

ABSTRACT
As the new millennium began, CDK12 and CDK13 were discovered as nucleotide sequences that encode protein kinases related to cell cycle CDKs. By the end of the first decade both proteins had been qualified as CTD kinases, and it was emerging that both are heterodimers containing a Cyclin K subunit. Since then, many studies on CDK12 have shown that, through phosphorylating the CTD of transcribing RNAPII, it plays critical roles in several stages of gene expression, notably RNA processing; it is also crucial for maintaining genome stability. Fewer studies on CKD13 have clearly shown that it is functionally distinct from CDK12. CDK13 is important for proper expression of a number of genes, but it also probably plays yet-to-be-discovered roles in other processes. This review summarizes much of the work on CDK12 and CDK13 and attempts to evaluate the results and place them in context. Our understanding of these two enzymes has begun to mature, but we still have much to learn about both. An indicator of one major area of medically-relevant future research comes from the discovery that CDK12 is a tumor suppressor, notably for certain ovarian and prostate cancers. A challenge for the future is to understand CDK12 and CDK13 well enough to explain how their loss promotes cancer development and how we can intercede to prevent or treat those cancers.
Abbreviations: CDK: cyclin-dependent kinase; CTD: C-terminal repeat domain of POLR2A; CTDK-I: CTD kinase I (yeast); Ctk1: catalytic subunit of CTDK-I; Ctk2: cyclin-like subunit of CTDK-I; PCAP: phosphoCTD-associating protein; POLR2A: largest subunit of RNAPII; SRI domain: Set2-RNAPII Interacting domain
KEYWORDS: RNA polymerase II, C-terminal repeat domain, CTD, CTD phosphorylation, co-transcriptional processes
1. Introduction
The C-terminal repeat domain (CTD) of eukaryotic RNA polymerase II (RNAPII) is phosphorylated by a set of “transcriptional CDKs” that are structurally related to the classical CDKs. For example, the catalytic subunit of the major transcription elongation-phase CTD kinase in yeast is Ctk1, which has CDK homology [1]. The corresponding cyclin-like subunit in yeast is Ctk2 [2]. During the “Early Studies” of transcriptional CDKs in eukaryotes, it was proposed that the metazoan ortholog of yeast Ctk1 was CDK9, the catalytic subunit of P-TEFb [3]. However, molecular evolutionists had found that CDK9 was actually most closely related to yeast Bur1, and that two human proteins, gi|14110386| and gi|20521690|, and one uncharacterized Drosophila protein, gi|24668141|, were most-closely related to yeast Ctk1 [4,5], as we pointed out in 2006 [6]. To begin the “Modern Era” of studies on the metazoan counterpart(s) of Ctk1, we focused first on the single gene/protein in Drosophila, now called CDK12 (NCBI Reference Sequence: NP_730643.1; flybase.org: CG7597). Subsequently we and others investigated the two human counterparts, now called CDK12 (NCBI Reference Sequence: NP_057591.2; Gene ID: 51755) and CDK13 (NCBI Reference Sequence: NP_003709.3; Gene ID: 8621), and much of this work is described below. One aside: I sometimes refer to CDK12/13 proteins (and their orthologs) as the “Ctk1 family” of CTD kinases, to distinguish them from the other families of transcriptional CDKs (e.g. CDK9 & CDK7). Before describing the Modern Era of CDK12/13 investigations, I give a brief summary of studies on metazoan Ctk1 counterparts carried out before 2010.
2. Early studies of CDK12 & CDK13
2.1. Primary structure & subunit composition
2.1.1. “CrkRS” (now, CDK12)
Pines and colleagues cloned a gene that encoded a protein of ~ 180 kDa that contained a cdc2-related protein kinase domain, along with numerous RS repeats, and they called it CrkRS [7]. Because CrkRS (now called CDK12) is fairly unusual for a CDK, as its 180,000 MW might suggest, I present an overview of its 1° structure along with those of other of Ctk1 family members in Figure 1(a). CrkRS/CDK12 contains a central kinase homology region of ~ 300 amino acids, from which extends an N-terminal “arm” about 700 residues long and a C-terminal arm about 500 residues long. Overall the arms contain many regions of low sequence complexity (see later), but the most noticeable feature is a stretch rich in RS dipeptides (RS domain), as found in a number of splicing factors. Also of note are two proline-rich regions (P), one in the N-terminal arm and one in the C-terminal arm. The presence of the RS domain and P regions suggest that the arms of CDK12 are likely to take part in numerous protein-protein interactions.
Figure 1.
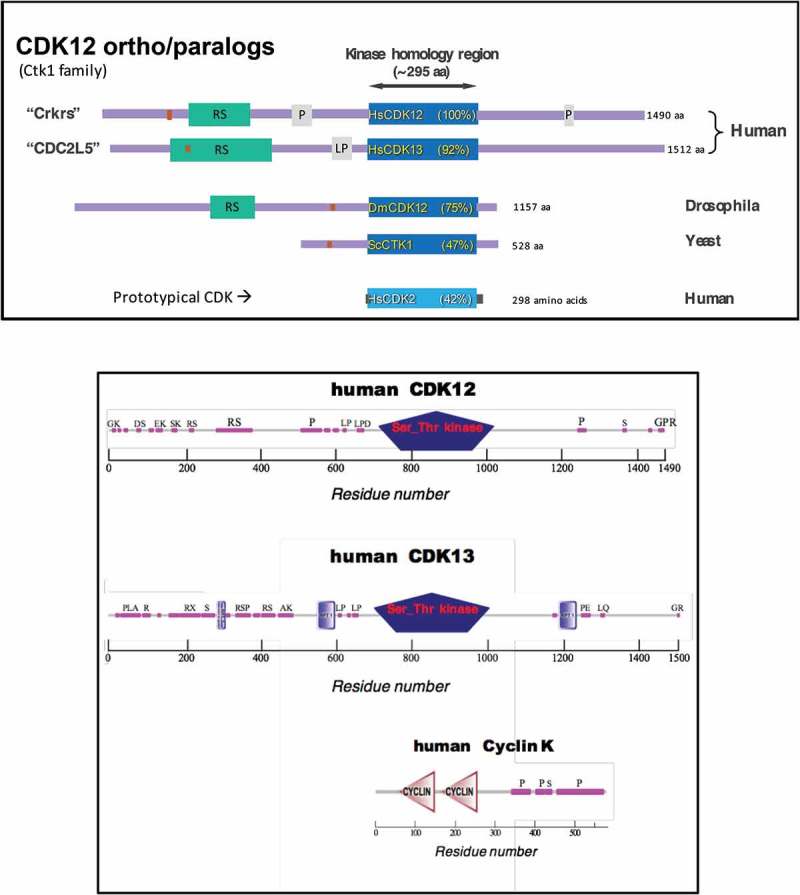
Primary Structures of CDK12 (Ctk1) family of transcriptional CDK kinases, and Cyclin K.
(a) “Ctk1 family” primary structures aligned by kinase homology domains (dark blue). Purple lines indicate protein “arms” extending from N- and C-termini of kinase domain. Green boxes = RS domains. Gray boxes = Pro- or LeuPro-rich domains. Rust-colored boxes = segments used as immunogens to raise antibodies. (b) Primary structures of human CDKs and Cyclin K (SMART database). Pink boxes = low sequence complexity regions. Letters above pink boxes indicate abundant residues therein. Drawn approx. to scale.
Fann and colleagues cloned a rat kinase that turned out to be homologous to human and mouse CrkRS, and they renamed this kinase CDK12 [8]. They speculated, based on previous in situ localization studies, that the cyclin associated with CDK12 might be Cyclin L. Using overexpression of tagged versions of CDK12 and differently-tagged versions of Cyclin L, they found that some Cyclin L co-IP’d with CDK12 and some CDK12 co-IP’d with Cyclin L. On the other hand, they did not show that native CDK12 interacted with native Cyclin L physically or functionally. From more recent work, described below, we now know that the cyclin associated with CDK12 is actually Cyclin K.
2.1.2. ‘CDC2L5ʹ (now, CDK13)
In 2000, Geneviére and colleagues had cloned cDNAs of sea urchin and human versions of a cdc2-related kinase they called CDC2L5 (now called CDK13), for which they had found presumed orthologs in other metazoa [9]. In addition, they found that humans contained another gene similar to CDC2L5, namely CrkRS (above). They described the 1° structures of this kinase family, showing that CDC2L5/CDK13 contains N-terminal and C-terminal arms reminiscent of those of CrkRS/CDK12 (Figure 1(a)). They also presented phylogenetic tree analysis that placed CrkRS (CDK12) and CDC2L5 (CDK13) on a branch that diverged from CDK9. Subsequently, this group described the nuclear localization of native human CDC2L5 using antibodies they generated [10].
In 2007, Fann and Geneviére groups collaborated to identify the cyclin subunit of the CDK13/CDC2L5 kinase [11], using overexpression of kinase and cyclin proteins. This approach led them to claim that Cyclin L is the partner of CDK13. However, as for CDK12, above, they did not test interactions using native, endogenous proteins. We now know that the subunit partner of CDK13 is actually Cyclin K, as it is for CDK12.
2.2. Activities
2.2.1. CrkRS (CDK12)
Pines and colleagues [7] showed that a CrkRS immunoprecipitate possessed CTD kinase activity. They used their antibodies to immunoprecipitate the CrkRS protein, and they showed that the immunoprecipitated material was able to phosphorylate a fusion protein carrying the yeast CTD. When they attempted to express and purify CrkRS in a baculovirus system, however, they were unable to produce soluble, purified protein, and thus were unable to test it for associated kinase activity. They conservatively, and properly, concluded that “ This also meant that we were unable to exclude the possibility that other kinases present in the anti-CrkRS immunoprecipitates are responsible for some of the phosphorylation activity.” Ironically, from what we now know, it is likely that they were in fact observing in vitro CTD phosphorylation by CrkRS (CDK12) for the first time.
Pines and colleagues also used their affinity-purified antibodies to show that endogenous CrkRS localized in the nucleus, largely in SC35 speckles. They also showed that the RS domain was mainly responsible for targeting overexpressed CrkRS constructs to speckles. Given the likelihood that CrkRS was a CTD kinase that carried RS domains and was targeted to speckles, they presciently speculated: “Thus, CrkRS could represent a novel, evolutionarily conserved RNA polymerase II CTD kinase that might directly link transcription with the splicing machinery.”
Fann et al. overexpressed versions of CDK12 in HEK293T cells and looked for effects on splice site selection of an E1a minigene construct [8]. While they did observe changes, it is not clear if this overexpression approach measures a physiologically relevant event.
2.2.2. ‘CDC2L5ʹ (CDK13)
In a collaboration the Fann and Geneviére groups used overexpression of tagged constructs to investigate CDC2L5/CDK13 [11] and observed overlap of CDC2L5 with nuclear speckles; they mapped the localization determinants to the N-terminal, RS-containing segment of the protein. As for CrkRS/CDK12 (above), they showed that overexpression of CDC2L5/CDK13 could alter splicing (so could overexpression of L-type cyclins). But, as for CrkRS/CDK12, it is not clear if this overexpression approach assesses physiologically relevant events.
SUMMARY of Early Studies. Two proteins (in mammals) were described that carry CDK-homologous kinase domains and two long protein arms composed of many low sequence complexity stretches and containing RS domains and Proline-rich regions. However, that these proteins, now called CDK12 and CDK13, were CTD kinases had not been rigorously demonstrated. Also, the cyclin subunit of the two CDKs had been mis-identified.
3. Beginning the modern era
While CDK12 and CDK13 had been identified and partially characterized before 2010, there still existed major gaps in our understanding of these two proteins. For example, neither had been rigorously shown to be a CTD kinase; also, the cyclin subunit partners for each CDK had not been correctly identified. Several groups began studies that would generate information to fill these knowledge gaps.
3.1. Primary structure & subunit composition
3.1.1. CDK12, CDK13 and Cyclin K
We generated affinity-purified rabbit antibodies against a peptide from the predicted amino acid sequence of the ‘CDK12ʹ protein of Drosophila melanogaster (cf, Figure 1(a)) and used them to immunopurify the protein using antibody beads [12]. The beads were washed with 0.4 M salt before eluting CDK12 off the beads with the immunogenic peptide; we showed that the peptide-eluted protein fractions possessed CTD kinase activity (see more below under POTENTIAL FUNCTIONS). Separately, we also washed bead-bound enzyme with 0.8 M salt, and showed that it retained CTD kinase activity. In terms of subunit composition of the kinase, we carried out MS analysis on proteins co-immunopurified using the antibody beads, and the only cyclin detected was Drosophila Cyclin K. We therefore suggested that the cyclin partner of CDK12 is Cyclin K, with the caveat that additional experiments were needed to prove this.
Peterlin and colleagues, approaching from the cyclin side, identified two proteins that bind human Cyclin K (Figure 1(b)), and determined that they were CDK12 and CDK13 [13]; their further experiments supported this finding and also showed that CDK12/Cyclin K and CDK13/Cyclin K were two distinct complexes.
Morin and colleagues used antibodies to human CDK12 (from J. Pines) and showed that the principal cyclin that co-IPs with endogenous CDK12 is Cyclin K. They went on to show that CDK12 interacts predominantly with Cyclin K and that Cyclin K interacts predominantly with CDK12 and CDK13. They also showed that knocking down Cyclin K expression reduced activity of subsequently immunopurified CDK12; this finding supports the functional significance of the CDK12–Cyclin K interaction.
Q. Li and co-workers found in mouse ES cells that Cyclin K associates with CDK12 and CDK13, but not with CDK9 [14], again consistent with the existence of two Cyclin K-containing CTD kinases.
Altogether these results indicate the existence of two Cyclin K-containing enzyme complexes in human and mouse cells: CDK12•Cyclin K and CDK13•Cyclin K. Evolutionary tree databases indicate that invertebrates contain just CDK12, whereas animals from bony vertebrates forward contain the paralogs CDK12 and CDK13.
3.2. Activities & functions
3.2.1. CTD kinase activity, by direct assay in vitro
One of our initial goals was to test the idea that metazoan CDK12 was actually a CTD kinase. Thus we used isolation conditions that presumably would preserve enzyme activity. From Drosophila nuclear “transcription” extracts we immunopurified CDK12, using antibodies directed against a peptide sequence outside the kinase catalytic region (see Figure 1(a)), and found that the IP indeed possessed CTD kinase activity in vitro [12]. Moreover, proteins selectively eluted from the antibody beads with the immunogenic peptide also directly phosphorylated the CTD in vitro [12]. These results persuasively support the idea that CDK12 has CTD kinase activity (presumably when bound to Cyclin K). Subsequent experiments have confirmed this idea.
At that time [12] we did not have useful antibodies to human CDK12, but subsequent tests in my and other labs showed that hCDK12 also has CTD kinase activity (see below). We also showed that antibodies to human CDK13 will immunopurify CTD kinase activity [12], supporting the idea that CDK13 is also a CTD kinase.
Blazek et al. [13] saw that knockdown of CDK12 and CDK13 expression resulted in instability of Cyclin K, whereas co-expression of Cyclin K with CDK12 and CDK13 stabilized these proteins; these results argue for existence of Cyclin K•CDK12 and Cyclin K•CDK13 complexes in the cell. They also pulled down tagged human CDK12 and CDK13 and showed that both pull-downs had CTD kinase activity; these results together with the knockdown/expression results suggest that the CTD phosphorylating activities in each case most likely contained Cyclin K as well as CDK12 or CDK13.
Morin and co-workers likewise adduced biochemical evidence that CDK12 is a CTD kinase, separate from CDK9, and they also performed experiments supporting the idea that CDK12 kinase activity depends on Cyclin K [15].
3.2.2. CTD phosphorylating activity in vivo
Following RNAi knockdown of CDK12 in Drosophila cells, the phosphorylation state of the CTD on bulk RNAPII changed dramatically, as revealed by Western blotting on whole cell extracts [12]. Analogously, in human cells RNAi knockdown of CDK12 resulted in alterations in CTD phosphorylation, as did knockdown of CDK13, although to a less obvious degree. These results support the proposed functions of CDK12 and CDK13 as CTD kinases.
I also want to mention some other relevant findings made using the affinity-purified IgG against Drosophila CDK12. First, on formaldehyde-fixed polytene chromosomes from Drosophila larvae, we showed that the genomic distribution of CDK12 very closely matches that of hyperphosphorylated (i.e. elongating) RNAPII (Figure 1 in paper) [12]. We also showed that the CDK12 distribution is distinct from that of CDK9 (Cyclin T) (Figure 2 in paper). In addition, some limited Ch-IP experiments confirmed that the CDK12 distribution is different from that of CDK9, with CDK12 signals (relative to RNAPII) tending to begin downstream of those for CDK9 and to be maintained throughout transcription units (Figure 3 in paper). These results support the argument that CDK9 and CDK12 kinases have distinct functions, and that CDK12 functions “downstream” of CDK9. Thus, CDK12 can be considered a transcription elongation-phase CTD kinase.
Figure 2.
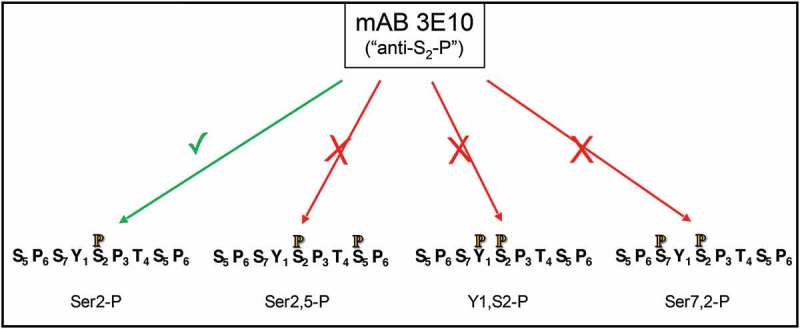
Inhibition of epitope detection by mAb 3E10 (“anti-Ser2-P”) due to phosphorylations on nearby residues.
Four differently-modified CTD repeat peptides are indicated along bottom, with phosphate indicated by orange, black-outlined P. All peptides carry Ser2-P, but the extra phosphate on the doubly-modified peptides inhibits antibody reactivity. Thus changes in amount of Ser2-P on the three doubly-modified repeat sequences cannot be determined by this antibody.
Figure 3.

Expression construct for human CDK12/Cyclin K in insect cells.
The protein breaks in the P2A sequence on the ribosome (cf. ref 26).
Peterlin and co-workers [13] found that knockdown of human CDK12 led to altered CTD phosphorylation in cells, and they also showed that knockdown of Cyclin K gave a similar result. Thus their results are consistent with the CDK12•Cyclin K complex having CTD kinase activity in vivo.
A genetics approach in another model organism also garnered support for CDK12 being a CTD kinase, distinct from CDK9. Kelly and colleagues [16] very interestingly found that in C. elegans (which has only one Ctk1-family kinase, CDK12), knockdown of CDK9 resulted in loss of “Ser2-P” in somatic cells but not in germline cells (immunofluorescence microscopy using 3E10 mAb, nominally anti-Ser2-P). In contrast, knockdown of CDK12 resulted in “complete” loss of the antibody signal in germ cells (as well as ~ 60% loss in somatic cells). After additional functional tests that firmly established the distinction between CDK9 and CDK12, Bowman et al. concluded that “…CTD phosphoepitopes may not always be accurate indicators of different stages of transcriptional regulation.” I agree with this conclusion, and I discuss problems with using antibodies to determine the phosphorylation state of the CTD in the next paragraph (“DIGRESSION”).
3.2.3. DIGRESSION – limitations of antibodies in analyzing CTD phospho-epitopes
In experiments that utilize antibodies raised against different CTD phospho-epitopes to assess the occurrence and amount of that phospho-epitope, the signal strength generated in the assay depends on the antibody used and on potentially-interfering nearby CTD modifications (previously discussed in [6]). As Eick and colleagues showed [17], the reactivity of the 3E10 mAb used by Bowman et al. to detect “Ser2-P” is reduced when any of several nearby residues is phosphorylated (Figure 2). Namely, if the upstream Tyr1 or Ser7 carry a phosphate, the 3E10 reactivity toward Ser2-P is inhibited. Or, if the downstream Ser5 is phosphorylated, the 3E10 signal will be decreased. Conversely, if phosphate is removed from one of these nearby residues, the 3E10 reactivity toward Ser2-P will increase. Thus, exactly what the 3E10 signal means is impossible to determine for this type of in vivo-derived sample. Similar phenomena influence the “Ser5-P” signal generated by the 3E8 mAb, etc. Thus, in observing an altered signal after an experimental manipulation, when using an anti-phosphoCTD mAb one can conclude that CTD phosphorylation is altered but not how it is altered. Because of these difficulties, I try to avoid making strong conclusions about which specific CTD phospho-residues are affected in vivo by various experimental manipulations when the phosphorylation state of the CTD is probed by antibodies (cf. also [6,17]).
3.2.4. Functions of CDK12 & CDK13 in vivo
Regulation of transcription of DNA damage/repair genes – ?? Blazek et al. [13] performed RNAi knockdowns on Cyclin K, CDK12 or CDK13 in HeLa cells, and they analyzed changes gene expression (after 72 hr) using total RNA and expression microarrays. They found that knockdown of CycK and CDK12 gave related, overlapping results, with about 4% and 2% of total genes being down-regulated. They concluded that the genes “down-regulated” after CycK or CDK12 knockdown tended to be long genes and genes with more exons. Unfortunately, the actual data were not presented (i.e. hybridization values for each gene), nor were the criteria (e.g. cutoffs) given for determining what genes were “down-regulated” (or “up-regulated”). It is therefore difficult to evaluate the significance of their findings.
Blazek et al. also concluded that knockdown of CycK led to down-regulation of 4 large functionally-clustered gene groups (their Figure 4(d)), labeled “DNA Replication, Recombination, and Repair;” “Lipid Metabolism;” “Nucleic Acid Metabolism;” and “Amino Acid Metabolism” (although they did not list the member genes of these groups). They focused on the “DNA Replication, Recombination, and Repair” group and ultimately concluded that “These data demonstrated that depletion of CycK/Cdk12 from cells resulted in disrupted expression of a small subset of genes and in the down-regulation of predominantly long, complex genes and a group of DDR genes.” However, as mentioned above, the actual data and details of classification schemes were not presented, and it is difficult to assess the significance of these conclusions.
Figure 4.
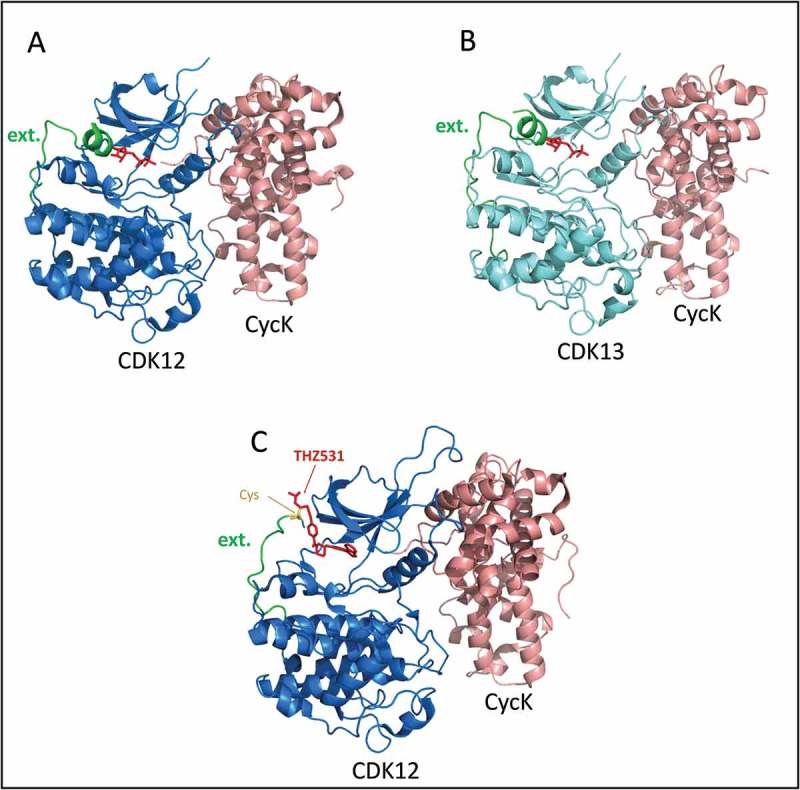
Crystal structures of recombinant human CDK12/13•Cyclin K complexes.
(a) Green color labels the segment of CDK12 chain that extends out from the back (C-term) of the well-folded kinase domain, as discussed in the text. The last residue of the short helix at the end of the green segment is Lys1046. The ADP in the active site is colored red. From PDB ID 4NST; image using PyMol. (b) Green color labels the segment of CDK13 chain that extends out from the back (C-term) of the well-folded kinase domain, as discussed in the text. The ADP in the active site is colored red. From PDB ID 5EFQ; image using PyMol. (c) Inhibitor THZ531 (red) in active site of CDK12 & extending out to react with Cys1039 (yellow). PDB ID 5ACB; image using PyMol.
Finally, Blazek et al. tested the possibility that CycK knockdown would increase sensitivity of cells to DNA damaging agents, and they reported results supporting this idea. On the other hand, whether the sensitivity was due to effects of the knockdown on transcription of DNA repair genes or on some other event/process could not be determined from these experiments.
I note here that results in two recent publications, discussed below in section 4 under “Humans – cancer,” are not in keeping with the idea that CDK12 is required for expression of DNA damage/repair genes [18,19].
Roles in ES cell self-renewal: Q. Li and colleagues [14] found that in mouse ES cells knockdown of CycK, CDK12, or CDK13 resulted in loss of self-renewal and led to differentiation. Interestingly, the differentiation markers expressed by the now-differentiating cells were different for CDK12 knockdown compared to CDK13 knockdown. Thus the functions of the two kinases in maintaining self renewal of mouse ES cells are distinct.
Role in chromatin modification: In C. elegans germline cells, Bowman, et al. [16] found that even though a major CTD phospho-epitope was lost when CDK12 was knocked down or absent, the loss of CDK12 did not seem to affect overall transcription, or germline development; CDK9 activity could apparently suffice for these processes. Remarkably, however, they found that loss of CDK12 activity in germline cells resulted in greatly reduced levels of H3K36me3 in transcribed regions, as generated by MET-1, the C. elegans ortholog of human SETD2; note that SETD2 is a phosphoCTD-associating protein (e.g. [20] and references therein). One role for for this CDK12-dependent H3K36me3 mark (and thus CDK12) is discussed next.
Aymard and colleagues [21], using a system that allows induction of ~ 150 double strand breaks in the genome of cultured human cells, reported the amazing finding that “ … transcriptionally active trimethylated histone H3 K36 (H3K36me3)-enriched chromatin is preferentially repaired by homologous recombination (HR) ….” The H3K36me3 mark is put on H3 by the histone methyltransferase SETD2, and (via the protein LEDGF) the H3K36me3 mark recruits RAD51 to actively transcribed genes, leading to HR repair. As pointed out just above, SETD2 is a phosphoCTD-associating protein (PCAP), binding to the PCTD of transcribing RNAPII through its SRI domain (e.g. [20] and references therein – also see Ref. 52 for SRI–cancer link). In yeast, either mutation of the SRI domain or absence of the yeast CDK12 ortholog (Ctk1) leads to loss of H3K36 trimethylation (e.g. [22]). In human cells, RNAi knockdown of SETD2 results in almost complete loss of H3K36me3 on chromatin and leads to almost complete abolishment of HR repair [21]. If SETD2 recruitment to transcribing RNAPII in human cells depends on CDK12, as it does in C. elegans, another in vivo function for CDK12 is to set up a chromatin modification state on active genes that, upon DNA damage (perhaps actually detected by an elongating RNAPII), will recruit the HR repair machinery !
4. Continuing the modern era
By 2014 it was clear that CDK12•CycK and CDK13•CycK were distinct complexes, both having CTD kinase catalytic activity, but also playing distinct roles in vivo. CDK12 had been shown to be distributed genome-wide in Drosophila with a chromosomal localization virtually identical to that of transcriptionally-active (hyperphosphorylated) RNAPII, suggesting roles for CDK12 during transcript elongation. Based on RNAi knockdowns, suggested functions for CDK12 included “regulation” of long genes and DNA damage response (DDR) genes; however, the soundness of this suggestion had not been tested rigorously. Of medical relevance, it had become clear that CDK12 is a tumor suppressor for ovarian and other cancers (discussed below).
On the other hand, neither CDK12 or CDK13 had been characterized structurally, and neither had been thoroughly characterized enzymatically, and such characterizations were badly needed. The Continuation of the modern era of CDK12/13 studies began with structures being solved. The structures, along with additional enzymatic characterizations led to an era of chemical genetics investigations. Together, these studies generated several leaps forward in understanding CDK12•CycK and CDK13•CycK as enzymes, and they contributed to new ideas and tests of their functions in vivo. The long “arms” of CDK12/13 were also preliminarily assessed for interactions with other nuclear proteins. Finally, investigations of ovarian and prostate cancer yielded remarkable insights into genetic consequences that can occur following the loss of CDK12 activity.
4.1. Structure
4.1.1. CDK12•CycK
In 2014 Geyer and colleagues published the X-ray crystal structure of a recombinant human CDK12•CycK complex, consisting of the kinase homology domain of CDK12 and the cyclin homology domain of Cyclin K {Cdk12 (696–1,082)/CycK (1–267)} [23] (see Figure 4). The structure was reminiscent of Cdk/Cyclin structures published up to that time, but it most resembled the structure of CDK9•Cyclin T. To facilitate crystallization, this group found it necessary to include about 30 residues C-terminal to the minimal kinase domain, and this led them to find a feature that is apparently conserved in CDK12 and CDK9 homologs in all eukaryotes. Namely, a stretch of 23 residues C-terminal to the well-folded kinase domain wraps back around the C-terminal lobe of the kinase domain to interact with the ATP in its binding pocket in the N-terminal lobe (Figure 4(a)). Just past this 23 residue extension, there is a region with several basic residues (1045KKRRRQR in CDK12) (Figure 4(a), extension terminates at residue 1046). This “polybasic cluster” is conserved in CDK12 and CDK9 homologs, along with other features of this extension. The authors speculate that this extension might help stabilize the binding of ATP in the active site. Consistent with a functional role for this extension, they found that when it was shortened down to residue 1044, removing the polybasic cluster, the kinase activity in vitro was reduced several-fold. Some catalytic features of this and other Ctk1 family members are discussed below (see Catalytic activity and inhibitors).
Dixon-Clarke et al. published another X-ray structural study of similar recombinant CDK12•CycK complexes in 2015 [24], with AMP-PNP co-crystallized with the ~ minimal enzyme complex {CDK12715–1052/CycK11–267}. Interestingly, they observed two different complexes in which the C-terminal extension of the kinase domain has different conformations. In one complex, the C-terminal extension wraps around to interact with the N-term lobe and the ATP analog (as in Geyer et al. above). In the other, the C-term extension wraps differently around the back of the kinase domain and does not approach the ATP binding pocket; remarkably, this second complex also does not contain AMP-PNP. This observation supports the idea that the extension affects ATP binding. This group also expressed differentially tagged, full-length CDK12 and CycK, and isolated soluble complexes by tandem (consecutive) affinity chromatography. Notably, the full-length versions were about 10-fold more active than the minimal recombinant complexes, and they showed tighter binding of both ATP and the CTD substrate. Thus, segments in the subunit outside the homology core region influence the activity of CDK12•CycK (more below under Catalytic activity and inhibitors).
4.1.2. CDK13•CycK
In 2016 Geyer and co-workers published the X-ray crystal structure of recombinant human CDK13•CycK [25]. As expected for proteins that are 92% identical in their kinase homology domains, the CDK13 and CDK12 recombinant protein structures are extremely similar (Figure 4(b)). Remember that CDK12/13 have long N- and C-terminal extensions that are not in these structures. In addition, Cyclin K has a long, Pro-rich, C-terminal extension that is also not in the structures.
4.2. Catalytic activity (and inhibitors)
4.2.1. CTD phosphorylation in vitro
Phosphates added mainly to Ser2 and Ser5 of CTD heptad repeats: Geyer and co-workers tested activity of recombinant CDK12•CycK on a GST-CTD fusion protein, using anti-phosphoepitope mAbs to assess the positions of phosphorylation along the CTD repeats [consensus sequence most commonly now written as (Y1S2P3T4S5P6S7)n]. They found relatively similar Ser2-P and Ser-5P reactivity by this Ab approach [23, Figure 5(e), lane 4]. A full-length, flag-tagged CDK12 expressed in mammalian cells (HCT116) and isolated with anti-flag beads showed a similar pattern, adding phosphates to ‘Ser2ʹ and ‘Ser5ʹ about equally (Figure 5(e), lane 2 in [23]). While other assays showed somewhat different results, overall the experiments of Bosken et al. support the idea that Ser5 and Ser2 are the principal targets of phosphorylation by CDK12•CycK in vitro.
Figure 5.
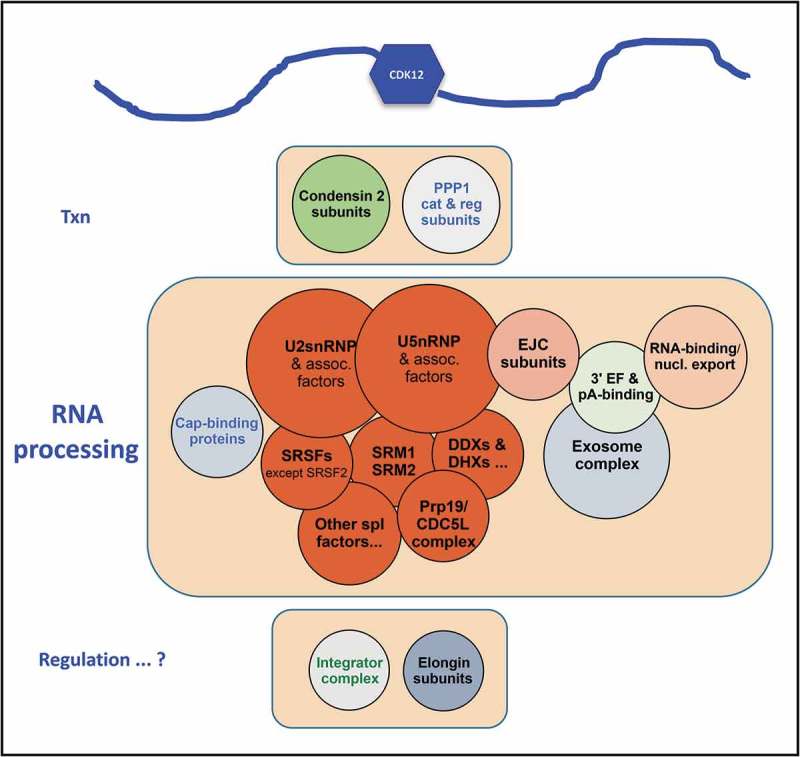
HeLa nuclear proteins that co-immunopurify with CDK12.
Most proteins co-IP’d with CDK12 are in or associated with structures/complexes indicated on the circles. Circle diameters indicate very approximately the relative number of proteins found in the IP’d sample (cf. [26]). A diagram of CDK12 is at the top as a reminder that its long arms provide many potential protein-interaction sites.
Hoping to biochemically characterize the kinase activity of full-length, “native” CDK12•CycK, we expressed full-length human CDK12 and CycK in insect cells and succeeded in purifying full-length, active enzyme to near homogeneity [26]. The trick was to express the two subunits in equimolar amounts, by having them encoded in one transcript (see Figure 3). The CDK12 and CycK were separated by a P2A sequence from porcine teschovirus, which leads to separation of the two proteins during translation. A His6 tag on the N-term of CDK12 enabled affinity purification on a Ni matrix, and size exclusion chromatography of the concentrated activity peak generated several fractions that contained virtually only CDK12 and CycK (Figure 3(a),lanes 11–12 in ref 26). We used purified full-length enzyme to phosphorylate a set of variant CTD fusion proteins carrying CTD segments ~ 15 repeats long. Specifically, we used Ser-substituted repeats: WT (YSPTSPS), S2A (YAPTSPS), S2E, S5A, S5E, and S7E. We found that all of these substrates were phosphorylated by the purified kinase, with the WT and the S7E repeats being the best templates in our assays (phosphorylated about equally). The S2A and S5A repeats were phosphorylated at about 70% and 50% of WT, consistent with phosphorylation on S5 and S2, respectively. Note that Jones et al., using the counterpart yeast kinase (Ctk1-containing kinase CTDK-I), showed that in vitro Ctk1 also phosphorylates Ser2 and Ser5 [27]. Jones et al. also showed that yeast CTDK-I can add phosphate to Ser2 when Ser5 is pre-phosphorylated, and vice versa; that is it can generate doubly-phosphorylated repeats (e.g. Ser2,5-P). We conclude that in our assays CDK12•CycK phosphorylates Ser2 and/or Ser5 of the CTD heptad repeats, and we suggest that it is able to generate doubly-phosphorylated “repeats” (i.e. CTD sequences of consecutive Ser2,5-P residues or consecutive Ser5,2-P residues).
Geyer and co-workers, who solved the structure of human recombinant CDK13•CycK [25], tested its specificity in vitro. Basically they found that CDK13•CycK behaves like CDK12•CycK (also [23] above).
Morin and colleagues [24] also compared recombinant and full-length versions of CDK12•CycK, and they found some interesting differences. For example, they found that the full length enzyme has a Km for ATP that is about 10-fold lower (i.e. tighter binding of ATP) than that of the recombinant enzyme (CDK12 715–1052/CycK11–267). Also, they found that the full length enzyme binds the CTD substrate more tightly than the truncated enzyme. These two effects presumably contribute to their observation that the specific activity of the full length kinase is about 10-fold greater than that of the recombinant enzyme. They made the important and reasonable conclusion that “These differences suggest that other domains within the full length proteins make important contributions to the substrate interactions.”
Pre-phosphorylation influences kinase specific activity: A very interesting observation of Geyer and co-workers is that in vitro both CDK12•CycK and CDK13•CycK are much more active toward CTD synthetic peptide repeats when Ser7 is pre-phosphorylated than when it is not [23,25]. One possibility is that this reflects the interaction of the Ser7-phosphate with the “polybasic” stretch of primary sequence C-terminal to the canonical kinase homology region (see above under Structure). While we found that, for full-length CDK12•CycK, a CTD comprising (YSPTSPE)16 (E being a poor Ser7-P mimic) is not a better substrate than WT [i.e. (YSPTSPS)16] [26], I note that we had found earlier for yeast CTDK-I that 3-repeat synthetic peptides pre-phosphorylated at either Ser2 or Ser5 are much better substrates than the unphosphorylated peptide [27]. It seems reasonable to conclude that the (pre) phosphorylation status of CTD repeats will influence the activity of an “incoming” CTD kinase. Also, for the Ctk1 family of kinases, this influence will likely occur at least in part through interactions of the phosphate groups with basic regions of the protein “arms” extending out from the canonical kinase homology domain.
Inhibition of kinase activity: A number of small-compound inhibitors have been used to study CTD kinases over the years, but during the same span of time our knowledge of the kinases and the kinase-specificity of these inhibitors has improved. I think a very brief summary/overview as related to CDK12/13, CDK9 and CDK7 is worth while.
“Off-the-shelf” inhibitors. Two inhibitors that have been used to inhibit CTD kinases are DRB and flavopiridol (e.g. [28] and refs therein). These inhibitors do show some kinase-specificity but using them to assign a particular kinase to a particular phosphorylation event in cells is problematic. For example, DRB was used in the past to “target” CDK7, but we now know it inhibits CDK9 almost as well as CDK7 [28]. In addition, it also inhibits CDK12 at higher concentrations (e.g. [26]).
Flavopiridol inhibits CDK9 (e.g. [29]), but it turns out that it also inhibits other kinases. Geyer and colleagues found that FVP inhibits CDK12, although less well than CDK9 (~ 10x difference under their assay conditions) [23]. We also found that FVP inhibits full length CDK12 at nM concentrations, and only about a 5x higher concentration was needed for CDK12 than for CDK9, under our assay conditions [26]. It is not known whether or how the relative FVP inhibition of CDK9 and CDK12/13 might be different in vivo. Thus from current information, one should assume that a concentration of FVP needed to “completely” inhibit CDK9 in vivo is likely also to significantly inhibit CDK12 and CDK13.
Novel inhibitors. Selective inhibitors of CDK12 have been sought in the last few years. Gray and colleagues leveraged their earlier work on the covalent CDK7 inhibitor, THZ1 [30], to develop a related inhibitor, THZ531, that is selective for CDK12 and CDK13 [31]. The unique feature of the THZs is that they form a covalent bond with a Cys residue that is actually not in the active site, but nearby, in these three CDKs. The inhibitory portion of a THZ molecule binds in the ATP binding site to prevent ATP binding, while a second part of the inhibitor reaches out of the ATP binding site and reacts with nearby, appropriately positioned Cys residues (Figure 4(c)). THZ531 inhibits in vitro kinase activity of both CDK12 and CDK13 with an IC50 around 100nM, whereas it is ~ 100-fold less effective in inhibiting CDK9 and CDK7. A feature of THZ531 that derives from its covalent mode of inhibition is that it takes some time to react and thus displays time-dependent inhibition. On the other hand, the inhibition is not reversible, and inhibitory effects in cells are not reversed even after inhibitor washout.
Recently, Astra-Zenica reported attempts to find noncovalent selective inhibitors of CDK12, particularly compounds with good inhibition in the presence of high ATP concentrations, as will occur in cells [32]. Several effective compounds were found, but the selectivity for CDK12 appears to be less that of THZ531.
Kinase mutant-selective inhibition. Another way to achieve kinase-selective inhibition is to mutate the kinase of interest so that it becomes sensitive to an otherwise “inactive” inhibitor. This elegant approach was pioneered by K. Shokat, and the inhibitors he developed are usually membrane-permeable analogs of an adenine nucleoside [33]. The Shokat approach is very useful for studying effects of inhibiting a kinase in a cellular context, and we used it to begin investigating attributes of human CDK12. We first generated a “Shokat” mutant version of CDK12 that was sensitive to inhibition by the analog 1-NM-PP1, which does not normally inhibit CDK12 (or CDK13). [26]. The IC50 for the analog-sensitive enzyme is approximately 100 nM. Since our goal for generating the mutant kinase was to use it to investigate CDK12 activity inside cells, we employed CRISPR/Cas and homologous recombination to introduce the same mutation into the CDK12 gene in HeLa cells; the recovered cell line carries one CDK12 allele expressing the desired analog-sensitive protein and one allele expressing a truncated, non-functional protein [34]. Initial uses of the CDK12 AS cell line are described briefly below (under IV “Into the future”).
4.3. Functions
What we really want to know is, “What roles do CDK12 and CDK13 play in vivo?” That is, what are their functions? If they do phosphorylate the CTD in vivo, what are the consequences of that phosphorylation? Also, do they phosphorylate other proteins, and with what outcomes? While some progress has been made in answering these questions, our understanding remains extraordinarily incomplete. Moreover, the literature contains apparently-conflicting claims for functional roles of CDK12, and these conflicts need to be resolved.
In this section 1 present various ideas that have been put forward, and along the way I also add some opinions. Hopefully these ideas and opinions will provoke discussion and stimulate further investigations.
4.3.1. Studies in cell culture & tissues
Inhibition & knockdown: As mentioned earlier, we used Drosophila polytene chromosomes to show that CDK12 is globally distributed on the genome in a pattern virtually identical to that of hyperphosphorylated RNAPII [12]. This result strongly suggests that CDK12 plays roles that are important during the elongation phase of transcription. More recently we showed, using the analog-sensitive CDK12AS HeLa cell line, that selective inhibition of CDK12 results in altered phosphorylation of the CTD on total cellular RNAPII after only 15 min of treatment by inhibitory analog [34]. Thus, as anticipated, it seems that the principal activity of CDK12 is indeed to phosphorylate the CTD of elongating RNAPII molecules. I would then say that using its catalytic activity to generate particular phosphorylation states/patterns on the CTD of transcribing RNA polymerases is CDK12’s main “role” or “function,” in that the CDK12-generated phosphorylation states will recruit specific functional sets of phosphoCTD-associating proteins (PCAPs) to the transcription elongation complex.
We also showed that selectively inhibiting the analog-sensitive CDK12AS resulted in relatively rapid inhibition of cell proliferation, although we have not investigated this phenomenon in any detail. From this result, along with others described elsewhere, we conclude that for many/most cells CDK12 activity is essential for cell proliferation.
Two groups have used RNAi knockdown of human CDK12 in studies focused on 3ʹ end formation on the short genes MYC and c-FOS [35,36]. In both studies the knockdown of CDK12 by RNAi led to impaired 3ʹ end processing.
Shilatifard and colleagues used RNAi to knock down expression of human CDK12 and CDK13 (and CDK9) in HCT116 cells, ultimately achieving about 80% knockdown [37]. Thus, at least 20% of the kinase presumably remains active in these experiments; the effects on CTD phosphorylation may therefore not be as great as in experiments using inhibitors. This group then used RNA-seq to analyze consequent changes in gene expression. For both kinases, a few genes showed increased transcript levels, but over 97% of the affected genes showed decreased transcript levels. Interestingly, although knockdown of CDK13 reduced expression of fewer genes (1510) than knockdown of CDK12 (3804), there was a large overlap in the affected genes (1141 gene overlap; thus ~ 75% of the CDK13- affected genes were also affected by CDK12 knockdown). In analyzing the functional categories of the genes affected, they also found overlap between CDK12 and CDK13 gene groups, in the categories of RNA processing, Ribonucleoprotein complex biogenesis, Translation, and Macromolecular complex assembly. On the other hand, CDK12-affected genes also fell into categories that included DNA metabolic complexes, Response to DNA damage stimulus, and DNA repair. In contrast, CDK13-affected genes also fell into categories that included Generation of precursor metabolites and energy, Oxidative phosphorylation, Ribosome biogenesis, and Cellular respiration. One interpretation of these results is that CDK12 and CDK13 influence a large set of common processes, but they also influence a smaller set of distinct processes.
Young, Gray and colleagues [31] used their inhibitor, THZ531, to look at in-cell effects of inhibiting CDK12 and CDK13 together. They found that THZ531 inhibited Jurkat cell proliferation with an IC50 of 50 nM, in the same concentration range as needed to inhibit CDK12 and CDK13 in vitro. At this concentration of inhibitor, the anti-proliferative effects seemed mostly due to cell death. These researchers found that at high concentrations of THZ531 (350–500 nM), there were more severe effects and a rapid induction of apoptosis. However, it seems likely that a number of effects at high doses of inhibitor “… may result from a combination of on-and off-target effects.” Therefore I will focus on effects seen at low doses (usually 50 nM, sometimes 200 nM).
Given the transcriptional connections of CDK12 and CDK13, Zhang et al. wanted to investigate potential transcriptional effects of adding THZ531 to cells. As a preliminary step, they performed ChIP-seq to determine the genomic distribution of CDK12. They found that CDK12 bound both to protein-coding genes and to enhancer regions. The CDK12 signal was very similar to the RNAPII signal both at promoters and gene bodies of active genes, and at active enhancers. Also, the intensity of CDK12 signal correlated with that of H3K27Ac, indicating that “… CDK12 is present at actively transcribed regions of the genome.” These results parallel those exploring CDK12 genome distribution in Drosophila tissues and cells [12].
Zhang et al. tested the effects of TZH531 on total RNAPII (and “Ser2-P”) presence on several selected genes that displayed high CDK12 signals; at 50 nM inhibitor they saw few effects. Higher doses caused some effects, but these may not be specific for CDK12 or CDK13 (see above). They also looked into inhibition of gene expression in Jurkat cells resulting from treatment with THZ531 for 6 hr. At low doses (50 nM) the inhibitor had modest effects (in the paper: Suppl. Table 5 in Suppl Dataset 3), but functional grouping of the top 2% of the genes with negative fold-change (~ 600 genes) yielded the top terms as “DNA metabolic process,” “DNA repair,” “Cellular response to stress,” and “Response to DNA damage stimulus” (using DAVID) [38]. This is somewhat reminiscent of Blazek et al. [13]; however, I think it should be kept in mind that more than half of the 600 genes in the clustering analysis actually displayed a log2 fold-change of < – 1 (less than 2-fold effect).
At 200 nM THZ531 many more genes showed significant changes, with about 1900 genes displaying a log2 fold-change of < – 2 (> 4-fold down-regulation) (in the paper: Suppl. Table 5 in Suppl Dataset 3), and analysis of functional groups showed that transcription factor-encoding genes were among the most affected. Interestingly, the most sensitive set of genes correlated with genes on which ChIP detected high amounts of CDK12, and with super-enhancers.
4.3.2. Proteins associated with CDK12 and CDK13
Determining the identity of proteins that associate with CDK12/13 can suggest processes and functions potentially linked to the kinases. Especially with the long N- and C-terminal arms, CDK12 and CDK13 are expected to take part in many protein-protein interactions. Thus, several groups have looked for proteins that co-purify with CDK12/13.
We took an immuno-purification approach to identify CDK12-associated proteins in a HeLa nuclear extract [26]. Using affinity-purified IgG directed at a peptide from near the N-terminus of human CDK12 (residues 201 to 220; see Figure 1(a)), we IP’d CDK12 at relatively high salt (0.4M NaCl) and identified co-IP’d proteins via mass spectrometry. The functional group with the largest number of CDK12-associated proteins turns out to be pre-mRNA processing (see Figure 5). Processing components that are very well-represented include the spliceosome (especially SF3/U2snRNP, U5snRNP, SRSFs & RNA helicases), the exon junction complex, HnRNP proteins, 5ʹ cap-binding proteins, 3ʹ end formation-involved proteins, the Integrator complex, and the Exosome complex. There are also proteins from other complexes with interesting and provocative functions; these proteins include Condensin 2 subunits, Elongin complex subunits, and presumptive CTD phosphatase catalytic and regulatory subunits. These identifications provide fuel for extensive speculations. Hopefully they will also propel future experiments.
Peterlin and colleagues used epitope-tagged CDK12 to IP it and associated proteins [36]. They found many RNA binding proteins involved in RNA processing, and their set of CDK12-associating proteins shows significant overlap with ours. In addition, they found that RNAi knockdown of CDK12 led to reduced recruitment of 3ʹ end-forming factors to the c-FOS gene.
Shilatifard and colleagues used epitope tagging and IP to identify proteins that associate with CDK12 as well as CDK13 [37]. Their findings for CDK12 overlap ours and Peterlin’s extensively; actually, both their CDK12- and CDK13-associated proteins include many that represent RNA processing, the spliceosome, and nuclear speckles.
Because IP approaches will pull down CDK12 molecules with different combinations of associated partners, it is not clear how many distinct complexes might exist, what the composition of each might be, or where along the transcription unit (or elsewhere?) they occur. Nevertheless, these results suggest that CDK12 (and CDK13) contributes extensively to gene expression, both through the catalytic activity of its kinase domain and through the protein interaction activities of its two extended arms.
4.3.3. Studies in human beings – cancer
CDK12: The first correlation between CDK12 and human cancers was reported in the TCGA study on ovarian carcinoma [39] and followed up by Carter et al. [40]. Thereafter, CDK12 was recognized as a tumor suppressor. Since then, a number of large sequencing efforts have found CDK12 alterations associated with several cancers, and some of these studies are discussed next. For recent reviews see, e.g. [41,42].
An extremely interesting observation was made by Popova and colleagues, who were investigating genomic instability associated with CDK12 loss in ovarian cancer [18]. They found that absence of active CDK12 (inactivating alterations in all alleles, occurring in around 4% of serous ovarian carcinomas) results in hundreds of tandem duplications (TDs), with a bi-modal size distribution centered on ~ 0.3 and ~ 3 MB lengths, spread quasi-randomly around the genome [18]. This “CDK12 TD-plus” phenotype so far appears to be specific to tumors that express disabled versions of the CDK12 protein; promoter methylation, leading to lack of expression of the CDK12 gene, did not correlate with the TD-plus phenotype (two cases to date). This research group also noticed that the CDK12 TD-plus phenotype occurred in 1% to 2% of prostate adenocarcinomas in the TCGA database, in line with results reported in 2018 (below). The molecular mechanisms by which CDK12 inactivation leads to the TD-plus phenotype are currently unknown (also see below).
Another finding of major interest in the study from Popova and colleagues is that gene expression changes in the tumors with CDK12 TD-plus phenotype did not show any significant functional clustering, in contrast to studies mentioned above that used RNAi to knock down CDK12 expression in cultured cells. In particular, Popova et al. did not find a correlation between CDK12 inactivation and down-regulation of expression of DNA damage-response genes. Also, they did not find that tumors with the CDK12 TD-plus phenotype were inactivated for BRCA1/2.
Chinnaiyan and colleagues [19] found, in a subtype of prostate cancers, that CDK12 inactivation leads to focal tandem duplications (FTDs), as found in ovarian carcinomas (above). They found that bi-allelic inactivation of CDK12 occurs in 6–7% of mCRPC (metastatic castration-resistant prostate cancer) and correlates with occurrence of FTDs (“CDK12-FTDs”). The frequency and size of the TDs are very similar in the CDK12-defective prostate and ovarian cancers.
A finding of major interest in the mCRPC studies is that, as in the CDK12 TD-plus ovarian tumor studies (above), there is no preferential down-regulation of DDR genes in CDK12-FTD prostate tumors. Instead, Wu et al. found that the most prominently-altered functional gene sets involved oxidative phosphorylation (down), inflammatory response (up), hormone receptor signaling (down) and epithelial dedifferentiation (down). They also found that expression of BRCA1 and BRCA2 was not affected, nor was expression of long genes preferentially affected (cf. [13]). In the opposite direction, they found that a small set of genes recurrently showed gains in gene number in the CDK12-FTD tumors, suggesting positive selection. As they commented, “Strikingly, candidate genes under positive selection, MCM7, RAD9A, CDK18, and CCND1, have crucial roles in DNA replication and genome stability.” It is not implausible to think that protein products of these genes may be involved in establishing the FTD phenotype.
These two large studies on tumors with “CDK12-TD” phenotypes are consistent in that they find no correlation between (presumably) complete loss of CDK12 activity and levels of expression of DDR genes. They also find that both sets of tumors still express both BRCA1 and BRCA2. In view of these findings, I think it is wise to reconsider the earlier notion that loss of CDK12 leads to oncogenesis because of down-regulation of DDR genes.
CDK13: There have been few reports of CDK13 influencing cancer development, although a recent paper describes an interesting connection that should be watched for future developments [43].
5. Into the future
A major goal of future work will be to determine what the in vivo activities and functions of CDK12/13 actually are. I present some unpublished work that hopefully will shed a bit of light on this goal. However, much more work is needed to understand CDK12/13 in enough detail to enable us to discern how their loss can lead to cancers and how we can intercede to prevent or treat those cancers.
5.1. Unpublished work
5.1.1. Selective inhibition of CDK12AS in HeLa cells
5.1.1.1. Effects on Pol II transcripts
To gain insights into how inhibiting CDK12 activity affects transcription and RNA processing, we carried out pulse and pulse-chase RNA labeling experiments, making use of our CDK12as HeLa cell line. In the “Bru-Seq” approach, nascent RNAs are labeled by Br-Uridine incorporation for 30 minutes (pulse) in the absence or presence of the CDK12AS inhibitory analog (1-NM-PP1), retrieved by anti-BrU antibodies, and subjected to RNA-Seq [44]. In the pulse-chase approach, called “BruChase-Seq” [44], RNAs are labeled for 30 min with BrU, the BrU is washed out and replaced with normal U, and incubation is continued for a chosen length of time (the chase). Subsequently, the surviving BrU-labeled RNAs are retrieved and sequenced. When inhibiting CDK12 activity, the inhibitory analog (1-NM-PP1) is added 15 min before the pulse (which is 30 min) and left in through the chase (which was 2 hr). Results from experiments using this approach are reproducible, novel, and sometimes surprising (e.g. [44,45]).
In the pulse-labeling (Bru-Seq) experiments, we found that in the presence of analog (1-NM-PP1) only ~ 700 transcripts showed a ≥ 2-fold reduction in RNA synthesis rate (Br-U incorporation), and ~ 200 showed an increase in rate, compared to absence of analog (Bartkowiak, Yan, Magnuson, Paulsen, Ljungman, & Greenleaf manuscript in preparation). Functional group analyses of the two sets of genes did not give any highly significant values. A very interesting feature was observed, however, in that for a significant fraction of the genes showing a reduction in transcription, that reduction did not begin until well into the transcription unit (e.g. Figure 6, “Txn defect”). A possible explanation emerged from the pulse-chase experiments.
Figure 6.
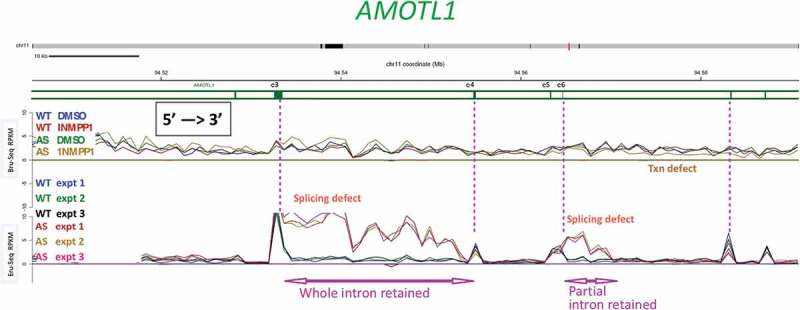
A subset of splice sites is affected by inhibiting CDK12AS for a short time.
Bru-Seq and BruChase-Seq were carried out as described in the text. An internal section of the AMOTL1 gene is shown. The top set of 4 traces (Bru-Seq) show the relative transcription rate (incorporation of BrU in 30 min pulse) in regular HeLa cells (WT) and in CDK12AS HeLa cells in the absence or presence of inhibitory analog 1-NM-PP1. The gold trace (CDK12AS cells + analog) begins to drop below the other three downstream of exon 6 (e6). The bottom set of 6 traces (BruChase-Seq) shows that in ‘WT cells in the presence of analog (blue, green, black), after a 30 min pulse of BrU and a 2 hour chase, exon signals predominate (four exons are indicated by vertical pink dotted lines). The introns are spliced out and degraded (valleys between WT peaks). In contrast, in CDK12AS cells treated identically, the intron following exon 3 (e3) is not spliced out and is “retained.” But the next intron is spliced out normally, and then intronic sequences past exon 6 (e6) are retained. Apparently only part of the intron is retained, since the pulse-chase (BruChase-Seq) traces return to normal about a third of the way through the intron. Farther downstream, splicing looks ~ normal.
In the pulse-chase (BruChase-Seq) experiments, inhibition of CDK12AS by the inhibitory analog led to some intriguing results. A major effect was that a subset of introns, spread throughout the transcriptome, showed inappropriate intron retention when CDK12 activity was inhibited (e.g. Figure 6). Frequently, a pre-mRNA displayed normal splicing-out of all introns except one or two (Figure 6). It appears that short-term inhibition of CDK12 activity selectively affects some splice sites such that they are used less efficiently. Another observation is that some “intron retention” events led to retention of only a 5ʹ segment of the intron, while the downstream segment of the intron was absent from the processed transcript (Figure 6 intron downstream of exon 6). One possible explanation for this type of event is that when a splice site is not recognized (due to short-term absence of CDK12 activity) an otherwise cryptic cleavage/poly-adenylation site in the intron becomes subject to cleavage (and poly-adenylation ?). While further analyses and experiments are needed to test this and other potential explanations, these behaviors are somewhat reminiscent of effects that follow depletion of U1snRNP [46].
5.2. Changes in phosphorylation of potential non-CTD substrates
We also utilized the HeLa CDK12AS cell line to ask if CDK12 might phosphorylate proteins besides the CTD of POLR2A; if CDK12 phosphorylates non-CTD proteins, the level of their phosphorylation is expected to decrease after CDK12 inhibition. We added (or not) the inhibitory analog 1-NM-PP1 to growing CDK12AS cell cultures (in triplicate) for 30 min, rapidly collected the cells and prepared total cell extracts for phospho-proteomic analysis at the Duke Proteomics facility (Bartkowiak, Yan, Turner, Moseley, Soderblom & Greenleaf manuscript in preparation). About fifty P-peptides (representing 40 proteins) decreased > 2-fold in abundance after inhibitor treatment; in contrast, only 3 P-peptides increased > 2-fold. Among the group of fifty P-peptides, half actually showed a decrease of 3-fold or more, with a dozen of those decreasing more than 5-fold. From inspecting the amino acid sequences of the affected phospho-peptides, we conclude that some are likely phosphorylated directly by CDK12, while others are probably phosphorylated by different kinases and affected indirectly by CDK12 inhibition; however, the normal phosphorylation levels at all of the sites depend on CDK12 catalytic activity. Candidate direct substrates include mainly proteins involved in either transcription or pre-mRNA processing/mRNA nuclear export, consistent with ideas on CDK12 functions. Further experimental tests are needed to determine which candidates are actual substrates of CDK12.
5.3. Human phosphoCTD-associating proteins highlight RNA processing and DNA repair
CDK12 is probably the major CTD kinase acting during transcription elongation by RNAPII, and it plays a large role in determining the patterns of phosphate groups on the CTD. Because different phosphoCTD-associating proteins (PCAPs) bind preferentially to different CTD phospho-epitopes, CDK12 undoubtedly plays a major role in orchestrating the identity and timing of PCAP associations with the CTD during transcription. Because many fewer PCAPs have been identified in human cells than in yeast, we felt that a systematic search through the human nuclear proteome would be profitable, potentially uncovering novel PCAPs and ultimately leading to a better understanding of CDK12-dependent, PCTD-linked functions. Therefore we applied a biochemical fractionation/affinity-purification protocol (cf. [47]) to proteins solubilized from a preparation of native human chromatin that retained transcriptionally-engaged RNAPII mega-complexes. Affinity purification on phospho-CTD peptide [(Ser2,5-P)3] beads followed by mass spectrometric analysis led to the identification of > 120 presumptive PCAPs (Bartkowiak, Lao, Yan, Turner, Moseley, Soderblom, & Greenleaf manuscript in preparation). The 120 human PCAPs we identified represent primarily two major functional categories.
As anticipated, we identified numerous PCAPs that are involved in RNA processing, with several of the current identifications reprising earlier findings. These identifications strengthen the idea that the PCTD helps organize a polymerase-associated processing “factory” or “assembly line” [48] that co-transcriptionally processes the pre-mRNA, from 5ʹ capping, through intron removal & splicing, to cleavage/polyadenylation (and transcription termination), to mRNA maturation and export out of the nucleus.
Perhaps surprisingly, the most significant functional group defined by these PCAPs was DNA damage/repair, a finding that suggests roles for the PCTD in responding to DNA damage and ultimately in maintaining genome stability. This may not be surprising, however, because results from a number of experimental approaches have implicated transcribing RNAPII as a good detector of DNA damage and suggest RNAPII-interacting factors are involved in DNA damage responses. Indeed, we had previously identified several human “DNA-directed” proteins as PCAPs in low-throughput experiments (e.g. PARP1, DNMT1, TOP1, RECQ5 [49,50]. Also, a fairly recent specific example, involving HR-mediated repair that is dependent on SETD2, a notable PCAP, was described above (Section 3, under Role in chromatin modification).
On the other hand, we previously showed that yeast Set2 plays a catalytic activity-independent role in response to DNA damage caused by MMS and other chemicals [[51]; note control experiment using H3[K36A]]. Moreover, we had ultimately realized that about 20% of the yeast PCAPs we identified by an approach similar to that used here [47] were needed for normal responses to DNA damaging agents [51]. This realization, along with further experiments, led us to propose the existence of a CTD-associated DNA damage response (“CAR”) system that reacts to damage detected by transcribing RNAPII [51]. Note that the CAR system is not the same as the TCR (transcription coupled repair) system. It seems sensible that association of a subset of damage/repair proteins with transcribing RNAPII (via PCTD binding) would greatly facilitate and accelerate responses to DNA lesions detected by the transcribing transcriptase.
6. Summary, conclusions, and speculation
Human CDK12 and CDK13, each complexed with the partner subunit Cyclin K, are “transcriptional” CDKs, and the main substrate for these kinases is the CTD of transcribing RNAPII. Because more is currently known about CDK12•CycK, this summary’s main focus will be on it.
CDK12 enzyme phosphorylates CTD repeats (Y1S2P3T4S5P6S7)n on Ser2 and Ser5 in vitro (as does its yeast ortholog, Ctk1). Counter to this, a number of studies using antibodies raised against different CTD phospho-epitopes have concluded that the main phosphorylation target of CDK12 in vivo is Ser2. However, antibody-based studies on in vivo samples suffer from confounding effects caused by the presence of currently-unknowable levels of interfering modifications on nearby residues (see “Digression” above, in Sect. II, under CTD phosphorylating activity in vivo). I suggest that we should consider that CDK12 actually phosphorylates Ser5 as well as Ser2 residues in vivo.
The major point of phosphorylating the CTD of elongating RNAPII is to enable recruitment of phosphoCTD-associating proteins (PCAPs) to the transcriptase’s tail. PCAPs include, for example, subsets of RNA-processing and DNA/chromatin-modifying factors, the proper functions of which require them to be positioned near the site of transcription. Because CDK12 is a major elongation-phase CTD phosphorylating activity and plays a key role in determining the phosphorylation status of the CTD, it is a major determinant of the protein makeup and functional capacity of the mega-complex of factors associated with elongating RNAPII. I would thus say that the overall primary role of CDK12, which of course depends on its catalytic activity, is to determine/maintain the phosphorylation status of the CTD of transcribing RNAPII ➔ Role No. 1.
CDK12 also plays secondary roles. Both CDK12 and CDK13 are large proteins, whose ~ 180 kDa molecular weights are due mostly to low-sequence complexity arms that extend out from the globular (~ 40 kDa) protein kinase domains. These arms can interact with numerous other nuclear proteins, notably RNA processing factors, recruiting them to the vicinity of RNAPII where they can participate in co-transcriptional processing of the transcript. Thus a second major role for CDK12, which depends on its structure, is to participate in numerous protein-protein interactions and help construct an RNA processing “factory” at the site of transcription ➔ Role No. 2.
Studies in progress suggest that human CDK12 phosphorylates some non-CTD proteins in vivo, as is already known for another CTD kinase, CDK9. The identities of non-CTD substrates of CDK12 are yet to be firmly established, and the consequences of their phosphorylation are not known; however, it appears that phosphorylating a set of non-CTD proteins is a third role for CDK12 ➔ Role No. 3.
Now that we know that loss of CDK12 activity can lead to ovarian and prostate cancers with unusual genome instabilities, what can we say about how CDK12 loss may lead to oncogenesis? Because only speculation is possible at this point, I present in that spirit a possible scenario for early events that may follow a loss of CDK12 catalytic activity. Because small in-frame deletions in the kinase catalytic domain can apparently lead to this kind of cancer, I suggest that losing CTD phosphorylating activity is the major factor leading to oncogenesis. Loss of CDK12 catalytic activity will presumably lead to alterations in gene expression after, for example, RNA processing PCAPs are no longer properly associated with the PCTD, transcripts are mis-processed, and critical mRNAs/proteins decrease in amount. However, because a deficit in CDK12 activity leads almost immediately to aberrant CTD phosphorylation, DNA damage-response and DNA repair PCAPs will quickly be lost from the CTD. This will decrease the cell’s ability to respond properly to RNAPII-detected DNA lesions. As non-repaired lesions build up in transcribed regions, the mutation burden will increase, and ultimately dire consequences will set in.
Finally, I would just say that CDK12/13 are complicated multi-tasking proteins of central importance to proper gene expression and to genome stability. Because there is so much left to learn about these enzymes, future investigations of CDK12 and CDK13 promise to be exciting and fulfilling.
Funding Statement
This work was supported by the National Institutes of Health [GM 040505].
Disclosure statement
No potential conflict of interest was reported by the author.
References
- [1].Lee J-M, Greenleaf AL.. CTD kinase large subunit is encoded by CTK1, a gene required for normal growth of Saccharomyces cerevisiae. Gene Expr. 1991;1:149–167. [PMC free article] [PubMed] [Google Scholar]
- [2].Sterner DE, Lee JM, Hardin SE, et al. The yeast carboxyl-terminal repeat domain kinase CTDK-I is a divergent cyclin-cyclin-dependent kinase complex. Mol Cell Biol. 1995;15:5716–5724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Wood A, Shilatifard A.. Bur1/Bur2 and the Ctk complex in yeast: the split personality of mammalian P-TEFb. Cell Cycle. 2006;5:1066–1068. [DOI] [PubMed] [Google Scholar]
- [4].Liu J, Kipreos ET. Evolution of cyclin-dependent kinases (CDKs) and CDK-activating kinases (CAKs): differential conservation of CAKs in yeast and metazoa. Mol Biol Evol. 2000;17:1061–1074. [DOI] [PubMed] [Google Scholar]
- [5].Guo Z, Stiller JW. Comparative genomics of cyclin-dependent kinases suggest co-evolution of the RNAP II C-terminal domain and CTD-directed CDKs. BMC Genomics. 2004;5:69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Phatnani HP, Greenleaf AL. Phosphorylation and functions of the RNA polymerase II CTD. Genes Dev. 2006;20:2922–2936. [DOI] [PubMed] [Google Scholar]
- [7].Ko TK, Kelly E, Pines J. CrkRS: a novel conserved Cdc2-related protein kinase that colocalises with SC35 speckles. J Cell Sci. 2001;114:2591–2603. [DOI] [PubMed] [Google Scholar]
- [8].Chen -H-H, Wang Y-C, Fann M-J. Identification and characterization of the CDK12/Cyclin L1 complex involved in alternative splicing regulation. Mol Cell Biol. 2006;26:2736–2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Marqués F, Moreau JL, Peaucellier G, et al. A new subfamily of high molecular mass CDC2-related kinases with PITAI/VRE motifs. Biochem Biophys Res Commun. 2000;279:832–837. [DOI] [PubMed] [Google Scholar]
- [10].Even Y, Durieux S, Escande M-L, et al. CDC2L5, a Cdk-like kinase with RS domain, interacts with the ASF/SF2-associated protein p32 and affects splicing in vivo. J Cell Biochem. 2006;99:890–904. [DOI] [PubMed] [Google Scholar]
- [11].Chen -H-H, Wong Y-H, Geneviere A-M, et al. CDK13/CDC2L5 interacts with L-type cyclins and regulates alternative splicing. Biochem Biophys Res Commun. 2007;354:735–740. [DOI] [PubMed] [Google Scholar]
- [12].Bartkowiak B, Liu P, Phatnani HP, et al. CDK12 is a transcription elongation-associated CTD kinase, the metazoan ortholog of yeast Ctk1. Genes Dev. 2010;24:2303–2316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Blazek D, Kohoutek J, Bartholomeeusen K, et al. The Cyclin K/Cdk12 complex maintains genomic stability via regulation of expression of DNA damage response genes. Genes Dev. 2011;25:2158–2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Dai Q, Lei T, Zhao C, et al. Cyclin K-containing kinase complexes maintain self-renewal in murine embryonic stem cells. J Biol Chem. 2012;287:25344–25352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Cheng SWG, Kuzyk MA, Moradian A, et al. Interaction of cyclin-dependent kinase 12/CrkRS with cyclin K1 is required for the phosphorylation of the c-terminal domain of RNA polymerase II. Mol Cell Biol. 2012;32:4691–4704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Bowman EA, Bowman CR, Ahn JH, et al. Phosphorylation of RNA polymerase II is independent of P-TEFb in the C. elegans germline. Development. 2013;140:3703–3713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Heidemann M, Hintermair C, Voß K, et al. Dynamic phosphorylation patterns of RNA polymerase II CTD during transcription. Biochim Biophys Acta. 2012;1829:55–62. [DOI] [PubMed] [Google Scholar]
- [18].Popova T, Manié E, Boeva V, et al. Ovarian cancers harboring inactivating mutations in CDK12 display a distinct genomic instability pattern characterized by large tandem duplications. Cancer Res. 2016;76:1882–1891. [DOI] [PubMed] [Google Scholar]
- [19].Wu Y-M, Cieślik M, Lonigro RJ, et al. Inactivation of CDK12 delineates a distinct immunogenic class of advanced prostate cancer. Cell. 2018;173:1770–1782.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Li M, Phatnani HP, Guan Z, et al. Solution structure of the Set2-Rpb1 interacting domain of human Set2 and its interaction with the hyperphosphorylated C-terminal domain of Rpb1. Proc Natl Acad Sci USA. 2005;102:17636–17641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Aymard F, Bugler B, Schmidt CK, et al. Transcriptionally active chromatin recruits homologous recombination at DNA double-strand breaks. Nat Struct Mol Biol. 2014;21:366–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Kizer KO, Phatnani HP, Shibata Y, et al. A novel domain in Set2 mediates RNA polymerase II interaction and couples histone H3 K36 methylation with transcript elongation. Mol Cell Biol. 2005;25:3305–3316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Bøsken CA, Farnung L, Hintermair C, et al. The structure and substrate specificity of human Cdk12/Cyclin K. Nat Commun. 2014;5:3505 EP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Dixon-Clarke SE, Elkins JM, Cheng S-WG, et al. Structures of the CDK12/CycK complex with AMP-PNP reveal a flexible C-terminal kinase extension important for ATP binding. Sci Rep. 2015;5:17122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Greifenberg AK, Hönig D, Pilarova K, et al. Structural and functional analysis of the Cdk13/Cyclin K Complex. Cell Rep. 2016;14:320–331. [DOI] [PubMed] [Google Scholar]
- [26].Bartkowiak B, Greenleaf AL. Expression, purification, and identification of associated proteins of the full-length hCDK12/CyclinK complex. J Biol Chem. 2015;290:1786–1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Jones JC, Phatnani HP, Haystead TA, et al. C-terminal repeat domain kinase I phosphorylates Ser2 and Ser5 of RNA polymerase II C-terminal domain repeats. J Biol Chem. 2004;279:24957–24964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Bensaude O. Inhibiting eukaryotic transcription. Which compound to choose? How to evaluate its activity? Transcription. 2014;2:103–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Chao SH, Fujinaga K, Marion JE, et al. Flavopiridol inhibits P-TEFb and blocks HIV-1 replication. J Biol Chem. 2000;275:28345–28348. [DOI] [PubMed] [Google Scholar]
- [30].Kwiatkowski N, Zhang T, Rahl PB, et al. Targeting transcription regulation in cancer with a covalent CDK7 inhibitor. Nature. 2014;511:616–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Zhang T, Kwiatkowski N, Olson CM, et al. Covalent targeting of remote cysteine residues to develop CDK12 and CDK13 inhibitors. Nat Chem Biol. 2016;12:876–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Johannes JW, Denz CR, Su N, et al. Structure-based design of selective noncovalent CDK12 inhibitors. ChemMedChem. 2018;13:231–235. [DOI] [PubMed] [Google Scholar]
- [33].Blethrow J, Zhang C, Shokat KM, et al. Design and use of analog-sensitive protein kinases. Curr Protoc Mol Biol. 2004;66:18.11.1–18.11.19. [DOI] [PubMed] [Google Scholar]
- [34].Bartkowiak B, Yan C, Greenleaf AL. Engineering an analog-sensitive CDK12 cell line using CRISPR/Cas. Biochim Biophys Acta. 2015;1849:1179–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Davidson L, Muniz L, West S. 3ʹ end formation of pre-mRNA and phosphorylation of Ser2 on the RNA polymerase II CTD are reciprocally coupled in human cells. Genes Dev. 2014;28:342–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Eifler TT, Shao W, Bartholomeeusen K, et al. Cyclin-dependent kinase 12 increases 3ʹ end processing of growth factor-induced c-FOS transcripts. Mol Cell Biol. 2015;35:468–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Liang K, Gao X, Gilmore JM, et al. Characterization of human cyclin-dependent kinase 12 (CDK12) and CDK13 complexes in C-terminal domain phosphorylation, gene transcription, and RNA processing. Mol Cell Biol. 2015;35:928–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. [DOI] [PubMed] [Google Scholar]
- [39].The Cancer Genome Atlas Research Network Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474:609–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Carter SL, Cibulskis K, Helman E, et al. Absolute quantification of somatic DNA alterations in human cancer. Nat Biotechnol. 2012;30:413–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Paculova H, Kohoutek J. The emerging roles of CDK12 in tumorigenesis. Cell Div. 2017;12:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Chilà R, Guffanti F, Damia G. Role and therapeutic potential of CDK12 in human cancers. Cancer Treat Rev. 2016;50:83–88. [DOI] [PubMed] [Google Scholar]
- [43].Dong X, Chen G, Cai Z, et al. CDK13 RNA over-editing mediated by ADAR1 associates with poor prognosis of hepatocellular carcinoma patients. Cell Physiol Biochem. 2018;47:2602–2612. [DOI] [PubMed] [Google Scholar]
- [44].Paulsen MT, Veloso A, Prasad J, et al. Use of Bru-Seq and BruChase-Seq for genome-wide assessment of the synthesis and stability of RNA. Methods. 2014;67:45–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Andrade-Lima LC, Veloso A, Paulsen MT, et al. DNA repair and recovery of RNA synthesis following exposure to ultraviolet light are delayed in long genes. Nucleic Acids Res. 2015;43:2744–2756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Kaida D, Berg MG, Younis I, et al. U1 snRNP protects pre-mRNAs from premature cleavage and polyadenylation. Nature. 2010;468:664–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Phatnani HP, Jones JC, Greenleaf AL. Expanding the functional repertoire of CTD kinase I and RNA polymerase II: novel phosphoCTD-associating proteins in the yeast proteome. Biochemistry. 2004;43:15702–15719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Bentley DL. The mRNA assembly line: transcription and processing machines in the same factory. Curr Opin Cell Biol. 2002;14:336–342. [DOI] [PubMed] [Google Scholar]
- [49].Carty SM, Greenleaf AL. Hyperphosphorylated C-terminal repeat domain-associating proteins in the nuclear proteome link transcription to DNA/chromatin modification and RNA processing. Mol Cell Proteomics. 2002;1:598–610. [DOI] [PubMed] [Google Scholar]
- [50].Kanagaraj R, Huehn D, Mackellar A, et al. RECQ5 helicase associates with the C-terminal repeat domain of RNA polymerase II during productive elongation phase of transcription. Nucleic Acids Res. 2010;38:8131–8140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Winsor TS, Bartkowiak B, Bennett CB, et al. A DNA damage response system associated with the phosphoCTD of elongating RNA polymerase II. PLoS One. 2013;8:e60909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Zhu X, He F, Zeng H, et al. Identification of functional cooperative mutations of SETD2 in human acute leukemia. Nat Genet. 2014;46:287–293. [DOI] [PMC free article] [PubMed] [Google Scholar]