

Conspectus
Protein kinases are enzymes that catalyze the covalent transfer of γ-phosphate of an adenosine triphosphate (ATP) molecule onto a tyrosine, serine, threonine, or histidine residue in the substrate, and thus send a chemical signal to networks of downstream proteins. They are important cellular signaling enzymes that regulate cell growth, proliferation, metabolism, differentiation and migration. Unregulated protein kinase activity is often associated with a wide range of diseases, therefore, making protein kinases major therapeutic targets. A prototypical system of central interest to understand the regulation of kinase activity is provided by tyrosine kinase c-Src, which belongs to the family of Src-related non-receptor tyrosine kinases (SFKs). Although the broad picture of autoinhibition via the regulatory domains and via the phosphorylation of the C-terminal tail is well characterized from a structural point of view, a detailed mechanistic understanding at the atomic-level is lacking. Advanced computational methods based on all-atom molecular dynamics (MD) simulations are employed to advance our understanding of tyrosine kinase activation. The computational studies suggest that the isolated kinase domain (KD) is energetically most favorable in the inactive conformation when the activation loop (A-loop) of the KD is not phosphorylated. The KD makes transient visits to a catalytically competent active-like conformation. The process of bimolecular trans-autophosphorylation of the A-loop eventually locks the KD in the active state. Activating point mutations may act by slightly increasing the population of the active-like conformation, enhancing the availability of the A-loop to be phosphorylated. The Src-homology 2 (SH2) and Src-homology 3 (SH3) regulatory domains, depending upon their configuration, either promote the inactive or the active state of the kinase domain. In addition to the roles played by the SH3, SH2 and KD, the Src-homology 4-Unique domain (SH4-U) region also serve as a key moderator of substrate specificity and kinase function. Thus, a fundamental understanding of the conformational propensity of the SH4-U region and how this affects the association to the membrane surface are likely to lead to the discovery of new intermediate states and alternate strategies for inhibition of kinase activity for drug discovery. The existence of a multitude of KD conformations poses a great challenge aimed at the design of specific inhibitors. One promising computational strategy to explore the conformational flexibility of the KD is to construct Markov state models from aggregated MD data.
Graphical Abstract
INTRODUCTION
A prototypical system of central interest to understand the regulation of kinase activity is provided by tyrosine kinase c-Src, which belongs to the SFKs. All nine members of SFKs (Src, Yes, Fyn, Lyn, Lck, Blk, Hck, Fgr, and Yrk) are multidomain proteins that are highly homologous sharing a similar regulatory mechanism (see Fig. 1).1 Src kinases function as complex allosteric molecular switches whose catalytic activity can be modulated in response to specific cellular signals, such as growth factors or cytokines binding to membrane-bound receptor proteins. Post-translational modifications such as phosphorylation are also crucial to the regulation of the activity. Phosphorylated Tyr527 (chicken c-Src numbering system) in the C-terminus tail of the KD plays a central role in down-regulating the kinase activity via interaction with the SH2 domain, while phosphorylation of Tyr416, a residue located in the A-loop (residue 404 to 424) near the active site in the KD, up-regulates the kinase activity.
Figure 1.

Primary structure and functional state of c-Src kinase. (a) The assembled autoinhibited conformation (pdb id:2SRC).2 (b) The so-called reassembled unphosphorylated active-like conformation (pdb id:1Y57).4 (c) The full-length structure comprises a short membrane-associating segment (SH4) and a “unique” (U) segment whose sequence varies widely, followed by two regulatory modules (SH2 and SH3), a linker region (L), and finally a catalytic kinase domain (KD or SH1). The domains are termed SH (src homology) domains, and are distinguished from one another by a numbering system beginning at the C-terminus.
X-ray crystallographic structures of the autoinhibited c-Src2 and Hck3 show that the SH3 and SH2 domains are assembled on the backside of the KD, with the SH2 domain binding to phosphorylated Tyr527 and the SH3 domain binding to the PxxP region of the linker connecting the SH2 and the KD (Fig. 1a). Another X-ray structure of c-Src shows the SH2 and SH3 “re-assembled” on the top of the KD instead of on the backside (Fig. 1b).4 While the A-loop is unphosphorylated, the KD is in an active-like state.5 The autoinhibited conformation of the enzyme is stabilized by the SH2-SH3 domains, which appear to act as a “clamp” that locks the KD in the inactive conformation. Disruption of either the SH2-pY527 or SH3-linker interaction destabilizes the autoinhibitory conformation, leading eventually to the activation of the enzyme. Conceivably, there could be additional inactive conformations, either by altering the configuration of critical residues in the active site, or by disrupting the binding of ATP or the accessibility of the substrate.
COMPUTATIONAL STUDIES OF TYROSINE KINASES
There have been numerous molecular dynamics (MD) studies of kinases from the Src family,6–17 as well as of protein kinase A (PKA),18,19 Abelson tyrosine kinase (c-Abl),20–24 epidermal growth factor receptor (EGFR),25–27 Janus kinase 2 (JAK2) tyrosine kinase,28 and hepatocyte growth factor receptor (HGFR, also known as c-Met).29 While unbiased MD trajectories can be informative, they are somewhat limited in their ability to quantitatively characterize complex processes taking place on very long time scales. However, the scope of MD can be considerably extended by various computational strategies such as umbrella sampling (US),9,15,30,31 metadynamics,21,27 and alchemical free energy perturbation MD simulations (FEP/MD).22–24 The string method has also been an effective approach to characterize large-scale conformational transition pathways.13,32,33 Lastly, a powerful strategy combining the information accumulated from a large number of MD simulations is provided by Markov state models (MSMs).16,19
a). Activating transition in the isolated KD
Converting the KD from an inactive to an active catalytically competent state requires an intricate conformational change comprising the opening the A-loop to an extended state and an inward rotation of the αC-helix (residue 304 to 318) resulting in the formation of a salt bridge between E310 and K295 (Fig. 2a). The rotation of the αC-helix also alters the hydrophobic regulatory (R-) spine (Fig. 2b and 2c).34 Characterizing the properties of the isolated KD is of considerable interest as it is constitutively active in solution according to experiment.5 Relying on a combination of MD simulations and kinetic assays in a study of Lyn kinase, Ozkirimli et al. first showed that an electrostatic network comprising of six polar residues act as a switch during activation.7,8 Further work on Hck kinase by Banavali and Roux highlighted the presence of charge asymmetry in the A-loop associated with the activation process.10,11 A multistate coarse-grained (CG) Gõ-like model was developed to simulate the activation of the KD of Hck kinase.35 A similar CG model of Lyn kinase was developed to search the optimal path for the same transition using the maximum flux transition path (MFTP) method.32
Figure 2.

Representation of the conformational changes in the KD of c-Src KD upon activation. (a) Comparison of the active (yellow) and the inactive (green) conformations. (b) The catalytic (yellow) and the regulatory (red) spines in the active (b) and inactive (c) state of the KD.
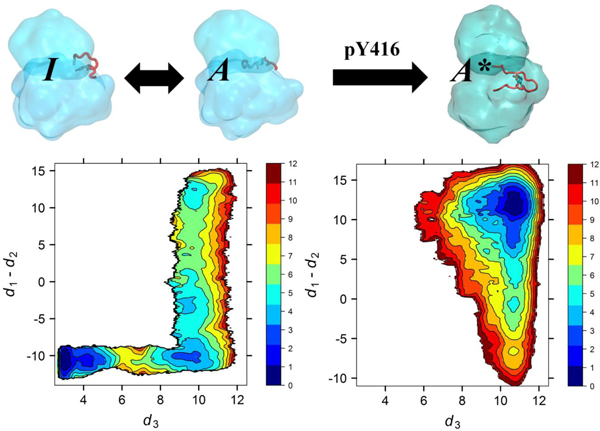
Yang et al constructed a low resolution free energy landscape for the activation of Hck kinase from all-atom MD simulations with explicit solvent, first pointing to the existence of intermediate conformations along the activation pathway.12 To gain further mechanistic insight at the atomic level about the inactive-active conformational transition, Gan et al.13 determine the optimal minimum free energy activation pathway of the isolated KD of wild-type (WT) c-Src kinase. The string pathway connects the inactive “I” conformation and the unphosporylated active-like “A” conformation. The pathway showed that the “I” to “A” conformational transition takes place as a two-step process in which the A-loop opens up first, followed by the rotation of the αC-helix.13 A MSM for the activation process of the same system was constructed using aggregated sampling generated from massively distributed MD,16 including several trajectories started from configurations taken along the transition pathway determined using the string method.13 The MSM confirmed the transition pathway and revealed specific intermediate conformations that could serve as potential targets for drug design.16 The complex nature of the activating transition from the MSM was analyzed using transition pathway theory, showing it takes place via a dense set of intermediate microstates distributed within a fairly broad multi-dimensional conformational “reaction tube” connecting the “I” and “A” conformations.36
The free energy landscape of the isolated KD of c-Src is also consistent with a two-step process for the activating transition (Fig. 3a).15 When the A-loop is unphosphorylated, the inactive conformation is more favorable even though rare occurrences of an active-like conformation are permitted (Fig. 3a), a behavior observed in other SFKs.12,32 Consistent with this result, the equilibrium probability of the unphosphorylated active-like “A” state deduced from the MSM of WT c-Src also suggests that this state is visited only transiently.16 It is noteworthy that the isolated KD (without its regulatory domains), is constitutively active according to experiment.5 Presumably, the KD needs to be trans-phosphorylated via a bimolecular encounter with another kinase in order to stabilize the fully active state “A*”. Indeed, the calculated free energy landscape shows that phosphorylation of Y416 in the A-loop essentially “locks” the kinase into its catalytically competent conformation (Fig. 3b).15 Nonetheless, while this conceptual framework is reasonable, it is unclear whether it can be reconciled with experimental observations: the timescale of the interconversion between the “I” and “A” state from the MSM is on the order of 100 μs whereas the experimentally observed timescale for Src-family tyrosine kinase autophosphorylation at residue Tyr416 is on the order of minutes. To relate these vastly disparate timescales, a simple kinetic model,
was constructed using the data extracted from atomistic simulations and a reasonable estimate for the bimolecular rate of kinase phosphorylation, ktrans-p.16 A similar conceptual framework was used to explain the functional behavior of the activating W260A mutant.37 At first glance, the computational result seems to be inconsistent with experiments: while the W260A mutation in c-Src is markedly up-regulated,38 the free energy cost to reach the active-like unphosphorylated state A is only slightly smaller in the W260A mutant compared with WT.37 Nonetheless, such a small difference in stability accounts for the large increase in the activity of the mutant that is observed experimentally.37
Figure 3.

Free energy landscapes of inactive-to-active transition in WT c-Src calculated from umbrella sampling simulations. The exact definition of the collective variables can be found in ref. 15; roughly, the x-axis tracks the opening of the A-loop from the inactive closed state (left) to the open active state (right), and the y-axis tracks the rotation of the αC helix from the outward inactive state (bottom) to inward active state (top). Shown are the free energy landscape for the (a) isolated KD with Y416 in A-loop unphosphorylated,15 (b) the isolated KD with Y416 in A-loop phosphorylated,15 (c) the assembled SH3-SH2-linker (Fig. 1a),17 and (d) the reassembled SH3-SH2-linker (Fig. 1b).17
The activating transition in the KD has also been studied in other tyrosine kinases, suggesting conservation in the conformational landscapes within the KD of tyrosine kinases. Shan et al. used long unbiased all-atom MD simulations for the unphosphorylated WT-EGFR, starting from the active-like conformation, and observed spontaneous transition to the inactive conformation.39 In retrospect, the spontaneous transition observed in unbiased MD is consistent with the free energy landscape of the isolated KD, which shows that the inactivated state is the most stable.16,31 Sutto and Gervasio used metadynamics simulations to investigate the impact of activating point mutations on the conformational equilibrium between the inactive and the active-like conformations of EGFR.27
The perspective emerging from these computational results about the structural factors associated with the up- and down-regulation of the enzyme, in which the unphosphorylated KD makes brief transient visits to an active-like state until it is locked a catalytically competent conformation via trans-phosphorylation of Y416, is tantalizing. Nevertheless, there are some discrepancies with experimental work, which shows that phosphorylation of Y416 only boosts kinase activity ~2.5-fold. Furthermore, the kinase with unphosphorylated Y416, and even the Y416F/A mutants, are active in solution.40,41 Explaining all differences between the computations, which are necessarily imperfect, and the experiments, which are subject to interpretation, is difficult. In part, it is possible that the presence of the substrate, a factor that is not taken into account in the computations, would affect the apparent population of the active and inactive enzyme in experiments. Alternatively, it is also possible that the computations, limited by the underlying force field, captured the relative conformational shift between the active and inactive states only qualitatively but not quantitatively.
b). Multidomain allosteric regulatory mechanism
Although the broad picture of autoinhibition of SFKs via the SH2 and SH3 regulatory domains is well characterized from a structural point of view, a detailed mechanistic understanding at the atomic-level is nonetheless still lacking. Young et al6 found that the activity of c-Src kinase is up-regulated by substitution of 3 residues by glycine in the connector between the SH3 and SH2 domains. To explain this puzzling observation, it was proposed that the connector acts as a structural “snap-lock” that is intrinsically rigid and required for the SH3 and SH2 modules to serve as an effective “clamp”, inhibiting the activity of the catalytic domain in the assembled state of the enzyme. This mechanism implicitly assumes that the mutations increasing the flexibility of the connector have little effect on the disassembled state. However, US computations on the SH2-SH3 tandem showed that the mutations negligibly affect the free energy of assembled states but caused a shift in the equilibrium from the assembled (inactive) state toward the disassembled (active) state.30 The wide range of multidomain configurational states of Hck in response to SH2- and SH3-binding peptides were further characterized from a Bayesian analysis combining small-angle X-ray solution scattering (SAXS) data with CG simulations.42 The conformational variability accessible to the multidomain kinase in solution was found to far exceed that reported through available X-Ray crystallographic structures. Thus, while one crystal structure of c-Src showing a re-assembled unphosphorylated active-like conformation is available,4 a multitude of active states might be accessible.
To elucidate the nature of the allosteric coupling between the regulatory SH2 and SH3 modules and the active or inactive conformation of the KD, the inactive-to-active transition pathway was recently determined in a SH3-SH2-linker-KD construct of c-Src using string method with swarms-of-trajectories.17 The calculated free energy landscapes show that the SH3-SH2-linker tandem, depending on its bound orientation, promotes either the inactive (Fig. 3c) or the active (Fig. 3d) state of the KD. Using the isolated c-Src KD as a baseline for comparison (Fig. 3a), the computations show that a significant thermodynamic penalty on the active conformation of the KD is imposed when the SH3-SH2-linker tandem is in the assembled conformation, which essentially inhibits the activation via a population shift mechanism (Fig. 3c). Similar to the effect of phosphorylation of the A-loop (Fig 3b), the active state is stabilized when the SH3-SH2-linker tandem is in the re-assembled conformation (Fig. 3d). Moreover, the computations showed that the regulatory structural information from the SH2-SH3 tandem is allosterically transmitted via the linker region connecting SH2 and KD.17 The SH2 and SH3 domains are essentially to stabilize the linker region in an “inhibiting conformation”, but it is the linker region itself that is actually responsible for locking the KD in an inactive conformation. These results are consistent an earlier computational study.9 The essential role of the linker region in the allosteric regulation of tyrosine kinase activity is well established from experiments.43
c). DFG motif and binding specificity of kinase inhibitors
Gleevec is a potent inhibitor of several tyrosine kinases including c-Abl and c-Kit but not of the homologous SFKs. The mechanism and molecular determinants of its specificity, however, still remain elusive. Initial results suggested that the drug binds to a distinctive inactive conformation of the activation loop unique to c-Abl in which an Asp-Phe-Gly structural motif (DFG-motif) along the A-loop adopts an uncommon “out” conformation.44 The idea that Gleevec can bind exclusively to a DFG-out conformation led to a “conformational selection” mechanism of binding specificity. The DFG-out conformation disrupts ATP binding and disassembles both the R-spine and the catalytic spine (Fig. 2b).45 The crystal structure of Gleevec in complex with the c-Src kinase domain subsequently showed that it also adopted the same inactive DFG-out conformation.46 This led to the view that there must be some “thermodynamic penalty” associated with the DFG-out conformation.47 Multiple structures of kinase show that Gleevec typically binds with the DFG-out, with the structure of Gleevec in complex with the Syk tyrosine kinase appears to be the only known exception to date (pdb id: 1XBB).48 The DFG-in and DFG-out conformations have been found to co-exist with multiple conformations of the αC-helix and the phosphate-binding (P-) loop in c-Abl.49 Analysis of structures from the PDB database shows that the DFG-out conformation is present in a large number of protein kinases.50
Because of its great importance, the conformational transition of the DFG motif and its impact on the binding specificity of inhibitors has been the subject of many computational studies.20–22,29,31,33,51,52 Generally, the results from computations support the concept of the thermodynamic penalty for the DFG flip. Early on, the molecular-mechanics Poisson-Boltzmann with surface area (MM/PBSA) approximation together with the relative binding free energies of Gleevec to the DFG-out conformation was used to indirectly estimate the relative free energy cost of the DFG-flip of c-Abl and c-Src.51 The observed MM/PBSA binding free energy was similar for c-Abl and c-Src,51 suggesting that the free energy cost for the DFG-out conformation must be larger in c-Src than in Abl. Lovera et al.21 determined the free energy landscape affecting the conformations of the DFG motif in WT c-Abl and c-Src using metadynamics simulations based on the Amber99SB-ILDN force field. Similarly, Lin et al.22 determined the free energy landscape affecting the conformations of the DFG motif in WT c-Abl and c-Src using US and MD simulations based on the CHARMM27-CMAP force field (Fig. 4). Despite differences in methodology and force fields, both studies showed that there is larger free energy cost on the order of 2 to 4 kcal/mol for the DFG flip in c-Src compared to c-Abl. The free energy landscapes for various c-Abl and c-Src mutants were also calculated using metadynamics simulations, showing that the DFG-out conformation is less favorable among those mutants.52 The thermodynamic penalty view is also supported by a structural propensity analysis of protein kinase using sequence-based methods.53
Figure 4.

Free energy profile of the DFG-flip in c-Src and in Abl kinases. The DFG-out conformation also displays a disassembled R-spine. Modified from ref. 31. Copyright (2015) American Chemical Society.
FEP/MD simulations were carried out to elucidate the binding specificity of Gleevec to c-Src, c-Abl, c-Kit and Lck tyrosine kinases.23 These FEP/MD calculations showed that the van der Waals dispersive interaction and the conformation of the DFG motif are important contributions to the binding specificity. In an effort to dissect the various thermodynamic contributions controlling binding specificity, a series of inhibitors based on the core scaffold of Gleevec was examined.54 One analog of Gleevec, G6G, was shown to bind equivalently to c-Abl as well as c-Src. FEP/MD calculations were used to shed light on the various hidden energetic contributions affecting the binding specificity of Gleevec and G6G.24 The results of the calculations, which agreed well with experiments, highlighted the importance the conformation of the P-loop in achieving the binding specificity of G6G (Fig. 5). However, unraveling all the hidden thermodynamic and kinetic factors affecting kinase inhibitors remains challenging. More recently, Kern and co-workers have argued that the origin of Gleevec’s binding specificity lies predominantly in a slow conformational change occurring after binding.55 They revisited the issue of binding specificity of Gleevec for c-Abl and c-Src using nuclear magnetic resonance (NMR) and stop-flow fluorescence experiments.55 A two-step binding process, comprising of a fast binding step followed by a slow step of global conformational change, was proposed based on their data. This “induced fit” view implies that the DFG-out conformation does not exist in the absence of the ligand, which is inconsistent with previous computational studies.21,22,31 The issue remains unresolved.
Figure 5.

An example of kinase inhibition by ligands. (a) Chemical structures of Gleevec and G6G. (b) Comparison of the binding mode of Gleevec and G6G in Abl and Src kinase.
SUMMARY AND OUTLOOK
SFKs regulation
The non-receptor tyrosine kinase c-Src, a prototypical model system and a representative member of the Src-family, functions as complex multidomain allosteric molecular switches. Although the broad picture of self-inhibition of c-Src via the SH2 and SH3 regulatory domains is well characterized from a structural point of view, a detailed molecular mechanism understanding is nonetheless still lacking. Advanced computational methods based on all-atom MD simulations are employed to advance our understanding of kinase activation. The computational studies suggest that the isolated KD makes transient short-lived visits to a catalytically competent active-like conformation. The process of bimolecular trans-autophosphorylation of the A-loop eventually locks the KD in the active state. Activating point mutations are able to increase the population of the active-like conformation, and thus increase the availability of A-loop to be phosphorylated in the trans-style. The SH3-SH2-linker tandem, depending upon its bound orientation, either promotes the inactive or the active state of the KD. The regulatory structural information from the SH2-SH3 tandem is allosterically transmitted via the N-terminal linker of the catalytic domain.
Conformational variants and the design of specific inhibitors
Large, allosteric proteins such as the SFKs often exhibit large-scale collective motions and can adopt multiple conformational states (e.g. Fig. 6). Knowing the conformational states themselves and the relative propensity of those states not only improves our mechanistic insights into the regulation/activation of tyrosine kinases but also facilitates designing kinase inhibitors. Therefore, the existence of multiple conformations of a target protein is a general problem that must be confronted, even when one or several X-ray structures of the target are available. Generally, our knowledge of all the relevant conformational states that can be accessed by any given target protein is either non-existent of very limited. Even when some structural information is available, it remains critical to assess the propensity of all accessible conformational variants. The issue of conformational variability is further compounded by the fact that a given inhibitor may be able to bind with different modes to different members of a family, where individual mode may be stabilized by local conformational variability. In order to be able to use a structure-based strategy with a flexible target, a number of critical questions must be answered, e.g. what is the ranking of multiple different conformations of the target protein? Which ones are energetically accessible, and thus possibly druggable? Are there conformations that could be exploited to design drug specificity? In real-world application, availability of crystallographic structures will always be helpful, but it does not alleviate all the issues. Only a computational method offers the hope to resolve this problem; a relevant conformation may be missed even when multiple crystal structures are available. One promising strategy to achieve this goal is to combine the information from a large number of relatively short simulations (100~400 ns/trajectory) to construct MSMs.
Figure 6.

Conformational variations of a few selected structural motifs in the KD taken from X-ray structures of SFKs. (a) P-loop. (b) αC-helix. (c) DFG motif. (d) A-loop.
Interaction of the SH4-Unique domain with the membrane and localization
In contrast to most catalytic enzymes, tyrosine kinases do not display a strong specificity toward their downstream substrate. For this reason, factors affecting the spatial localization of the protein kinases are most certainly of paramount importance to maintain the integrity of the biological signals. In this context, it is of special importance to note that SFKs are often associated with the cell membrane. It has long been recognized that acylation of the SH4 domain is critical for membrane association of SFKs,56 but recent evidence suggests that the U domain may also play important roles in this process.57 Furthermore, there are also indications that SH4-U region may make important contacts with the regulatory domains,58 raising the possibility that additional moderators of SFK substrate specificity and function have yet to be discovered. The N-terminal cap region in c-Abl also plays a crucial role in down-regulating the kinase activity.59 The scarcity of information about the three-dimensional (3D) configuration of full-length enzyme in its membrane-associated form raises a host of questions about the sensitivity of the signaling event to the local structural features of the SH4-U region and how this might impact the accessibility to a specific phosphorylation site in a downstream target protein (Fig. 7). Thus, a fundamental understanding of the conformational propensity of the SH4-U region and how this affects the association to the membrane surface are likely to lead to the discovery of new intermediate states and alternate strategies for inhibition of kinase activity for drug discovery.
Figure 7.

Schematic representation of a membrane-associated SFK in putative inactive and active conformations, and how the active conformation may hypothetically position the catalytic site of the kinase domain toward a specific phosphorylation site in the downstream target protein.
Acknowledgment
This work is supported by the National Cancer Institute (NCI) of the National Institutes of Health (NIH) through grant R01-CAO93577. The computations are supported in part by NIH through resources provided by the Computation Institute and the Biological Sciences Division of the University of Chicago and Argonne National Laboratory under grant S10 RR029030-01, by the Extreme Science and Engineering Discovery Environment (XSEDE) grant number OCI-1053575, and by the University of Chicago Research Computing Center. The authors declare no conflict of interest.
Biographical Information
Yilin Meng received a B.E. in 2004 in chemical engineering from Dalian University of Technology, China. He obtained a Ph.D. in chemistry from the University of Florida in 2010 under the supervision of Prof. Adrian E. Roitberg. He moved to the University of Chicago and has worked with Prof. Benoît Roux since then. His general research interest lies in protein kinases and computer-aided drug design.
Matthew Pond received a B.S. in chemistry from Pepperdine University in 2005. He then began NMR studies of hemoprotein structure and dynamics under the direction of Juliette Lecomte, for which he earned a Ph.D. in biophysics from Johns Hopkins University in 2012. He currently works at the University of Chicago in the lab of Benoît Roux where he investigates Src-family kinase regulatory mechanisms using both experimental techniques and MD simulations.
Benoît Roux was born in the city of Montreal, Canada, in 1958. In 1981, he received a B.Sc. in physics from the University of Montreal, followed by a M.Sc. in Biophysics in 1985 under the supervision of Remy Sauvé. In 1990, he obtained a Ph.D. in Biophysics from Harvard University under the direction of Martin Karplus. In the past decade, he has held positions at the University of Montréal and the Weill Medical College of Cornell University. Since 2005, he has been at the University of Chicago, where he is the Amgen Professor of Biochemistry and Molecular Biology and Professor in the Chemistry Department. He also has a joint appointment at Argonne National Laboratory, where he is Senior Computational Biologist.
References
- (1).Boggon TJ; Eck MJ Structure and regulation of Src family kinases. Oncogene 2004, 23, 7918–7927. [DOI] [PubMed] [Google Scholar]
- (2).Xu WQ; Doshi A; Lei M; Eck MJ; Harrison SC Crystal structures of c-Src reveal features of its autoinhibitory mechanism. Mol. Cell 1999, 3, 629–638. [DOI] [PubMed] [Google Scholar]
- (3).Sicheri F; Moarefi I; Kuriyan J Crystal structure of the Src family tyrosine kinase Hck. Nature 1997, 385, 602–609. [DOI] [PubMed] [Google Scholar]
- (4).Cowan-Jacob SW; Fendrich G; Manley PW; Jahnke W; Fabbro D; Liebetanz J; Meyer T The crystal structure of a c-Src complex in an active conformation suggests possible steps in c-Src activation. Structure 2005, 13, 861–871. [DOI] [PubMed] [Google Scholar]
- (5).Yamaguchi H; Hendrickson WA Structural basis for activation of human lymphocyte kinase Lck upon tyrosine phosphorylation. Nature 1996, 384, 484–489. [DOI] [PubMed] [Google Scholar]
- (6).Young MA; Gonfloni S; Superti-Furga G; Roux B; Kuriyan J Dynamic coupling between the SH2 and SH3 domains of c-Src and hck underlies their inactivation by C-terminal tyrosine phosphorylation. Cell 2001, 105, 115–126. [DOI] [PubMed] [Google Scholar]
- (7).Ozkirimli E; Post CB Src kinase activation: A switched electrostatic network. Protein Sci 2006, 15, 1051–1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Ozkirimli E; Yadav SS; Miller WT; Post CB An electrostatic network and long-range regulation of Src kinases. Protein Sci 2008, 17, 1871–1880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Banavali NK; Roux B The N-terminal end of the catalytic domain of Src kinase Hck is a conformational switch implicated in long-range allosteric regulation. Structure 2005, 13, 1715–1723. [DOI] [PubMed] [Google Scholar]
- (10).Banavali NK; Roux B Anatomy of a structural pathway for activation of the catalytic domain of Src kinase Hck. Proteins: Struct., Funct., Bioinf 2007, 67, 1096–1112. [DOI] [PubMed] [Google Scholar]
- (11).Banavali NK; Roux B Flexibility and charge asymmetry in the activation loop of Src tyrosine kinases. Proteins: Struct., Funct., Bioinf 2009, 74, 378–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Yang S; Banavali NK; Roux B Mapping the conformational transition in Src activation by cumulating the information from multiple molecular dynamics trajectories. Proc. Natl. Acad. Sci. U.S.A 2009, 106, 3776–3781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Gan W; Yang S; Roux B Atomistic view of the conformational activation of Src kinase using the string method with swarms-of-trajectories. Biophys. J 2009, 97, L8–L10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Shan YB; Kim ET; Eastwood MP; Dror RO; Seeliger MA; Shaw DE How Does a Drug Molecule Find Its Target Binding Site? J. Am. Chem. Soc 2011, 133, 9181–9183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Meng Y; Roux B Locking the Active Conformation of c-Src Kinase through the Phosphorylation of the Activation Loop. J. Mol. Biol 2014, 426, 423–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Shukla D; Meng Y; Roux B; Pande VS Activation pathway of Src kinase reveals intermediate states as targets for drug design. Nat. Commun 2014, 5, 3397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Fajer M; Meng Y; Roux B The Activation of c-Src Tyrosine Kinase: Conformational Transition Pathway and Free Energy Landscape. J. Phys. Chem. B 2016, [Epub ahead of print]. [DOI] [PMC free article] [PubMed]
- (18).Masterson LR; Cembran A; Shi L; Veglia G: Allostery and Binding Cooperativity of the Catalytic Subunit of Protein Kinase A by NMR Spectroscopy and Molecular Dynamics Simulations. In Adv. Protein Chem. Struct. Biol; Christo C, Tatyana K-C, Eds.; Academic Press, 2012; Vol. 87; pp 363–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Boras BW; Kornev A; Taylor SS; McCulloch AD Using Markov State Models to Develop a Mechanistic Understanding of Protein Kinase A Regulatory Subunit RI alpha Activation in Response to cAMP Binding. J. Biol. Chem 2014, 289, 30040–30051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Shan YB; Seeliger MA; Eastwood MP; Frank F; Xu HF; Jensen MO; Dror RO; Kuriyan J; Shaw DE A conserved protonation-dependent switch controls drug binding in the Abl kinase. Proc. Natl. Acad. Sci. U.S.A 2009, 106, 139–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Lovera S; Sutto L; Boubeva R; Scapozza L; Dolker N; Gervasio FL The Different Flexibility of c-Src and c-Abl Kinases Regulates the Accessibility of a Druggable Inactive Conformation. J. Am. Chem. Soc 2012, 134, 2496–2499. [DOI] [PubMed] [Google Scholar]
- (22).Lin YL; Meng Y; Jiang W; Roux B Explaining why Gleevec is a specific and potent inhibitor of Abl kinase. Proc. Natl. Acad. Sci. U.S.A 2013, 110, 1664–1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Lin YL; Roux B Computational Analysis of the Binding Specificity of Gleevec to Abl, c-Kit, Lck, and c-Src Tyrosine Kinases. J. Am. Chem. Soc 2013, 135, 14741–14753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Lin YL; Meng Y; Huang L; Roux B Computational Study of Gleevec and G6G Reveals Molecular Determinants of Kinase Inhibitor Selectivity. J. Am. Chem. Soc 2014, 136, 14753–14762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Arkhipov A; Shan YB; Das R; Endres NF; Eastwood MP; Wemmer DE; Kuriyan J; Shaw DE Architecture and Membrane Interactions of the EGF Receptor. Cell 2013, 152, 557–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Shan YB; Eastwood MP; Zhang XW; Kim ET; Arkhipov A; Dror RO; Jumper J; Kuriyan J; Shaw DE Oncogenic Mutations Counteract Intrinsic Disorder in the EGFR Kinase and Promote Receptor Dimerization. Cell 2012, 149, 860–870. [DOI] [PubMed] [Google Scholar]
- (27).Sutto L; Gervasio FL Effects of oncogenic mutations on the conformational free-energy landscape of EGFR kinase. Proc. Natl. Acad. Sci. U. S. A 2013, 110, 10616–10621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Shan YB; Gnanasambandan K; Ungureanu D; Kim ET; Hammaren H; Yamashita K; Silvennoinen O; Shaw DE; Hubbard SR Molecular basis for pseudokinase-dependent autoinhibition of JAK2 tyrosine kinase. Nat. Struct. Mol. Biol 2014, 21, 579–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Yan M; Wang H; Wang Q; Zhang Z; Zhang C Allosteric inhibition of c-Met kinase in sub-microsecond molecular dynamics simulations induced by its inhibitor, tivantinib. Phys. Chem. Chem. Phys 2016, 18, 10367–10374. [DOI] [PubMed] [Google Scholar]
- (30).Faraldo-Gomez JD; Roux B On the importance of a funneled energy landscape for the assembly and regulation of multidomain Src tyrosine kinases. Proc. Natl. Acad. Sci. U.S.A 2007, 104, 13643–13648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Meng Y; Lin YL; Roux B Computational study of the “DFG-flip” conformational transition in c-Abl and c-Src tyrosine kinases. J. Phys. Chem. B 2015, 119, 1443–1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Huang H; Zhao RJ; Dickson BM; Skeel RD; Post CB alpha C Helix as a Switch in the Conformational Transition of Src/CDK-like Kinase Domains. J. Phys. Chem. B 2012, 116, 4465–4475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Vashisth H; Maragliano L; Abrams CF “DFG-Flip” in the Insulin Receptor Kinase Is Facilitated by a Helical Intermediate State of the Activation Loop. Biophys. J 2012, 102, 1979–1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Kornev AP; Haste NM; Taylor SS; Ten Eyck LF Surface comparison of active and inactive protein kinases identifies a conserved activation mechanism. Proc. Natl. Acad. Sci. U.S.A 2006, 103, 17783–17788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Yang S; Roux B Src kinase conformational activation: Thermodynamics, pathways, and mechanisms. PLoS Comput. Biol 2008, 4, e1000047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Meng Y; Shukla D; Pande VS; Roux B Transition path theory analysis of c-Src kinase activation. Proc. Nat. Acad. Sci. U.S.A 2016, 113, 9193–9198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Meng Y; Roux B Computational study of the W260A activating mutant of Src tyrosine kinase. Protein Sci 2016, 25, 219–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).LaFevre-Bernt M; Sicheri F; Pico A; Porter M; Kuriyano J; Miller WT Intramolecular regulatory interactions in the Src family kinase Hck probed by mutagenesis of a conserved tryptophan residue. J. Biol. Chem 1998, 273, 32129–32134. [DOI] [PubMed] [Google Scholar]
- (39).Shan YB; Arkhipov A; Kim ET; Pan AC; Shaw DE Transitions to catalytically inactive conformations in EGFR kinase. Proc. Natl. Acad. Sci. U.S.A 2013, 110, 7270–7275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Moarefi I; LaFevre-Bernt M; Sicheri F; Huse M; Lee CH; Kuriyan J; Miller WT Activation of the Src-family tyrosine kinase Hck by SH3 domain displacement. Nature 1997, 385, 650–653. [DOI] [PubMed] [Google Scholar]
- (41).Porter M; Schindler T; Kuriyan J; Miller WT Reciprocal regulation of Hck activity by phosphorylation of Tyr(527) and Tyr(416) - Effect of introducing a high affinity intramolecular SH2 ligand. J. Biol. Chem 2000, 275, 2721–2726. [DOI] [PubMed] [Google Scholar]
- (42).Yang S; Blachowicz L; Makowski L; Roux B Multidomain assembled states of Hck tyrosine kinase in solution. Proc. Natl. Acad. Sci. U.S.A 2010, 107, 15757–15762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Gonfloni S; Williams JC; Hattula K; Weijland A; Wierenga RK; Superti-Furga G The role of the linker between the SH2 domain and catalytic domain in the regulation and function of Src. Embo J 1997, 16, 7261–7271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Schindler T; Bornmann W; Pellicena P; Miller WT; Clarkson B; Kuriyan J Structural mechanism for STI-571 inhibition of Abelson tyrosine kinase. Science 2000, 289, 1938–1942. [DOI] [PubMed] [Google Scholar]
- (45).Kornev AP; Taylor SS Defining the conserved internal architecture of a protein kinase. BBA-Proteins Proteom 2010, 1804, 440–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Seeliger MA; Nagar B; Frank F; Cao X; Henderson MN; Kuriyan J c-Src binds to the cancer drug imatinib with an inactive Abl/c-Kit conformation and a distributed thermodynamic penalty. Structure 2007, 15, 299–311. [DOI] [PubMed] [Google Scholar]
- (47).Cowan-Jacob SW; Mobitz H; Fabbro D Structural biology contributions to tyrosine kinase drug discovery. Curr. Opin. Cell Biol 2009, 21, 280–287. [DOI] [PubMed] [Google Scholar]
- (48).Atwell S; Adams JM; Badger J; Buchanan MD; Feil IK; Froning KJ; Gao X; Hendle J; Keegan K; Leon BC; Muller-Dieckmann HJ; Nienaber VL; Noland BW; Post K; Rajashankar KR; Ramos A; Russell M; Burley SK; Buchanan SG A novel mode of Gleevec binding is revealed by the structure of spleen tyrosine kinase. J. Biol. Chem 2004, 279, 55827–55832. [DOI] [PubMed] [Google Scholar]
- (49).Levinson NM; Kuchment O; Shen K; Young MA; Koldobskiy M; Karplus M; Cole PA; Kuriyan J A Src-like inactive conformation in the Abl tyrosine kinase domain. PLoS Biol 2006, 4, 753–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Vijayan RSK; He P; Modi V; Duong-Ly KC; Ma HC; Peterson JR; Dunbrack RL; Levy RM Conformational Analysis of the DFG-Out Kinase Motif and Biochemical Profiling of Structurally Validated Type II Inhibitors. J. Med. Chem 2015, 58, 466–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Aleksandrov A; Simonson T Molecular Dynamics Simulations Show That Conformational Selection Governs the Binding Preferences of Imatinib for Several Tyrosine Kinases. J. Biol. Chem 2010, 285, 13807–13815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Lovera S; Morando M; Pucheta-Martinez E; Martinez-Torrecuadrada JL; Saladino G; Gervasio FL Towards a Molecular Understanding of the Link between Imatinib Resistance and Kinase Conformational Dynamics. PLoS Comput. Biol 2015, 11, e1004578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Haldane A; Flynn WF; He P; Vijayan RSK; Levy RM Structural propensities of kinase family proteins from a Potts model of residue co-variation. Protein Sci 2016, 25, 1378–1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Seeliger MA; Ranjitkar P; Kasap C; Shan YB; Shaw DE; Shah NP; Kuriyan J; Maly DJ Equally Potent Inhibition of c-Src and Abl by Compounds that Recognize Inactive Kinase Conformations. Cancer Res 2009, 69, 2384–2392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Agafonov RV; Wilson C; Otten R; Buosi V; Kern D Energetic dissection of Gleevec’s selectivity toward human tyrosine kinases. Nat. Struct. Mol. Biol 2014, 21, 848–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Patwardhan P; Resh MD Myristoylation and Membrane Binding Regulate c-Src Stability and Kinase Activity. Mol. Cell. Biol 2010, 30, 4094–4107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Amata I; Maffei M; Pons M Phosphorylation of unique domains of Src family kinases. Front. Genet 2014, 5, 181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Maffei M; Arbesú M; Le Roux A-L; Amata I; Roche S; Pons M The SH3 Domain Acts as a Scaffold for the N-Terminal Intrinsically Disordered Regions of c-Src. Structure 2015, 23, 893–902. [DOI] [PubMed] [Google Scholar]
- (59).Nagar B; Hantschel O; Seeliger M; Davies JM; Weiss WI; Superti-Furga G; Kuriyan J Organization of the SH3-SH2 unit in active and inactive forms of the c-Abl tyrosine kinase. Mol. Cell 2006, 21, 787–798. [DOI] [PubMed] [Google Scholar]