

ABSTRACT
Gene transcription is regulated with distinct sets of regulatory factors at multiple levels. Transcriptional and post-transcriptional regulation constitute two major regulation modes of gene expression to either activate or repress the initiation of transcription and thereby control the number of proteins synthesized during translation. Disruptions of the proper regulation patterns at transcriptional and post-transcriptional levels are increasingly recognized as causes of human diseases. Consequently, identifying the differential gene expression at transcriptional and post-transcriptional levels respectively is vital to identify potential disease-associated and/or causal genes and understand their roles in the disease development. Here, we proposed a novel method with a linear mixed model that can identify a set of differentially expressed genes at transcriptional and post-transcriptional levels. The simulation and real data analysis showed our method could provide an accurate way to identify genes subject to aberrant transcriptional and post-transcriptional regulation and reveal the potential causal genes that contributed to the diseases.
KEWORDS: transcriptional regulation, post-transcriptional regulation, gene expression, causal gene, regulation pattern
Introduction
It is well known that gene expression involves a number of steps starting with the transcription of a gene into precursor mRNA (pre-mRNA), which is usually processed into a mature mRNA molecule that is exported from the nucleus for translation by cytoplasmic ribosomes [1]. Gene transcription is regulated with distinct sets of regulatory factors at multiple levels [2]. Transcriptional regulation (e.g. by transcription factors) and post-transcriptional regulation (e.g. by microRNAs) constitute two major regulation modes of gene expression to either activate or repress the initiation of transcription and thereby control the number of proteins synthesized during translation [3]. In the case of transcriptional regulation, a single gene can be regulated during the conversion of DNA to pre-mRNA in a range of ways, for example from altering the number of copies of RNA that are transcribed, to the control of transcription factors when the gene is transcribed. Post-transcriptional regulation is brought about by small non-coding RNAs (small RNAs/microRNAs) which bind the mature RNA of the target gene via the RNA-induced silencing complex, resulting in the degradation of the mRNAs and/or the inhibition of translation [4,5]. Generally, the relevant biological processes of diseases depend on protein levels, and mRNA levels are merely proxies for protein levels [6]. If a gene is regulated mostly transcriptionally, its pre-mRNA level is a good proxy for its protein level. Conversely, post-transcriptional regulation can set protein levels independently from pre-mRNA levels [7–10]. Disruptions of the proper regulation patterns at transcriptional and post-transcriptional regulation levels are increasingly recognized as some of the causes of human diseases [11]. Understanding the respective contributions of transcriptional and post-transcriptional regulation on gene expression dysregulation is of central importance for identifying the causal genes and understanding the fundamental molecular mechanisms of disease pathogenesis, as well as generating effective new treatments or interventions for these disorders [12].
Current technological advances have enabled quantitative measurements of thousands of genes with altered transcriptional regulation in a condition of interest [13]. RNA sequencing and exon array have been the most widely used technologies for transcriptome analysis [14]. These technologies have enabled the use of total RNA measurements to capture both intronic and exonic expression changes. For a given target gene, reads/probes mapping to its introns were used to investigate its pre-mRNA dynamics, and reads/probes mapping to exons represented a composite measure of its pre-mRNA and mature mRNA [15]. Comparing intronic and exonic expression changes across different experimental conditions allows the separation of transcriptional and post-transcriptional contributions to observed changes in RNA levels [15], thereby providing information pertinent to gene functions and regulation patterns.
In this study, we proposed a computational approach to separately quantify the differential expression of pre-mRNA and mature mRNA, which we defined as differential expression at transcriptional and post-transcriptional levels, respectively. Meanwhile, one important challenge with differential expression analysis is to distinguish causal changes from reactive ones in the context of disease, which means some differentially expressed genes have causal roles in disease development, and other genes show differential expression as responses to disease [16]. Our approach allowed one to examine differential expression at transcriptional and post-transcriptional levels to elucidate both potential causal candidate and reactive genes in an effective manner [17]. The simulation results suggested that our method can reliably identify the differential expression at transcriptional and post-transcriptional levels and provided evidence to reveal the potential causal genes. When applied our method to the gene expression profile of gastric cancer, we identified a subset of genes that exhibit differential expression at transcriptional or post-transcriptional level. The results also indicated that only a small proportion of differentially expressed genes may have direct causal roles for the development of gastric tumor by altering their protein expressions. Taken together, our method provided insight into the impact of gene regulatory patterns on aberrant gene expression and had implications for treatment and for the identification of novel therapeutic targets.
Method
Let yijgk denote the expression measurement from ith subject, jth treatment, kth region of gth gene. To account for the multiple sources of variation in expression observation of genes, consider the following gene-specific model [18]:
where Gg is the overall mean expression level for gene g, (VG)jg represents the interaction between jth treatment and gth gene, Ai represents the effect of the ith subject following a Normal distribution of N(0, ), and the error terms ϵijkg are assumed to be independent and identically distributed as N(0,). (VG)jg is the term that is of primary interest in our analysis, and it captures gene expression alterations across different treatments.
To distinguish differential expression at transcriptional and post-transcriptional levels, here Gg can be divided into two parts, GgT and GgPT, which represent the expression levels of gene g after the transcriptional and post-transcriptional regulations, respectively. Thus, the model can be rewritten as:
where and represent variations in the expression levels of gene g across different treatments under study at transcriptional and post-transcriptional levels. Both terms are of primary interest in our analysis. When the kth region is located in the intron region, reduced to not including post-transcriptional effects, GgPT and. When the kth region is located in the exon region, includes effects of both transcriptional and post-transcriptional levels. Typically, the effects of, and both terms and in the mixed model will be modeled as fixed effects.
To identify differential expression at transcriptional and post-transcriptional levels is equivalent to test if the effects of and are equal to zero. We exploit these standard mixed-model normality assumptions by using the method of restricted maximum likelihood (REML) to estimate the components of interest. REML also produces estimates of all effects in the model along with appropriate standard errors. The estimates of primary interest are those of and, which measure the treatment effects for each gene. We test these effects using mixed-model-based t-tests for each gene. All estimates are performed in the statistical package R using the lmer() function from the R-package lme4 [19]. In the following, we would further distinguish the potential causal genes from reactive ones by investigating if the gene can potentially cause the differential expression of the corresponding protein. Those genes shown differential expression at the post-transcriptional level would potentially alter the protein expression, since mature mRNA is translated into proteins. Thus, these genes can potentially contribute to the phenotype of interest as “candidate causal genes”. On the contrary, for the genes shown differential expression only at transcriptional level, their expressions of mature mRNA don’t show significant difference across different treatment groups. This implies that their expressions would not lead to their protein expression alterations. So these genes will be treated as “candidate reactive genes” in this study.
Results
Simulation study
To assess the performance of the proposed method, we performed a series of simulation studies under several different settings. We generated gene expression datasets in a case-control experiment by assuming the following model:
Yijgk was the observed expression value of kth probe/region of gene g for ith subject, where j indicated the treatment group (j = 1 for case group and j = 2 for the control group), ajg and bjg were respectively the mean expression levels at transcriptional and post-transcriptional levels for gene g in group j. γig ~ N(0, 5) was the effect of the ith subject for gene g, ϵijgk~ N(0, 2) represented the measurement error. For example, if one probe or region was located in the intron region of a gene, its expression level would include the expression level of this gene at transcriptional level ajg as well as subject effect γig and random error ϵijgk. If one probe or region was located in exon region of the gene, its expression level would include the expression level of this gene at both transcriptional and post-transcriptional levels (ajg and bjg), as well as subject effect and random error. We defined the intron probe/region as strictly intron region that is not part of a processed mRNA in any isoforms.
For simplicity, we simulated gene expression data of one gene in this study, and sample size in each group was set as 100. The simulations were designed under different scenarios shown as following:
No differential expression at both transcriptional and post-transcriptional levels;
Differential expression only at transcriptional level;
Differential expression only at post-transcriptional level;
Differential expression at both transcriptional and post-transcriptional levels, but regulation directions were opposite; and
Differential expression at both transcriptional and post-transcriptional levels and regulation directions were same.
Table 1 summarized the different scenarios with the individual mean expression levels and magnitude of differential expression at two levels across different groups (ajg and bjg), as well as the corresponding values at intron and exon regions. Among 5 scenarios, scenario 2 mimicked the potential reactive genes and scenario 3–5 mimicked different types of potential causal genes. For each scenario, we generated gene expression data with 5 probes in intron regions and 3 probes in exon regions. The simulation script can be freely accessed at https://github.com/xu1912/PostTxn.git.
Table 1.
Summary of parameter settings across different 5 scenarios.
| Transcriptional level |
Post-Transcriptional level |
Intron region |
Exon region |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Scenario | Case | Control | DE | Case | Control | DE | Case | Control | DE | Case | Control | DE |
| 1 | 1 | 1 | 0 | 1 | 1 | 0 | 1 | 1 | 0 | 2 | 2 | 0 |
| 2 | 2 | 1 | 1 | 1 | 1 | 0 | 2 | 1 | 1 | 3 | 2 | 1 |
| 3 | 1 | 1 | 0 | 1 | 0 | 1 | 1 | 1 | 0 | 2 | 1 | 1 |
| 4 | 2 | 1 | 1 | 0 | 1 | −1 | 2 | 1 | 1 | 2 | 2 | 0 |
| 5 | 2 | 1 | 1 | 2 | 1 | 1 | 2 | 1 | 1 | 4 | 2 | 2 |
Note: DE is differential expression level
We compared the powers to identify differentially expressed genes for probe-based t-test and our method in each simulation scenario. The simulation results were presented in Table 2, and in each scenario, gene/or probe was considered to be significantly differentially expressed if p-value< 0.05. In scenario 1, both methods performed well in controlling type I error. In scenario 2, probe-based t test detected the differential expression in both intron and exon regions with ~ 75% power, implying that this gene is a potential causal gene whose protein may show differential expression. However, results of our method revealed that this gene had no differential expression at the post-transcriptional level, thus it was a potential reactive gene. In scenario 3, both probe-based t-test and our method detected the differential expression with ~ 75% and 100% powers respectively. Our method indicated that this gene was a potential causal gene due to the differential expression at the post-transcriptional level. Probe-based t-test gave conflictive results that only exon regions were detected expressed. In scenario 4, we observed similar results as scenario 3. In scenario 5, probe-based t-test and our method delivered similar results, suggesting that only at the situation as scenario 5 two methods show consistent results. Meanwhile, it should be noted that in scenario 5, probe-based t-test in exon regions would overestimate the gene effect on the phenotype. The impact of one gene expression on the phenotype mainly depends on the expression level of mature mRNA. The probes in intron regions represented the expression level of pre-mRNA and the probes in exon regions represented the expression levels from pre- and mature mRNAs. This would cause the incorrect estimation of effect on phenotype for the gene.
Table 2.
Comparison of identification of differentially expressed genes between two methods.
| Probe-based t test |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| intron |
exon |
Proposed method |
||||||||
| Scenario | prob_1 | prob_2 | prob_3 | prob_4 | prob_5 | prob_6 | prob_7 | prob_8 | test-pre | test-post |
| 1 | 0.050 | 0.051 | 0.049 | 0.049 | 0.05 | 0.052 | 0.050 | 0.051 | 0.049 | 0.049 |
| 2 | 0.751 | 0.754 | 0.747 | 0.755 | 0.753 | 0.758 | 0.754 | 0.758 | 0.85 | 0.051 |
| 3 | 0.051 | 0.049 | 0.051 | 0.049 | 0.049 | 0.759 | 0.753 | 0.759 | 0.048 | 1 |
| 4 | 0.763 | 0.762 | 0.768 | 0.764 | 0.761 | 0.044 | 0.047 | 0.048 | 0.865 | 1 |
| 5 | 0.746 | 0.754 | 0.757 | 0.754 | 0.751 | 0.999 | 0.999 | 0.999 | 0.849 | 1 |
It was noted that the simulation studies demonstrated that probe-based t-test was prone to present conflictive results at intron and exon regions due to not distinguishing transcriptional and post-transcriptional levels when identifying differential expression. For example in scenario 2, our method correctly identified that there was no differential expression at the post-transcriptional level, which indicated that the gene expression change has no direct impact on the phenotype. But the probe-based t test showed the reversed conclusion. In scenario 4, since regulation directions of differential expression were opposite between transcriptional and post-transcriptional levels, probe-based t-test identified differential expression only in intron region with about 76% power, however our method could identify the differential expression at both levels with high power (86.5% and ~100%). Thus, through simulation studies, our method presented more consistent results than probe-based t test due to distinguishing differential expression at transcriptional and post-transcriptional levels respectively.
We designed more scenarios to illustrate the model performance under varying sample size, level of noise, overall intronic/exonic levels, and gene expression levels. In Figure 1(a), our method was more robust to the change of sample size in identifying the differential post-transcriptional expression. When the sample size reduced to 25 per group, our method can still yield a power of ~93.4%, comparing to the method using exon probes with a power of 74.5%. When the level of measurement error changed, the proposed method performed similarly with the method using exon probes in the power for post-transcriptional expression change. But the proposed method was more powerful at identifying the pre-transcriptional expression level. Using scenario 5 as a reference panel, we increased the pre- and post-transcriptional expression level respectively, while keeping their differential expression level the same as that in the reference panel. The different overall intronic vs exonic levels did not affect the performance (Figure 1(c)). We then added the overall gene expression level, which did not influence the model performance (Figure 1(d)).
Figure 1.
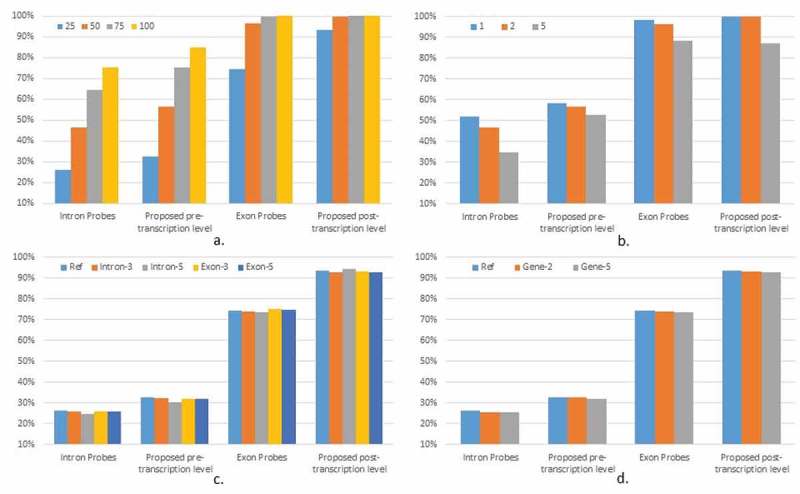
Power change under scenario 5 with: (a) sample size (per group); (b) measurement error (50 subjects per group); c) intronic and exonic levels; d) gene expression level.
However, the number of intron and exon probes do affect the power especially for the detection of post-transcriptional change. When we fixed the number of intron probes, the power of detecting post-transcriptional change increased greatly with the increase of the number of exon probes, while the power for pre-transcriptional difference did not vary (Figure 2(a)). When we fixed the number of exon probes, the more intron probes led to a power increase for not only post-transcriptional but also the pre-transcriptional test (Figure 2(b)). All the simulation result were listed in the supplementary material.
Figure 2.
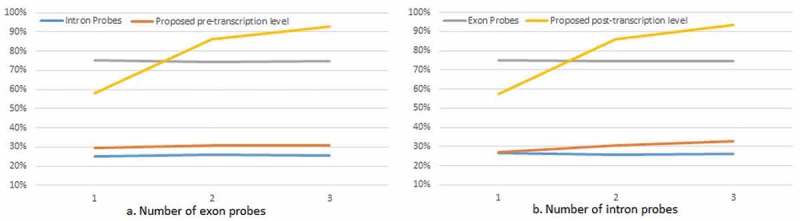
Power change under scenario 5 with the increase of: (a) exon probes; (b) intron probes.
Application on real data
We also applied the proposed method to a gene expression profile of gastric cancer (Expression data from 45 paired of gastric cancer tissues and gastric normal tissues, GEO accession number GSE63089) using the Affymetrix Gene Chip Exon Arrays 1.0 ST. We analyzed about 270,000 high confidence probes from the core probe sets of the Exon Array. Probes were mapped to a transcript database compiled from CCDS (Consensus coding sequences). The CCDS project is a collaborative effort to identify a core set of human protein-coding regions that are consistently annotated and of high quality. After mapping, 3,108 genes were identified for our analysis, which had both intron and exon region probes.
After Bonferroni correction (p < 1.60E-05), 11 genes were identified differentially expressed at post-transcriptional level, which could be candidate causal genes. And 136 genes were identified at transcriptional level. Table 3 listed the genes with differential expression at transcriptional and post-transcriptional levels (top 15 genes) respectively. It was interesting that the differentially expressed genes identified at transcriptional level were much more than those identified at the post-transcriptional level. This might indicate that only a small number of genes have effects on the development of gastric cancer through gene expression changes, and most gene differential expressions could be caused by gastric cancer. Further, we found the expression alterations of most of the genes at transcriptional level could be “remedied” by post-transcriptional regulation. Among 136 genes, there were 133 candidate reactive genes that showed expression alterations across two groups only at the transcriptional level. In line with existing evidence in the literature, reactive genes usually outnumber the causative genes [20]. Although these candidate reactive genes may not contribute to the development of gastric cancer, they can be used as biomarkers to classify the gastric cancer tissues from normal tissues. For example, previous studies revealed that COL1A1 mRNA expression was significantly upregulated in premalignant and malignant tissues of gastric cancer than in normal tissues. However, COL1A1 expression was unrelated to clinical pathological parameters. Our analysis could give a reasonable interpretation of this phenomenon. The expression change of COL1A1 only occurs at transcriptional level, but not at post-transcriptional level. Therefore, COL1A1 might have its potential as a monitoring factor to screen early gastric cancer [21].
Table 3.
P-values of differentially expressed genes identified in gastric cancer data.
| DE genes at Post-transcriptional level |
DE genes at Transcriptional level |
||||
|---|---|---|---|---|---|
| Symbols | TL | PTL | Symbols | TL | PTL |
| ATP4A | 2.00E-14 | 2.82E-14 | COL1A1 | <1.00E-15 | 0.04 |
| SLC25A22 | 6.13E-3 | 6.98E-09 | CAD | <1.00E-15 | 1.06E-04 |
| C2orf57 | 1.09E-10 | 4.99E-08 | SPP1 | 1.78E-15 | 2.54E-03 |
| DGAT1 | 0.26 | 2.47E-07 | ITGA2 | 4.66E-15 | 0.24 |
| BRD8 | 9.68E-09 | 2.98E-07 | TOP2A | 1.64E-14 | 0.74 |
| F2RL2 | 0.17 | 7.49E-07 | ATP4A | 2.00E-14 | 2.82E-14 |
| ZBTB16 | 0.06 | 1.50E-06 | SULF2 | 1.08E-13 | 0.25 |
| CCL20 | 2.06E-4 | 3.07E-06 | PRKDC | 2.30E-13 | 0.50 |
| APOBEC2 | 0.25 | 3.28E-06 | LAPTM4B | 5.83E-13 | 0.03 |
| PGA3 | 6.47E-05 | 5.07E-06 | IQGAP3 | 1.66E-12 | 4.79E-04 |
| ZNRF2 | 0.02 | 9.06E-06 | TNFSF15 | 1.49E-11 | 0.05 |
| MMP3 | 1.97E-11 | 0.26 | |||
| NIP7 | 4.09E-11 | 0.45 | |||
| C2orf57 | 1.09E-10 | 4.99E-08 | |||
| KIF20A | 1.11E-10 | 0.09 | |||
Note: DE: differential expression; TL: transcriptional level; and PTL: post-transcriptional level.
When comparing the differentially expressed genes between two levels, we found 1) there were 3 genes (ATP4A, C2orf57, and BRD8) showing expression changes at both levels, which might be dominated by transcriptional regulation; 2) there were 8 genes showing expression changes only at the post-transcriptional level, which were dominated by post-transcriptional regulation. Since mature mRNAs were translated into proteins, these 11 genes could potentially alter the protein expression, which could be used to elucidate the pathogenesis of gastric cancer. One example is ATP4A gene, which encodes the catalytic subunit of a gastric proton pump that uses the hydrolysis of ATP to generate an acid environment in the stomach and it is an important serum biomarker for gastric cancer as well as plays a critical role in gastric neuroendocrine tumor [22–24]. A mutation in this gene could inhibit gastric acid production and explain the achlorhydria and hypergastrinemia that were associated with the development of gastric cancer [25]. Besides, it has been reported that the ATP4A mRNA is down-regulated in gastric tumors relative to normal gastric tissues, and the consistent methylation pattern of ATP4A could be observed in tumor tissue samples [22]. Also the relevant evidence could be found for other genes, e.g. PGA3 [26–28] CCL20 [29–31] and ZBTB16 [32,33].
We applied our approach to a second real dataset (GEO no. GSE21034), which includes 29 pairs of prostate cancer and normal tissues profiled using Affymetrix Gene Chip Exon Arrays 1.0 ST. After similar pre-processing, we analyzed 3,539 genes with both intron and exon probes. We found 369 and 7 significant differentially expressed genes at pre- and post-transcriptional level respectively after Bonferroni correction. The top 10 genes were listed in Table 4. For the 7 genes significant at the post-transcriptional level, three genes, MUC13, VSIG10L, FAM221B, were also differentially expressed at pre-transcriptional levels and ranked top 3, which might be dominated by transcriptional regulation. The rest 4 genes were probably dominated by post-transcriptional regulation. The MUC13 promoter region contains binding sites for a transcription factor plays a role in the pathogenesis of prostate cancer [34]. PLIN2 hinders lipolytic pathways, which results in the accumulation of lipid droplets and associated with prostate cancer [35]. The other genes potentially alter the protein expression related to the prostate cancer are worth further investigation.
Table 4.
P-values of differentially expressed genes identified in prostate cancer data.
| DE genes at Post-transcriptional level |
DE genes at Transcriptional level |
||||
|---|---|---|---|---|---|
| Symbols | TL | PTL | Symbols | TL | PTL |
| TRAF3 | 6.91E-01 | 9.94E-31 | MUC13 | 2.91E-11 | 2.20E-10 |
| MUC13 | 2.91E-11 | 2.20E-10 | VSIG10L | 1.25E-07 | 8.93E-07 |
| PYM1 | 1.46E-05 | 8.10E-10 | FAM221B | 1.08E-12 | 1.46E-06 |
| VSIG10L | 1.25E-07 | 8.93E-07 | USF3 | 2.50E-29 | 6.66E-05 |
| FAM221B | 1.08E-12 | 1.46E-06 | POMGNT1 | 7.38E-14 | 7.22E-05 |
| PLIN2 | 1.79E-01 | 6.18E-06 | RIMS4 | 7.73E-07 | 5.97E-04 |
| LOC729966 | 7.14E-01 | 1.22E-05 | PRKCG | 7.61E-27 | 6.37E-04 |
| CACNA1C | 8.97E-29 | 6.45E-04 | |||
| UPK2 | 1.65E-07 | 6.57E-04 | |||
| ARHGAP44 | 1.39E-18 | 1.44E-03 | |||
Note: DE: differential expression; TL: transcriptional level; and PTL: post-transcriptional level.
Discussion
In this study, we proposed a method to respectively identify the differential expression for a given gene at transcriptional and post-transcriptional levels across different experimental conditions. Unlike traditional strategies, our method can be used to determine whether expression changes were caused by transcriptional or post-transcriptional mechanisms, pinpointing the candidate disease-associated and/or causal genes from a large number of differentially expressed genes under study. Also, we can obtain an estimate of the relative importance of transcriptional and posttranscriptional mechanisms in regulating gene expression. As shown in our simulation and real data study, our method can gain insights into the regulatory mechanism responsible for the observed expression changes and elucidate potential causative changes that lead to disease. However, the current simulation study mainly served as a sanity check. The biological significance of the proposed model in practice needs further investigation.
Identifying disease causal genes and characterizing functional contributions to complex diseases is challenging [36]. With our analysis, genes shown differential expression at post-transcriptional level may have more impact on phenotype than other genes by directly contributing to the differential expression of their proteins [37,38]. However, for those genes shown differential expression only at the transcriptional level, they didn’t show differential expression due to post-transcriptional regulation. Consequently, those genes may not influence the phenotype by their expression changes. As shown in the results of real data analysis, the 11 genes shown differential expression at post-transcriptional level may have more important functions in the development of gastric cancer. As to genes shown differential expression only at the transcriptional level, many of them have been identified as biomarkers for gastric cancer [21,39,40]. These results highlighted the importance of distinguishing differential expression at individual transcriptional and post-transcriptional levels [41]. Both of the two real datasets identified a small number of differential post-transcriptional genes, which may be due to the limited number of exon probes. Our simulation study also showed the number of exon probes was correlated with the test power (Figure 2).
Estimating the contributions of transcriptional and post-transcriptional regulation was essential for inferring their biological regulation patterns and mechanisms via biological networks [36]. Transcriptional and post-transcriptional regulation both can cause differential expression of genes, but their relative contributions remain contested. Identification of differential expression with distinguishing transcriptional and post-transcriptional levels could benefit downstream analyses, including the inference of regulation pattern on gene expression. For the genes shown only differential expression at the transcriptional level, their expression changes across different groups were dominated by transcriptional regulation, such as transcriptional factors, methylation level. For the genes shown only differential expression at post-transcriptional level, their expression changes were dominated by post-transcriptional regulation, such as miRNA-mediated regulation [42–44]. For example, previous studies showed that miR-16 acts as a tumor suppressor and significantly inhibits cell proliferation and migration [45–47]. In our study SLC25A22 was identified as a potential causal factor for gastric cancer, and SLC25A22 has been a validated target of mir-16 [48]. Since expression change of SLC25A22 is due to post-transcriptional regulation, we can infer that miR-16 acts as a tumor suppressor by mediating expression changes of SLC25A22. Thus, one of the important features of our method was that it can distinguish direct miRNA-mediated effects from transcriptional effects. For genes shown differential expression at both transcriptional and post-transcriptional levels, we could infer the individual contributions of transcriptional and post-transcriptional regulation by comparing the magnitudes of differential expression at both levels. In the results of the real data analysis, there were 133 genes that showed expression changes across two groups only at transcriptional level. This indicated that the expression alterations of these genes were attributed to transcriptional regulation. Meanwhile, it suggested that cells use some feedback mechanism feedback to eliminate the expression changes of these genes by post-transcriptional regulation, and its mechanism likely relies on sensing the levels of pre-mRNA. This result underscored the role of post-transcriptional regulation on mediating gene expression [5].
In summary, we have shown here that our proposed approach can separate differentially expressed genes that were under transcriptional control from those that were regulated predominantly on a post-transcriptional level. The insights gained by our modeling approach provided a consistent framework toward the elucidation of operational and molecular strategies used to regulate transcriptional responses. Therefore, our method increases the value of many existing and future gene expression data sets and provides a tool to study transcriptional and post-transcriptional contributions to expression changes.
Supplementary Material
Funding Statement
This work was supported by the National Institutes of Health [U19 AG055373, R01AR057049, R01AR059781, R01AR069055, D43TW009107, P20 GM109036, R01MH107354, R01MH104680, R01GM109068]; Tulane University [Edward G. Schlieder Endowment fund].
Acknowledgments
The investigators were partially supported by grants from the National Institutes of Health [U19 AG055373, R01AR057049, R01AR059781, R01AR069055, D43TW009107, P20 GM109036, R01MH107354, R01MH104680, R01GM109068], and the Edward G. Schlieder Endowment fund from Tulane University.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- [1].Ravi S, Schilder RJ, Kimball SR.. Role of precursor mRNA splicing in nutrient-induced alterations in gene expression and metabolism. J Nutr. 2015. May;145(5):841–846. PubMed PMID: 25761502; PubMed Central PMCID: PMC4408736. Eng. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Halbeisen RE, Galgano A, Scherrer T, et al. Post-transcriptional gene regulation: from genome-wide studies to principles. Cell Mol Life Sci. 2008. March;65(5):798–813. PubMed PMID: 18043867; PubMed Central PMCID: PMC2771128. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Zhao BS, Roundtree IA, He C.. Post-transcriptional gene regulation by mRNA modifications. Nat Rev Mol Cell Biol. 2017. January;18(1):31–42. PubMed PMID: 27808276; PubMed Central PMCID: PMC5167638. eng. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Hamzeiy H, Allmer J, Yousef M. Computational methods for microRNA target prediction. Methods Mol Biol. 2014;1107:207–221. PubMed PMID: 24272439; eng. . [DOI] [PubMed] [Google Scholar]
- [5].Franks A, Airoldi E, Slavov N. Post-transcriptional regulation across human tissues. PLoS Comput Biol. 2017. May;13(5):e1005535 PCOMPBIOL-D-16-02064 [pii]. PubMed PMID: 28481885; PubMed Central PMCID: PMC5440056. eng. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Edfors F, Danielsson F, Hallstrom BM, et al. Gene-specific correlation of RNA and protein levels in human cells and tissues. Mol Syst Biol. 2016. October 20;12(10):883 PubMed PMID: 27951527; PubMed Central PMCID: PMC5081484. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Kuersten S, Goodwin EB. The power of the 3ʹ UTR: translational control and development. Nat Rev Genet. 2003. August;4(8):626–637. PubMed PMID: 12897774; eng. . [DOI] [PubMed] [Google Scholar]
- [8].Carpenter S, Ricci EP, Mercier BC, et al. Post-transcriptional regulation of gene expression in innate immunity. Nat Rev Immunol. 2014. June;14(6):361–376. PubMed PMID: 24854588; eng. [DOI] [PubMed] [Google Scholar]
- [9].Diab T, Hanoun N, Bureau C, et al. The role of the 3ʹ untranslated region in the post-transcriptional regulation of KLF6 gene expression in hepatocellular carcinoma. Cancers (Basel). 2013. December 19;6(1):28–41. PubMed PMID: 24378751; PubMed Central PMCID: PMC3980593. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Matoulkova E, Michalova E, Vojtesek B, et al. The role of the 3ʹ untranslated region in post-transcriptional regulation of protein expression in mammalian cells. RNA Biol. 2012. May;9(5):563–576. PubMed PMID: 22614827; eng. [DOI] [PubMed] [Google Scholar]
- [11].Cooper TA, Wan L, Dreyfuss G. RNA and disease. Cell. 2009. February 20;136(4):777–793. S0092-8674(09)00148-2 [pii]. PubMed PMID: 19239895; PubMed Central PMCID: PMC2866189. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Bonifer C, Cockerill PN. Chromatin mechanisms regulating gene expression in health and disease. Adv Exp Med Biol. 2011;711:12–25. PubMed PMID: 21627039; Eng. [DOI] [PubMed] [Google Scholar]
- [13].Goncalves E, Raguz NZ, Zampieri M, et al. Systematic analysis of transcriptional and post-transcriptional regulation of metabolism in yeast. PLoS Comput Biol. 2017. January;13(1):e1005297 PCOMPBIOL-D-16-01445 [pii]. PubMed PMID: 28072816; PubMed Central PMCID: PMC5224888. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Zhao S, Fung-Leung WP, Bittner A, et al. Comparison of RNA-Seq and microarray in transcriptome profiling of activated T cells. PLoS One. 2014;9(1):e78644 PONE-D-13-21649 [pii]. PubMed PMID: 24454679; PubMed Central PMCID: PMC3894192. eng. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Gaidatzis D, Burger L, Florescu M, et al. Analysis of intronic and exonic reads in RNA-seq data characterizes transcriptional and post-transcriptional regulation. Nat Biotechnol. 2015. July;33(7):722–729. nbt.3269 [pii]. PubMed PMID: 26098447; Eng. [DOI] [PubMed] [Google Scholar]
- [16].Deo RC, Musso G, Tasan M, et al. Prioritizing causal disease genes using unbiased genomic features. Genome Biol. 2014. December 03;15(12):534 s13059-014-0534-8 [pii]. PubMed PMID: 25633252; PubMed Central PMCID: PMC4279789. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Lee E, Cho S, Kim K, et al. An integrated approach to infer causal associations among gene expression, genotype variation, and disease. Genomics. 2009. October;94(4):269–277. S0888-7543(09)00134-7 [pii]. PubMed PMID: 19540336; eng. [DOI] [PubMed] [Google Scholar]
- [18].Cui X, Churchill GA. Statistical tests for differential expression in cDNA microarray experiments. Genome Biol. 2003;4(4):210 PubMed PMID: 12702200; PubMed Central PMCID: PMC154570. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Lamprianou I. Application of single-level and multi-level Rasch models using the lme4 package. J Appl Meas. 2013;14(1):79–90. PubMed PMID: 23442329; eng. [PubMed] [Google Scholar]
- [20].Hasin Y, Seldin M, Lusis A. Multi-omics approaches to disease. Genome Biol. 2017. May 05;18(1):83 10.1186/s13059-017-1215-1 [pii]. PubMed PMID: 28476144; PubMed Central PMCID: PMC5418815. eng. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Li J, Ding Y, Li A. Identification of COL1A1 and COL1A2 as candidate prognostic factors in gastric cancer. World J Surg Oncol. 2016. November 29;14(1):297 10.1186/s12957-016-1056-5 [pii]. PubMed PMID: 27894325; PubMed Central PMCID: PMC5126984. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Raja UM, Gopal G, Rajkumar T. Intragenic DNA methylation concomitant with repression of ATP4B and ATP4A gene expression in gastric cancer is a potential serum biomarker. Asian Pac J Cancer Prev. 2012;13(11):5563–5568. PubMed PMID: 23317218; eng. [DOI] [PubMed] [Google Scholar]
- [23].Calvete O, Herraiz M, Reyes J, et al. A cumulative effect involving malfunction of the PTH1R and ATP4A genes explains a familial gastric neuroendocrine tumor with hypothyroidism and arthritis. Gastric Cancer. 2017. May 04 PubMed PMID: 28474257; eng DOI: 10.1007/s10120-017-0723-8. [DOI] [PubMed] [Google Scholar]
- [24].Judd LM, Andringa A, Rubio CA, et al. Gastric achlorhydria in H/K-ATPase-deficient (Atp4a(-/-)) mice causes severe hyperplasia, mucocystic metaplasia and upregulation of growth factors. J Gastroenterol Hepatol. 2005. August;20(8):1266–1278. JGH3867 [pii] PubMed PMID: 16048577; eng. [DOI] [PubMed] [Google Scholar]
- [25].Calvete O, Reyes J, Zuniga S, et al. Exome sequencing identifies ATP4A gene as responsible of an atypical familial type I gastric neuroendocrine tumour. Hum Mol Genet. 2015. May 15;24(10):2914–2922. PubMed PMID: 25678551; eng. [DOI] [PubMed] [Google Scholar]
- [26].Tu H, Sun L, Dong X, et al. Temporal changes in serum biomarkers and risk for progression of gastric precancerous lesions: a longitudinal study. Int J Cancer. 2015. January 15;136(2):425–434. PubMed PMID: 24895149; PubMed Central PMCID: PMC4354768. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Shen S, Jiang J, Yuan Y. Pepsinogen C expression, regulation and its relationship with cancer. Cancer Cell Int. 2017;17:57 426 [pii]. PubMed PMID: 28546787; PubMed Central PMCID: PMC5442862. eng. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Li H, Yu B, Li J, et al. Characterization of differentially expressed genes involved in pathways associated with gastric cancer. PLoS One. 2015;10(4):e0125013 PONE-D-14-44651 [pii]. PubMed PMID: 25928635; PubMed Central PMCID: PMC4415781. eng. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Han G, Wu D, Yang Y, et al. CrkL meditates CCL20/CCR6-induced EMT in gastric cancer. Cytokine. 2015. December;76(2):163–169. S1043-4666(15)00189-1 [pii]. PubMed PMID: 26044596; eng. [DOI] [PubMed] [Google Scholar]
- [30].Ohtani H, Nakayama T, Yoshie O. In situ expression of the CCL20-CCR6 axis in lymphocyte-rich gastric cancer and its potential role in the formation of lymphoid stroma. Pathol Int. 2011. November;61(11):645–651. PubMed PMID: 22029675; eng. . [DOI] [PubMed] [Google Scholar]
- [31].Wu YY, Tsai HF, Lin WC, et al. Upregulation of CCL20 and recruitment of CCR6+ gastric infiltrating lymphocytes in Helicobacter pylori gastritis. Infect Immun. 2007. September;75(9):4357–4363. IAI.01660-06 [pii] PubMed PMID: 17562763; PubMed Central PMCID: PMC1951156. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Chang W, Ma L, Lin L, et al. Identification of novel hub genes associated with liver metastasis of gastric cancer. Int J Cancer. 2009. December 15;125(12):2844–2853. PubMed PMID: 19569046; eng. [DOI] [PubMed] [Google Scholar]
- [33].Song W, Liu YY, Peng JJ, et al. Identification of differentially expressed signatures of long non-coding RNAs associated with different metastatic potentials in gastric cancer. J Gastroenterol. 2016. February;51(2):119–129. 10.1007/s00535-015-1091-y [pii]. PubMed PMID: 26045391; PubMed Central PMCID: PMC4742487. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Macher DM, Gupta BK, Nagata S, et al. Mucin 13: structure, function, and potential roles in cancer pathogenesis. Mol Cancer Res. 2011;9(5):531–537. PMID: 21450906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Tirinato L, Pagliari F, Limongi T, et al. An overview of lipid droplets in cancer and cancer stem cells. Stem Cells Int. 2017;Article ID 1656053. PMID: 28883835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Schadt EE. Molecular networks as sensors and drivers of common human diseases. Nature. 2009. September 10;461(7261):218–223. nature08454 [pii]. PubMed PMID: 19741703; eng. . [DOI] [PubMed] [Google Scholar]
- [37].Jovanovic M, Rooney MS, Mertins P, et al. Immunogenetics. Dynamic profiling of the protein life cycle in response to pathogens. Science. 2015. March 06;347(6226):1259038 PubMed PMID: 25745177; PubMed Central PMCID: PMC4506746. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Csardi G, Franks A, Choi DS, et al. Accounting for experimental noise reveals that mRNA levels, amplified by post-transcriptional processes, largely determine steady-state protein levels in yeast. PLoS Genet. 2015. May;11(5):e1005206 PGENETICS-D-14-01701 [pii]. PubMed PMID: 25950722; PubMed Central PMCID: PMC4423881. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Epplein M, Zheng W, Xiang YB, et al. Prospective study of Helicobacter pylori biomarkers for gastric cancer risk among Chinese men. Cancer Epidemiol Biomarkers Prev. 2012. December;21(12):2185–2192. PubMed PMID: 23035179; PubMed Central PMCID: PMC3518572. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Zhuo C, Li X, Zhuang H, et al. Elevated THBS2, COL1A2, and SPP1 expression levels as predictors of gastric cancer prognosis. Cell Physiol Biochem. 2016;40(6):1316–1324. PubMed PMID: 27997896; eng. . [DOI] [PubMed] [Google Scholar]
- [41].Laloo B, Simon D, Veillat V, et al. Analysis of post-transcriptional regulations by a functional, integrated, and quantitative method. Mol Cell Proteomics. 2009. August;8(8):1777–1788. PubMed PMID: 19411282; PubMed Central PMCID: PMC2722765. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Gosline SJ, Gurtan AM, JnBaptiste CK, et al. Elucidating microRNA regulatory networks using transcriptional, post-transcriptional, and histone modification measurements. Cell Rep. 2016. January 12;14(2):310–319. S2211-1247(15)01455-2 [pii]. PubMed PMID: 26748710; PubMed Central PMCID: PMC4831719. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Agarwal V, Bell GW, Nam JW, et al. Predicting effective microRNA target sites in mammalian mRNAs. Elife. 2015. August 12;4 PubMed PMID: 26267216; PubMed Central PMCID: PMC4532895. eng DOI: 10.7554/eLife.05005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Chiu HS, Llobet-Navas D, Yang X, et al. Cupid: simultaneous reconstruction of microRNA-target and ceRNA networks. Genome Res. 2015. February;25(2):257–267. PubMed PMID: 25378249; PubMed Central PMCID: PMC4315299. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Li S, Zhang H, Wang X, et al. Direct targeting of HGF by miR-16 regulates proliferation and migration in gastric cancer. Tumour Biol. 2016. November;37(11):15175–15183. PubMed PMID: 27683052; eng. [DOI] [PubMed] [Google Scholar]
- [46].Xia L, Zhang D, Du R, et al. miR-15b and miR-16 modulate multidrug resistance by targeting BCL2 in human gastric cancer cells. Int J Cancer. 2008. July 15;123(2):372–379. PubMed PMID: 18449891; eng. [DOI] [PubMed] [Google Scholar]
- [47].Shin VY, Jin H, Ng EK, et al. NF-kappaB targets miR-16 and miR-21 in gastric cancer: involvement of prostaglandin E receptors. Carcinogenesis. 2011. February;32(2):240–245. PubMed PMID: 21081469; eng. [DOI] [PubMed] [Google Scholar]
- [48].Selbach M, Schwanhausser B, Thierfelder N, et al. Widespread changes in protein synthesis induced by microRNAs. Nature. 2008. September 04;455(7209):58–63. PubMed PMID: 18668040; eng. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.