

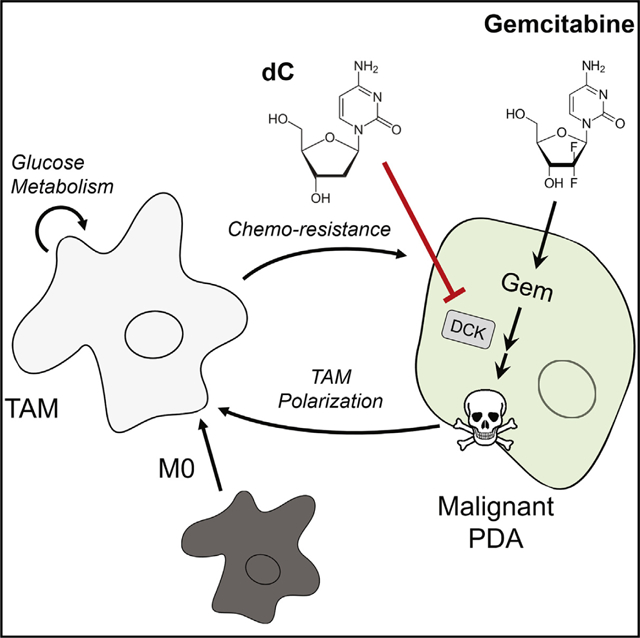
SUMMARY
Pancreatic ductal adenocarcinoma (PDA) is characterized by abundant infiltration of tumor-associated macrophages (TAMs). TAMs have been reported to drive resistance to gemcitabine, a frontline chemotherapy in PDA, though the mechanism of this resistance remains unclear. Profiling metabolite exchange, we demonstrate that macrophages programmed by PDA cells release a spectrum of pyrimidine species. These include deoxycytidine, which inhibits gemcitabine through molecular competition at the level of drug uptake and metabolism. Accordingly, genetic or pharmacological depletion of TAMs in murine models of PDA sensitizes these tumors to gemcitabine. Consistent with this, patients with low macrophage burden demonstrate superior response to gemcitabine treatment. Together, these findings provide insights into the role of macrophages in pancreatic cancer therapy and have potential to inform the design of future treatments. Additionally, we report that pyrimidine release is a general function of alternatively activated macrophage cells, suggesting an unknown physiological role of pyrimidine exchange by immune cells.
In Brief
Macrophages are present in high abundance in pancreatic ductal adenocarcinoma. Halbrook et al. identify that alternatively activated macrophages release a spectrum of pyrimidine nucleosides that are consumed by pancreatic cancer cells. Among these, deoxycytidine can directly compete with gemcitabine, hindering its efficiency as a chemotherapy.
Graphical Abstract
INTRODUCTION
Pancreatic ductal adenocarcinoma (PDA) has emerged as one of the most lethal human cancers (Siegel et al., 2018). It is characterized by a dense matrix rich with activated fibroblasts and tumor-associated macrophages (TAMs). This inhibits vascularization and/or vessel function and thus presumably delivery of therapeutic agents (Feig et al., 2012; Olive et al., 2009; Provenzano et al., 2012; Rhim et al., 2014). Patients respond poorly to current treatments, where the degree of therapeutic resistance correlates with the fibroinflammatory response (Koay et al., 2014) and the composition is predictive of survival (Mahajan et al., 2018).
Nutrient acquisition and metabolic pathways are rewired in PDA cells to support survival and growth in this environment (Halbrook and Lyssiotis, 2017; Perera and Bardeesy, 2015). TAMs constitute a large proportion of the overall cellularity and are important regulators of the tumor microenvironment (Di Caro et al., 2016). TAM abundance correlates with a worse response to therapy in PDA (Di Caro et al., 2016), and systemic TAM depletion can block pancreatic tumorigenesis and regress established PDA tumors (Mitchem et al., 2013; Zhang et al., 2017a).
In physiological settings, inflammatory and anti-inflammatory properties of macrophages can be directed and mediated by cellular metabolism programs (Van den Bossche et al., 2017). Similarly, TAM functions are also shaped by cell-intrinsic metabolism and the functional consequences of this metabolism on the tumor microenvironment (Lyssiotis and Kimmelman, 2017; Murray, 2016). Based on this and the abundance of TAMs in PDA tumors, we hypothesized that TAMs may influence therapeutic response in PDA tumors through metabolic crosstalk with cancer cells.
RESULTS
Macrophage-Released Pyrimidines Confer Gemcitabine Resistance to PDA Cells
To study metabolite crosstalk between macrophages and PDA cells, we generated tumor-educated macrophages (TEMs) by culturing murine bone-marrow-derived macrophages (BMDMs) in PDA conditioned media (CM) (Figure S1A) (Zhang et al., 2017a, 2017b). In parallel, BMDMs were directed to a classically activated phenotype (M1) with lipopolysaccharide (LPS) or polarized to an alternatively activated phenotype (M2) with interleukin 4 (IL4) (Van den Bossche et al., 2017). CM from these cultures were profiled using liquid chromatography-coupled tandem mass spectrometry metabolomics (Figure 1A), which revealed accumulation of pyrimidine nucleosides and nucleobases in TEM CM (Figure 1B). In contrast, we did not observe a similar profile for release of purine species beyond adenosine. When PDA cells were then incubated in TEM CM, many of the accumulated pyrimidines were depleted (Figure 1C), suggesting a directional transfer of these metabolites.
Figure 1. TEMs Confer Gem Resistance to PDA Cells through dC Release.
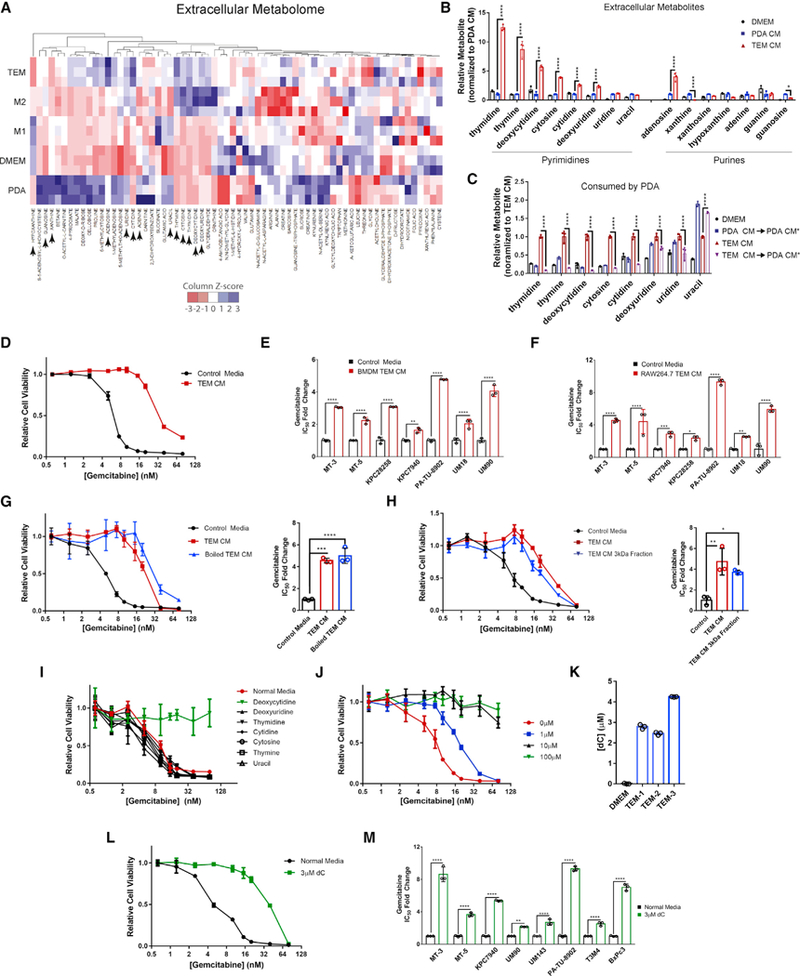
(A) Heat map of metabolites in the CM of TEM, M2, M1, iKras*3 PDA cell line, and DMEM. Blue represents higher relative metabolite, red represents lower relative metabolite. Metabolites with arrow are presented in (B) (n = 3).
(B) Relative nucleoside species found in DMEM, PDA CM, and TEM CM, normalized to PDA CM (n = 3).
(C) Relative pyrimidine nucleosides in DMEM, TEM CM, or PDA CM after 24 h of culture with iKras*3 PDA cells, normalized to initial TEM CM (n = 3). PDA CM* denotes post-culture with PDA cells.
(D) Relative viability of MT3-KPC cells treated with Gem in the presence of 75% TEM CM versus control (n = 3).
(E) Relative fold change of Gem IC50 between control or 75% CM from bone-marrow-derived macrophages (BMDM) polarized to TEMs (n = 3).
(F) Relative fold change of Gem IC50 between control or 75% CM from RAW 264.7 macrophages polarized to TEMs (n = 3).
(G) Relative viability and IC50 of MT3-KPC cells treated with Gem in the presence of 75% TEM CM, heat denatured TEM CM, or control (n = 3).
(H) Relative viability and IC50 of MT3-KPC cells treated with Gem in the presence of 75% TEM CM, 75% TEM CM passed through a 3 kDa filter, or control (n = 3).
(I) Relative viability of MT3-KPC cells treated with Gem in the presence of 100 μM of the indicated pyrimidine in DMEM or DMEM alone (n = 3).
(J) Relative viability of MT3-KPC cells treated with Gem in the presence of the indicated concentration of dC in DMEM (n = 3).
(K) Calculated abundance of dC from TEM CM generated by 3 independent TEM preparations, or DMEM, determined via LC-MS/MS (n = 3).
(L) Relative viability of MT3-KPC cells treated with Gem in the presence of 3 μM dC versus DMEM (n = 3).
(M) Relative fold change of Gem IC50 for cells treated with 3 μM dC versus DMEM (n = 3).
Error bars represent mean ± SD, *p ≤ 0.05; **p ≤ 0.01; ***p < 0.001; ****p < 0.0001. See also Figure S1; Table S1.
This observation was provocative for several reasons. First, pyrimidine release and their transfer among cells have neither been previously reported nor characterized. Further, gemcitabine (Gem), a pyrimidine anti-nucleoside, has long served as a core component of chemotherapy treatments available to patients. Gem resistance has been linked to TAMs in PDA (Mitchem et al., 2013), although the mechanism behind this link remains unclear. Thus, we hypothesized that pyrimidine nucleosides released by TEMs may directly confer Gem resistance to PDA cells.
Supporting this, we observed a shift in Gem sensitivity with TEM CM across a large panel of cell lines including primary human patient-derived cultures (Figures 1D, 1E, and S1B) and with TEM CM generated from a murine macrophage cell line (Figures 1F and S1C). TEM CM retained the ability to confer Gem resistance after boiling or passage through a 3 kDa cutoff filter (Figures 1G and 1H), providing evidence for a metabolite factor driving the chemoresistance phenotype. To determine if pyrimidines found in TEM media confer Gem resistance, we treated PDA cells with Gem in the presence of a high concentration (100 μM) of these nucleosides (Figure 1l). Among these, we found deoxycytidine (dC) uniquely blocked the cytotoxic activity of Gem in PDA cells and this response was dose dependent (Figure 1J). We quantitated dC and other pyrimidines in TEM CM by mass spectrometry and observed them to be in the micromolar range (Figure 1K; Table S1). Importantly, we were able to phenocopy the Gem-resistance activity of TEM CM by simply supplementing growth media with 3 μM dC (Figures 1L, 1M, and S1D), the concentration observed in TEM CM.
Pyrimidine Release Is a Property of Alternatively Activated Macrophage Metabolism
In addition to TEMs, M2 macrophages also released dC into CM (Figure 1A), which conferred Gem resistance to PDA cells (Figure 2A), neither of which was observed for M1 macrophages. Given this dichotomy, we were interested in understanding how their metabolic programs were wired to facilitate pyrimidine release. Consistent with previous reports, M2 macrophages preferentially utilize mitochondrial respiration and fatty acid oxidation (FAO) (Jha et al., 2015; Vats et al., 2006), which we also observed in TEMs (Figures 2B–2E and S2A–S2C). Examination of macrophage intracellular metabolomic profiles showed a correlation between M2 macrophages and TEMs (Figure 2F), and pathway enrichment analysis indicated that nucleoside metabolism distinguished TEMs and M2 macrophages from M1 macrophages (Table S2).
Figure 2. Oxidative Metabolism of TEMs and M2 Macrophages Promote Pyrimidine Biosynthesis from Glucose.
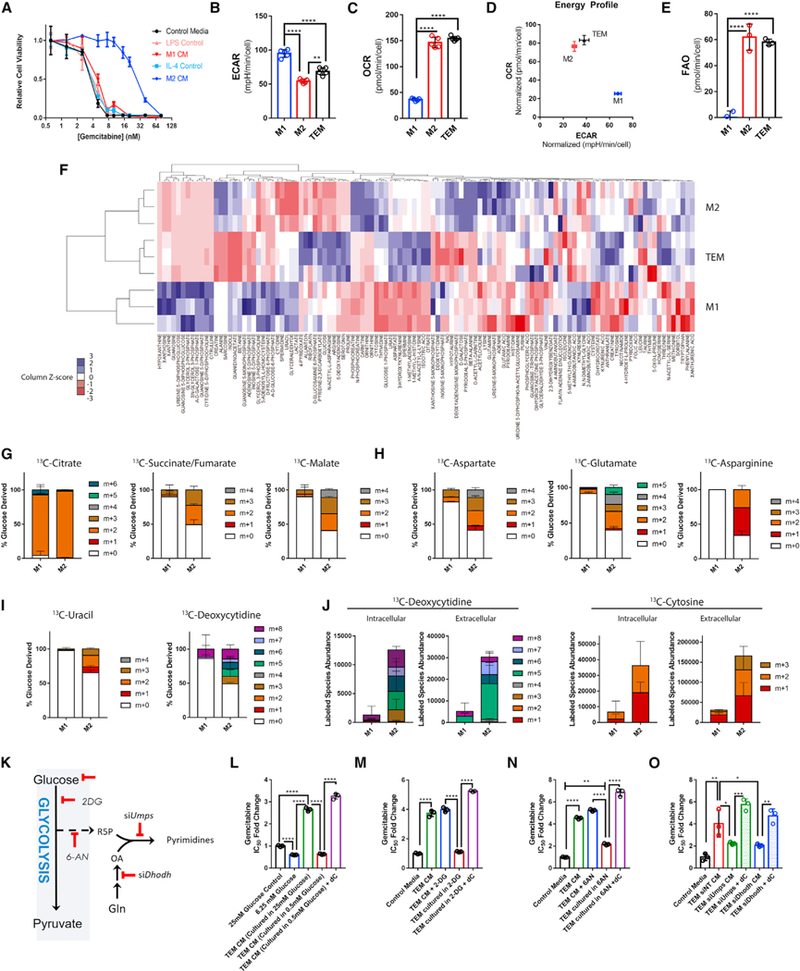
(A) Relative viability of MT3-KPC cells treated with gemcitabine (Gem) in the presence of M1 or M2 conditioned media (CM) versus control media (n = 3).
(B) Basal extracellular acidification rates (ECAR) of TEM, M1, and M2 macrophages (n = 5).
(C) Basal oxygen consumption rates (OCR) of TEM, M1, and M2 macrophages (n = 5).
(D) Comparative energy profile of TEM, M1, and M2 macrophages comparing ECAR versus OCR.
(E) Basal rate of exogenous fatty acid oxidation (FAO) of TEM, M1, and M2 macrophages (n = 5).
(F) Heatmap representation of intracellular metabolites (replicate CV < 0.4) found in TEM, M1, and M2 macrophages by metabolomics profiling (n = 3).
(G) Fractional labeling patterns from uniformly 13C-glucose of TCA cycle metabolites after 16 h in M1 versus M2 macrophages (n = 3).
(H) Fractional labeling patterns from uniformly 13C-glucose of amino acids after 16 h in M1 versus M2 macrophages (n = 3).
(I) Fractional labeling patterns from uniformly 13C-glucose of pyrimidines after 16 h in M1 versus M2 macrophages (n = 3).
(J) Intra and extracellular abundance of pyrimidine isotopologues labeled as in (G–I) (n = 3).
(K) Simplified pyrimidine biosynthesis pathway diagram. 2-DG, 2-deoxyglucose; 6-AN, 6-aminonicotinamide; Dhodh, dihydroorotate dehydrogenase; Gln, glutamine; R5P, ribose 5-phosphate; OA, Orotate; Umps, uridine 5′-monophosphate synthase.
(L) Relative Gem IC50 of KPC-MT3 cells treated in normal DMEM (25 mM), 75% glucose restricted media (6.25 mM final), 75% CM from TEMs grown in normal DMEM, 75% CM from TEMs grown in glucose restricted media, or 75% CM from TEMs grown in glucose restricted media + 3 μM dC (n = 3).
(M) RelativeGem IC50 in KPC-MT3 cells treated in DMEM, 75% TEM CM, TEM CM + 200 μM 2-DG, 75% CM from TEMs grown in 200 μM 2-DG, or 75% CM from TEMs grown in 200 μM 2-DG + 3 μMdC(n = 3).
(N) RelativeGem IC50cin KPC-MT3cellstreated in DMEM,75% TEM CM,TEM CM + 1 μM 6-AN, 75% CM from TEMs grown in 1 μM 6-AN, or75% CM from TEMs grown in 1 μM 6-AN + 3 μM dC (n = 3).
(O) Relative Gem IC50 in KPC-MT3 cells treated in normal DMEM, or 75% CM from TEMs transfected with siRNA targeting Dhodh, Umps, or nontargeting (NT) siRNA, or siDhodh, or siUmps CM + 3 μM dC (n = 3).
Error bars represent mean ± SD, *p ≤ 0.05; **p ≤ 0.01; ****p < 0.0001. See also Figures S2 and S3; Table S2.
To gain further insight, we carried out stable isotope tracing using uniformly labeled 13C-Glucose. The necessity of using PDA CM to polarize TEMs prevented sufficient label incorporation into TEM biosynthetic pathways, limiting our tracing analysis to M1 and M2 macrophages. Consistent with our bioenergetic profiling analysis (Figures 2B–2E), 13C-Glucose fractional labeling patterns revealed increased glucose carbon incorporation into the tricarboxylic acid (TCA) cycle in M2 macrophages (Figure 2G), which facilitated TCA cycle anaplerosis (Figure 2H) and, ultimately, pyrimidine biosynthesis (Figures 2I and S2D). Furthermore, M2 macrophages demonstrated an increased abundance of intracellular and extracellular de novo synthesized pyrimidines as compared to M1 macrophages (Figure 2J), supporting the idea that pyrimidine release is a property of alternatively activated macrophages. Overall, M2 macrophages exhibited a vastly increased biosynthetic capacity from glucose, relative to M1 macrophages (Figure S2E).
We next sought to determine if targeting pyrimidine production by inhibiting nodes of glucose metabolism would impair the ability of TEMs to confer Gem resistance (Figure 2K). Limiting the availability of glucose, inhibiting glycolysis with 2-deoxyglucose (2-DG), or blocking glucose incorporation into the oxidative arm of the pentose phosphate pathway with 6-aminonicotinamide (6-AN) all inhibited the ability of TEMs to produce dC and modulate Gem sensitivity in PDA cells (Figures 2L–2N and S3A–S3D). Importantly, these treatments had a minimal impact on TEM proliferative capacity (Figure S3E), and Gem resistance could be restored with the addition of exogenous dC to these media (Figures 2L–2N and S3A–S3C). Similar results were obtained by directly disrupting key pyrimidine biosynthesis enzymes, Dhodh and Umps, using a genetic knockdown strategy (Figures 2O and S3F–S3H). These results demonstrate the requirement of de novo pyrimidine biosynthesis for TEM CM to inhibit Gem activity in PDA cells.
dC Competitively Inhibits Gem Uptake and Metabolism
Gem is a prodrug that requires activation. This is initiated upon uptake through phosphorylation by deoxycytidine kinase (DCK) (Figure 3A) (Parker, 2009). Given the structural similarity between dC and Gem, we hypothesized that dC could act to inhibit Gem activity through molecular competition (Shukla et al., 2017). To examine this, we measured levels of intra- and extracellular Gem in the presence or absence of dC and observed that dC treatment resulted in the accumulation of Gem both inside and outside the cell (Figures 3B and S3I). The intracellular accumulation suggested a lack of Gem processing and incorporation into DNA. To test this, we further treated PDA cells with 3H-radiolabeled Gem in the presence or absence of dC and observed that dC treatment prevented Gem incorporation into DNA (Figure 3C). These results suggest that dC is acting to decrease the effective concentration of Gem and its activated species experienced by the cell.
Figure 3. dC Blocks the Uptake and Incorporation of Gem and Other DCK-Activated Pyrimidine-Based Chemotherapies.
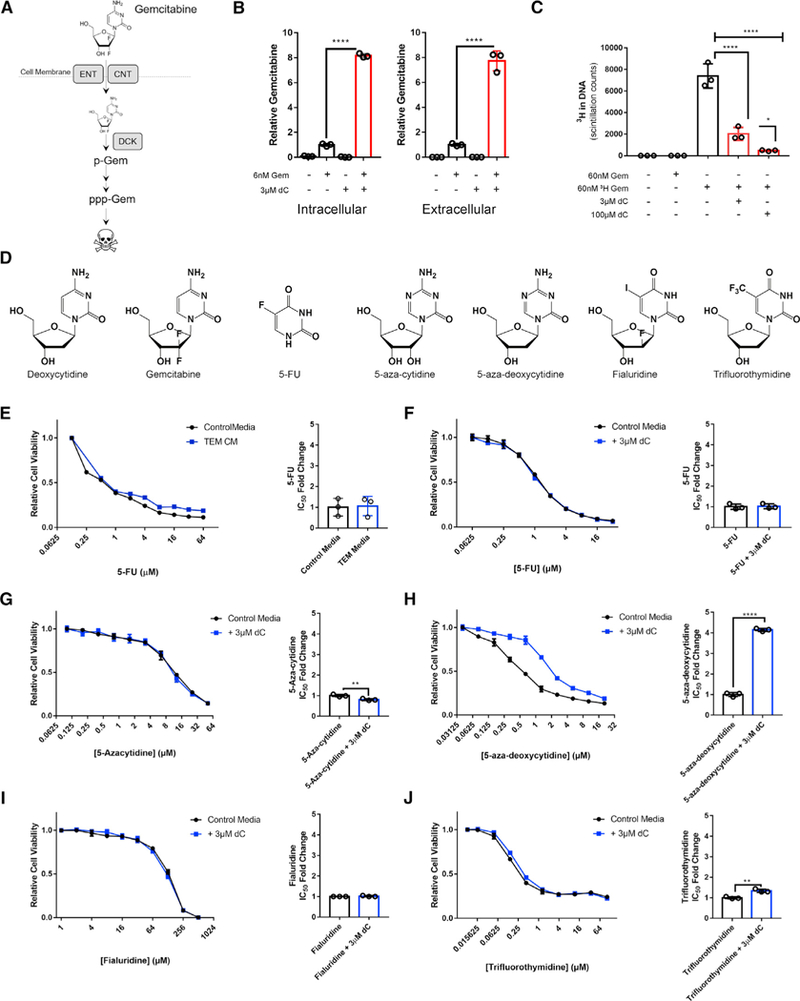
(A) Schematic representation of the mechanism of Gem uptake and metabolism. ENT, equilibrative nucleoside transporter; CNT, concentrative nucleoside transporter; DCK, deoxycytidine kinase; p-Gem, Gem monophosphate; ppp-Gem, Gem triphosphate.
(B) Relative intra and extracellular abundance of Gem in KPC-MT3 cells or media, respectively, after16h of treatment with 6 nM Gem in the presence or absence of 3 μM dC, as measured by LC-MS/MS (n = 3).
(C) Incorporation of Gem into the DNA in KPC-MT3 cells treated with 60 nM 3H-labeled Gem in the presence or absence of dC (n = 3).
(D) Chemical structures of dC and pyrimidine chemotherapies.
(E) Relative viability and IC50 of MT3-KPC cells treated with 5-FU in the presence of 75% TEM CM versus control (n = 3).
(F) Relative viability and IC50 of MT3-KPC cells treated with 5-FU in the presence of 75% 3 μM dC versus control (n = 3).
(G–J) Relative viability and IC50 of MT3-KPC cells treated with 5-aza-cytidine(G), 5-aza-deoxycytidine(H), fialuridine (I), or trifluorothymidine (J) in the presence of 3 μM dC or control (n = 3).
Error bars represent mean ± SD, *p ≤ 0.05; **p ≤ 0.01; ****p ≤ 0.0001. See also Figure S3; Table S3.
Pyrimidine-based chemotherapies represent a large and well-characterized class of drugs (Figure 3D) (Parker, 2009). Based on the model proposed and the data in Figures 3B and 3C, the protective mechanism(s) of dC should be limited to those pyrimidine-based chemotherapies that share uptake or metabolism properties. We first examined 5-fluorouracil (5-FU), a different class of pyrimidine nucleoside-based chemotherapy for PDA treatment with distinct transport and activation properties from Gem (Table S3) (Conroy et al., 2011). We found that cytotoxic activity of 5-FU in PDA cells is not impacted by TEM CM or dC (3E, 3F, and S3J). We then tested a panel of other pyrimidine-based chemotherapies (Figure 3D) and observed the cytotoxic activity of 5-aza-deoxycytidine, but not 5-aza-cytidine, fialuridine, or trifluorothymidine, was inhibited by concurrent treatment with 3 μM dC (Figures 3G–3J). The transporters used by dC and the ribose-bearing chemotherapies tested share a high degree of overlap (Table S3), and intracellular activation by DCK differentiates compounds that are inhibited by dC from those that are not. The results from this chemical profiling strategy suggest that TEM-derived dC is inhibiting the cytotoxic activity of Gem in PDA cells by reducing the effective concentration through molecular competition at DCK. To verify that TEM CM and exogenous dC treatment was not inhibiting Gem activation via downregulation of DCK, we verified these treatments resulted in no differences in DCK protein expression across a panel of PDA cell lines (Figure S3K).
Macrophages Modulate Gem Response
We next sought to determine if macrophages contribute to Gem resistance in vivo using a syngeneic PDA tumor transplantation model in mice expressing a diphtheria toxin (DT) receptor under the CD11b promoter (Zhang et al., 2017a). This allows for selective, temporary depletion of myeloid cells within PDA tumors (Figures 4A, S4A, and S4B). We found the size of tumors treated with Gem after depletion of myeloid cells to be decreased dramatically, as compared to single treatment or vehicle-treated arms (Figure 4B). The increased efficiency of Gem in myeloid depleted tumors resulted in increased DNA damage, marked by γH2AX immunostaining (Figures 4C and 4D). In contrast, differences in proliferation, apoptosis, and DCK expression patterns were not evident among the groups (Figure S4A). Macrophages have been suggested to modulate the abundance of tumor-initiating cells (TICs) (Jinushi et al., 2011), which are resistant to chemotherapy with increased mitochondrial content (Sancho et al., 2015; Viale et al., 2014). However, we saw no changes among our treatments (Figure S4A).
Figure 4. Macrophages Inhibit Gem Treatment; Burden Predicts Treatment Response.
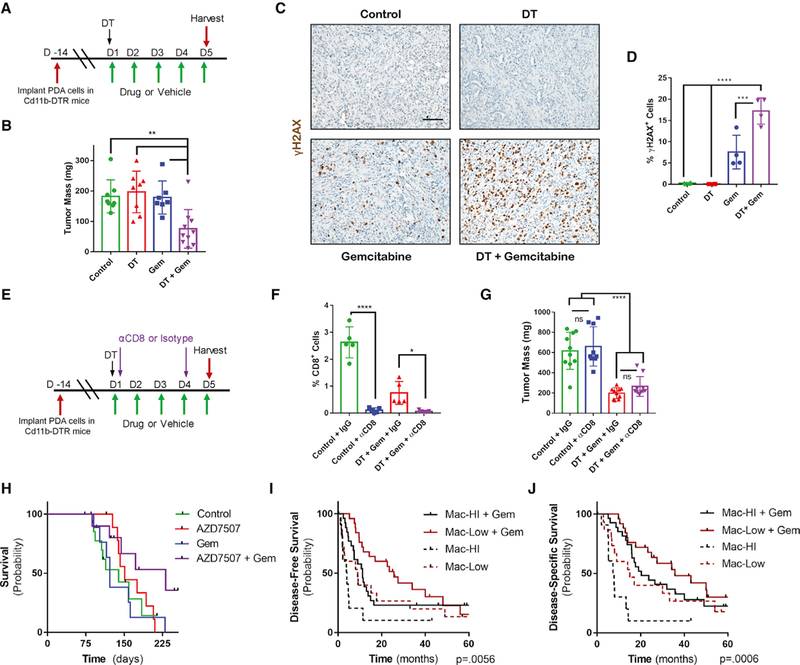
(A) Schematic of CD11b-DTR macrophage depletion tumor model with Gem treatment schedule.
(B) Mass of vehicle- (n = 8), Gem- (n = 7), diphtheria toxin (DT) (n = 8), or DT + Gem (n = 10) KPC-MT3 tumors at endpoint.
(C) Immunostaining of γH2AX in tumor tissue from (B) (n = 4).
(D) Quantification of γH2AX in tumor tissue from (B) (n = 4).
(E) Schematic of CD11b-DTR macrophage depletion tumor model with Gem treatment and CD8 depletion schedule.
(F) Quantification of CD8 cells in vehicle + isotype control, vehicle + αCD8 antibody, DT + Gem + isotype control, or DT + Gem + αCD8 KPC-MT3 tumors at endpoint (n = 5).
(G) Mass of vehicle + isotype control, vehicle + αCD8 antibody, DT + Gem + isotype control, or DT + Gem + αCD8 KPC-MT3 tumors at endpoint (n = 10).
(H) Kaplan-Meier survival curve of control KPC mice (n = 15), KPC mice treated with AZD7507 (n = 11), KPC mice treated with Gem (n = 9), or KPC mice treated with AZD7507 + Gem (n =1 0).
(I) Kaplan-Meier disease-free survival of human PDA patients with either high (n = 12) or low macrophage (n = 15) burden, compared to those treated with adjuvant Gem therapy (high n = 26, low n = 25), as determined by gene-expression signature.
(J) Disease-specific survival of human PDA patients with either high (n = 12) or low macrophage (n = 15) burden, compared to treated with adjuvant Gem therapy (high n = 26, low n = 25), as determined by gene-expression signature.
Error bars represent mean ± SD, *p ≤ 0.05 **p ≤ 0.01; ***p ≤ 0.001; ****p ≤ 0.0001. Scale bar, 100 μM. See also Figure S4; Table S4.
As we have previously also demonstrated that myeloid cell depletion can promote the immune response against PDA tumors, we assessed the levels of cytotoxic CD8 T cell infiltration into these tumors (Figure S4A). Myeloid depletion alone resulted in a significant increase in the number of CD8 T cells within the tumor; however, this difference was lost when combined with Gem treatment. To further rule out an immune response, we depleted CD8 T cells concurrent with myeloid depletion and found that Gem remained effective at reducing tumor size in mice (Figures 4E–4G and S4C). Lastly, 5-FU-based chemotherapy demonstrated no benefit from myeloid depletion (Figure S4D).
To test the clinical benefit of targeting macrophages in combination with Gem treatment, we treated a genetically engineered, autochthonous pancreatic cancer model (Kras+/LSL-G12D; Trp53+/LSL-R172H; Pdx1-Cre, known as KPC) with Gem in combination with the colony stimulating factor receptor 1 (CSFR1) inhibitor AZD7507, which blocks the recruitment and activation of monocytes in KPC tumors (Candido et al., 2018). In this model, we observed prolonged survival compared to single arm treatments or control-treated mice (Figure 4H). We additionally found a significant correlation between pathological response to Gem and the abundance of myeloid cells in a small cohort of neoadjuvant Gem treated patients (Figures S4E and S4F).
To determine if long-term response to Gem correlates with the abundance of macrophages present in primary human tumors, we examined a cohort of patients with PDA who underwent surgery followed by adjuvant Gem. We found significantly better survival in patients with a low macrophage signature compared with those with a high macrophage signature (median disease-specific survival 35.8 months and 20.3 months, respectively) (Figures 4I and 4J). Of note, macrophage-high patients fared worse than macrophage-low patients in the absence of adjuvant therapy, indicating additional challenges that may be posed by macrophages, such as immunosuppression (Zhang et al., 2017a; Zhu et al., 2014).
In summary, our data demonstrate a previously undescribed mechanism of intra-tumoral metabolic crosstalk that promotes therapeutic resistance. In pancreatic tumors, malignant PDA cells recruit and polarize macrophages into TAMs (Liou et al., 2015; Zhang et al., 2017b). We found that TAMs impair the cytotoxic activity of the front-line chemotherapy Gem through the release of dC. In the tumor, dC competes with Gem based on their molecular similarity and thereby reduces the therapeutic efficacy. Accordingly, since Gem remains a backbone of the standard chemotherapy regimen for pancreatic cancer patients (Von Hoff et al., 2013), strategies to inhibit the recruitment of macrophages or reprogram alternatively activated macrophages have the potential to increase the utility of this well tolerated and mainstay therapy, as has already been seen with FOLFIRONOX (Nywening et al., 2016).
DISCUSSION
Pyrimidine biosynthesis is energetically costly, suggesting that release is a regulated process. Purines are well-established signaling molecules (Di Virgilio and Adinolfi, 2017), and release of pyrimidine nucleosides at micromolar levels may suggest a similar function. Alternatively, metabolites and breakdown products of metabolism can act in a paracrine fashion to regulate metabolism and have important roles in physiology. Further, we and others have reported on examples of such processes among the cells in tumors (Lyssiotis and Kimmelman, 2017; Murray, 2016). We propose that the mechanism we discovered using a tumor model system is a previously undescribed physiological process, in which alternatively activated macrophages provide another cell type with pyrimidines during normal development. Indeed, a potential insight for the role of pyrimidines comes from a system-wide genetic deletion of the dC salvage enzyme dCK in mice. In these animals, development of lymphocytes is grossly impaired, demonstrating a requirement for pyrimidine salvage during lymphocyte development (Austin et al., 2012; Toy et al., 2010). Our data suggest that the source of the salvaged pyrimidines may be from macrophages. Furthermore, as TAMs release a panel of pyrimidine nucleosides, they may promote chemoresistance to other therapies. Accordingly, these insights may shift the paradigm of how we consider the evolution of chemoresistance to nucleoside-like drugs, not only in PDA but, potentially, in many other tumor types where TAMs play an important role.
Limitations of the Study
Pancreatic tumors have an intrinsically complex microenvironment complicating metabolic analysis in bulk tumors (Halbrook et al., 2018). As we utilized a system in which cell types interacted in an isolated circuit, further studies including the contributions of other cells, such as fibroblasts, will be important to supplement these findings. In addition, it remains to be determined if pyrimidine release is limited to macrophages or if multiple myeloid lineages share this feature given the Gem resistance data generated with the Cd11b-DTR model. Finally, it is important to note that pyrimidine release is one of the many overall pro-tumor roles TAMs mediate in response to Gem treatment, a further and more detailed understanding of which may provide additional therapeutic opportunities.
STAR★METHODS
Detailed methods are provided in the online version of this paper and include the following:
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Costas A. Lyssiotis (clyssiot@med.umich.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human Subjects and Data
Deidentified patient data from the neoadjuvant gemcitabine cohort from the University of Michigan Department of Pathology was provided without any additional previous procedure information. All protocols and procedures for the human studies were approved by the university Institutional Review Board. Patient data from the Australian Pancreatic Genome Initiative (APGI) has been previously described (Dreyer et al., 2018), and patients were treated post-surgery as described in Table S4.
Mice
Kras+/G12D; Pdx7-Cre; Trp53+/R172H (KPC) mice have been previously described (Hingorani et al., 2005). KPC animal experiments were performed under UK Home Office license and approved by the University of Glasgow animal welfare and ethical review committee. Mice were bred in house on a mixed background, maintained in conventional cages with access to standard diet and water ad libitum at constant ambient temperature and a 12-hour light cycle, and genotyped by Transnetyx (Cordoba, TN, USA). Mice of both sexes were randomly assigned to treatment cohorts at 10 weeks of age.
B6.FVB-Tg(ITGAM-DTR/EGFP)34Lan/J (CD11b-DTR) mice were maintained in a mixed C57BL/6J x FVB/NJ background and experimental mice were generated through an F1 cross with pure background C57BL/6J and maintained in SPF housing with access to standard diet and water ad libitum at constant ambient temperature and a 12-hour light cycle. Adult mice (≤ 8 weeks of age) of both sexes within 4 weeks of age were used for tumor implantation experiments and randomized onto treatment arms, and all experiments were conducted in accordance with the Office of Laboratory Animal Welfare and approved by the Institutional Animal Care and Use Committees of the University of Michigan.
Cell Culture
C57B6/J PDA lines KPC MT-3 and MT-5, were kind gifts from Dr. David Tuveson, KPC28258 from Dr. Sunil Hingorani, and KPC7940 from Dr. Gregory Beatty and all derived from mice of unknown sex. iKras*3 PDA cells were previously described (Zhang et al., 2017a) and derived from mice of unknown sex. MiaPaCa2 (male), Panc1 (male), BXPC3 (female), L929 (male), and RAW264.7 (male) cell lines were purchased from ATCC. PA-TU-8902 (female) was purchased from DSMZ. UM patient-derived cell lines were generated from de-identified patient tumor samples of unknown sex (Li et al., 2007), with the approval of the Institutional Review Board of the University of Michigan. All commercial cell lines were validated by STR profiling. All cells were maintained in high-glucose DMEM (Gibco) supplemented with 10% FBS (Corning) at 37C, and routinely tested for mycoplasma contamination using MycoAlert (Lonza). iKras*3 PDA cells were additionally maintained in 1 μg/mL doxycycline, and 1 μg/mL doxycycline was added as a control to other conditions in experiments in which this cell line was used.
METHOD DETAILS
Conditioned Medium
Conditioned medium was produced by changing cell media of >50% confluent plates, then removing the medium from cells after 48 hours of growth and filtering through a 0.45μm polyethersulfone membrane (VWR). Boiled conditioned media was warmed to 100°C for 15 minutes and the precipitate was filtered out. Size cutoff columns (EMD Millipore, UFC900308) were used to remove species larger than 3kDafrom the medium. The filtered conditioned medium was then resuspended in DMEM to the original volume. Conditioned media was used at a 3:1 ratio with fresh media to avoid effects of nutrient/metabolite exhaustion.
L929 conditioned media was prepared by incubating confluent L929 cells in fresh media for 48 hours, after which it was filtered through a 0.45μm polyethersulfone membrane.
Bone-Marrow Derived Macrophage Differentiation
Bone marrow was extracted from the femurs of C57B6/J mice as described (Celada et al., 1984), and cultured in macrophage differentiation media (i.e. high-glucose DMEM (Gibco) supplemented with 10% FBS (Corning), Sodium Pyruvate (Gibco), Penicillin/Streptomycin (Gibco), and 30% L929 conditioned media) for 5 days, refreshing on day 3.
Macrophage Polarization
Bone-marrow derived macrophages and RAW264.7 cells were polarized using either murine 10ng/mL M-CSF (Peprotech), 10ng/mL LPS (Enzo, ALX-581–011-L001), 10ng/mL murine IL-4 (Peprotech), or 75% PDA conditioned media. Macrophage subtypes were polarized from matched biological replicates. They were maintained in the presence of polarization factors for the duration of media conditioning (48 hours), overnight for metabolic flux assays, or 24 hours for cell viability assays.
Metabolite Sample Preparation
Metabolite extraction from medium samples was done by adding 1 mL of conditioned medium to 4 mL of cold (−80°C) 100% methanol, then clarified by centrifugation. Intracellular metabolite fractions were prepared from cells that were lysed with cold (−80°C) 80% methanol, then clarified by centrifugation. Metabolite pellets from intracellular fractions were normalized to the protein content of a parallel sample, and all samples were lyophilized via speed vac. Dried metabolite pellets from cells or media were re-suspended in 35 μL 50:50 MeOH: H2O mixture for metabolomics analysis.
13C-Glucose tracing was performed using glucose free DMEM (Gibco) supplemented with 10% dialyzed FBS (Gibco) and either 25mM 12C glucose (Sigma) or uniformly labeled 13C-glucose (Cambridge Isotopes). BMDM cultures were polarized with appropriate stimuli for 24 hours in normal media, then the media was replaced with 13C or 12C glucose labeling media and incubated overnight. Samples were then harvested and prepared as above.
Metabolomics
For steady state metabolite profiling, an Agilent 1290 UHPLC-6490 Triple Quadrupole (QqQ) tandem mass spectrometer (MS/MS) system was used. For negative ion acquisition, a Waters Acquity UPLC BEH amide column (2.1 × 100mm, 1.7μm) was used with the mobile phase (A) consisting of 20 mM ammonium acetate, pH 9.6 in water, and mobile phase (B) acetonitrile. The following gradient was used: mobile phase (B) was held at 85% for 1 min, increased to 65% at 12 min, then to 40% at 15 min and held for 5 min before going to initial condition and held for 10 min. For positive ion acquisition, a Waters Acquity UPLC BEH TSS C18 column (2.1 × 100mm, 1.7μm) was used with mobile phase (A) consisting of 0.5 mM NH4F and 0.1% formic acid in water; mobile phase (B) consisting of 0.1% formic acid in acetonitrile. The following gradient was used: mobile phase (B) was held at 1% for 1.5 min, increased to 80% in 15 min, then to 99% in 17 min and held for 2 min before going to initial condition and held for 10 min. The column was kept at 40°C and 3 μL of sample was injected into the LC-MS/MS with a flow rate of 0.2 mL/min. Tuning and calibration of the QqQ was achieved through Agilent ESI Low Concentration Tuning Mix.
Optimization was performed on the 6490 QqQ in negative or positive mode individually for each of 220 standard compounds to get the best fragment ion and other MS parameters for each standard. Retention time for each standard of the 220 standards was measured from pure standard solution or a mix standard solution. The LC-MS/MS method was created with dynamic (d)MRMs with RTs, RT windows and MRMs of all the 220 standard compounds.
In both acquisition modes, key parameters of AJS ESI were: Gas temp 275°C, Gas Flow 14 L/min, Nebulizer at 20 psi, Sheath Gas Heater 250°C, Sheath Gas Flow 11 L/min, Capillary 3000 V. For negative mode MS: Delta EMV was 350 V, Cycle Time 500 ms and Cell accelerator voltage was 4 V, whereas for positive acquisition mode MS: Delta EMV was set at 200 V with no change in cycle time and cell accelerator voltage.
The QqQ data were pre-processed with Agilent MassHunter Workstation Quantitative Analysis Software (B0700). Additional analyses were post-processed for further quality control in the programming language R. Each sample was normalized by the total intensity of all metabolites. Finally, each metabolite abundance level in each sample was divided by the median of all abundance levels across all samples for proper comparisons, statistical analyses, and visualizations among metabolites. The statistical significance test was done by a two-tailed t-test with a significance threshold level of 0.05.
To quantitate deoxycytidine concentrations, a standard curve of deoxycytidine in 1:1 MeOH:H2O was run in parallel to TEM conditioned media samples, with a concentration range of 4.4×10−3 M to 1.28×10−13 M in twofold dilutions. Samples were verified to be in a linear range of the standard curve.
For the 13C glucose tracing studies, an Agilent 1260 UHPLC combined with a 6520 Accurate-Mass Q-TOF LC/MS was utilized. Agilent MassHunter Workstation Software LC/MS Data Acquisition for 6200 series TOF/6500 series QTOF (B.06.01) was used for calibration and data acquisition. A Waters Acquity UPLC BEH amide column (2.1 × 100mm, 1.7μm) was used with mobile phase (A) consisting of 20 mM NH4OAc in water pH 9.6, and mobile phase (B) consisting of acetonitrile. The following gradient was used: mobile phase (B) was held at 85% for 1 min, increased to 65% at 12 min, then to 40% at 15 min and held for 5 min before going to initial condition and held for 10 min. The column was at 40°C and 3 μL of sample was injected into the LC-MS with a flow rate of 0.2 mL/min. Calibration of TOF MS was achieved through Agilent ESI Low Concentration Tuning Mix.
Key parameters for both acquisition modes were: mass range 100–1200 da, Gas temp 350°C, Fragmentor 150 V, Skimmer 65 V, Drying Gas 10 L/min, Nebulizer at 20 psi and Vcap 3500 V, Ref Nebulizer at 20 psi. For negative mode the reference ions were at 119.0363 and 980.01637 m/z whereas for positive acquisition mode, reference ions at 121.050873 and 959.9657 m/z
For 13C-labeling data analysis, we used Agilent MassHunter Workstation Software Profinder B.08.00 with Batch Targeted Feature Extraction and Batch Isotopologue Extraction and Qualitative Analysis B.07.00. Various parameter combinations, e.g. mass and RT tolerance, were used to find best peaks and signals by manual inspection. Key parameters were: mass tolerance = 20 or 10 ppm and RT tolerance = 1 or 0.5 min. Isotopologue ion thresholds and the anchor ion height threshold was set to 250 counts and the threshold of the sum of ion heights to 500 counts. Coelution correlation threshold was set to 0.3.
All other bioinformatics analyses including graphs and plots were done using R/Bioconductor.
Chemicals
Gemcitabine, 5-azacytidine, decitabine, fialuridine, trifluorouridine, capecitabine, and 5-fluorouracil were sourced from Cayman Chemical. 6-Aminonicotinamide, 2-deoxyglucose, and all nucleosides and nucleobases were obtained from Sigma-Aldrich. Compounds were dissolved into PBS (Gibco) and added to media to treat cells. AZD7507 CSF1R has been previously described (Scott et al., 2013). FCCP, oligomycin, rotenone, antimycin A were from Sigma-Aldrich and stocks were prepared in DMSO; etomoxir, carnitine, palmitate, were from Sigma-Aldrich and prepared in water; BSA fraction V was from Roche and prepared in water.
Cell Viability Assay
PDA cells were grown on white walled 96-well plates (Costar 3917, Corning) at 1000 cells/well in triplicate for four days. Cell viability was measured using the Cell Titer Glo 2.0 luminescence assay (Promega G9243). Luminescence was measured for 500ms using a SpectraMax M3 Microplate reader (Molecular Devices). IC50 values were calculated using GraphPad Prism 7.
Metabolic Flux Assay
To estimate the rate of glycolysis and mitochondrial respiration, a Seahorse Metabolic Flux Analyzer e96 XF instrument (Agilent) was used according to the manufacturer’s suggestion. 50,000 cells/well were seeded in the respective polarization culture media the day prior to the assay. The next day media was exchanged to the Seahorse assay media, containing 25 mM glucose, adjusted to pH~7.4. The plate was allowed to equilibrate for 1 hour in a non-CO2, 37°C incubator, followed by 3 sequential measurements for the basal respiration. The mitostress assay was performed by sequentially injecting 2μM oligomycin, 5μM FCCP, 0.5μM rotenone/0.5μM anti-mycin A. The cell number adjustment/normalization was performed using CyQuant NF (Thermo) after the assay.
Seahorse Fatty Acid Oxidation Assay
Macrophages were polarized as described above. BSA-palmitate conjugates were prepared, and the FAO assay was performed according to the manufacturer’s application notes with the following modifications: 50,000 cells/well supplied with the corresponding polarization factors in 5 replicates were seeded the night before the assay. Incubation with the substrate-limiting media was omitted. The next day cells were washed twice with FAO media and incubated for 1 hour in a non-CO2 incubator. Etomoxir was added to final concentration 40μM to the corresponding control wells, cells were incubated for 15 mins, BSA-Palmitate or BSA were added immediately before the assay. The level of exogenous FAO was calculated as a difference between BSA-Palmitate and Etomoxir-Palmitate, and the respiration attributed to the proton leak was removed.
Metabolic Pathway Analysis
Metabolic pathway enrichment analysis was performed using metabolites determined to be significant between M1 macrophages, M2 macrophages, and TEMs by two-tailed t-test with a significance threshold level of 0.05 using Metabolanalyst online software (http://www.metaboanalyst.ca/).
RNAi
ON-TARGETplus siRNAs targeting murine Dhodh, Umps, or a nontargeting sequence were purchased from Dharmacon. siRNAs were transfected into RAW246.7 cells using Lipofectamine RNAiMAX (ThermoFisher) per the manufacturer’s instructions. Media was changed to iKras*3 PDA conditioned media after 48 hours and allowed to condition for 48 additional hours. Media was then collected and filtered through a 0.45μm filter prior to use.
Western Blotting
Cells were lysed in RIPA buffer (Sigma Aldrich), supplemented with cOmplete EDTA-free protease inhibitor (Sigma Aldrich). Lysates were quantified by BCA assay (Thermo Fisher Scientific Inc., Waltham, MA) and equal protein amounts were run onto SDS-PAGE gels. Proteins were transferred from SDS-PAGE gel to Immobilon-FL PVDF membrane, blocked, then incubated with primary antibodies at a 1:1000 dilution. After washing, membranes were then incubated in secondary antibody, washed, then exposed on auto-radiography film (Bioexpress) with West Pico ECL (Thermo Fisher Scientific).
3H-Gemcitabine DNA Incorporation Assay
PDA cells were grown at near confluence for 12 hours in 6 or 60nM 3H-gemcitabine (14.8Ci/mmol) (Moravek Biochemicals, MT1572) plus or minus 3 or 100 μM dC. DNA was harvested using a Qiagen DNeasy Blood and Tissue Kit and normalized according to concentration. The samples were diluted with 5mL of scintillation fluid and measured for 30 minutes using a Beckman LS6500 Scintillation Counter.
Mouse Treatments
Kras+/LSL-G12D; Pdx1-Cre; Trp53+/LSL-R172H (KPC) mice of both sexes were randomly assigned to treatment cohorts at 10 weeks of age and treated unblinded with Gemcitabine (LC Labs) at 100mg/kg i.p. twice weekly +/− AZD7507 CSF1R inhibitor (AstraZeneca) at 100mg/kg p.o. twice daily; vehicle p.o. twice daily, or AZD7507 alone. Mice were culled at ethical endpoint by schedule 1 methods. For CD11b-DTR experiments, 1×106 PDA cells in 200μL of 1:1 Matrigel-DMEM + 10%FBS (Corning) were injected subcutaneously into the flanks of F1 C57BL/6J CD11b-DTR mice and allowed to establish tumors for 14 days. For myeloid cell depletion, mice were then treated unblinded with DT (Enzo Life Science) at a concentration of 25 ng/g or vehicle IP, and then daily with gemcitabine at 100mg/kg IP, or capecitabine at 500mg/kg PO. For CD8+ T-cell depletion, anti-CD8 monoclonal antibody (BioXcell #BE0061; clone 2.43; 200 μg/mouse) was injected IP twice per week, unblinded.
Histology
Mice were sacrificed by CO2 asphyxiation then tissue was quickly harvested and fixed overnight at room temperature with Z-fix solution (Anatech LTD). Tissues were processed using a Leica ASP300S Tissue Processor, paraffin embedded, and cut into 5 μm sections. Immunohistochemistry was performed on Discovery Ultra XT autostainer (Ventana Medical Systems Inc) and counterstained with hematoxylin. The following antibody dilutions were used: γH2AX 1:300, VDAC1 1:250, DCK1 1:200, CD8 1:500, Cleaved caspase 3 1:100, Ki67 1:500. IHC slides were scanned on a Pannoramic SCAN slide scanner (Perkin Elmer), and annotation regions en-compassing greater than 1mm of tissue were processed using Halo software (Indica Labs).
Flow Cytometry
Mouse tumors were quickly excised then mechanically dissociated with scissors in sterile PBS. Tumor fragments were pelleted under centrifugation and then resuspended in 1mg/mL Collagenase V (Sigma), digested for 30 minutes at 37°C, quenched with DMEM + 10% FBS, then filtered through a series of 500μm, 100μm, and 40μm filters (Corning). The cells were then subject to an RBC lysis, washed in PBS, then blocked and stained with antibodies at a 1:100 dilution in 100% FBS. The cells were then washed in FACS buffer (1% BSA, 1mM EDTA in PBS), and run on a MoFlo Astrios flow cytometer (Beckman Coulter).
Multiplex Fluorescent Immunohistochemistry Staining
Formalin fixed paraffin embedded pancreas cancer specimens from patients who underwent neoadjuvant gemcitabine treatment for pancreas cancer were cut with a thickness of 5 μm onto polarized slides. The slides were then baked at 60°C for one hour followed by xylene for ten minutes in triplicate. Slides were then immersed in ethanol at 100%, 95% and 70% followed by water for 2 minutes then neutral buffered formalin for 30 min. After washing with water, the slides underwent multiple rounds of multiplex fluorescent immunohistochemistry with the Opal Multiplex Fluorescent Immunohistochemistry (Perkin Elmer) as previously described (Lazarus et al., 2018). The following primary antibodies and subsequent Opal TSA combinations were used: CD3 (1:400) and Opal 520, CD8 (1:400) and Opal 620, CD163 (1:400) and Opal 570, pancytokeratin (1:500) and Opal 690, and DAPI. Slides were mounted with coverslips using Prolong Diamond (ThermoFisher).
Imaging, Phenotyping, and Data Analysis of Multiple-IF
Slides were imaged using the Mantra Quantitative Pathology Work Station (Perkin Elmer). One to four images were taken per slide at 20x magnification. All filter cubes were used for each image (DAPI, CY3, CY5, CY7, Texas Red, Qdot). Analysis was performed using inForm Cell Analysis Software (Perkin Elmer). A standard library was used in the analysis for unmixing and showed no overlap between fluorophores. Cells were segmented into the cytoplasm, nucleus and membrane components as previously described (Lazarusetal., 2018). After cell segmentation, the cells were phenotyped using the trainable inForm software to detect T cells, epithelial cells, and macrophages. Using R, the mean signal intensity of the cytoplasm of Opal 520 and 620 were used to make a complex phenotype: i.e. CD3+CD8+T cells. After all cells were phenotyped using inForm trainable software and R, cells numbers and percentages were calculated in Excel.
Patient Survival Data
Patients were identified from the Australian Pancreatic Genome Initiative’s (APGI) contribution to the International Cancer Genome Consortium’s Pancreatic Cancer project (Dreyer et al., 2018). Patients with sufficient clinical data that underwent RNA sequencing (RNAseq) and analysis were included. All patients included underwent primary surgical resection for PDA and were defined as receiving Gemcitabine-based adjuvant therapy if completing 3 cycles or more. RNAseq and analysis was performed as previously described (Bailey et al., 2016). Macrophage infiltration signature was determined by gene enrichment analysis that defined upregulated gene expression associated with macrophage infiltration (Bailey et al., 2016; Rooney et al., 2015). Genes used for the macrophage infiltration signature were FUCA1, MMP9, LGMN, HS3ST2, TM4SF19, CLEC5A, GPNMB, C11orf45, CD68, and CYBB, based on previously described methodology (Rooney et al., 2015). Patients were dichotomized as high or low signature based on ranking the relative signature score from highest to lowest (Bailey et al., 2016). Clinical variables were determined using the AJCC 7th staging system. Deidentified patient data are provided in Supplemental Data Table 4.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistics were performed using Graph Pad Prism 7 (Graph Pad Software Inc). Groups of 2 were analyzed with two-tailed students t test, groups greater than 2 with a single variable were compared using one-way ANOVA analysis with Tukey post hoc test, and groups greater than two multiple variables were compared with two-way ANOVA with Tukey post hoc test. Survival statistics were calculated using a Log-rank Mantel-Cox test. All error bars represent mean with standard deviation, all group numbers and explanation of significant values are presented within the figure legends. Sample-sizes for mouse tumor models were based on previous studies (Candido et al., 2018; Zhang et al., 2017a), and human studies used all available samples. No data was excluded from these studies.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-CD3 | Dako | Cat#:A0452; RRID: AB_2335677 |
| Rabbit monoclonal clone SP239 anti-CD8 (for human IF) | Spring Biosciences | Cat#:M5390; RRID: AB_2756374 |
| Mouse monoclonal ant-CD163 | Leica | Cat#:NCL-L-CD163; RRID: AB_2756375 |
| Mouse monoclonal Clone AE1/AE3 anti-PanCK | Dako | Cat#:M3515; RRID: AB_2132885 |
| Rabbit polyclonal anti-DHODH | Proteintech | Cat#:14877-1-AP; RRID: AB_2091723 |
| Rabbit polyclonal anti-UMPS | Proteintech | Cat#:14830-1-AP; RRID: AB_2212392 |
| Rabbit polyclonal anti-VDAC1 | Abcam | Cat#:ab15895; RRID: AB_2214787 |
| Rabbit polyclonal anti-DCK | ThermoFisher Scientific | Cat#:PA5-21846; RRID: AB_11152040 |
| Rat M1/70 monoclonal anti-CD11b APC Cy7 | BD Pharmingen | Cat#:557657; RRID: AB_396772 |
| Rat 30-F11 monoclonal anti-CD45 Pacific Orange | ThermoFisher Scientific | Cat#:mcd4530; RRID: AB_2539700 |
| Rat BM8 monoclonal F4/80 PE-Cy5 | eBioscience | Cat#:15-4801-82; RRID: AB_468798 |
| Rabbit polyclonal anti-Ki67 | Abcam | Cat#:ab15580; RRID: AB_443209 |
| Rabbit monoclonal 5A1E anti-Cleaved Caspase 3 | Cell Signaling | Cat#:9664; RRID: AB_2070042 |
| Rabbit monoclonal 20E3 Histone H2A.X phosphoS139 | Cell Signaling | Cat#:9718; RRID: AB_2118009 |
| Rabbit monoclonal D4W2Z anti-CD8 (for mouse IHC) | Cell Signaling | Cat#:98941; RRID: AB_2756376 |
| Rabbit monoclonal E1E9V anti-Vinculin | Cell Signaling | Cat#:13901; RRID: AB_2728768 |
| Rat monoclonal 2.43 anti-CD8 (for in vivo depletion) | BioXcell | Cat#:BE0061; RRID: AB_1125541 |
| Anti-Rabbit IgG HRP | Cell Signaling | Cat# 7074; RRID: AB_2099233 |
| Biological Samples | ||
| Neoadjuvant Gemcitabine Treated PDA Patient FFPE Blocks | University of Michigan Department of Pathology | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| AR9 buffer | Perkin Elmer | Cat#:AR9001KT |
| 3H-Gemcitabine | Moravek Biochemicals | Cat#:MT1572 |
| Uniformly labeled 13C-glucose | Cambridge Isotopes | Cat#:CLM-1396 |
| Gemcitabine HCl | Cayman Chemical | Cat#:9003096 |
| RIPA buffer | Sigma Aldrich | Cat#:R0278 |
| cOmplete EDTA-Free | Sigma Aldrich | Cat#:11873580001 |
| Doxycycline Hyclate | Sigma Aldrich | Cat#:D9891 |
| 5-azacytidine | Cayman Chemical | Cat#:11164 |
| Decitabine | Cayman Chemical | Cat#:11166 |
| Fialuridine | Cayman Chemical | Cat#:15867 |
| Trifluridine | Cayman Chemical | Cat#:21366 |
| Capecitabine | Cayman Chemical | Cat#:10487 |
| 5-fluorouracil | Cayman Chemical | Cat#:14416 |
| 6-Aminonicotinamide | Sigma Aldrich | Cat#:A68203 |
| 2-deoxyglucose | Sigma Aldrich | Cat#:D3179 |
| FCCP | Sigma Aldrich | Cat#:C2920 |
| Oligomycin | Sigma Aldrich | Cat#:75351 |
| Rotenone | Sigma Aldrich | Cat#:R8875 |
| Antimycin A | Sigma Aldrich | Cat#:A8674 |
| Etomoxir | Sigma Aldrich | Cat#:E19G5 |
| AZD7507 | AstraZeneca | N/A |
| LPS | Enzo Life Sciences | Cat#:ALX-581-G11-LGG1 |
| IL6 | Peprotech | Cat#:216–16 |
| IL4 | Peprotech | Cat#:214–14 |
| MCSF | Peprotech | Cat#:315-G2 |
| Critical Commercial Assays | ||
| Vectra Stain ABC Kit | Vector Labs | Cat#:PK-61GG; RRID: AB_2336819 |
| DAB | Vector Labs | Cat#:SK-41GG; RRID: AB_2336382 |
| Opal 7-Color Manual IHC Kit | Perkin Elmer | Cat#:NEL811GG1KT |
| Lipofectamine RNAiMAX | ThermoFisher Scientific | Cat#:13778G75 |
| MycoAlert | Lonza | Cat#:LTG7–318 |
| Cell Titer Glo 2.0 | Promega | Cat#:G9241 |
| Experimental Models: Cell Lines | ||
| Mouse: KPC MT-3 | Gift from Dr. David Tuveson | N/A |
| Mouse: KPC MT-5 | Gift from Dr. David Tuveson | N/A |
| Mouse: KPC28258 | Gift from Dr. Sunil Hingorani | N/A |
| Mouse: KPC 7940 | Gift from Dr. Gregory Beatty | N/A |
| Human: MiaPaCa2 | ATCC | Cat#:CRL-142G; RRID: CVCL_0428 |
| Human: Panc1 | ATCC | Cat#:CRL-1469; RRID: CVCL_0480 |
| Human: BXPC3 | ATCC | Cat#:CRL-1687; RRID: CVCL_0186 |
| Mouse: L929 | ATCC | Cat#:CCL-1; RRID: CVCL_0462 |
| Mouse: RAW264.7 | ATCC | Cat#:SC-6003 |
| Human: PA-TU-8902 | DSMZ | Cat#:ACC 179; RRID: CVCL_1845 |
| Human: UM18 | Generated as in Li et al., 2007 | N/A |
| Human: UM90 | Generated as in Li et al., 2007 | N/A |
| Human: UM143 | Generated as in Li et al., 2007 | N/A |
| Mouse: iKras*3 | Zhang et al., 2017a | N/A |
| Experimental Models: Organisms/Strains | ||
| Mouse: C57BL/6J | Jackson Laboratory | Cat#: 000664; RRID: IMSR_JAX:000664 |
| Mouse: B6.FVB-Tg(ITGAM-DTR/EGFP)34Lan/J | Jackson Laboratory | Cat#: 006000; RRID: IMSR_JAX:006000 |
| Mouse: Kras+/LSL-G12D;Trp53+/LSL-R172H;Pdx1-Cre | Hingorani et al. (2005) | N/A |
| Oligonucleotides | ||
| ON-TARGETplus siNT | Dharmacon | Cat#:D-001810–10-05 |
| ON-TARGETplus siDhodh | Dharmacon | Cat#:L-063269–01-0005 |
| ON-TARGETplus siUmps | Dharmacon | Cat#:L-042834–01-0005 |
| Software and Algorithms | ||
| Metabolanalyst | http://www.metaboanalyst.ca/ | N/A |
| Halo | Indica Labs | http://www.indicalab.com/halo/ |
| Bioconductor | https://www.bioconductor.org/ | RRID:SCR_006442 |
| Prism 7 | GraphPad | https://www.graphpad.com/scientific-software/prism/; RRID: SCR_002798 |
| MassHunter Workstation Quantitative Analysis Software | Agilent | https://www.agilent.com/en/products/software-informatics/masshunter-suite/masshunter-quantitative-analysis |
| MassHunter Workstation Software Profinder | Agilent | https://www.agilent.com/en/products/software-informatics/masshunter-suite/masshunter-for-life-science-research/profinder-software |
Highlights.
Macrophages polarized by pancreatic cancer cells release pyrimidine nucleosides
Pyrimidine release is a property of alternatively activated macrophage metabolism
Deoxycytidine from macrophages inhibits gemcitabine treatment of cancer cells
Targeting macrophages enhances gemcitabine treatment of pancreatic cancer
ACKNOWLEDGMENTS
The authors would like to thank Devon Pendlebury, Drs. Luigi Franchi, Filip Bednar, Andrea Viale, Vinee Purohit, and the Lyssiotis lab for scientific feedback and Daniel Long for histology support. C.J.H. was supported by UL1TR000433, T32CA009676, and F32CA228328; L.L. by Pancreatic Cancer UK; S.K. by T32GM113900; B.S.N. by T32CA009676 and T32DK094775; J.S. by NCI 1K08CA234222; H.C.C. by the Sky Foundation; J.P.M. by Cancer Research UK; H.S.H. byT32AI007413; C.A.L., H.C.C., and M.P.d.M. by a Cancer Center Support Grant (P30CA046592) and U01 CA224145; M.P.d.M. by the American Cancer Society; C.A.L. by the Pancreatic Cancer Action Network/AACR (13–70-25-LYSS), Damon Runyon Cancer Research Foundation (DFS-09–14), V Foundation for Cancer Research (V2016–009), Sidney Kimmel Foundation for Cancer Research (SKF-16–005), and the AACR (17–20-01-LYSS). Metabolomics studies were supported by DK097153, an Agilent ACT-UR grant mechanism, the Charles Woodson Research Fund, and the UM Pediatric Brain Tumor Initiative.
Footnotes
DECLARATION OF INTERESTS
C.A.L. is an inventor on patents pertaining to Kras-regulated metabolic pathways, redox control pathways in pancreatic cancer, and targeting GOT1 as a therapeutic approach.
SUPPLEMENTAL INFORMATION
Supplemental Information includes four figures and four tables and can be found with this article online at https://doi.org/10.1016/j.cmet.2019.02.001.
REFERENCES
- Austin WR, Armijo AL, Campbell DO, Singh AS, Hsieh T, Nathanson D, Herschman HR, Phelps ME, Witte ON, Czernin J, et al. (2012). Nucleoside salvage pathway kinases regulate hematopoiesis by linking nucleotide metabolism with replication stress. J. Exp. Med. 209, 2215–2228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey P, Chang DK, Nones K, Johns AL, Patch AM, Gingras MC, Miller DK, Christ AN, Bruxner TJ, Quinn MC, et al. (2016). Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 531, 47–52. [DOI] [PubMed] [Google Scholar]
- Candido JB, Morton JP, Bailey P, Campbell AD, Karim SA, Jamieson T, Lapienyte L, Gopinathan A, Clark W, McGhee EJ, et al. (2018). CSF1R+ macrophages sustain pancreatic tumor growth through T cell suppression and maintenance of key gene programs that define the squamous subtype. Cell Rep. 23, 1448–1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celada A, Gray PW, Rinderknecht E, and Schreiber RD (1984). Evidence for a gamma-interferon receptor that regulates macrophage tumoricidal activity. J. Exp. Med. 160, 55–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conroy T, Desseigne F, Ychou M, Bouché O, Guimbaud R, Bécouarn Y, Adenis A, Raoul JL, Gourgou-Bourgade S, de la Fouchardière C, et al. (2011). FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N. Engl. J. Med. 364, 1817–1825. [DOI] [PubMed] [Google Scholar]
- Di Caro G, Cortese N, Castino GF, Grizzi F, Gavazzi F, Ridolfi C, Capretti G, Mineri R, Todoric J, Zerbi A, et al. (2016). Dual prognostic significance of tumour-associated macrophages in human pancreatic adenocarcinoma treated or untreated with chemotherapy. Gut 65, 1710–1720. [DOI] [PubMed] [Google Scholar]
- Di Virgilio F, and Adinolfi E (2017). Extracellular purines, purinergic receptors and tumor growth. Oncogene 36, 293–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreyer SB, Jamieson NB, Upstill-Goddard R, Bailey PJ, McKay CJ, Pancreatic Cancer Genome Initiative, Australian, Biankin AV, and Chang DK (2018). Defining the molecular pathology of pancreatic body and tail adenocarcinoma. Br. J. Surg. 105, e183–e191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feig C, Gopinathan A, Neesse A, Chan DS, Cook N, and Tuveson DA (2012). The pancreas cancer microenvironment. Clin. Cancer Res. 18, 4266–4276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halbrook CJ, and Lyssiotis CA (2017). Employing metabolism to improve the diagnosis and treatment of pancreatic cancer. Cancer Cell 31, 5–19. [DOI] [PubMed] [Google Scholar]
- Halbrook CJ, Pasca di Magliano M, and Lyssiotis CA (2018). Tumor cross-talk networks promote growth and support immune evasion in pancreatic cancer. Am. J. Physiol. Gastrointest. Liver Physiol. 315, G27–G35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hingorani SR, Wang L, Multani AS, Combs C, Deramaudt TB, Hruban RH, Rustgi AK, Chang S, and Tuveson DA (2005). Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell 7, 469–483. [DOI] [PubMed] [Google Scholar]
- Jha AK, Huang SCC, Sergushichev A, Lampropoulou V, Ivanova Y, Loginicheva E, Chmielewski K, Stewart KM, Ashall J, Everts B, et al. (2015). Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity 42, 419–430. [DOI] [PubMed] [Google Scholar]
- Jinushi M, Chiba S, Yoshiyama H, Masutomi K, Kinoshita I, Dosaka-Akita H, Yagita H, Takaoka A, and Tahara H (2011). Tumor-associated macrophages regulate tumorigenicity and anticancer drug responses of cancer stem/initiating cells. Proc. Natl. Acad. Sci. USA 108, 12425–12430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koay EJ, Truty MJ, Cristini V, Thomas RM, Chen R, Chatterjee D, Kang Y, Bhosale PR, Tamm EP, Crane CH, et al. (2014). Transport properties of pancreatic cancer describe gemcitabine delivery and response. J. Clin. Invest. 124, 1525–1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarus J, Maj T, Smith JJ, Perusina Lanfranca MP, Rao A, D’Angelica MI, Delrosario L, Girgis A, Schukow C, Shia J, et al. (2018). Spatial and phenotypic immune profiling of metastatic colon cancer. JCI Insight 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li CW, Heidt DG, Dalerba P, Burant CF, Zhang LJ, Adsay V, Wicha M, Clarke MF, and Simeone DM (2007). Identification of pancreatic cancer stem cells. Cancer Res. 67, 1030–1037. [DOI] [PubMed] [Google Scholar]
- Liou GY, Döppler H, Necela B, Edenfield B, Zhang L, Dawson DW, and Storz P (2015). Mutant KRAS-induced expression of ICAM-1 in pancreatic acinar cells causes attraction of macrophages to expedite the formation of precancerous lesions. Cancer Discov. 5, 52–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyssiotis CA, and Kimmelman AC (2017). Metabolic interactions in the tumor microenvironment. Trends Cell Biol. 27, 863–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahajan UM, Langhoff E, Goni E, Costello E, Greenhalf W, Halloran C, Ormanns S, Kruger S, Boeck S, Ribback S, et al. (2018). Immune cell and stromal signature associated With progression-free survival of patients With resected pancreatic ductal adenocarcinoma. Gastroenterology 155, 1625–1639. [DOI] [PubMed] [Google Scholar]
- Mitchem JB, Brennan DJ, Knolhoff BL, Belt BA, Zhu Y, Sanford DE, Belaygorod L, Carpenter D, Collins L, Piwnica-Worms D, et al. (2013). Targeting tumor-infiltrating macrophages decreases tumor-initiating cells, relieves immunosuppression, and improves chemotherapeutic responses. Cancer Res. 73, 1128–1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray PJ (2016). Amino acid auxotrophy as a system of immunological control nodes. Nat. Immunol. 17, 132–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nywening TM, Wang-Gillam A, Sanford DE, Belt BA, Panni RZ, Cusworth BM, Toriola AT, Nieman RK, Worley LA, Yano M, et al. (2016). Targeting tumour-associated macrophages with CCR2 inhibition in combination with FOLFIRINOX in patients with borderline resectable and locally advanced pancreatic cancer: a single-centre, open-label, dose-finding, non-randomised, phase 1b trial. Lancet Oncol. 17, 651–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olive KP, Jacobetz MA, Davidson CJ, Gopinathan A, McIntyre D, Honess D, Madhu B, Goldgraben MA, Caldwell ME, Allard D, et al. (2009). Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science 324, 1457–1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker WB (2009). Enzymology of purine and pyrimidine antimetabolites used in the treatment of cancer. Chem. Rev. 109, 2880–2893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perera RM, and Bardeesy N (2015). Pancreatic cancer metabolism: breaking it down to build it back up. Cancer Discov. 5, 1247–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Provenzano PP, Cuevas C, Chang AE, Goel VK, Von Hoff DD, and Hingorani SR (2012). Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell 21, 418–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhim AD, Oberstein PE, Thomas DH, Mirek ET, Palermo CF, Sastra SA, Dekleva EN, Saunders T, Becerra CP, Tattersall IW, et al. (2014). Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell 25, 735–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rooney MS, Shukla SA, Wu CJ, Getz G, and Hacohen N (2015). Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell 160, 48–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sancho P, Burgos-Ramos E, Tavera A, Bou Kheir TB, Jagust P, Schoenhals M, Barneda D, Sellers K, Campos-Olivas R, Graña O, et al. (2015). MYC/PGC-1 alpha balance determines the metabolic phenotype and plasticity of pancreatic cancer stem cells. Cell Metab. 22, 590–605. [DOI] [PubMed] [Google Scholar]
- Scott DA, Dakin LA, Daly K, Del Valle DJ, Diebold RB, Drew L, Ezhuthachan J, Gero TW, Ogoe CA, Omer CA, et al. (2013). Mitigation of cardiovascular toxicity in a series of CSF-1R inhibitors, and the identification of AZD7507. Bioorg. Med. Chem. Lett. 23, 4591–4596. [DOI] [PubMed] [Google Scholar]
- Shukla SK, Purohit V, Mehla K, Gunda V, Chaika NV, Vernucci E, King RJ, Abrego J, Goode GD, Dasgupta A, et al. (2017). MUC1 and HIF-1alpha signaling crosstalk induces anabolic glucose metabolism to impart gemcitabine resistance to pancreatic cancer. Cancer Cell 32, 71–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel RL, Miller KD, and Jemal A (2018). Cancer statistics, 2018. CA Cancer J. Clin. 68, 7–30. [DOI] [PubMed] [Google Scholar]
- Toy G, Austin WR, Liao HI, Cheng D, Singh A, Campbell DO, Ishikawa TO, Lehmann LW, Satyamurthy N, Phelps ME, et al. (2010). Requirement for deoxycytidine kinase in T and B lymphocyte development. Proc. Natl. Acad. Sci. USA 107, 5551–5556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van den Bossche J, O’Neill LA, and Menon D (2017). Macrophage immunometabolism: where are we (going)? Trends Immunol. 38, 395–406. [DOI] [PubMed] [Google Scholar]
- Vats D, Mukundan L, Odegaard JI, Zhang L, Smith KL, Morel CR, Wagner RA, Greaves DR, Murray PJ, and Chawla A (2006). Oxidative metabolism and PGC-1beta attenuate macrophage-mediated inflammation. Cell Metab. 4, 13–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viale A, Pettazzoni P, Lyssiotis CA, Ying H, Sanchez N, Marchesini M, Carugo A, Green T, Seth S, Giuliani V, et al. (2014). Oncogene ablation-resistant pancreatic cancer cells depend on mitochondrial function. Nature 574, 628–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Von Hoff DD, Ervin T, Arena FP, Chiorean EG, Infante J, Moore M, Seay T, Tjulandin SA, Ma WW, Saleh MN, et al. (2013). Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N. Engl. J. Med. 369, 1691–1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Velez-Delgado A, Mathew E, Li D, Mendez FM, Flannagan K, Rhim AD, Simeone DM, Beatty GL, and Pasca di Magliano M (2017a). Myeloid cells are required for PD-1/PD-L1 checkpoint activation and the establishment of an immunosuppressive environment in pancreatic cancer. Gut 66, 124–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Yan W, Mathew E, Kane KT, Brannon A 3rd, Adoumie M, Vinta A, Crawford HC, and Pasca di Magliano M (2017b). Epithelial-Myeloid cell crosstalk regulates acinar cell plasticity and pancreatic remodeling in mice. Elife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Knolhoff BL, Meyer MA, Nywening TM, West BL, Luo J, Wang-Gillam A, Goedegebuure SP, Linehan DC, and DeNardo DG (2014). CSF1/CSF1R blockade reprograms tumor-infiltrating macrophages and improves response to T-cell checkpoint immunotherapy in pancreatic cancer models. Cancer Res. 74, 5057–5069. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.