the direction of causality. Furthermore, direct effects may be confounded by indirect effects due to cascades of interactions (2,4).
There are several bioinformatics tools developed to prioritize TFs given a gene set, or a gene expression signature, as an input. These tools include: VIPER (5), DoRothEA (6,7), BART (8), TFEA.ChIP (9), oPOSSUM (10) and MAGICACT (11). VIPER compares a gene expression signature against putative TF regulons inferred from tissue-specific gene expression data using the tool ARACNe (4,5). DoRothEA v2 uses the VIPER tool in conjunction with a set of TF regulons derived from motif data, GTEx data (12), public ChIP-seq data, and the literature, to predict TFs mostly associated with a gene expression signature (7). Binding Analysis for Regulation of Transcription (BART) performs enrichment analysis against published ChIP-seq studies listed in the Cistrome Data Browser (13). BART infers a cis-regulatory profile of a query gene set using the tool MARGE (14). BART then predicts transcription factors associated with the cis-regulatory elements based on publicly available TF ChIP-seq data (8). oPOSSUM (10) detects over-representation of conserved TF binding-site-combinations given gene sets. MAGICACT (11) uses TF ChIP-seq data to determine whether the peak signals for a query gene set as a whole are greater than what would be expected by chance for a given TF. Similarly, TFEA.ChIP performs gene set enrichment analysis against published ChIP-seq data using either the Fisher's exact test (FET) or the Gene Set Enrichment Analysis (GSEA) method (9,15).
In this domain, we previously published ChEA (16) and ChEA2 (17), which are ChIP-seq and ChIP-chip TF enrichment analysis tools that utilize gene set libraries from published ChIP data extracted from multiple sources. Similar to ChEA and ChEA2, ChEA3 is a web-server application developed to conduct transcription factor enrichment analysis. ChEA3 integrates data about TF/target–gene associations from multiple assay types and other sources of evidence. TFs are prioritized based on the overlap between user-inputted gene sets and annotated sets of TF targets stored within the ChEA3 database. ChEA3 builds upon the prior versions of ChEA by including many more libraries from various types of omics assays and integrating libraries for improved TF ranking.
ChEA3 is accessible via a web interface and an API that enable users to submit gene sets for analysis. We systemically evaluated the predictive performance of each of the six primary ChEA3 libraries, and the two integration approaches for their ability to rank the perturbed TFs from gene sets derived from 946 single-TF perturbation followed by expression experiments mined from the Gene Expression Omnibus (GEO) (18–20). This single-TF perturbation followed by expression dataset is also used to compare the predictive performance of ChEA3 with other similar tools. For benchmarking against existing tools that require queries from a consistent cell line, we use a dataset generated from 49 TF shRNA knockdowns in a B-cell line (2). We demonstrate that integrating multiple independent omics resources improves TF prioritization, and ChEA3 performs well compared with other tools. By combining TF perturbations followed by expression with other sources of evidence, we infer whether a given TF is generally an activator or a repressor.
MATERIALS AND METHODS
Generating the ChEA3 primary TF-Target gene set libraries
ChEA3 contains six primary reference gene set libraries created from multiple resources. Below is a brief description of each library and the processing procedure to create the library from each resource. To harmonize gene names across libraries, all gene symbols were mapped to 2019 HGNC-approved gene symbols (21) using an R package we developed for the project called genesetr (https://github.com/MaayanLab/genesetr). Gene symbols that could not be mapped using synonyms or aliases were discarded. The set of 1634 unique HGNC-mappable human site-specific transcription factors that are used were previously defined by Lambert et al. (1).
GTEx co-expression
All RNA-seq samples at the read counts level with their associated metadata were downloaded from the GTEx portal on 6 January 2018 (12). Samples were quantile-normalized. Duplicate genes were removed by retaining the genes with the highest variance. For each TF, the set of putative targets was composed by retaining the 300 genes with the greatest absolute Pearson correlation coefficient between the TF and the putative target gene.
ARCHS4 co-expression
50,000 samples from human were randomly selected for creating a co-expression matrix from the ARCHS4 resource (20). Read counts and metadata were downloaded from the ARCHS4 website on 27 April 2018. These samples were processed as described above for the GTEx data.
ENCODE ChIP-seq
The ENCODE (22) TF–target gene-set library was initially generated for Enrichr (23,24) using uniformly reprocessed ENCODE TF ChIP-seq experiments. Peak calling was applied to the aligned files with MACS (25). Peaks were then sorted by distance to the transcription start site (TSS). The top 2000 target genes with the closest peaks to their TSS were retained for each experiment. Each gene set corresponds to a specific ChIP-seq experimental condition. Therefore, there are multiple gene sets corresponding to some of the same TFs.
Gene sets from individual ChIP-seq publications
The literature-based ChIP-seq TF target library is derived from TF ChIP-seq and ChIP-chip experiments mined from publications found within the biomedical research literature. Previous versions of this library were used in ChEA (16), ChEA2 (17), and Enrichr (23,24). The metadata of the gene set library includes the TF that was profiled, the PubMed ID of the publication from which the experiment originated, as well as the species, the assay type, and the cell- or tissue-type. If only the BED file was provided by the authors of the original study, peaks were mapped to genes using a custom script. Each gene set corresponds to a specific ChIP-seq experimental condition in a specific study. Therefore, there are multiple gene sets corresponding to some of the same TFs.
ReMap ChIP-seq
BED files from the ReMap resource were batch downloaded to a local server (26). A peak score si,j,k was generated for each ChIP-seq peak i corresponding to a TF j within a 50 kb window around the TSS k where si,j,k = 1 – distancei,k/50 000. We let the distancei,k be the distance of the peak i summit from the TSS k. For each TSS k and TF j pair, peak scores were summed to produce a score tj,k to each TSS for each TF. TF targets were then assigned to the top 5% nonzero TSS scores with a cap of 1500 top targets per TF.
Enrichr Queries
User-submitted lists to the Enrichr tool (23,24) were dumped from the Enrichr database on 27 October 2017. The collection of queries totaled 1 097 157 unique lists. Lists used for internal testing, lists with >2000 genes, lists with fewer than two genes, and lists from IP addresses that submitted >1000 lists were discarded. Co-occurrence analysis was performed on the remaining 293 747 lists as follows: For each TF i, the probability of co-occurrence of the transcription factor TFi with a gene gj, P(TFi ∩ gj), was computed for all genes in G. The top 300 co-occurring genes with each TFi were used as putative TFi targets.
Transcription factor enrichment analysis
The significance of the overlap between two gene sets is computed using the FET. The background is set to 20 000 genes by default. This value was selected as an estimate to reflect the typical number of genes in most analyses. It also produces a reasonable amount of TFs passing a significant threshold. The background number does not affect the ranks of the results but does have an influence on the P-values. ChEA3 only accepts Human Genome Organisation (HUGO) Gene Nomenclature Committee (HGNC)-approved gene symbols. Therefore, ChEA3 accepts genes from other species that have orthologs with gene symbols that directly map to human gene symbols. False discovery rates are computed with the Benjamini–Hochberg correction method for each library separately. An integer rank is generated for each gene set in a library, where 1 indicates the gene set in the library has the lowest corrected FET P-value, and k is the rank of the gene set in the library with the highest P-value where k is the number of unique TFs in the library. Ties are broken by random assignment. For those libraries containing multiple gene sets corresponding to the same TF, the gene set with the lowest P-value is used. A scaled rank is computed by dividing each integer rank by k. Therefore, for a single query, there is one TF gene set ranking for each gene set library in ChEA3. The six sets of TF gene set rankings are integrated by two methods: MeanRank and TopRank. For MeanRank, the mean rank of each TF across all libraries containing that TF is the score by which a composite list of TFs is re-ranked. For TopRank, the best scaled rank of each TF across all libraries is used as the score by which a composite list of TFs is re-ranked.
Generating the benchmarking datasets
Single-TF perturbation experiments including knockdowns, knockouts, overexpression, and chemical inhibition followed by genome-wide microarray profiling were manually curated by the crowd for the CREEDS resource (18). The automatically extracted CREEDS signatures were not used in the benchmarking of ChEA3. Full signatures were computed as described in Wang et al. (18) using the Characteristic Direction method (27). Of the 786 TF LOF/GOF experiments from CREEDS, 283 are from human and 503 are from mouse. These 786 signatures contain 275 unique site-specific TFs. An additional 160 human single-TF perturbation RNA-seq experiments were manually curated from the Gene Expression Omnibus (GEO) by first programmatically searching the ARCHS4 resource metadata for potential studies that contain relevant TF-related signatures. Only studies with at least two perturbation samples and at least two control samples were retained. The uniformly reprocessed GEO samples were downloaded from ARCHS4, quantile normalized and signatures were generated with the Characteristic Direction method (27).
We generated three types of benchmarking query gene sets from the 946 signatures created from CREEDS and ARCHS4: (i) gene sets containing the top 600 differentially expressed genes either from CREEDS or ARCHS4 (top 300 upregulated and top 300 downregulated genes), which we term TFpertGEOupdn; (ii) gene sets containing only the upregulated genes from TFpertGEOupdn, which we call TFpertGEOup; and (iii) gene sets containing only the downregulated genes from TFpertGEOupdn, which we call TFpertGEOdn. Up- and downregulated genes in these sets were determined from the Characteristic Direction method's coefficients. To examine the effect of gene set size on performance, we also generated TFpertGEO200 and TFpertGEO1000 in the same manner as TFpertGEOupdn, but with gene set sizes of 200 and 1000, respectively. We segregated the TFpertGEOupdn benchmarking set into human and mouse benchmarking sets: hsTFpertGEOupdn and mmTFpertGEOupdn. Finally, we refer to the 443 single-TF LOF/GOF full signatures from human that were used to benchmark other TF prediction tools as hsTFpertGEOsig. The TFpertGEO benchmarking datasets contain 443 human TF LOF/GOF and 503 mouse TF LOF/GOF experiments.
In addition to the TFpertGEO benchmarking datasets, another benchmarking dataset was derived from Cusanovich et al. (2). Of the 59 knockdowns of TFs and chromatin modifiers available in this dataset, 49 were mappable to the set of site-specific TFs within ChEA3. To generate the Cusanovich benchmarking dataset, probe-level estimates (log2-transformed) from GSE50588 were downloaded from GEO (2). Illumina probes were then mapped to HGNC-approved gene symbols using the illuminaHumanv4.db R package. For probes mapping to the same gene, the probe with the highest variance across samples was retained. The data were then quantile-normalized across samples. Forty-nine TF shRNA vs. control signatures were generated by the t-test according to the VIPER R package vignette. This dataset of full gene signatures, which we term sigCusanovich, was used to benchmark the published VIPER B-cell regulon available in the viperbcell R package and a B-cell regulon we generated from the GSE50588 expression dataset with ARACNe-AP (28). The top 300 upregulated and the top 300 downregulated genes from each sigCusanovich signature were used to generate TF shRNA-associated gene sets for the setCusanovich benchmarking dataset, which was used to benchmark ChEA3 for comparison to the VIPER B-cell regulons.
Benchmarking metrics
Each gene set from the TFpertGEOupdn, TFpertGEOup, TFpertGEOdn, hsTFpertGEOupdn, mmTFpertGEOupdn and setCusanovich benchmarking datasets was submitted to ChEA3. Transcription factors were ranked within each library according to the returned FET P-values. Ranks within each library were then scaled between 1/n and 1, where n is the number of unique TFs in the library, to accommodate different library sizes. The R package PRROC was used to compute the area under the Receiver-Operating Characteristic (ROC) curve and Precision-Recall (PR) curve for each library. The positive class consists of ranks of the perturbed TF. The negative class consists of the ranks of all other TFs that were not perturbed in the experiment. To generate PR curves and ROC curves, we down-sampled the negative class to the same size as the positive class, similarly to the way it is described by Garcia-Alonso et al. (7). Each library has a different number of TFs and therefore has a different ‘random classifier’ PR curve. By down-sampling the negative class to the same size as the positive class, a random classifier would have a PR AUC of 0.5. For consistency, we also down-sampled the negative class in the same manner to generate the ROC curves. ROC and PR curves were bootstrapped in this manner 5000 times and then the mean ROC and PR AUCs were reported. The base R function approx() was used to linearly interpolate between all points from the 5000 ROC curves and the 5000 PR curves in order to generate composite ROC and PR curves for each library and tool for visualization. We also employed an additional metric of performance as follows. The set of rank values of the perturbed transcription factors were identified for all gene set queries. We then examined the cumulative distribution function of this set of ranks, D(r). If the perturbed TFs do not display preferentially low or high ranks, then we expect a uniform distribution D(r) = r. We therefore examine D(r) – r for significant deviations from zero in order to evaluate different libraries and methods. Anderson-Darling tests were used to evaluate the null hypothesis, D(r) = r, and were performed using the goftest R package.
Benchmarking existing tools
BART
To generate TF predictions from BART, cis-regulatory profiles for each gene set were obtained by submitting each gene set in the TFpertGEOupdn benchmarking dataset to MARGE (14) running on Python 3. These enhancer predictions were then ported to BART running on Python 3 to generate TF predictions for each gene set. All site-specific TFs were ranked according to the order that they were ranked by BART, which is based on a composite score.
TFEA.ChIP
Gene sets from TFpertGEOupdn were queried according to the TFEA.ChIP R package vignette FET example. All site-specific TFs were ranked according to their P-values.
VIPER
VIPER was benchmarked using full signatures from the sigCusanovich benchmarking dataset according to the VIPER R package vignette. Two input regulon objects were used: the published B-cell VIPER regulon available in the bcellviper R package, and a Cusanovich dataset-specific regulon that we generated. The Cusanovich dataset-specific regulatory network was generated in ARACNe-AP using all 200 GSE50588 samples with a P-value threshold of 1 × 10−8. The list of regulatory proteins inputted to ARACNE-AP were all TFs defined by Lambert et al. (1) that were also present in the probe set of Cusanovich et al. (2), which totaled 731 TFs. One-hundred bootstraps were consolidated to form the final network. The VIPER R package was used to generate a regulon object from the ARACNe-generated network according to the package vignette example. The VIPER Master Regulator Inference Analysis (MARINA) was conducted for each of the 49 TF shRNA signatures. One-thousand permutations on the data were used to generate a null model. All site-specific TFs were ranked according to their P-values. VIPER was also benchmarked with the hsTFpertGEOsig signatures using an ARACNe-AP regulatory network generated from GTEx data. The GTEx ARACNe network was generated using 200 quantile-normalized random samples from the GTEx RNA-seq data with a P-value threshold of 1 × 10−8. The set of regulatory TFs given to ARACNe consisted of 1607 HGNC-mappable TFs defined by Lambert et al. (1) that were also present in the GTEx RNA-seq data. MARINA was conducted for each of the 443 signatures in the hsTFpertGEOsig dataset. All site-specific TFs were ranked according to the absolute value of their normalized enrichment scores (NES).
DoRothEA
VIPER MARINA was conducted with the hsTFpertGEOsig signatures using the DoRoTHEa v2 A, B, C, D, E and TOP10score regulon R objects available at https://github.com/saezlab/DoRothEA (7). All site-specific TFs were ranked according to the absolute value of their NES.
MAGICACT
All TFPertGEOupdn gene sets were submitted to the MAGICACT (11) executable for MacOSX. All site-specific TFs were ranked according to their composite scores returned by MAGICACT.
Determining whether a TF is an activator or a repressor
Odds ratios were computed for all TFi perturbation and TFi ChIP-seq pairs using the TFpertGEOup, TFpertGEOdn, ReMap, ENCODE and Literature ChIP-seq datasets. Odds ratios were computed using the equation below where for a given TFi ChIP-seq/TFi perturbation experiment pair, ‘a’ is the intersection between upregulated genes and ChIP-seq targets, ‘b’ is the intersection between downregulated genes and ChIP-seq targets, ‘c’ is the set of upregulated genes not found to be ChIP-seq targets, and ‘d’ is the set of downregulated genes not found to be ChIP-seq targets. Odds ratio P-values were computed with the hypergeometric test.
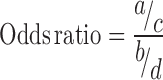 |
(1) |
To compare our analysis to an independent source, we downloaded the raw human and mouse TF–target interactions from the TRRUST database. Mouse genes and TFs were mapped to HGNC-approved symbols, and the human and mouse data were combined. TF–target interactions with unknown directionality were discarded. TFs that had at least 20 targets with known directionality were retained for the analysis.
ChEA3 web server application
The server-side of ChEA3 was written in Java and runs on Tomcat 9. Java servlets process gene list submissions from the front end. The user interface of ChEA3 is implemented with jQuery (29), the templating application Mobirise 4.8.1, and Bootstrap v4 (30). The interactive TF network visualization is implemented with D3.js v4 (31). The front and back end components are compiled and assembled together into a JAR file. The web application is running in a Docker container (32) and the Docker image is deposited in Docker Hub (https://hub.docker.com/r/maayanlab/chea3). ChEA3 also provides API access to the service. The results from the API are returned in JavaScript Object Notation (JSON) format. The complete ChEA3 web service code is available on GitHub at https://github.com/maayanlab/chea3web.
Transcription factor coexpression network visualization
To create an interactive global view of the human TF regulatory network, Weighted Gene Co-expression Network Analysis (WGCNA) (33) was applied on GTEx (12), ARCHS4 (20) and TCGA expression data. The quantile-normalized GTEx gene expression dataset was filtered to only include TFs. WGCNA was applied on the reduced TF GTEx matrix using the WGCNA R package with default parameters. Similarly, 100 random RNA-seq samples for each of 18 tissue types were pulled from the ARCHS4 database and were quantile normalized. The expression dataset was filtered to include only TFs, and WGCNA was applied with default parameters. To generate the TCGA network, TCGA primary tumor samples were randomly sampled such that we obtained a set of 26 cancer types with 100 samples for each type. The expression dataset was quantile-normalized, filtered to include only TFs, and WGCNA was applied with default parameters. The three resulting networks were visualized using Cytoscape (34) with the Allegro Edge-Repulsive Strong Clustering plugin. Node positions were exported from Cytoscape and visualized on the ChEA3 results page using D3.js.
To annotate the GTEx network, module eigengenes were correlated to GTEx tissue sample labels. Nodes were colored by the most significant tissue correlation to their parent module. GO Biological Pathway enrichment was conducted on the network-module-gene-members using the topGO R package (35) with the set of TFs as the background gene universe. Nodes were colored by the most significant result from this enrichment analysis. To annotate the TCGA network, module eigengenes were correlated to TCGA tumor sample types. Nodes were colored by the most significant tumor correlation to their parent module. To annotate the ARCHS4 network, module eigengenes were correlated to ARCHS4 tissue sample labels. Nodes were colored by the most significant tissue correlation to their parent module.
Transcription factor co-regulatory network visualization
A transcription factor co-regulatory network was constructed from all TF–TF interactions described by the six ChEA3 primary libraries. Edges that were supported by evidence from two or more different libraries were retained in the network. Edges are directed where ChIP-seq evidence supports the interaction and are undirected in the case of co-occurrence or co-expression evidence only. The network is subset based on the top TF results from a user query and is visualized using D3.js.
Clustergrammer visualization
From the results of each query, a binary matrix with the top 5 TFs returned by each library on the columns and query genes on the rows is populated according to whether the query gene appears within the target gene set of the library TF. This matrix is submitted to the Clustergrammer API (36) which returns a URL to an interactive clustergram of the matrix. This URL is displayed in an iframe as part of the ChEA3 results visualizations.
RESULTS AND DISCUSSION
ChEA3 libraries and web tool
ChEA3 performs TF target overrepresentation analysis against six TF target set libraries covering 1,632 unique TFs (Table 1). Site-specific DNA-binding TFs were included in ChEA3 as defined in the seminal publication by Lambert et al. (1). Non-specific transcription factors, cofactors, and chromatin modifiers are excluded. Genes that are highly co-expressed with transcription factors were pulled from the GTEx (12) RNA-seq data, and from the uniformly reprocessed GEO RNA-seq data from ARCHS4 (20). Uniformly reprocessed publicly available ChIP-seq data were collected from ENCODE (22) and ReMap (26). ChIP-seq data were also manually mined from the literature by curating target lists from supporting materials of individual TF studies as an expansion of the work done for ChEA and ChEA2 (16,17). Finally, we used a wisdom of the crowd based approach to identify TF targets by mining user-submitted queries to the web tool Enrichr (23,24) to identify genes that frequently co-occur in submitted gene list queries with all human TFs. ChEA3 uses the pairwise FET to compare a user-submitted gene set query to each gene set in each ChEA3 library. Results are returned for each library separately as a list of TFs ranked by their FET P-value. TFs are given both an integer rank, with 1 corresponding to the most significant matching TF associated gene set, and a scaled rank from 1/n to 1 where n is the number of unique TFs in the library.
Table 1.
Transcription factor target gene set libraries included in ChEA3. The TF coverage heatmap spans the 1634 human site-specific TFs in (1) with 1632 of those factors covered by ChEA3
| Library | Unique TFs | Unique TF Interactions | Gene Sets |
|---|---|---|---|
| ARCHS4 Coexpression | 1628 | 480 504 | 1628 human |
| ENCODE ChIP-seq | 118 | 392 667 | 552 (470 human, 82 mouse) |
| Enrichr Queries | 1404 | 409 279 | 1404 (unknown species) |
| GTEx Coexpression | 1607 | 468 672 | 1607 human |
| Literature ChIP-seq | 164 | 340 547 | 307 (138 human, 164 mouse, 5 rat) |
| ReMap ChIP-seq | 297 | 417 025 | 297 human |
We hypothesized that integrating TF target overrepresentation results from across libraries to generate a composite ranking of TFs might overcome unique biases associated with each library and improve the predictive performance of ChEA3. To this end, we developed two integration techniques: MeanRank and TopRank. For each of the 1632 TFs covered by ChEA3, we take the mean of the integer ranks across all libraries and re-rank based on this mean to generate a composite ranking that we term MeanRank. For TopRank, we take the maximum scaled rank assigned to each TF across all libraries and re-rank to generate a composite ranking. By benchmarking the quality of the ranking using an independent TF–target dataset, we demonstrate that the MeanRank and TopRank approaches indeed outperform the original six TF–target libraries using the benchmarking strategy described below.
Benchmarking the ChEA3 libraries
To benchmark the predictive performance of ChEA3, gene expression signatures were extracted from 946 single-TF loss-of-function (LOF) and gain-of-function (GOF) human and mouse experiments from GEO. Relevant studies were identified, then control and perturbation samples were tagged, and then signatures were extracted using a uniform pipeline. This was first achieved for microarray studies by contributors to a microtask crowdsourcing project (18) and then for RNA-seq utilizing the ARCHS4 resource (20). We generated up, down, and combined up/down gene sets from these signatures and queried the ChEA3 API with each gene set to determine how well each ChEA3 library recovers the perturbed TF. ROC and PR curves were generated from the rankings of the experimentally perturbed TFs which compose the positive class, and sampled rankings of the unperturbed TFs that compose the negative class (Figure 1A-C). We also looked at the empirical cumulative probability density (ECPD) of the ranks of the positive class D(r) for each library as compared to the ECPD of a uniform rank distribution r, which is D(r) – r (Figure 1D). A greater deviation from zero indicates better recovery of the perturbed TFs. Integrating results across libraries yielded improved predictive performance by multiple metrics that assess the global distribution of ranks. By these metrics, the MeanRank approach performs the best. Interestingly, the Enrichr ‘wisdom of the crowd’ library displays the best performance of the six ChEA3 TF target libraries.
Figure 1.

Performance of the ChEA3 libraries and integration techniques in recovering the perturbed TFs from 946 TF LOF and GOF experiments from the TFpertGEOupdn benchmark dataset. (A) Mean ROC AUC and mean PR AUC over 5000 bootstrapped ROC and PR curves; (B) composite ROC curves generated from 5000 boostrapped curves; (C) composite PR curves generated from 5000 bootstrapped curves; (D) the deviation of the cumulative distribution from uniform of the scaled rankings of each perturbed TF in the benchmarking dataset. Anderson-Darling test of uniformity: MeanRank P = 6.34 × 10−7; TopRank P = 6.34 × 10−7; ARCHS4 P = 6.34 × 10−7; ENCODE P = 2.06 × 10−6; Enrichr Queries P = 6.83 × 10−7; GTEx P = 6.45 × 10−7; Literature ChIP-seq P = 1.28 × 10−6; ReMap P = 1.02 × 10−6.
Arguably, a ChEA3 user is interested only in the top-ranked TFs returned by the tool. Therefore, for each rank percentile, we examined the fraction of the benchmarking dataset TF perturbations that are recovered. We computed this fraction in two ways: one where we considered the entire benchmarking dataset, and one where we considered only the subset of the benchmarking dataset where the perturbed TFs are covered by the library (Supplementary Figure S1). When we examined the fraction of the benchmarking subset recovered in the top percentile of the TF ranks, we observed that the integrated libraries perform comparably to the ChIP-seq libraries, but with much greater TF coverage (Figure 2, Supplementary Figure S2).
Figure 2.
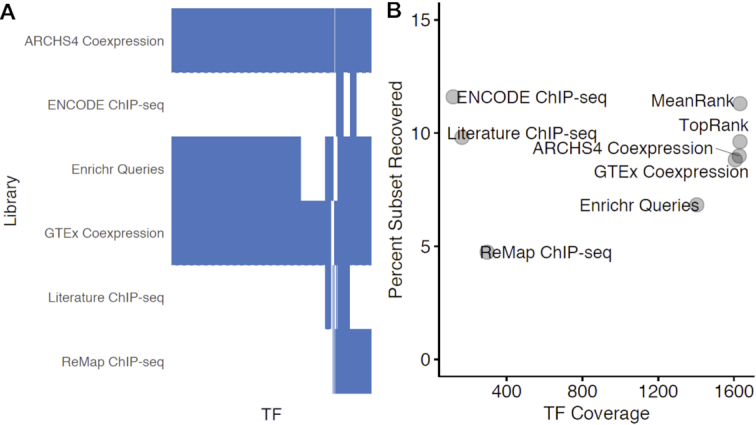
Fraction of the TFpertGEOupdn benchmarking dataset subset recovered in the top one percentile of rankings compared to the library TF coverage. (A) A heatmap visualizing transcription factor coverage for the ChEA3 libraries. (B) The fraction of the TFpertGEOupdn subset TFs recovered in the top percentile of ranks for each ChEA3 library. Only the TFpertGEOupdn gene sets where the perturbed TF was covered by the library were considered when computing the ‘Percent Subset Recovered’.
In our global assessment of the TF rankings returned by the ChEA3 libraries, the ChIP-seq libraries displayed the lowest performance when assessing the global ranks. In contrast, in the subset of the benchmarking experiments where there is ChIP-seq data for the perturbed TF, the ChIP-seq libraries performed well in recovering the perturbed TFs (Figure 2B). This may reflect that some TF target sets may be more amenable to determination by ChIP-seq analysis than others. This could be due to several factors. Others have reported a high rate of non-functional binding sites (2). Further, ‘hyper-ChIPable’ regions of the genome exist near highly expressed genes and these regions show binding of many TFs (37), possibly further diminishing the specificity of the TF target gene sets in these libraries. Some TFs and their target sets may be more or less tissue- or context-specific than others (38), and some TFs may regulate their targets more distally (39).
In order to assess aspects of a signature that could be contributing to better predictions, the benchmarking dataset was separated into four groups of TF perturbation gene sets. These four groups consist of: (i) upregulated sets of genes from TF GOF experiments (Figure 3A; Supplementary Figures S3A and S4A); (ii) downregulated sets of genes from TF GOF experiments (Figure 3B; Supplementary Figures S3B and S4B); (iii) upregulated sets of genes from TF LOF experiments (Figure 3C; Supplementary Figures S3C and S4C), and (iv) downregulated sets of genes from TF LOF experiments (Figure 3D; Supplementary Figures S3D and S4D). For queries where the perturbed upstream TF had a loss of function, the ChIP-seq libraries perform best when queried with the downregulated genes. Conversely, for signatures where the upstream TFs were over-expressed, the ChIP-seq libraries perform best when queried with the upregulated genes. This observed behavior aligns with the notion that most transcription factors are activators. The co-expression and co-occurrence libraries recover the upstream TFs comparably well across TF perturbation and query types. We also assessed human and mouse TF LOF/GOF experiment-associated gene sets separately using the human hsTFpertGEOupdn and mouse mmTFpertGEOupdn datasets and found comparable ChEA3 performance for both species (Supplementary Figures S5–S8). Finally, we assessed the effect of input size on predictive performance using the TFpertGEO200 and TFpertGEO1000 benchmark sets and found that the performance of ChEA3 is robust to a range of input gene set sizes (Supplementary Figures S9–S14).
Figure 3.

Effect of input type on ChEA3 performance. The deviation of the cumulative distribution from uniform of the scaled rankings of perturbed TFs in the benchmarking dataset for: (A) TF overexpression or chemical activation experiments from TFpertGEOup; (B) TF overexpression or chemical activation experiments from TFpertGEOdn; (C) TF knockdown, knockout or chemical inactivation experiments from TFpertGEOup; and (D) TF knockdown, knockout or chemical inactivation experiments from TFpertGEOdn.
Comparing ChEA3 with similar TF ranking tools
There are several existing tools that perform TF prioritization given gene sets or signatures as input (Table 2). Since these tools were built with human data, we used the human TF LOF and GOF experiments for benchmarking them. For tools that accept discrete gene sets as input, which include BART, TFEA.ChIP and MAGICACT, we used the 443 single TF GOF and LOF experiments from the hsTFpertGEOupdn benchmarking dataset. VIPER and DoRothEA v2 require full gene expression signatures as input. We benchmarked DoRothEA regulons and an ARACNe-AP regulon generated from GTEx data on the 443 full signatures in the hsTFpertGEOsig benchmarking dataset. These results were compared to ChEA3 benchmarked on the hsTFpertGEOupdn dataset (Figure 4–5, Supplementary Figure S15). We show that both ChEA3 integration strategies, MeanRank and TopRank, outperform all the tools we tested when benchmarked against the hsTFpertGEO benchmarking datasets and also have greater TF coverage.
Table 2.
Summary of tools benchmarked against ChEA3
| Tool | TF coveragea | Required input | Method | Data used to make predictions | Availability |
|---|---|---|---|---|---|
| TFEA.ChIP (9) | 271 | Gene set or sorted list of DEGs | FET or GSEA (15) | ENCODE (22) and GEO (19) ChIP-seq experiments | R package, Web Server https://www.iib.uam.es/TFEA.ChIP/ |
| BART (8) | 273 | Gene set | Correlates cis-regulatory profile derived from query gene set with TF genomic binding profiles | DNAse I hypersensitivity, TF ChIP-seq | Standalone Application, Web Server http://bartweb.uvasomrc.io |
| VIPER (5) | 454b, 731c, 1,607d | Gene signature | aREA (analytic Rank-based Enrichment Analysis) (5) | ARACNe-generated gene regulatory network in same tissue type as query | R package |
| DoRothEA v2 (7) | Reg. A: 163 Reg. B: 188 Reg. C: 313 Reg. D: 416 Reg. E: 1306 Top10Score: 1306 | Gene signature | aREA (5) | Literature, ReMap ChIP-seq (26), TF motif (47,48), GTEx co-expression (12) | R object for use with VIPER R package |
| MAGICACT (11) | 109 | Gene set | Mann-Whitney test | ENCODE (22) ChIP-seq | Standalone Application |
aHGNC-mappable TFs that are considered site-specific TFs as defined by Lambert (1). Some tools contain additional general transcription factors, co-factors, or chromatin modifiers.
bPublished B-cell regulatory network available in the bcellviper R package.
dARACNe-AP built network using expression data from GTEx (12).
Figure 4.

Comparison of available TF prediction tools with ChEA3 with the hsTFpertGEO benchmarking dataset. (A) Composite ROC curves generated from 5000 bootstrapped curves; (B) composite PR curves generated from 5000 bootstrapped curves; (C) the deviation of the cumulative distribution from uniform of the scaled rankings of each perturbed TF in the benchmarking dataset; Anderson–Darling test of uniformity: VIPER GTEx Regulon P = 1.39 × 10−6, MAGICACT P = 6.58 × 10−5, TFEA.ChIP P = 2.47 × 10−6, BART P = 2.34 × 10−6, DoRothEA Regulon A P = 2.39 × 10−6; DoRothEA Regulon B P = 2.22 × 10−6, DoRothEA Regulon C P = 1.92 × 10−6, DoRothEA Regulon D P = 1.71 × 10−6, DoRothEA Regulon E P = 1.46 × 10−6, DoRothEA Regulon TOP10score P = 1.46 × 10−6; (D) mean ROC AUC and mean PR AUC over 5000 bootstrapped ROC and PR curves for available TF prediction tools as compared with ChEA3 benchmarked with hsTFpertGEO.
Figure 5.

Comparison of available TF prediction tools with ChEA3. (A) The percent of the perturbed TFs recovered by the tool in the top one percentile of ranks as compared to TF coverage of the tool. For the ‘Percent Subset Recovered’ metric, we consider only the subset of the hsTFpertGEO TF perturbation experiments where the TF is covered by the tool. (B) The percent of the perturbed TFs recovered by the tool in the top one percentile of ranks as compared to TF coverage of the tool. For the ‘Percent Total Recovered’ metric, we consider all 443 TF perturbation experiments in the hsTFpertGEO benchmarking datasets. (C) Mean AUROC over 5000 bootstrapped curves compared to tool TF coverage. (D) Mean AUPR over 5000 bootstrapped curves compared to tool TF coverage.
VIPER was designed for a gene regulatory network inferred from the same cell or tissue type as the query. Therefore, we generated signatures from 49 TF shRNA experiments applied to B-lymphoblastoid cell line (2) as described in the methods. We tested two regulatory networks for VIPER: a published B-cell ARACNe regulatory network (40,41), and a network we built from all expression data described in the 49 TF shRNA study (2) using ARACNe-AP (28). We also derived discrete gene sets from the same TF shRNA dataset for input into ChEA3 for comparison (Supplementary Figure S16 and S17). We show that both ChEA3 integration strategies, MeanRank and TopRank, outperform both B-cell regulons tested. Notably, TF-regulons derived from ARCHS4 and GTEx co-expression data, which includes many disparate cell and tissue types, performs better on the benchmarking dataset than the VIPER A GM19238 B-cell regulon. It should be noted that the VIPER A regulon was derived from the same gene expression dataset that was also used to generate the benchmarking query signatures.
Global analysis of ChEA3 libraries to identify activator and repressor TFs
Integrating libraries of putative TF targets allows for global analyses of transcription factor activity. To understand the general repressing or activating characteristics of the TFs in the ChEA3 libraries, we computed odds ratios (ORs) using gene sets from the TFpertGEOupdn benchmarking dataset and the ChEA3 ChIP-seq libraries. For the odds ratio computation, we define the numerator as the number of genes that are both upregulated upon perturbation of the TF and are ChIP-seq targets of the TF divided by the number of upregulated genes that are not ChIP-seq targets. We define the denominator as the number of genes that are both downregulated on perturbation of the TF and are ChIP-seq targets of the TF, divided by the number of downregulated genes that are not ChIP-seq targets. If the numerator is greater, then it can be said that the targets tend to be upregulated and the OR > 1. If the denominator is greater, then it can be said that the targets tend to be downregulated and the OR < 1. We then consider whether the perturbation increased or decreased the activity of the TF. If the TF acts as an activator and the experiment was a GOF perturbation, then we expect the OR > 1. Conversely if the TF acts as an activator and the experiment was a LOF perturbation, then we expect the OR < 1. If the TF acts as a repressor and the experiment was a GOF perturbation, then we expect the OR < 1. Finally, if the TF acts as a repressor and the experiment was a LOF perturbation, then we expect the OR > 1. Therefore, the negative log(OR) was considered for TF LOF experiments, while the positive log(OR) was considered for TF GOF experiments (Figure 6A, B). These values will be positive if the TF is an activator and negative if the TF is a repressor. We recover known activators and repressors in this analysis, for example, REST is a known repressor of neuronal genes in non-neuronal cell types. REST is shown to predominately be downregulating its targets. CTCF is predominantly considered an insulator (42), but also shown to be an activator (43). In our analysis, CTCF appears to be predominately activating its putative targets. This observation points to a potential role in tethering distant enhancers to their promoters (44). MYC, while predominately shown to be an activating TF, also has significant ORs that suggest a repressor role in some contexts, which is supported by previous studies (45). We also compared our analysis to mouse and human TF–target interactions mined from literature in the TRRUST v2 reference database (Figure 6C) (46). The TRRUST database contained signed and directed connections mined from the literature. Overall, our automated analysis agreed with the trends observed from TRRUST.
Figure 6.

Scatterplots showing activating/repressing activity across TFs. Significant ORs (P< 0.05) are plotted. For uniformity, when examining loss-of-function TF perturbations, we consider –log(OR), as this will be positive if the TF acts as an activator of its targets and negative if it acts as a repressor. Conversely, we consider log(OR) for gain-of-function perturbations, which will be positive if the TF is an activator and negative if the TF acts as a repressor. Red arrows indicate TFs discussed in the results. (A) ORs from gain-of-function TF perturbations; (B) ORs from loss-of-function TF perturbations; (C) TF–target interactions from the TRRUST v2 database. For each TF, the percent of activating TF–target interactions (red) or repressive TF–target interactions (blue) from the subset of TF–target interactions in TTRUST v2 for which directionality is available.
The ChEA3 web interface
The ChEA3 landing page contains an input form for users to submit their gene list. Following submission of a gene set, searchable, sortable and exportable results tables appear for each of the six ChEA3 libraries, and for the two integration methods: MeanRank and TopRank. These tables appear in the order of how well the library, or the integration technique, performed in our benchmark. This is implemented to aid users with deciding which table is most relevant for hypotheses generation. We project the results from these tables onto three global edgeless TF co-expression networks, and also generate local TF co-regulatory networks for each library with the top TF results. A clustergram tab shows the overlapping query gene targets among the top library results (36), and a bar chart shows the contributions of each library to the top TF rankings from the MeanRank integration method. The global co-expression networks serve to provide context for the user about how the most enriched TFs fit within the larger TF co-regulatory network. The local TF co-regulatory networks contain directed and undirected edges to communicate how the top returned TFs may co-regulate one another. The clustergram provides visualization of consensus targets and enriched TFs across libraries. The networks and diagrams are exportable as publication-quality figures in vector graphics format. The ChEA3 landing page also contains information about the methods, benchmarking results, a brief tutorial, and example code for demonstrating how to submit queries through the ChEA3 API. These informational sections are accessible using the navigation bar at the top of the page, or by scrolling.
SUMMARY
ChEA3 is a web server application that predicts TFs associated with user-submitted gene sets using data from multiple orthogonal omics sources. Other sources for TF–target association are also available from the ChEA3 site, and this collection is expected to continually grow. We benchmarked the performance of six primary libraries within ChEA3 and show that integrating enrichment analyses from multiple libraries improves the recovery of the ‘correct’ upstream TFs associated with a user gene set. This data integration approach highlights the strength of combining evidence from independent sources. Interestingly, the ‘wisdom-of-the-crowd’ gene set library created from the Enrichr queries outperformed all other libraries in the global analysis of rankings. This passive form of discovery, resulting from the usage of a bioinformatics tool, can be applied to other tasks, for example, gene function prediction.
We also show that ChEA3 outperformed TF ranking when compared with other existing tools. Such a benchmarking approach should be challenged, tested by others, and used in future studies to compare similar tools. We also demonstrate how integrating data from two assay types, namely ChIP-seq and genome-wide mRNA expression, enables global analysis that can determine whether a TF is mostly an activator or a repressor. Integrating ChIP-seq and genome-wide mRNA expression from TF perturbation studies can be used to construct signed directed networks that can be further analyzed to better understand the topology of the human TF regulatory network. Overall, ChEA3 can guide many future experimental and computational studies that aim to explore gene expression regulatory mechanisms in mammalian cells.
Supplementary Material
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
NIH [U54-HL127624 (LINCS-DCIC), U24-CA224260 (IDG-KMC), T32-GM062754 (Pharmacological Sciences Training Program), and OT3-OD025467 (NIH Data Commons)]. Funding for open access charge: NIH [U54-HL127624 (LINCS-DCIC)].
Conflict of interest statement. None declared.
REFERENCES
- 1. Lambert S.A., Jolma A., Campitelli L.F., Das P.K., Yin Y., Albu M., Chen X., Taipale J., Hughes T.R., Weirauch M.T.. The human transcription factors. Cell. 2018; 172:650–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Cusanovich D.A., Pavlovic B., Pritchard J.K., Gilad Y.. The functional consequences of variation in transcription factor binding. PLos Genet. 2014; 10:e1004226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Jolma A., Yan J., Whitington T., Toivonen J., Nitta K.R., Rastas P., Morgunova E., Enge M., Taipale M., Wei G. et al.. DNA-binding specificities of human transcription factors. Cell. 2013; 152:327–339. [DOI] [PubMed] [Google Scholar]
- 4. Margolin A.A., Nemenman I., Basso K., Wiggins C., Stolovitzky G., Dalla Favera R., Califano A.. ARACNE: an algorithm for the reconstruction of gene regulatory networks in a mammalian cellular context. BMC Bioinformatics. 2006; 7(Suppl. 1):S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Alvarez M.J., Shen Y., Giorgi F.M., Lachmann A., Ding B.B., Ye B.H., Califano A.. Functional characterization of somatic mutations in cancer using network-based inference of protein activity. Nat. Genet. 2016; 48:838–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Garcia-Alonso L., Iorio F., Matchan A., Fonseca N., Jaaks P., Peat G., Pignatelli M., Falcone F., Benes C.H., Dunham I. et al.. Transcription factor activities enhance markers of drug sensitivity in cancer. Cancer Res. 2018; 78:769–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Garcia-Alonso L., Ibrahim M.M., Turei D., Saez-Rodriguez J.. Benchmark and integration of resources for the estimation of human transcription factor activities. 2018; bioRxiv doi: 10.1101/337915, 18 June 2018, preprint: not peer reviewed. [DOI] [PMC free article] [PubMed]
- 8. Wang Z., Civelek M., Miller C.L., Sheffield N.C., Guertin M.J., Zang C.. BART: a transcription factor prediction tool with query gene sets or epigenomic profiles. Bioinformatics. 2018; 34:2867–2869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Puente-Santamaria L., del Peso L.. TFEA.ChIP: a tool kit for transcription factor binding site enrichment analysis capitalizing on ChIP-seq datasets. 2018; bioRxiv doi: 10.1101/303651, 18 April 2018, preprint: not peer reviewed. [DOI] [PubMed]
- 10. Kwon A.T., Arenillas D.J., Worsley Hunt R., Wasserman W.W.. oPOSSUM-3: advanced analysis of regulatory motif over-representation across genes or ChIP-Seq datasets. G3 (Bethesda). 2012; 2:987–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Roopra A. MAGICACT: A tool for predicting transcription factors and cofactors driving gene sets using ENCODE data. 2019; bioRxiv doi: 10.1101/492744, 07 January 2019, preprint: not peer reviewed. [DOI] [PMC free article] [PubMed]
- 12. Consortium G.T. The genotype-tissue expression (GTEx) project. Nat. Genet. 2013; 45:580–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mei S., Qin Q., Wu Q., Sun H., Zheng R., Zang C., Zhu M., Wu J., Shi X., Taing L.. Cistrome Data Browser: a data portal for ChIP-Seq and chromatin accessibility data in human and mouse. Nucleic Acids Res. 2017; 45:D658–D662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wang S., Zang C., Xiao T., Fan J., Mei S., Qin Q., Wu Q., Li X., Xu K., He H.H. et al.. Modeling cis-regulation with a compendium of genome-wide histone H3K27ac profiles. Genome Res. 2016; 26:1417–1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S. et al.. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U.S.A. 2005; 102:15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lachmann A., Xu H., Krishnan J., Berger S.I., Mazloom A.R., Ma’ayan A.. ChEA: transcription factor regulation inferred from integrating genome-wide ChIP-X experiments. Bioinformatics. 2010; 26:2438–2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kou Y., Chen E.Y., Clark N.R., Duan Q., Tan C.M., Ma’ayan A.. CD-ARES 2013: Availability, Reliability, and Security in Information Systems and HCI. 2013; Berlin, Heidelberg: Springer; 416–430. [Google Scholar]
- 18. Wang Z., Monteiro C.D., Jagodnik K.M., Fernandez N.F., Gundersen G.W., Rouillard A.D., Jenkins S.L., Feldmann A.S., Hu K.S., McDermott M.G. et al.. Extraction and analysis of signatures from the Gene Expression Omnibus by the crowd. Nat. Commun. 2016; 7:12846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Edgar R., Domrachev M., Lash A.E.. Gene expression omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002; 30:207–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lachmann A., Torre D., Keenan A.B., Jagodnik K.M., Lee H.J., Wang L., Silverstein M.C., Ma’ayan A.. Massive mining of publicly available RNA-seq data from human and mouse. Nat. Commun. 2018; 9:1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Braschi B., Denny P., Gray K., Jones T., Seal R., Tweedie S., Yates B., Bruford E.. Genenames.org: the HGNC and VGNC resources in 2019. Nucleic Acids Res. 2019; 47:D786–D792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Davis C.A., Hitz B.C., Sloan C.A., Chan E.T., Davidson J.M., Gabdank I., Hilton J.A., Jain K., Baymuradov U.K., Narayanan A.K. et al.. The encyclopedia of DNA elements (ENCODE): data portal update. Nucleic Acids Res. 2018; 46:D794–D801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chen E.Y., Tan C.M., Kou Y., Duan Q., Wang Z., Meirelles G.V., Clark N.R., Ma’ayan A.. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics. 2013; 14:128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kuleshov M.V., Jones M.R., Rouillard A.D., Fernandez N.F., Duan Q., Wang Z., Koplev S., Jenkins S.L., Jagodnik K.M., Lachmann A. et al.. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016; 44:W90–W97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhang Y., Liu T., Meyer C.A., Eeckhoute J., Johnson D.S., Bernstein B.E., Nusbaum C., Myers R.M., Brown M., Li W. et al.. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008; 9:R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cheneby J., Gheorghe M., Artufel M., Mathelier A., Ballester B.. ReMap 2018: an updated atlas of regulatory regions from an integrative analysis of DNA-binding ChIP-seq experiments. Nucleic Acids Res. 2018; 46:D267–D275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Clark N.R., Hu K.S., Feldmann A.S., Kou Y., Chen E.Y., Duan Q., Ma’ayan A.. The characteristic direction: a geometrical approach to identify differentially expressed genes. BMC Bioinformatics. 2014; 15:79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lachmann A., Giorgi F.M., Lopez G., Califano A.. ARACNe-AP: gene network reverse engineering through adaptive partitioning inference of mutual information. Bioinformatics. 2016; 32:2233–2235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. De Volder K. JQuery: a generic code browser with a declarative configuration language. PADL 2006: Practical Aspects of Declarative Languages. 2006; 88–102.Lect. Notes Comput. Sci.. [Google Scholar]
- 30. Spurlock J. Bootstrap: Responsive Web Development. 2013; Sebastopol: O’Reilly Media. [Google Scholar]
- 31. Bostock M., Ogievetsky V., Heer J.. D3 data-driven documents. IEEE Trans. Vis. Comput. Graph. 2011; 17:2301–2309. [DOI] [PubMed] [Google Scholar]
- 32. Merkel D. Docker: lightweight Linux containers for consistent development and deployment. Linux J. 2014; 2014:2. [Google Scholar]
- 33. Langfelder P., Horvath S.. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008; 9:559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Shannon P., Markiel A., Ozier O., Baliga N.S., Wang J.T., Ramage D., Amin N., Schwikowski B., Ideker T.. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003; 13:2498–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lachmann A., Xie Z., Ma’ayan A.. Elysium: RNA-seq alignment in the cloud. 2018; bioRxiv doi: 10.1101/382937, 02 August 2018, preprint: not peer reviewed. [DOI]
- 36. Fernandez N.F., Gundersen G.W., Rahman A., Grimes M.L., Rikova K., Hornbeck P., Ma’ayan A.. Clustergrammer, a web-based heatmap visualization and analysis tool for high-dimensional biological data. Sci. Data. 2017; 4:170151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Teytelman L., Thurtle D.M., Rine J., van Oudenaarden A.. Highly expressed loci are vulnerable to misleading ChIP localization of multiple unrelated proteins. Proc. Natl. Acad. Sci. U.S.A. 2013; 110:18602–18607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sonawane A.R., Platig J., Fagny M., Chen C.Y., Paulson J.N., Lopes-Ramos C.M., DeMeo D.L., Quackenbush J., Glass K., Kuijjer M.L.. Understanding tissue-specific gene regulation. Cell Rep. 2017; 21:1077–1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bulger M., Groudine M.. Functional and mechanistic diversity of distal transcription enhancers. Cell. 2011; 144:327–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Alvarez M.J. 2018; 1.18.0 ed. Bioconductor, pp. R package. [Google Scholar]
- 41. Lefebvre C., Rajbhandari P., Alvarez M.J., Bandaru P., Lim W.K., Sato M., Wang K., Sumazin P., Kustagi M., Bisikirska B.C. et al.. A human B-cell interactome identifies MYB and FOXM1 as master regulators of proliferation in germinal centers. Mol. Syst. Biol. 2010; 6:377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chung J.H., Whiteley M., Felsenfeld G.. A 5′ element of the chicken beta-globin domain serves as an insulator in human erythroid cells and protects against position effect in Drosophila. Cell. 1993; 74:505–514. [DOI] [PubMed] [Google Scholar]
- 43. Vostrov A.A., Quitschke W.W.. The zinc finger protein CTCF binds to the APBbeta domain of the amyloid beta-protein precursor promoter. Evidence for a role in transcriptional activation. J. Biol. Chem. 1997; 272:33353–33359. [DOI] [PubMed] [Google Scholar]
- 44. Ren G., Jin W., Cui K., Rodrigez J., Hu G., Zhang Z., Larson D.R., Zhao K.. CTCF-mediated enhancer-promoter interaction is a critical regulator of cell-to-cell variation of gene expression. Mol. Cell. 2017; 67:1049–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wiese K.E., Walz S., von Eyss B., Wolf E., Athineos D., Sansom O., Eilers M.. The role of MIZ-1 in MYC-dependent tumorigenesis. Cold Spring Harb. Perspect. Med. 2013; 3:a014290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Han H., Cho J.W., Lee S., Yun A., Kim H., Bae D., Yang S., Kim C.Y., Lee M., Kim E. et al.. TRRUST v2: an expanded reference database of human and mouse transcriptional regulatory interactions. Nucleic Acids Res. 2018; 46:D380–D386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kulakovskiy I.V., Vorontsov I.E., Yevshin I.S., Sharipov R.N., Fedorova A.D., Rumynskiy E.I., Medvedeva Y.A., Magana-Mora A., Bajic V.B., Papatsenko D.A. et al.. HOCOMOCO: towards a complete collection of transcription factor binding models for human and mouse via large-scale ChIP-Seq analysis. Nucleic Acids Res. 2018; 46:D252–D259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Khan A., Fornes O., Stigliani A., Gheorghe M., Castro-Mondragon J.A., van der Lee R., Bessy A., Cheneby J., Kulkarni S.R., Tan G. et al.. JASPAR 2018: update of the open-access database of transcription factor binding profiles and its web framework. Nucleic Acids Res. 2018; 46:D1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.